

Abstract
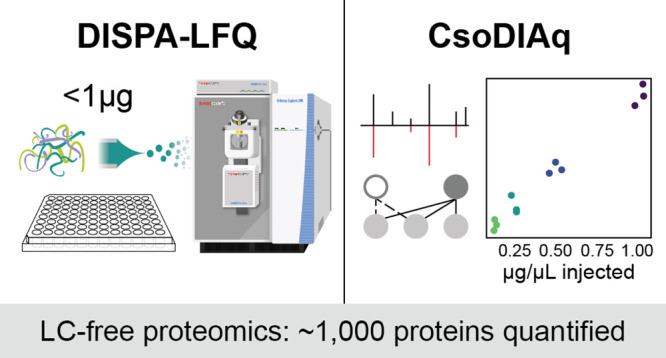
Large-scale proteome analysis requires rapid and high-throughput analytical methods. We recently reported a new paradigm in proteome analysis where direct infusion and ion mobility are used instead of liquid chromatography (LC) to achieve rapid and high-throughput proteome analysis. Here, we introduce an improved direct infusion shotgun proteome analysis protocol including label-free quantification (DISPA-LFQ) using CsoDIAq software. With CsoDIAq analysis of DISPA data, we can now identify up to ∼2000 proteins from the HeLa and 293T proteomes, and with DISPA-LFQ, we can quantify ∼1000 proteins from no more than 1 μg of sample within minutes. The identified proteins are involved in numerous valuable pathways including central carbon metabolism, nucleic acid replication and transport, protein synthesis, and endocytosis. Together with a high-throughput sample preparation method in a 96-well plate, we further demonstrate the utility of this technology for performing high-throughput drug analysis in human 293T cells. The total time for data collection from a whole 96-well plate is approximately 8 h. We conclude that the DISPA-LFQ strategy presents a valuable tool for fast identification and quantification of proteins in complex mixtures, which will power a high-throughput proteomic era of drug screening, biomarker discovery, and clinical analysis.
Introduction
Rapid technological progress in liquid chromatography separation, mass spectrometry detection, and bioinformatics has enabled large-scale detection and quantitation of proteins from cells, tissues, or biofluids.1 Proteome analysis by liquid chromatography mass spectrometry (LC–MS) creates new opportunities for biology research.2 The desire to expand the utility of proteomics for clinical diagnosis3,4 and drug development5 leads to a need for faster mass spectrometry-based proteomic methods. Separation by LC is a rate-limiting step in the speed of proteome analysis; LC traditionally required at least 1 h to separate a proteomic sample. With the emergence of new technologies in recent years, researchers have pushed LC separation times below 1 h.6−8
The logical extreme of this trend is to remove LC entirely and perform direct infusion mass spectrometry (DI-MS).9,10 Theoretically, there are two key challenges that limit proteome coverage by direct infusion methods: (1) ion suppression due to different ionization efficiency and/or different abundance of ionizable analytes and (2) ion competition in the mass analyzer where the number of accumulated ions for measurement is a finite value, which conceals low abundance ions. Ion suppression may be addressed by improving the sampling efficiency and increasing the ionization efficiency with low flow11−13 or mobile phase additives like DMSO.14 Ion competition is a problem for trap-type instruments. Quadrupole selection before mass analysis partly eliminates the ion competition effect, but quadrupole selection alone is not enough for extremely complex peptide mixtures from the human proteome, which contain well over 100,000 distinct peptide sequences including over 1000 unique peptide precursors within a 4 m/z window.10,15 Ion mobility spectrometry (IMS) technologies have recently been commercialized as a complementary mode of ion separation that could reduce peptide signal complexity.16,17 High-field asymmetric waveform ion mobility spectrometry (FAIMS) is one such technology that effectively acts as an ion filter. Other types of ion mobility like structures for lossless ion manipulation (SLIM)18,19 and trapped ion mobility20 provide continuous separation based on collisional cross section.
We recently described direct infusion shotgun proteomic analysis (DISPA) which achieves expeditious quantitative proteome analysis using gas-phase separations by ion mobility and quadrupole selection instead of LC.10 In the original publication of this method, we identified over 500 proteins and quantified over 300 proteins from 132 samples in under 3 min of data collection per sample. In a subsequent paper, we showed how DISPA can enable fast targeted quantification for specific proteins.21 We further developed software called CsoDIAq to enable analysis of DISPA data.22 There are several earlier examples of direct infusion proteome analysis,9,23−26 including the creative use of tandem ion mobility spectrometry (IMS),27 but none of these examples could compete with the depth of LC-MS-based proteomics until recently.
Here, we significantly improve the whole DISPA workflow applied to human proteome analysis (Figure 1). We evaluated how the protein identification results are influenced by various experimental parameters including mass resolution, sample concentration, dissolution solvents, and ion mobility (FAIMS). Using our recent software CsoDIAq along with optimized data acquisition, we identified up to 35 peptides from a single scan, and a total of over 2000 human protein groups from HeLa samples in one injection of less than 1 μg of protein. The proteins we can now follow by DISPA participate in diverse cellular pathways including central carbon metabolism, DNA replication, RNA transport, protein synthesis, and endocytosis. Additionally, a new label-free quantitative (LFQ) strategy founded on fragment ion intensity enabled accurate quantification of over 2000 peptides and ∼1000 proteins.DISPA-LFQ was applied to a high-throughput drug response experiment with 293T cells grown and processed in a 96-well plate. This work provides a complete guide from experimental procedures to data analysis software, leading researchers to easily perform direct infusion proteomics in their own labs. We expect the DISPA-LFQ workflow for large-scale, high-throughput proteome analysis will find widespread utility in a diverse array of applications.
Figure 1.

Overview of DISPA-LFQ using CsoDIAq software for peptide and protein group identification and quantification. (A) Scheme showing our strategy for peptide analysis by DISPA with FAIMS. (B) Schematic of a representative targeted experiment with actual data. Peptides were directly infused into the mass spectrometer over the duration of the experiment. Six FAIMS CVs were selected sequentially, and within each CV, specific m/z regions were selected with Q1 before fragmentation to produce chimeric MS/MS spectra. (C) Overview showing identification and label-free quantification of multiple peptides from one chimeric spectrum using the CsoDIAq software package with the same real data example in (B).
Materials and Methods
Peptide Standards and Chemicals
Angiotensin I (Sigma, A9650-1MG), QCAL Peptide Mix (Sigma, MSQC2), and HeLa digest standard (Thermo Fisher Scientific, Catalog number: 88328) were dissolved into different concentrations with 50% acetonitrile (ACN) in 0.2% formic acid (FA). Dimethyl sulfoxide (DMSO) and deferoxamine mesylate salt (D9533) were purchased from Sigma.
Cell Culture, Lysis, Digestion, and Desalting
For deferoxamine treatment, 293T cells were cultured in Dubelco's modified Eagle Medium (DMEM) containing 25 mM glucose, 4 mM glutamine, 1 mM pyruvate, 10% fetal bovine serum, and antimycotic-antibiotic (Gibco 15240096). Thirty thousand cells were plated per well in a 96-well plate and then allowed to grow for 24 hours before washing with phosphate buffered saline and lysis by the addition of 8 M urea with 50 mM TEAB buffer at pH 8.5. The plate was vortexed until homogeneous with lysis buffer and sonicated for 5 min in a Covaris water bath maintained at 4 °C. After sonication, TCEP (Thermo Fisher Scientific 77720) and chloroacetamide were added to a final concentration of 10 mM to reduce protein disulfide bonds and alkylate the free cysteines in the dark for 30 min. Then, lysis buffer was diluted to 2 M urea using 50 mM TEAB, and catalytic hydrolysis of proteins was initiated by trypsin (Promega Sequencing Grade Modified Trypsin, Frozen) at a weight ratio of 1:100 protease/substrate. Then, proteome proteolysis was incubated overnight at room temperature. Peptides were desalted using a 96-well μElution plate from Waters (Oasis HLB 96-well μElution plate, 2 mg sorbent per well, 30 μm) and then dried completely in Speed-Vac. Peptides were resuspended in ACN/Water/FA (50%/49.9%/0.1%, volume ratio) for DISPA. For the preparation of the 293T proteome used for experimental parameter analysis, lysis and digestion were conducted with the same protocol, but cells were cultured in a 9 cm plate and desalted with Strata reversed-phase cartridges (Phenomenex Strata-X 33 μm Polymeric Reversed Phase 30 mg / 1 mL, Tubes, 8B-S100-TAK).
DISPA Method
DISPA of the HeLa proteome was performed on an Orbitrap Explois 240 (Thermo Fisher Scientific) mass spectrometer coupled with the FAIMS Pro Interface. DISPA of the 293T proteome and deferoxamine treatment were performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) coupled with the FAIMS Pro Interface. A nano-ESI source (“Nanospray Flex”) was used for sample ionization, and the nano-tip was pulled with a laser puller (Model, P2000, Sutter instrument Co). Samples were directly infused into a 50 μm inner diameter capillary tip that was packed with approximately 2 mm of C8 particles (ESI Source Solutions, 5 μm particle size) to prevent clogging of the tip. The ultimate 3000 HPLC system (Thermo Fisher Scientific, USA) was used to control the automated sample loading, flow rate, and mobile phase composition. A flow rate of 1.4 μL/min was maintained for the first 0.75 min to quickly deliver the sample to the nanoESI emitter, and then, the flow rate was dropped to 0.20 μL/min at 0.8 min for data collection. An isocratic flow consisting of 50% ACN in 0.2% FA was maintained during the whole acquisition time.
The working state of the nanospray emitter, FAIMS module, and mass spectrometry was tested with 1 fmol/μL angiotensin I using a compensation voltage (CV) of −55 V with targeted m/z: 433 (charge state: +3) and HeLa proteome infusion. The normalized TIC signal from two samples should be at least 1E5 and 1E7, respectively. More method details for scouting and targeted experiments are provided in the Supporting Information. The scan segment time and targeted window list for HeLa are shown in Table S1.
Peptide and Protein Identification
Peptides and proteins were identified with CsoDIAq software version 1.2 we developed previously.22 CsoDIAq is a python software package designed to enhance the usability and sensitivity of the projected spectrum–spectrum match scoring concept.28 CsoDIAq works by comparing spectra in a library to experimental spectra and scoring the presence of potential peptides. CsoDIAq also infers proteins from peptides and estimates the false discovery rate of peptide and protein identifications, and our new version also quantifies peptides and proteins. CsoDIAq thus simplifies DISPA data analysis, enabling anyone to perform DISPA experiments. The original Thermo .RAW files were converted to mzXML files using msconvert.29 mzXML files were input to CsoDIAq GUI with the default settings except the initial fragment mass tolerance was set at 20 ppm. Spectra were searched against human protein library developed with SpectraST in the TraML tsv format. Spectral libraries were generated from the MS-Fragger8 database search of data-dependent acquisition (DDA) files data as described previously.22 CsoDIAq produces three output files for each input mzXML file that report spectra, peptides, and proteins filtered to <1% FDR. In each case, CsoDIAq sorts peptide identifications by match count and cosine (MaCC) score, calculates the FDR for each identification using a modification of the target-decoy approach where FDR at score S = number of decoys/number of targets, and removes SSMs below a 0.01 FDR threshold. The peptide FDR calculations only use the highest-scoring match among all SSMs for each peptide. CsoDIAq uses the IDPicker algorithm to identify protein groups from the list of discovered peptides and adds them as an additional column in the output. Detailed description about data processing, FDR calculation, and protein inference was listed in our previous paper.22
Peptide and Protein Quantification
MSconvertGUI (part of ProteoWizard) was used to create mzXML files for quantification analysis with a new LFQ version of CsoDIAq. Angiotensin and MS-QCAL peptides were quantified using python to extract representative y-ion fragment intensity. Peptides from the HeLa dilution series were quantified using CsoDIAq to extract the sum of all detected fragment ion intensities of common peptides in all input files.
Pathway Enrichment Analysis
The UniProt IDs from CsoDIAq outputs were converted to gene IDs with DAVID.30 The KEGG_2021_Human gene set library was applied, and pathway enrichment analysis was done in Cytoscape31 with the plugin ClueGO.32 GO term/pathway network connectivity (Kappa score) was set at 0.5. GO term grouping and two-sided hypergeometric test were used and leading group term ranked based on the highest significance.
Supporting Methods and FAQ Online
Supporting Information contains more details about how to generate a spectral library, how to monitor the instrument for quality control, and a frequently asked questions section that addresses the purpose of FAIMS, the volume of sample required, what is a projected spectrum, how CsoDIAq determines matches, and the identification rate of MS/MS spectra collected with DISPA.
Data Availability
All raw mass spectrometry data are available from massive.ucsd.edu under MassIVE MSV000089546 (ftp://MSV000089546@massive.ucsd.edu).
Results and Discussion
Peptide and Protein Identification with DISPA + CsoDIAq
DISPA involves nanoflow direct infusion of peptides and electrospray ionization followed by gas-phase separation with ion mobility, quadrupole m/z selection, dissociation of peptides using higher-energy collisional activation (HCD), and fragment ion detection by DIA with high-resolution MS/MS (Figures 1A and S1). The performance of the nanospray emitter, FAIMS module, and mass spectrometer are key metrics for a successful DISPA experiment. The system was tested with both 1 fmol/μL angiotensin I and 0.125 μg/μL HeLa proteome (Figures S2 and S3). The normalized TIC signal from two samples should be at least 1 × 105 and 1 × 107, respectively. After verification of system performance with angiotensin infusion, we performed a peptide discovery experiment where the isolation width of the quadrupole was set at 2 m/z and stepped across the m/z range of interest at each CV. Once we identify peptide targets from a scouting experiment, we perform a targeted experiment only isolating the combinations of FAIMS/Q1 isolation that produced peptide identifications (Figure 1B). With DISPA, only MS/MS spectra are collected, which are then identified by CsoDIAq software including spectra–spectra match scoring, FDR filtering, and protein inference (Figure 1C).
To better understand and optimize protein identifications from DISPA with CsoDIAq, the effects of various experiment parameters were tested with HeLa and 293T proteomes, including FAIMS CV, mass spectrometer resolution, sample concentration, and ESI solvent. As shown in Figure 2A, more peptides were identified in CVs between −40 and −60 V for both samples. Identified protein groups demonstrate the same trend as peptides in all CVs (Figure 2B). Theoretically, applying more CVs can enable us to identify more peptides and protein groups. However, closer voltages, such as −45 and −50 V, usually produce more duplicated peptides and proteins, while voltages with a longer distance like −50 and −80 V exhibit less duplicated peptides and proteins (Figure S4). A parallel LC–MS/MS experiment of the 293T sample with different CVs verified this conclusion as well (Figure S5). Therefore, to reduce duplicate detection of same peptides, we selected CVs from −30 to −80 V with steps of 10 V for all experiments in this article.
Figure 2.

Protein identifications from HeLa and 293T proteomes by DISPA analysis across different experimental parameters. (A,B) Peptide (A) and protein (B) identifications at various FAIMS CVs using DISPA for 293T and HeLa proteomes. Mass resolution: 120 K. Concentration: 293T: 1 μg/μL. HeLa: 0.5 μg/μL. (C,D) Number of protein identifications as a function of mass resolution in HeLa (C) and T293 (D) with and without FAIMS. (E,F) Number of protein identifications as a function of sample concentration in HeLa (E) and 293T (F) with and without FAIMS. (G,H) Number of protein identifications as a function of resuspension solvent in HeLa (G) and 293T (H) with and without FAIMS.
Increasing mass resolution from 30 to 240 k with constant peptide concentration increases the number of protein group identifications three-fold in both HeLa and 293T proteomes (Figure 2C,D). This is likely due to an increased ability to resolve fragment ions and the increased ion accumulation time allowed while waiting for longer ion detection times. Similarly, the number of identified protein groups increases as the sample concentration increases in both samples (Figure 2E,F). However, the number of protein groups does not increase infinitely as the concentration increases and the recommended optimal concentration range for DISPA is 1–4 μg/μL (Figure 2F). It is noteworthy that we achieved ∼2000 protein group identifications for both sample types with appropriate concentration and mass resolution (1 μg/μL, 240 k), which is almost quadruple the 552 protein identifications reported in the original DISPA paper.
More interestingly, up to 35 peptides were identified in a single scan of the HeLa proteome (scan #302, m/z = 700.5, FAIMS CV = −40 V, 240 K, 1 μg/μL,Table S2), and nearly all of them showed excellent agreement between the projected spectrum and library spectrum (Figures S6 and S7). This demonstrates the staggering amount of information carried by a single scan, even after separations by the quadrupole and FAIMS. We believe the combination of optimal data acquisition settings with CsoDIAq software contributed to this improvement in protein identifications.
Previous studies suggested that different solvents are responsible for altering the charge state distribution of peptides for ESI, which may alter protein identifications.14 Here, the DISPA results showed that there are no significant statistical differences among four kinds of tested solvents in both HeLa and 293T proteomes (Figure 2G,H).
Protein identifications of DISPA with FAIMS and without FAIMS were also compared across all experimental parameters including different mass resolutions, sample concentration, and resuspended solvent. Results demonstrated that the identified protein groups increased at least two times with FAIMS as an additional gas-phase separation (Figure 2C–H).
The peptide identifications of different experiment parameters demonstrated the same pattern with proteins, namely, they increase as the mass resolution and concentration increase, they decrease with the removal of FAIMS, and they show no difference among ESI solvents (Figure S8). The average peptides per protein are at least three peptides per protein for both samples (Figure S9). Based on these results, we concluded that the peptide and protein identifications benefit substantially from a higher mass resolution, appropriate concentration range, and FAIMS.
In addition to quantities of the protein, the rate and robustness of protein identifications are also key factors of this method. Specially, we realized a maximum identification rate of 8.2 proteins per second, but note that the total acquisition time varies greatly according to different mass resolutions, which leads to a decrease in the protein identification rate with the increase of mass resolution (Figures S10 and S11). Thus, there is a trade-off between the number of protein identifications and the rate of protein identification with our approach. Nevertheless, a compromise condition is taken as an example, 1 μg/μL and 120 k, and a protein identification rate of 6.3 proteins per second was achieved (Figure S10).
Next, the reproducibility of peptide and protein identifications was tested with replicates of all conditions. We tested the coefficient of variation of each peptide across different concentrations, and the majority peptides and proteins are below 30% (Figure S12). Moreover, the TIC figures showed great similarity between two replicates (Figure S13, HeLa, 120 K, 1 μg/μL), and the mass spectrum of 12 randomly selected scans also exhibited excellent reproducibility (Figure S14). We also compared the peptide identification results of each target m/z scan segment in two repeats, and results proved that 175 out of 769 scan segments demonstrate 100% repeatability (calculated by shared peptides/sum of peptides) and 90% scan segments show more than 50% repeatability (Figure S15). The ratio of repeatedly detected proteins to the average number of proteins in three replicates exceeds 80% in all experimental conditions and some even higher than 90% (Figures S16 and S17). Detailed protein identifications and repeatable proteins across different resolution and concentration are shown in Figure S18. To conclude, we believe that our method successfully balanced protein quantities, speed, and robustness, but there is still space for improvement in software and instrumentation.
KEGG Pathway Enrichment Analysis of Identified Proteins
Speed and quantity of protein identification are not the only relevant metrics; in what biological processes do these proteins function? Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed on the 1628 common proteins found in three replicates (1 μg/μL concentration, 240 k resolution). The result revealed numerous important cellular pathways, including central carbon metabolism (i.e., tricarboxylic acid cycle, glycolysis, pyruvate metabolism, pentose phosphate pathway, glutathione metabolism), protein synthesis and degradation (i.e., splicosome, ribosome, proteasome, and proteome processing in the endoplasmic reticulum), and nucleic acid replication and transport (DNA replication, RNA transport) and endocytosis (Figures 3A,B and S19, S20). Interestingly, with increasing mass resolution (and therefore also ion accumulation time and sensitivity), the identified proteins in the above pathways also increased (Figure 3C,D). In contrast, the gain in proteins for most pathways depended less on the concentration of the peptide infused (Figure 3E,D). Protein identifications from the less abundant pathways like endocytosis and protein export did increase slightly as the concentration increased (Figure 3D). Our approach detected almost two-thirds of the proteins in the TCA cycle and proteasome pathways. This is nearly 3 times more TCA cycle proteins than the original publication of DISPA, and we believe this significant progress will expand the potential applications of this strategy.
Figure 3.

KEGG pathway enrichment analysis of proteins identified by DISPA from HeLa cells. (A,B) The number in pie charts indicates how many proteins in the pathway were identified, and the colored proportion of each circle reflects the coverage of the proteins in each pathway using a mass resolution of 240 K and a concentration of 1 μg/μL. (C,D) Variation in the number of detected protein groups of various pathways with different resolution settings using a sample concentration of 0.5 μg/μL. (E,F) Variation of detected protein groups of various pathways as a function of peptide concentration using a mass resolution of 240 K.
Label-Free Quantification Strategy for DISPA
The DISPA strategy produces only MS/MS spectra without retention time or elution profiles to use for peak integration. The previous quantitative DISPA approaches relied on stable isotope-labeled peptides, either spike in or SILAC. Approaches that require stable isotopes are not ideal for DISPA because they complicate sample preparation and require extra time and effort, which is counter to the goal of high-throughput proteomic analysis. To overcome these obstacles, we explored a label-free quantitative strategy based on fragment ion intensity. DISPA showed excellent reproducibility as measured by fragment ion intensity stability of angiotensin (Figure S21), MS-QCAL peptides33 (R2 = 0.979), and HeLa peptides (R2 = 0.997) (Figure 4A,B). Specifically, four fragment ions from angiotensin I (y4+, b4+, b5+, b6+) demonstrated outstanding stability during the whole acquisition time, and the fragment intensity was proportional to the injected concentration (Figure S22). Further quantitative evaluation (using fragment ion b4+) with concentrations across a 107-fold (100 amol/μL–1 nmol/μL) range showed a great linear curve before reaching an extreme high concentration (1 nmol/μL), and the quantification curve was not significantly affected after the addition of 0.2 μg/μL MQCAL peptides as an interference (Figure 4C). Similarly, 10 QCAL peptides across a series of dilutions (15,625-fold, over four orders of magnitude, 320 pg/μL–0.5 μg/μL) illustrated an exceptional linear curve (Figure 4D). The quantitative result was also tested with more fragment ions from angiotensin I, and the results demonstrated that the quantitative curve was not affected by the number of fragments used (Figure S23). These results suggested that LFQ is possible from DISPA even from only one measurement per peptide.
Figure 4.

Label-free quantification with DISPA using standards. (A,B) Scatterplot of peptide fragment intensities from two injections of the MS-QCAL protein (A) and HeLa peptides (B). (C,D) Quantification curve of a standard peptide angiotensin I (C) and MS-QCAL protein (D) at different concentrations. (E,F) Peptide (E) and protein (F) quantities determined by DISPA-LFQ from a dilution series of the HeLa proteome. The shaded area represents one standard deviation from the mean in the middle. Only peptides or proteins with positive slopes were plotted in either set, which excludes 60 peptides or 18 proteins before the Pearson test for good slope (p value < 0.05) or 3 peptides and zero proteins after the Pearson test for slope (p value < 0.05). Significant peptides or proteins were defined as those with a p value from the Pearson correlation of less than 0.05. (G,H) Examples of well-quantified peptide (G) and protein (H).
Encouraged by the above results, the DISPA-LFQ method was further assessed with a more complex human proteome sample analysis. DISPA data was collected from HeLa peptides at four diluted concentrations. Various parameters that influence the quality of peptide quantitation were explored. The number of fragments used to quantify was important; the most peptides were quantified with a good slope using at least five fragments (Figure S24). We also found the repeatability of identification to be a key predictor of quantification quality. A total of 2321 peptides from CsoDIAq results were identified in all 12 replicates from the dilution experiment, and CsoDIAq automatically quantified them all. Of these, 1799 peptides had a positive slope and p values less than 0.05 from Pearson correlation, or 78% of the repeatedly identified peptides (Figures 4E and S25). The shared peptides correspond to 1061 proteins, of which 905 exhibited good linearity with p values less than 0.05 from Pearson correlations (Figures 4F and S26). This indicates that over 85% of repeatedly identified proteins were well quantified. Raw values of significant peptides and proteins are provided in Table S3. The representative peptides and proteins from triplicate injections are shown in Figure 4G–H. Based on these results, we further added this LFQ logic and function into our identification software, CsoDIAq LFQ. In the new version, CsoDIAq will automatically calculate the intensity of all fragment ions from the identified peptides and then export two quantitative files of peptides and corresponding proteins common to all input files. The new LFQ module was added into our new graphical user interface (GUI) (Figure S27). In this way, even researchers unfamiliar with programming can easily use it for rapid LFQ analysis of DISPA data.
Rapid Screening of Human Cellular Responses to Toxins with DISPA-LFQ
To demonstrate the capability of DISPA-LFQ for biological applications, an example of a high-throughput dose–response assay with chemical treatment (deferoxamine, DFO) was conducted using human 293T cells. Notably, to match the high-throughput data acquisition method, we designed an integrated sample preparation workflow including cell culture, drug treatment, cell lysis, and digestion in a 96-well plate. We applied six treatment groups with different concentrations of DFO (from 10 nM to 200 μM) and two different control groups (with and without DMSO). An average of ∼1100 proteins were identified per well. Data collection from all samples required only 8 h of mass spectrometry time (Figure 5A). Due to the extreme stress of high concentrations of DFO, only 218 common proteins were quantified in all wells of the plate that passed the quality control threshold of at least 1300 peptide identifications. CsoDIAq analysis automatically generated a quantitative report for those proteins (Figure 5B). DFO has been considered to be a chelating agent used to remove excess iron or aluminum, which previously reported altered quantities of glycolytic proteins. Quantities of the 10 proteins from the glycolysis pathway were compared across groups. As shown in Figure 5C–L, all 10 glycolytic proteins exhibit a significantly upregulated due to DFO stress versus controls when the concentration reached 100 μM. Interestingly, the upregulated phenomenon of glycolytic proteins is not significant in low-concentration DFO treatments (lower than 10 μM). Overall, together with the high-throughput sample preparation workflow in a 96-well plate, the rapid DISPA-LFQ method is a promising method for drug and biomarker discovery studies.
Figure 5.

Rapid screening of human cellular responses to toxins with DISPA-LFQ. (A) Scheme of the high-throughput experimental workflow of cytotoxic stress response to deferoxamine in a 96-well plate. (B) Heatmap of 218 common proteins detected in 82 wells. (C–L) Glycolytic proteins quantified by CsoDIAq from DISPA-LFQ across different concentrations of deferoxamine (DFO) vs controls. Con: No DFO treatment without DMSO, Con (+): No DFO treatment with DMSO. Protein quantities were corrected by the intensity ratio to histone H2A. p values are from one-way ANOVA, followed by Tukey’s post hoc test. *P < 0.05, **P < 0.01. The number of replicates for each group was between 6 and 12.
Conclusions
There are multiple competing fast proteomics methods using LC–MS that can achieve more than 2000 protein identifications with minutes.7,8,34−38 However, time spent on sample loading, column equilibration, and column wash may not be included in reported times. If all time is counted, usually the maximum throughput for 24 h is less than 300 samples. For our method, the total time including sampling and data acquisition is less than 4.5 min, meaning that DISPA-LFQ can collect proteomes from a whole 96-well plate of drug screening application in 8 h (Figure 5) or approximately 300 samples/day. Moreover, taking advantage of no LC column, the equilibration time and potential carry-over were eliminated, and robustness was improved. More importantly, for certain applications, once the target peptides or proteins are known, we can easily conduct a target analysis of these peptide in seconds of time per sample without waiting for elution of these peptides from LC.21
In conclusion, the integrated DISPA-LFQ strategy using CsoDIAq software described here provides a complete high-throughput direct infusion proteomic data acquisition and analysis platform. This method enables up to 2000 human protein identifications and LFQ of up to 1000 human proteins with less than 1 μg sample consumption in only a few minutes. This is nearly 4x identified and over 2x quantified proteins compared to our original report.10 As compared to the traditional LC-based methodology, this approach has the advantages of speed and simplicity and has the potential for higher throughput than competing LC–MS approaches. Finally, an example application to high-throughput chemical response screening in human cells grown in a 96-well plate validated quantities reported from DISPA-LFQ by CsoDIAq.
In future iterations, we believe this approach will benefit from more efficient ionization sources, different types of ion mobility selection, and faster, more sensitive mass spectrometers. We expect this direct infusion proteomics platform will continue to improve contributing to the high-throughput era of proteomics. We also expect that with these developments in LFQ and software solutions, using this work as a complete guide for direct infusion proteomic analysis, DISPA can now be routinely applied in more practical applications by researchers across the globe, such as clinical research and drug discovery where analysis of thousands of samples is required.
Acknowledgments
This work was supported by the United States National Institute of Health (NIH) NIGMS R35 GM142502. The authors thank other lab members Quinn Dickinson and Amanda Momenzadeh for helpful discussions. The authors also thank Ali Haghani, Simion Kreimer, Oleg Karpov, Saeed Seyedmohammad, Oliver Wang, Sarah Parker, and Jennifer Van Eyk for helpful discussions and assistance related to mass spectrometry and cell culture. We thank Dasom Hwang for graphic design help.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c02249.
Experimental details of DISPA methods; CsoDIAq library build procedures; frequently asked questions (FAQ); and DISPA platform, CsoDIAq GUI, peptide identifications, FAIMS function, reproducibility and robustness assessment, LFQ analysis, and pathway enrichment analysis (PDF)
Scan segment time and m/z window list for each compensation voltage (XLSX)
CsoDIAq results of over 2000 protein groups, 35 peptides in Scan 302, and explanation of columns of CsoDIAq outputs (XLSX)
Raw quantified values of significant peptides and protein groups (XLSX)
CsoDIAq library creation commands for SpectraST (TXT)
The authors declare the following competing financial interest(s): JGM has filed a patent related to this technology.
Supplementary Material
References
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M. C.; Yates J. R. Chem. Rev. 2013, 113, 2343. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr S. A.; Abbatiello S. E.; Ackermann B. L.; Borchers C.; Domon B.; Deutsch E. W.; Grant R. P.; Hoofnagle A. N.; Hüttenhain R.; Koomen J. M.; Liebler D. C.; Liu T.; MacLean B.; Mani D. R.; Mansfield E.; Neubert H.; Paulovich A. G.; Reiter L.; Vitek O.; Aebersold R.; Anderson L.; Bethem R.; Blonder J.; Boja E.; Botelho J.; Boyne M.; Bradshaw R. A.; Burlingame A. L.; Chan D.; Keshishian H.; Kuhn E.; Kinsinger C.; Lee J. S.; Lee S. W.; Moritz R.; Oses-Prieto J.; Rifai N.; Ritchie J.; Rodriguez H.; Srinivas P. R.; Townsend R. R.; Van Eyk J.; Whiteley G.; Wiita A.; Weintraub S. Mol. Cell. Proteomics 2014, 13, 907. 10.1074/mcp.m113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberring J.; Ciborowski P. Trends Analyt Chem 2010, 29, 128. 10.1016/j.trac.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischak H.; Apweiler R.; Banks R. E.; Conaway M.; Coon J.; Dominiczak A.; Ehrich J. H.; Fliser D.; Girolami M.; Hermjakob H.; Hochstrasser D.; Jankowski J.; Julian B. A.; Kolch W.; Massy Z. A.; Neusuess C.; Novak J.; Peter K.; Rossing K.; Schanstra J.; Semmes O. J.; Theodorescu D.; Thongboonkerd V.; Weissinger E. M.; Van Eyk J. E.; Yamamoto T. Proteomics Clin Appl 2007, 1, 148. 10.1002/prca.200600771. [DOI] [PubMed] [Google Scholar]
- Schirle M.; Bantscheff M.; Kuster B. Chem. Biol. 2012, 19, 72. 10.1016/j.chembiol.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Kelstrup C. D.; Bekker-Jensen D. B.; Arrey T. N.; Hogrebe A.; Harder A.; Olsen J. V. J. Proteome Res. 2018, 17, 727. 10.1021/acs.jproteome.7b00602. [DOI] [PubMed] [Google Scholar]
- Bache N.; Geyer P. E.; Bekker-Jensen D. B.; Hoerning O.; Falkenby L.; Treit P. V.; Doll S.; Paron I.; Müller J. B.; Meier F.; Olsen J. V.; Vorm O.; Mann M. Mol. Cell. Proteomics 2018, 17, 2284. 10.1074/mcp.tir118.000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner C. B.; Demichev V.; Bloomfield N.; Yu J. S. L.; White M.; Kreidl M.; Egger A. S.; Freiwald A.; Ivosev G.; Wasim F.; Zelezniak A.; Jürgens L.; Suttorp N.; Sander L. E.; Kurth F.; Lilley K. S.; Mülleder M.; Tate S.; Ralser M. Nat. Biotechnol. 2021, 39, 846. 10.1038/s41587-021-00860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S.; Kori Y.; Lopes M.; Yuan Z. F.; Kim H. J.; Kulej K.; Janssen K. A.; Agosto L. M.; Cunha J.; Andrews A. J.; Garcia B. A. Genome Res. 2019, 29, 978. 10.1101/gr.247353.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. G.; Niemi N. M.; Pagliarini D. J.; Coon J. J. Nat Methods 2020, 17, 1222. 10.1038/s41592-020-00999-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca L. S.; Gargano A. F. G.; Schoenmakers P. J. Anal. Chim. Acta 2021, 1156, 338349. 10.1016/j.aca.2021.338349. [DOI] [PubMed] [Google Scholar]
- Cong Y.; Motamedchaboki K.; Misal S. A.; Liang Y.; Guise A. J.; Truong T.; Huguet R.; Plowey E. D.; Zhu Y.; Lopez-Ferrer D.; Kelly R. T. Chem. Sci. 2020, 12, 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faserl K.; Kremser L.; Müller M.; Teis D.; Lindner H. H. Anal. Chem. 2015, 87, 4633. 10.1021/acs.analchem.5b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J. G.; Komives A. K. J. Am. Soc. Mass Spectrom. 2012, 23, 1390. 10.1007/s13361-012-0404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A.; Cox J.; Mann M. J. Proteome Res. 2011, 10, 1785. 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- Pfammatter S.; Bonneil E.; McManus F. P.; Thibault P. J. Proteome Res. 2019, 18, 2129. 10.1021/acs.jproteome.9b00021. [DOI] [PubMed] [Google Scholar]
- Hebert A. S.; Prasad S.; Belford M. W.; Bailey D. J.; McAlister G. C.; Abbatiello S. E.; Huguet R.; Wouters E. R.; Dunyach J. J.; Brademan D. R.; Westphall M. S.; Coon J. J. Anal. Chem. 2018, 90, 9529. 10.1021/acs.analchem.8b02233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt J. R.; Wormwood Moser K. L.; Van Aken G.; Doyle R. M.; Talamantes T.; DeBord D.; Maxon L.; Stafford G.; Fjeldsted J.; Miller B.; Sherman M. J. Am. Soc. Mass Spectrom. 2021, 32, 2019. 10.1021/jasms.0c00434. [DOI] [PubMed] [Google Scholar]
- May J. C.; Leaptrot K. L.; Rose B. S.; Moser K. L. W.; Deng L.; Maxon L.; DeBord D.; McLean J. A. J. Am. Soc. Mass Spectrom. 2021, 32, 1126. 10.1021/jasms.1c00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgeway M. E.; Lubeck M.; Jordens J.; Mann M.; Park M. A. Int. J. Mass Spectrom. 2018, 425, 22. 10.1016/j.ijms.2018.01.006. [DOI] [Google Scholar]
- Trujillo E. A.; Hebert A. S.; Rivera Vazquez J. C.; Brademan D. R.; Tatli M.; Amador-Noguez D.; Meyer J. G.; Coon J. J. Rapid Targeted Quantitation of Protein Overexpression with Direct Infusion Shotgun Proteome Analysis (DISPA-PRM).. Anal. Chem. 2022, 94, 1965–1973. 10.1021/acs.analchem.1c03243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranney C. W.; Meyer J. G. CsoDIAq Software for Direct Infusion Shotgun Proteome Analysis. Anal. Chem. 2021, 93, 12312. 10.1021/acs.analchem.1c02021. [DOI] [PubMed] [Google Scholar]
- Kretschy D.; Gröger M.; Zinkl D.; Petzelbauer P.; Koellensperger G.; Hann S. Int. J. Mass Spectrom. 2011, 307, 105. 10.1016/j.ijms.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Medrano A. G.; Sterling A.; Snijders A. P.; Reardon K. F.; Wright P. C. J. Am. Soc. Mass Spectrom. 2007, 18, 1714. 10.1016/j.jasms.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Chen S. Proteomics 2006, 6, 16. 10.1002/pmic.200500043. [DOI] [Google Scholar]
- Xiang Y.; Koomen J. M. Anal. Chem. 2012, 84, 1981. 10.1021/ac203011j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine S. J.; Kurulugama R. T.; Bohrer B. C.; Merenbloom S. I.; Sowell R. A.; Mechref Y.; Clemmer D. E. Int. J. Mass Spectrom. 2009, 283, 149. 10.1016/j.ijms.2009.02.030. [DOI] [Google Scholar]
- Wang J.; Tucholska M.; Knight J. D.; Lambert J. P.; Tate S.; Larsen B.; Gingras A. C.; Bandeira N. Nat. Methods 2015, 12, 1106. 10.1038/nmeth.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers M. C.; Maclean B.; Burke R.; Amodei D.; Ruderman D. L.; Neumann S.; Gatto L.; Fischer B.; Pratt B.; Egertson J.; Hoff K.; Kessner D.; Tasman N.; Shulman N.; Frewen B.; Baker T. A.; Brusniak M. Y.; Paulse C.; Creasy D.; Flashner L.; Kani K.; Moulding C.; Seymour S. L.; Nuwaysir L. M.; Lefebvre B.; Kuhlmann F.; Roark J.; Rainer P.; Detlev S.; Hemenway T.; Huhmer A.; Langridge J.; Connolly B.; Chadick T.; Holly K.; Eckels J.; Deutsch E. W.; Moritz R. L.; Katz J. E.; Agus D. B.; MacCoss M.; Tabb D. L.; Mallick P. Nat. Biotechnol. 2012, 30, 918. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitsky L. I.; Klein J. A.; Ivanov M. V.; Gorshkov M. V. J. Proteome Res. 2019, 18, 709. 10.1021/acs.jproteome.8b00717. [DOI] [PubMed] [Google Scholar]
- Huang W.; Sherman B. T.; Lempicki R. A. Nat. Protoc. 2009, 4, 44. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Genome Res. 2003, 13, 2498. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers C. E.; Simpson D. M.; Wong S. C.; Beynon R. J.; Gaskell S. J. J. Am. Soc. Mass Spectrom. 2008, 19, 1275. 10.1016/j.jasms.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Zhang F.; Xu L.; Yue L.; Kon O. L.; Zhu Y.; Guo T. Adv Drug Deliv Rev 2021, 176, 113844. 10.1016/j.addr.2021.113844. [DOI] [PubMed] [Google Scholar]
- Meier F.; Brunner A. D.; Frank M.; Ha A.; Bludau I.; Voytik E.; Kaspar-Schoenefeld S.; Lubeck M.; Raether O.; Bache N.; Aebersold R.; Collins B. C.; Röst H. L.; Mann M. Nat. Methods 2020, 17, 1229. 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- Ivanov M. V.; Bubis J. A.; Gorshkov V.; Tarasova I. A.; Levitsky L. I.; Lobas A. A.; Solovyeva E. M.; Pridatchenko M. L.; Kjeldsen F.; Gorshkov M. V. Anal. Chem. 2020, 92, 4326. 10.1021/acs.analchem.9b05095. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen D. B.; Martínez-Val A.; Steigerwald S.; Rüther P.; Fort K. L.; Arrey T. N.; Harder A.; Makarov A.; Olsen J. V. Mol. Cell. Proteomics 2020, 19, 716. 10.1074/mcp.tir119.001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M.; Konno R.; Nakajima D.; Gotoh M.; Fukasawa K.; Sato H.; Nakamura R.; Ohara O.; Kawashima Y. J. Proteome Res. 2022, 21, 2085. 10.1021/acs.jproteome.2c00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw mass spectrometry data are available from massive.ucsd.edu under MassIVE MSV000089546 (ftp://MSV000089546@massive.ucsd.edu).