

Abstract
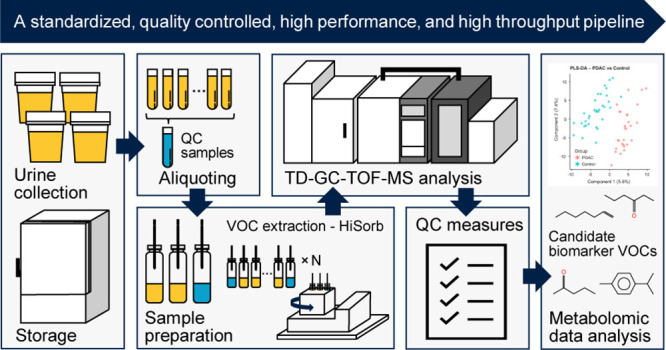
Volatolomics offers an opportunity for noninvasive detection and monitoring of human disease. While gas chromatography–mass spectrometry (GC–MS) remains the technique of choice for analyzing volatile organic compounds (VOCs), barriers to wider adoption in clinical practice still exist, including: sample preparation and introduction techniques, VOC extraction, throughput, volatolome coverage, biological interpretation, and quality control (QC). Therefore, we developed a complete pipeline for untargeted urinary volatolomic profiling. We optimized a novel extraction technique using HiSorb sorptive extraction, which exhibited high analytical performance and throughput. We achieved a broader VOC coverage by using HiSorb coupled with a set of complementary chromatographic methods and time-of-flight mass spectrometry. Furthermore, we developed a data preprocessing strategy by evaluating internal standard normalization, batch correction, and we adopted strict QC measures including removal of nonlinearly responding, irreproducible, or contaminated metabolic features, ensuring the acquisition of high-quality data. The applicability of this pipeline was evaluated in a clinical cohort consisting of pancreatic ductal adenocarcinoma (PDAC) patients (n = 28) and controls (n = 33), identifying four urinary candidate biomarkers (2-pentanone, hexanal, 3-hexanone, and p-cymene), which can successfully discriminate the cancer and noncancer subjects. This study presents an optimized, high-throughput, and quality-controlled pipeline for untargeted urinary volatolomic profiling. Use of the pipeline to discriminate PDAC from control subjects provides proof of principal of its clinical utility and potential for application in future biomarker discovery studies.
Volatile organic compounds (VOCs), produced within the body as a consequence of normal and aberrant metabolism, offer an opportunity for noninvasive detection and monitoring of disease states.1−4
While previous studies have tended to focus on the VOC signature of exhaled breath, other matrices such as urine have garnered less attention. Recent systematic reviews found promising evidence supporting urinary VOC analysis for cancer detection.5−7 Like exhaled breath, urine analysis is entirely noninvasive and widely accepted by patients.6,7 Additional advantages of urine include ease of collection and storage, VOC abundance, and the ability to generate pooled samples for quality control (QC) processes. Regarding sample preparation and introduction techniques for urinary volatolomics, the most common methods are direct headspace analysis, solid phase microextraction (SPME), and stir bar sorptive extraction (SBSE).8 Headspace approaches suffer from low sensitivity, while SPME and SBSE require manual handling, which can potentially introduce contamination and limit high-throughput analysis. Furthermore, the cost and fragility of SPME fibers further limit their use in large-scale clinic studies.9 The development of novel alternative sorptive extraction techniques, such as HiSorb sorptive extraction, offers the opportunity for broader profiling with adaptability for high-throughput analysis of clinical samples.10
Herein, we present a methodology for urinary volatolomics using the HiSorb. Dual polar and nonpolar column methodologies coupled with high resolution time-of-flight mass spectrometry (GC-TOF-MS) and strict QC framework are described for reliable VOC detection and identification.11 The performance of the optimized method was assessed using the urine of patients with pancreatic ductal adenocarcinoma (PDAC) and control patients.
Experimental Section
Urine Samples
A cohort of 15 healthy volunteers (8 males and 7 females, age 32 ± 5 years) was recruited for method development (REC reference 04/Q0403/119). All subjects provided informed written consent prior to participation. No special dietary restrictions were required prior to enrollment. Subjects were asked to pass urine into standard 50 mL urine specimen vials, which were immediately sealed. All samples were aliquoted into smaller 15 mL vials before being stored at −80 °C.
The methodology established using urine from healthy volunteers was subsequently evaluated in a clinical cohort consisting of 28 patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) and 33 controls (REC reference 17/WA/016 and 14/LO/1136). PDAC patients and controls were recruited from Hammersmith Hospital (London, UK) between March 2016 and March 2020. Patients were recruited at the time of routine investigation or treatment of a known PDAC. Controls were recruited during routine oesophago-gastro-duodenoscopy (OGD). Inclusion criteria were as follows: (i) PDAC patients aged >18 years with biopsy confirming PDAC and (ii) controls aged >18 years presenting with or without upper gastrointestinal symptoms but with normal upper gastrointestinal tract on endoscopy. Where available, additional investigations (e.g., imaging studies) were used to verify the absence of PDAC. Patients’ medical records were also reviewed at least one year after the time of recruitment to ensure that a diagnosis of cancer had not been made during this period. Exclusion criteria were as follows: (i) patients with nonadenocarcinoma pancreatic cancers (e.g., neuroendocrine tumors), (ii) patients with benign gastrointestinal diseases, (iii) patients diagnosed with other synchronous cancers, (iv) patients with either a suspected or confirmed active infection, liver failure and/or renal failure, and finally, (v) patients who were unable to provide informed written consent or who were not able to provide a >5 mL urine sample. Patient demographics are presented in Table S1.
Chemicals and Consumables
Analytical grade sodium chloride, methanol, hydrochloride acid (HCl), hexane, alkane standards (n-C8 to n-C20 in hexane, 40 mg/L), isotopically labeled analytical standards, including toluene-d8, acetone-d6, butyraldehyde-d2, phenol-d6, benzene-d6, and acetophenone-d8, and laser cryo-tags were purchased from Sigma-Aldrich (Gillingham, UK). Deionized nanopure water was produced by Millipore Direct-Q 3 water purification system (Merck Millipore, Watford, UK). 1.5 and 2 mL cryovials with safe lock, as well as 15 and 50 mL centrifuge tubes were purchased from Scientific Laboratory Supplies, Ltd. (Nottingham, UK). Pipettes and pipette tips were purchased from Fisher Scientific UK, Ltd. (Loughborough, UK). Clear glass 20 mL headspace vials with crimp top and round bottom, caps with HiSorb septa, HiSorb septum plugs, HiSorb handle for manual probe extraction, HiSorb agitator, HiSorb probes coated with polydimethylsiloxane (PDMS), empty inert-coated thermal desorption (TD) tubes with stainless and inert-coated stainless steel DiffLok caps, Tenax TA/Carbograph stainless sorbent TD tubes, and TC-20 TD tube conditioner were supplied from Markes International (Llantrisant, UK). Urine osmolality was measured with an OsmoPRO multisample micro-osmometer from Advanced Instruments (Horsham, UK).
VOC Extraction
Prior to extraction, urine samples were removed from the −80 °C freezer and thawed overnight at 4 °C. Pooled volunteer urine samples were generated by mixing equal amounts (10 mL) of each healthy sample. Three technical replicates were created for each test condition. A variable volume (see Optimization of Urinary VOC Extraction section below) of pooled urine or water (blank) was transferred to glass 20 mL headspace vials. Internal standards, including toluene-d8, acetone-d6, butyraldehyde-d2, phenol-d6, benzene-d6, and acetophenone-d8 (20 mg/L in MeOH-H2O 1:1) were spiked, and NaCl (0.4 g) were added to each sample. The vials were sealed and left for 15 min to equilibrate.
Extraction of urinary VOCs was carried out using HiSorb sorptive extraction. TD tubes with HiSorb probes loaded were preconditioned using the TC-20 TD tube conditioner at 260 °C for 180 min under N2 stream at 20 psi head pressure. A single HiSorb probe was inserted into the glass headspace vial through a septum, exposing the extracting phase in the urine headspace while avoiding contact with the urine or the wall of the vial. After HiSorb probe insertion, vials were placed in a HiSorb agitator, which allowed 16 glass headspace vials to be agitated at the same time. Following the extraction, the HiSorb probes were removed from the vials using the HiSorb handle and placed back into their corresponding TD tubes, which were immediately sealed using DiffLok caps.
Optimization of Urinary VOC Extraction
Urinary extraction conditions that were evaluated included the following: (i) extraction temperature and time; (ii) sample acidification; (iii) sample volume; (iv) sample dilution; and (v) headspace versus immersive analysis. Baseline conditions were as follows: 2 mL, undiluted, unacidified urine samples extracted at 37 °C/30 min with headspace analysis.
Extraction Temperature and Time
VOC extraction from a standard volume of urine (2 mL) was assessed under the following conditions: 37 °C/30 min, 37 °C/120 min, 60 °C/30 min, and 60 °C/120 min.
Sample Acidification
HCl (5 M) was slowly added to urine samples (5 mL) at room temperature monitored by a pH meter, until a pH of 2.0 was reached. Unacidified urine samples (5 mL) served as a control.
Sample Volume
Urine samples of different volumes (1, 2, 3, and 5 mL) were assessed.
Sample Dilution
Undiluted urine samples (2 mL) were compared to urine samples that were diluted at a ratio of 1:1 (2 mL of urine:2 mL of deionized nanopure water), 1:2 (2 mL of urine:4 mL of water), 1:3 (2 mL of urine:6 mL of water), and 1:4 (2 mL of urine:8 mL of water).
Headspace Versus Immersive Analysis
VOCs in a mixture of VOC standards spiked in water and VOCs in urine were extracted using both headspace and immersive analyses.
Method performance was evaluated based on comparisons of the total peak area/chemical class of 14 selected VOCs, which belong to potential cancer biomarker chemical classes,6 including alcohols (1-butanol), ketones (acetone, 2-butanone, 2-pentanone, and 4-heptanone), aldehydes (butanal, pentanal, hexanal, heptanal, octanal, and nonanal), and heterocyclic (furan, 2-methyl) and organosulfur compounds (dimethyl sulfide and dimethyl disulphide). For experiments comparing headspace versus immersive sampling, pooled healthy volunteer urine, standard VOC mixture, and blanks (nanopure water) were analyzed in five replicates per condition. Blank levels were subtracted and average peak areas of headspace versus immersive analysis were compared (Figure S1). All comparisons between test conditions were performed by unpaired t-test, with statistical significance assigned to p values of <0.05.
Instrumentation
The principal analytical platform used for VOC analysis was a TOF-based Markes TD100XR thermal desorber coupled with an Agilent 7890B GC and a Markes BenchTOF select MS (Markes International, Llantrisant, UK). A Markes TD100 thermal desorber (Markes International, Llantrisant, UK) coupled with an Agilent 7890B GC and a quadrupole 5977A MS system served as a reference assay. Chromatographic separation was performed for the polar TOF assay using a Mega WAX-HT, (20 m × 0.18 mm × 0.18 μm, MEGA S.r.l., Legnano, Italy), for the nonpolar TOF assay using a DB5-MS UI (30 m × 0.25 mm × 1.00 μm, Agilent Technologies, Santa Clara, USA) and for the reference quadrupole MS assay using a Zebron ZB-624UI, 60 m × 0.25 mm × 1.40 μm (Phenomenex, Torrance, USA) capillary column.
TD-GC-MS Settings
TD tubes with HiSorb probes loaded were initially prepurged for 1 min with He flow at 50 mL/min. Primary desorption was performed at 250 °C for 5 min to desorb the VOCs onto a cold trap (material emissions, Markes International, Llantrisant, UK) at 25 °C in split mode (1:10). Trap (secondary) desorption was performed at 250 °C (ballistic heating at 60 °C/s) for 3 min, with the flow path onto GC heated constantly at 200 °C. For sample recollection, conditioned Tenax/Carbograph-5 TD tubes were used (Markes International, Llantrisant, UK).
For the optimized TOF-based polar assay, the He flow was set at 0.7 mL/min. The oven temperature was initially set at 35 °C for 2 min and was increased to 240 °C (4 °C/min with 2 min hold). For the optimized TOF-based nonpolar assay, the He flow was set at 2 mL/min. The oven temperature was initially set at 35 °C for 4.5 min and was increased to 300 °C (10 °C/min with 4 min hold). The MS transfer line was maintained at 260 °C, and the ion source (70 eV electron impact) was at 260 °C. The MS analyzer was set to acquire over the range of 30 to 600 m/z, with the data acquisition rate at 6 Hz.
For the reference quadrupole-based method, the column flow was set at 1.0 mL/min of He. The oven temperature was initially set at 40 °C for 4 min, increased to 150 °C (15 °C/min without hold), and finally increased to 240 °C (10 °C/min with 15 min hold). The MS transfer line was maintained at 230 °C; the ion source (70 eV electron impact) was at 240 °C, and the MS quadrupole was held at 150 °C. The quadrupole was set to acquire over the range of 35 to 250 m/z, with the data acquisition rate at 6 Hz.
Sample Recollection
Using pooled urine samples and the optimized nonpolar GC-TOF-MS method developed in the preceding section, sample recollection was evaluated. During recollection, the split flow (90% of the sample) was transferred on to a preconditioned sorbent TD tube. Using the same GC-TOF-MS method, this TD tube was analyzed and recollected a further four times.
Urine Density Correction
Urine samples from 11 healthy volunteers (undiluted and diluted 1:1 with nanopure water) were extracted and analyzed using the optimized nonpolar GC-TOF-MS method. VOC profiles were compared with supervised multivariate statistical analysis (orthogonal projections to latent structures–discriminant analysis, OPLS-DA) before and after osmolality correction in SIMCA 15 (Sartorius, Malmö, Sweden).
Quality Control
Pooled urine QC samples were generated for both the method development (healthy volunteers) and clinical (PDAC patients and controls) cohorts by mixing an equal amount (0.8 mL) of each study sample. For method development and the comparison of different experimental conditions, healthy volunteer pooled QC urine and blanks (deionized nanopure water) were used. For the PDAC cohort samples, one blank sample and six pooled QC samples were analyzed with every 25 clinical samples, resulting in an analytical batch of 32 samples (Table 1). Furthermore, a dilution series (0.625, 1.25, 2.5, 5, 10, 25, 50, and 75% of QC) was analyzed prior to the analysis of clinical batches.11 Metabolic features with a coefficient of variation (CV) of <30% were considered reproducible, and features with a 1-tailed Spearman’s rho of >0.7 and a q value of <0.05 after Benjamini–Hochberg correction12,13 were considered linear. VOC features with blank average levels of <30% in nanopure water compared to their corresponding levels in the pooled QC samples were considered to be uncontaminated. Siloxanes (artifacts generated either from chromatographic columns or extraction sorbents), features with a signal to noise ratio (S/N) of <3, or annotated features whose reverse matched factor (RMF) < 800 versus NIST 17 Mass Spectral and Retention Index Libraries (NIST, Gaithersburg, USA) penalized with retention index (RI) were removed after peak integration from downstream analysis.14
Table 1. Run Order in a Typical Analytical Batcha.
| no. of run | 1 | 2 | 3–7 | 8 | 9–13 | 14 | 15–19 | 20 | 21–25 | 26 | 27–31 | 32 |
| sample | B | QC1 | S1–5 | QC2 | S6–10 | QC3 | S11–15 | QC4 | S16–20 | QC5 | S21–25 | QC6 |
B: blank; QC: pooled quality control sample; S: study sample.
Data Extraction, Preprocessing, and Statistical Analysis
Data acquisition was performed with ChromSpace (Markes International, Llantrisant, UK) and quadrupole data with MassHunter (Agilent Technologies, Santa Clara, USA). ChromSpace followed by Gavin15 was used for deconvolution, peak picking, and integration. All data files were dynamically baseline corrected (DBC) in ChromSpace before further analysis. Mass spectra with their corresponding RI were searched in NIST 17. High-quality data were acquired by deleting nonlinear, irreproducible, or contaminated metabolic features, performing best-matched internal standard normalization16 and performing QC normalization, which can correct interbatch analytical variabilities.11 Details of data normalization are presented in the Supporting Information. Univariate statistical analyses were performed with SPSS 25 (SPSS Inc., Chicago, USA) and MATLAB R2022a (MathWorks, Natick, USA). Figures were generated with Prism 9 (GraphPad Software Inc., La Jolla, USA). Multivariate statistical analysis was performed on R 4.1.2 (R Foundation for Statistical Computing, Vienna, Austria). Total area normalization was applied to each sample. Principal component analysis (PCA) was used to investigate the structure of the data and remove possible outliers using the “mixOmics” package.17 Polar and nonpolar datasets were then combined before generating partial least square–discriminant analysis (PLS-DA) models. PLS-DA model performance was evaluated using leave-one-out (loo) cross-validation and models with a classification error rate (CER) of <0.5 were considered informative. Metabolites with a variable importance projection score (VIP) of >1.5 were considered relevant for group separation. Loadings were extracted from the different models to identify group contribution. Four endogenous metabolites with the highest VIP scores were identified versus authentic standards (Figure S4) and used for the generation of biomarker-specific PLS-DA models. The dataset was split into training and test data (3/4 and 1/4 of the data, respectively) using the “tidymodels” package before evaluating model performance.18 The “auroc” function from the mixOmics package was used to generate the final receiver operating characteristic (ROC) curve on the test data. Additionally, ROC curves for each biomarker were generated from the full dataset using MetaboAnalyst 5.0 (Xia Lab @ McGill, Ste. Anne de Bellevue, Canada).19 The code used for multivariate data analysis is available on GitHub (https://github.com/simonezuffa/Manuscript_HiSorb).
Results and Discussion
VOC Extraction
A summary of the findings of VOC extraction optimization experiments is presented in Figure 1. With the exception of heterocyclic organic compound and aldehydes (37 °C/120 min) and organosulfur compounds and ketones (60 °C/120 min), there was no statistically significant difference among the four temperature and time conditions (Figure 1a). This indicated that for most compounds, extraction temperature and time did not have a significant impact on VOC detection when using the chosen HiSorb extraction method. Therefore, in the interest of time efficiency, 30 min is proposed to be a suitable extraction time for future clinical trials. In addition, extracting by HiSorb probes at 60 °C provided higher total peak area in most chemical groups.
Figure 1.

Optimization of HiSorb extraction conditions. Effects of (a) extraction temperature and time, (b) acidification, (c) sample volume, and (d) dilution were evaluated. The total peak area of 14 selected VOCs, which belong to five potential cancer biomarker chemical classes, including alcohols (1-butanol), ketones (acetone, 2-butanone, 2-pentanone, and 4-heptanone), aldehydes (butanal, pentanal, hexanal, heptanal, octanal, and nonanal), heterocyclic (furan, 2-methyl) and organosulfur compounds (dimethyl sulfide and dimethyl disulphide), was compared. Unpaired t-test was used for condition-by-condition comparison. Boxplots represent lower, upper quartile, and interquartile range (IQR); whiskers represent minimum and maximum values; *: p < 0.05, **: p < 0.01, ***: p < 0.001.
For all chemical groups, samples acidified with HCl had significantly higher total peak area when compared to unacidified samples (Figure 1b). This finding suggests that adding acid to urine samples increases total peak area and should therefore be carried out routinely. This is supported by other studies, which showed that higher numbers and concentration of urinary VOCs are detected in an acid environment compared to a neutral or alkaline environment.20−22 Although there are limited examples within the literature, HCl is the most common acid used for urine acidification.21 Changing the pH can alter the urine sample matrix by increasing the decomposition and degradation of selected compounds, especially when a powerful oxidizing acid is used. It may also cause more compounds to transition from liquid to gas phase by increasing activity coefficients and decreasing partition coefficients.21 Therefore, testing pH adjustment effects before adopting it in the volatile profiles is recommended.
For the majority of chemical classes, sample volume did not appear to have a significant effect on total peak area (Figure 1c). For 1 mL samples, higher variability was however observed, raising doubts about extraction stability. Thus, a minimum sample volume of 2 mL is considered adequate for urinary VOC analysis to achieve optimal extraction efficiency.
Urine is considered to be a rich source of VOCs. In such circumstances, the large number of components may negatively impact on analysis of trace compounds, establishing a matrix effect.23 The dilution of urine samples has the potential to compensate for such matrix effects. In the current study, the total peak area of all chemical groups except alcohols for undiluted samples were higher than diluted ones, and the variation of undiluted samples were often higher (Figure 1d). As a result, sample dilution is not recommended to ensure optimal extraction efficiency.
Based on the above observations, the selected HiSorb extraction conditions were: 2 mL of undiluted and acidified urine at 60 °C for 30 min. These conditions appear to offer both reliable VOC detections with the opportunity for high-throughput analysis that is desired within clinical trials.
GC–MS Optimization
Two GC-TOF-MS methods were evaluated, one with a thin phase polar column designed for separation of acidic VOCs and a second with a general-purpose thick phase nonpolar column that provided coverage for remaining VOC classes.
The performance of the four polar column assays is shown in Table 2. The highest number of urinary VOCs (n = 121) (Table S2) that were linear, reproducible, considered noncontaminant, and library matched was observed with 2 mL of sample volume, 0.7 mL/min column flow (close to optimum linear velocity), and a slow temperature gradient (57 min). Remarkably, for lower sample volume (0.25 mL), greater performance was observed with the high flow rate method where in just 17 min, 85 VOCs were identified. This can be explained by the highly retentive nature of the WAX columns, which in long gradients tend to generate narrow peaks with a lower signal to noise ratio.
Table 2. Four-Assay Panel of GC Methods (Polar Column).
| instrument | TD-GC-TOF-MS (polar column) | |||
| urine volume | 2 mL | 0.25 mL | 2 mL | 0.25 mL |
| GC column flow | 0.7 mL/min | 0.7 mL/min | 1.2 mL/min | 1.2 mL/min |
| GC oven gradients | initial temperature at 35 °C hold for 2 min, ramp to 240 °C at 4 °C/min hold for 2 min | initial temperature at 35 °C hold for 1.9 min, ramp to 240 °C at 20 °C/min hold for 0.2 min | ||
| GC cycle time | 57 min | 57 min | 17 min | 17 min |
| no. of VOCs | 121 | 33 | 96 | 85 |
For the nonpolar assays, a longer column with a thicker phase was chosen to achieve a broad VOC coverage. The highest VOC yield (n = 167) (Table S3) was observed with 2 mL of sample volume, 2 mL/min column flow, and a faster temperature gradient (37 min) (Table 3).
Table 3. Four-Assay Panel of GC Methods (Nonpolar Column).
| instrument | TD-GC-TOF-MS (nonpolar column) | |||
| urine volume | 2 mL | 0.25 mL | 2 mL | 0.25 mL |
| GC column flow | 1.6 mL/min | 1.6 mL/min | 2 mL/min | 2 mL/min |
| GC oven gradients | initial temperature at 35 °C hold for 4.5 min, ramp to 300 °C at 8 °C/min hold for 4 min | initial temperature at 35 °C hold for 4.5 min, ramp to 300 °C at 10 °C/min hold for 4 min | ||
| GC cycle time | 44 min | 44 min | 37 min | 37 min |
| no. of VOCs | 154 | 85 | 167 | 103 |
The overlap between “reliable” urinary VOCs detected by the highest performing polar and nonpolar methods was less than 20% (n = 23) (Tables S2 and S3). While some annotations may be inaccurate due to the nature of untargeted analysis, it is evident that the two methods are highly complementary. Furthermore, it must be noted that this was observed even with the nonpolar PDMS sorbent, which prioritizes the absorption of the less polar VOCs. Thus, in a potential multibed sorbent approach, the complementarity of these methods would be expected to be even greater. In total, 455 urinary VOCs were observed using these methodologies. Forty-one (9% of the total) of these compounds have previously been detected in human urine, according to a recent review by Drabińska et al.5 Importantly, unlike in previous studies, VOCs reported by this study were also subject to QC filtering measures.5 The methodology proposed therein therefore offers both a deep and high-quality profile of the human urinary volatolome, highlighting its potential applicability in future biomarker discovery studies.
Sample Recollection
Results of sample recollection experiments are presented in Table 4. After one recollection cycle, 147/167 (94.6%) VOCs were retained, falling to 133 (79.6%) VOCs by the fourth recollection. Accordingly, in clinical studies, where sample volumes maybe limited, sample recollection enables the option for multiplatform analyses of a single sample.
Table 4. Performance of Sample Recollection.
| cycle | original run | recollection 1 | recollection 2 | recollection 3 | recollection 4 |
| instrument | TD-GC-TOF-MS (nonpolar column) | ||||
| urine volume | 2 mL | ||||
| no. of VOCs | 167 | 158 | 147 | 147 | 133 |
| % of VOC recovered | 100% | 94.6% | 88.0% | 88.0% | 79.6% |
Urine Density Correction
Urine concentration is regulated by renal function and is subject to wide variation both in healthy and disease states. Under normal physiological conditions, urine volume has been found to fluctuate by up to 15-fold, resulting in a significant variation in urinary metabolite levels.24 Therefore, in order to improve data quality, several strategies have been investigated.25 To account for this effect, creatinine adjustment is the most widely applied, but its use is controversial since creatinine levels are affected by muscle mass and coexisting disease states. Thus, osmolality correction, which accounts for the number of dissolved particles per unit of water in urine,24 was investigated in the presented volatolomic assays, as a more independent method of accounting for urine dilution. Osmolality normalization did not appear to improve data quality and, counterintuitively, no correlation between sample total peak areas and osmolality was observed (Figure S2).
Application in PDAC Patients and Controls
Urine samples from 61 patients, 15 pooled QC samples, and 3 blanks were analyzed in three batches. Urine (2 mL) was profiled both with the optimal 57 min polar and the optimal 37 min nonpolar assay (see Tables 2 and 3). Internal standard normalization and QC normalization yielded the highest number of reliable urinary VOC features (Supporting Information and Table S4). Therefore, they were adopted for data preprocessing.
Extracted and preprocessed metabolic profiles, both polar and nonpolar, obtained with Gavin were assessed with PCA. PCA plots revealed a tight cluster of QC samples that were surrounded by study samples (Figure 2), suggesting high data quality.11
Figure 2.
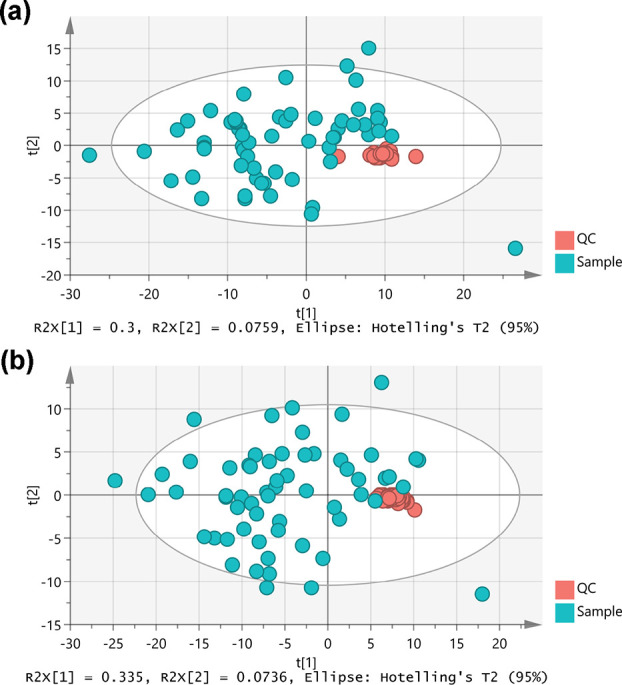
PCA score plots for (a) polar and (b) nonpolar datasets. QC data points clustered tightly, in comparison to the total variance in the projection, indicating high data quality.
Polar and nonpolar datasets were then combined to generate a single PLS-DA model (CER 0.23) (Figure 3). Seventy metabolites had a VIP score of >1.5 (Table S5) and of these, 9 were present in higher abundance in PDAC patients, while 61 were higher in controls. The top 25 most discriminant and annotated metabolites were then further investigated. Four endogenous metabolites (2-pentanone, hexanal, 3-hexanone, and p-cymene, Table 5) were then retained and used to rebuild a final PLS-DA model (CER 0.18), for which an ROC curve with area under the curve (AUC) 0.82 (p value 0.037) was generated (Figure S3). ROC curves for each individual target metabolites are also presented in Figure S3. These findings demonstrated that using the proposed analytical pipeline may successfully differentiate PDAC patients from control subjects based on four urinary VOCs.
Figure 3.
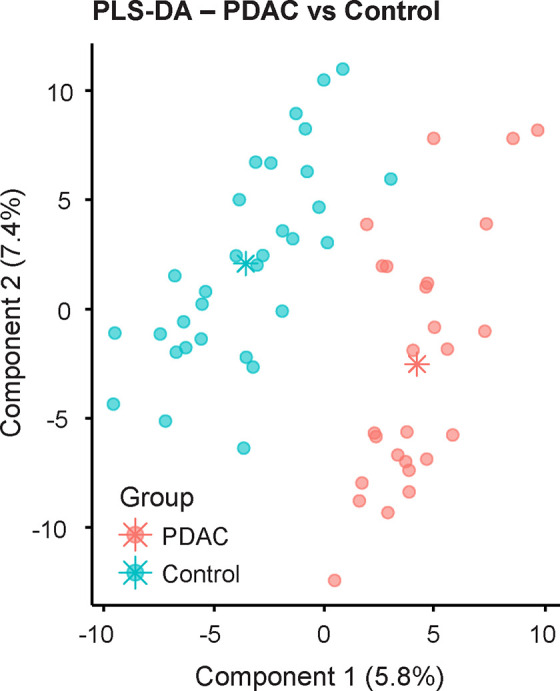
Score plot of the PLS-DA model generated on the whole dataset, combining polar and nonpolar datasets. The classification error rate (CER) of the model was 0.23, indicating informative classification.
Table 5. Candidate Biomarker VOCs for PDAC Diagnosis.
| top ions | compound name | CAS no. | chemical class | increase/decrease in PDAC |
|---|---|---|---|---|
| 43, 86, 41 | 2-pentanone | 107-87-9 | ketone | ↓ |
| 44, 56, 41 | hexanal | 66-25-1 | aldehyde | ↓ |
| 43, 57, 71 | 3-hexanone | 589-38-8 | ketone | ↓ |
| 119, 134, 91 | p-cymene | 99-87-6 | aromatic | ↓ |
Due to a paucity of mechanistic studies focusing on human VOC metabolism,6 the biological significance of the identified metabolites remains largely unclear. Only one of the identified VOCs, 2-pentanone, has previously been linked to pancreatic cancer in the literature. Daulton et al. reported that urinary 2-pentanone is able to separate urine of PDAC versus chronic pancreatitis (CP) patients and CP versus healthy subjects, with ROC-AUC 0.75.26 2-pentanone has also been found to be associated with ulcerative colitis,27 celiac disease,28 nonalcoholic fatty liver disease,29 and Crohn’s disease,27 suggesting that it may serve as a more general biomarker of inflammation.
Hexanal has previously been reported as urinary biomarker of prostate,30 bladder,31 and lung cancer.32 As a short chain aldehyde, it can be produced by peroxidation of unsaturated fatty acids in many parts of the body33 and also by oxidation of 2,2,6-trimethyl-cyclohexanone and 3-hexanone.5
Previous studies have also linked urinary 3-hexanone with lung, breast, and colon cancer34 and p-cymene with colorectal cancer, lymphoma, leukemia, and breast cancer.35,36 Further studies are now needed to both independently validate those biomarkers in a larger patient cohort and to explore the underlying biology of their production in PDAC.
The optimized urinary VOC analytical pipeline worked in a high-throughput, automated, and reliable manner, allowing for 25 clinical samples to be analyzed in a single day. This analysis capability could be increased on demand if an adequate supply of HiSorb probes and GC–MS instruments is available, highlighting its potential for large-scale clinical adoption.
The use of HiSorb probes coated with PDMS that extract mainly nonpolar compounds was an acknowledged limitation of this study. It is anticipated that the ability to explore the contribution of polar compounds to PDAC detection would further improve model performance. A multibed HiSorb probe will aid in the detection of VOCs ranging from polar to nonpolar. A further limitation of this study was that observations were made from a relatively small number of healthy subjects and patients both in terms of method development and clinical application stages. A follow-up study enrolling the appropriate number of patients is needed, which will enable the conducting of a cohort for VOC biomarker discovery and an independent validation cohort to verify the results.
Conclusions
The study has presented an optimized and quality-controlled pipeline for untargeted urinary volatolomic profiling with sorptive extraction and GC-TOF-MS. The clinical utility of this pipeline was demonstrated by its ability to differentiate the urinary volatile profiles of PDAC patients and controls.
Findings underly a potential future role for urinary VOCs as a noninvasive method for disease detection and monitoring. Inclusion of QC measures within a standardized pipeline offers the opportunity for analytical reliability with multicenter trials and support wider clinical adoption.
Acknowledgments
This work was supported by Rosetrees Trust, National Institute of Health Research (NIHR) London In-Vitro Diagnostics Co-operative (LIVD), and NIHR Leadership. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c02873.
Supplemental figures and tables detailing study cohort characteristics, method development, evaluation of osmolality normalization, identified urinary VOCs, data preprocessing, PDAC related metabolites, and ROC curves (PDF)
Author Present Address
⊥ Australian National Phenome Centre, Murdoch, Western Australia 6150, Australia (S.-T.C.)
Author Present Address
∥ Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093–0751, United States (S.Z.).AQ2: Please check if the present addresses were linked to the correct authors
Author Contributions
# Q.W. and A.M. contributed equally to this paper. Study conception and obtaining fund were done by G.H. Study design was done by Q.W., A.M., P.B., and G.H. Patient recruitment was done by S.H. and S.M. Instrumental analysis was done by Q.W., A.M., A.P., and ST.C. Data analysis was done by Q.W., A.M., and S.Z. Manuscript editing and final approval were done by all authors. G.H. was the study custodian. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Markar S. R.; Wiggins T.; Antonowicz S.; Chin S.-T.; Romano A.; Nikolic K.; Evans B.; Cunningham D.; Mughal M.; Lagergren J.; Hanna G. B. Assessment of a Noninvasive Exhaled Breath Test for the Diagnosis of Oesophagogastric Cancer. JAMA Oncol. 2018, 4, 970–976. 10.1001/jamaoncol.2018.0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi D. K.; Sinclair E.; Xu Y.; Sarkar D.; Walton-Doyle C.; Liscio C.; Banks P.; Milne J.; Silverdale M.; Kunath T.; Goodacre R.; Barran P. Discovery of Volatile Biomarkers of Parkinson’s Disease from Sebum. ACS Cent. Sci. 2019, 5, 599–606. 10.1021/acscentsci.8b00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal F.; Kumar S.; Edwards M.; Veselkov K.; Belluomo I.; Kebadze T.; Romano A.; Trujillo-Torralbo M.-B.; Faiez T.; Walton R.; Ritchie A.; Wiseman D.; Laponogov I.; Donaldson G.; Wedzicha J.; Johnston S.; Singanayagam A.; Hanna G. Virus-induced Volatile Organic Compounds are Detectable in Exhaled Breath During Pulmonary Infection. Am. J. Respir. Crit. Care Med. 2021, 204, 1075. 10.1164/rccm.202103-0660OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield G.; Belluomo I.; Boshier P. R.; Waller A.; Fayyad M.; von Wagner C.; Cross A. J.; Hanna G. B. Feasibility and acceptability of breath research in primary care: a prospective, cross-sectional, observational study. BMJ Open 2021, 11, e044691 10.1136/bmjopen-2020-044691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabińska N.; Flynn C.; Ratcliffe N.; Belluomo I.; Myridakis A.; Gould O.; Fois M.; Smart A.; Devine T.; Costello B. D. L. A literature survey of all volatiles from healthy human breath and bodily fluids: the human volatilome. J. Breath Res. 2021, 15, 034001 10.1088/1752-7163/abf1d0. [DOI] [PubMed] [Google Scholar]
- Wen Q.; Boshier P.; Myridakis A.; Belluomo I.; Hanna G. B. Urinary Volatile Organic Compound Analysis for the Diagnosis of Cancer: A Systematic Literature Review and Quality Assessment. Metabolites 2021, 11, 17. 10.3390/metabo11010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna G. B.; Boshier P. R.; Markar S. R.; Romano A. Accuracy and Methodologic Challenges of Volatile Organic Compound-Based Exhaled Breath Tests for Cancer Diagnosis: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 1069. 10.1001/jamaoncol.2019.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David F.; Sandra P. Stir bar sorptive extraction for trace analysis. J. Chromatogr. A 2007, 1152, 54–69. 10.1016/j.chroma.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Yang C.; Wang J.; Li D. Microextraction techniques for the determination of volatile and semivolatile organic compounds from plants: A review. Anal. Chim. Acta 2013, 799, 8–22. 10.1016/j.aca.2013.07.069. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Mannion D. T.; O’Sullivan M. G.; Miao S.; Kerry J. P.; Kilcawley K. N., Comparison of Automated Extraction Techniques for Volatile Analysis of Whole Milk Powder. Foods 2021, 10 (), 10.3390/foods10092061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadhurst D.; Goodacre R.; Reinke S. N.; Kuligowski J.; Wilson I. D.; Lewis M. R.; Dunn W. B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. 10.1007/s11306-018-1367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg Y.; Benjamini Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B. Stat. Methodol. 1995, 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Karaman I.; Climaco Pinto R.; Graça G., Metabolomics Data Preprocessing: From Raw Data to Features for Statistical Analysis. In Comprehensive Analytical Chemistry, Jaumot J.; Bedia C.; Tauler R., Eds. Elsevier: 2018; Vol. 82, pp. 197–225. [Google Scholar]
- Behrends V.; Tredwell G. D.; Bundy J. G. A software complement to AMDIS for processing GC-MS metabolomic data. Anal. Biochem. 2011, 415, 206–208. 10.1016/j.ab.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Boysen A. K.; Heal K. R.; Carlson L. T.; Ingalls A. E. Best-Matched Internal Standard Normalization in Liquid Chromatography–Mass Spectrometry Metabolomics Applied to Environmental Samples. Anal. Chem. 2018, 90, 1363–1369. 10.1021/acs.analchem.7b04400. [DOI] [PubMed] [Google Scholar]
- Rohart F.; Gautier B.; Singh A.; Lê Cao K.-A. mixOmics: An R package for omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M.; Wickham H., Tidymodels: a collection of packages for modeling and machine learning using tidyverse principles. Boston, MA, USA: Accessed December 2020. [Google Scholar]
- Chong J.; Xia J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. 10.1093/bioinformatics/bty528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggio R. B. M.; Mayor A.; Coyle S.; Reade S.; Khalid T.; Ratcliffe N. M.; Probert C. S. J. Freeze-drying: an alternative method for the analysis of volatile organic compounds in the headspace of urine samples using solid phase micro-extraction coupled to gas chromatography - mass spectrometry. Chem. Cent. J. 2016, 10, 9. 10.1186/s13065-016-0155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal P.; Baker J.; Boyd M. T.; Coyle S.; Probert C.; Chapman E. A. Optimisation of Urine Sample Preparation for Headspace-Solid Phase Microextraction Gas Chromatography-Mass Spectrometry: Altering Sample pH, Sulphuric Acid Concentration and Phase Ratio. Metabolites 2020, 10, 482. 10.3390/metabo10120482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natalia D.; Starowicz M.; Krupa-Kozak U. Headspace Solid-Phase Microextraction Coupled with Gas Chromatography–Mass Spectrometry for the Determination of Volatile Organic Compounds in Urine. J. Anal. Chem. 2020, 75, 792–801. 10.1134/S1061934820060088. [DOI] [Google Scholar]
- Silvestro L.; Tarcomnicu I.; Savu S. R., Matrix Effects in Mass Spectrometry Combined with Separation Methods — Comparison HPLC, GC and Discussion on Methods to Control these Effects. Tandem Mass Spectrometry - Molecular Characterization 2013, 10.5772/55982. [DOI] [Google Scholar]
- Chetwynd A. J.; Abdul-Sada A.; Holt S. G.; Hill E. M. Use of a pre-analysis osmolality normalisation method to correct for variable urine concentrations and for improved metabolomic analyses. J. Chromatogr. A 2016, 1431, 103–110. 10.1016/j.chroma.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Rosen Vollmar A. K.; Rattray N. J. W.; Cai Y.; Santos-Neto Á. J.; Deziel N. C.; Jukic A. M. Z.; Johnson C. H., Normalizing Untargeted Periconceptional Urinary Metabolomics Data: A Comparison of Approaches. Metabolites 2019, 9 (), 10.3390/metabo9100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulton E.; Wicaksono A. N.; Tiele A.; Kocher H. M.; Debernardi S.; Crnogorac-Jurcevic T.; Covington J. A. Volatile organic compounds (VOCs) for the non-invasive detection of pancreatic cancer from urine. Talanta 2021, 221, 121604 10.1016/j.talanta.2020.121604. [DOI] [PubMed] [Google Scholar]
- Ahmed I.; Greenwood R.; Costello B.; Ratcliffe N.; Probert C. S. Investigation of faecal volatile organic metabolites as novel diagnostic biomarkers in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2016, 43, 596–611. 10.1111/apt.13522. [DOI] [PubMed] [Google Scholar]
- Di Cagno R.; De Angelis M.; De Pasquale I.; Ndagijimana M.; Vernocchi P.; Ricciuti P.; Gagliardi F.; Laghi L.; Crecchio C.; Guerzoni M. E.; Gobbetti M.; Francavilla R. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol. 2011, 11, 219. 10.1186/1471-2180-11-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M.; Ahmed I.; Gillevet P. M.; Probert C. S.; Ratcliffe N. M.; Smith S.; Greenwood R.; Sikaroodi M.; Lam V.; Crotty P.; Bailey J.; Myers R. P.; Rioux K. P. Fecal Microbiome and Volatile Organic Compound Metabolome in Obese Humans With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e3. 10.1016/j.cgh.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Lima A. R.; Pinto J.; Azevedo A. I.; Barros-Silva D.; Jerónimo C.; Henrique R.; de Lourdes Bastos M.; Guedes de Pinho P.; Carvalho M. Identification of a biomarker panel for improvement of prostate cancer diagnosis by volatile metabolic profiling of urine. Br. J. Cancer 2019, 121, 857–868. 10.1038/s41416-019-0585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchi M.; Weber C. M.; Bolt B. J.; Spratt P. B.; Bessant C.; Turner D. C.; Willis C. M.; Britton L. E.; Turner C.; Morgan G. Evaluation of gas chromatography mass spectrometry and pattern recognition for the identification of bladder cancer from urine headspace. Anal. Methods 2016, 8, 4037–4046. 10.1039/C6AY00400H. [DOI] [Google Scholar]
- Saalberg Y.; Wolff M. VOC breath biomarkers in lung cancer. Clin. Chim. Acta 2016, 459, 5–9. 10.1016/j.cca.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Ratcliffe N.; Wieczorek T.; Drabińska N.; Gould O.; Osborne A.; De Lacy Costello B. A mechanistic study and review of volatile products from peroxidation of unsaturated fatty acids: an aid to understanding the origins of volatile organic compounds from the human body. J. Breath Res. 2020, 14, 034001 10.1088/1752-7163/ab7f9d. [DOI] [PubMed] [Google Scholar]
- da Costa B. R. B.; De Martinis B. S. Analysis of urinary VOCs using mass spectrometric methods to diagnose cancer: A review. Clin. Mass Spectrom. 2020, 18, 27–37. 10.1016/j.clinms.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva C. L.; Passos M.; Câmara J. S. Investigation of urinary volatile organic metabolites as potential cancer biomarkers by solid-phase microextraction in combination with gas chromatography-mass spectrometry. Br. J. Cancer 2011, 105, 1894–1904. 10.1038/bjc.2011.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva C. L.; Passos M.; Câmara J. S. Solid phase microextraction, mass spectrometry and metabolomic approaches for detection of potential urinary cancer biomarkers—A powerful strategy for breast cancer diagnosis. Talanta 2012, 89, 360–368. 10.1016/j.talanta.2011.12.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.