

Abstract
The recent breakthroughs in structure prediction, where methods such as AlphaFold demonstrated near‐atomic accuracy, herald a paradigm shift in structural biology. The 200 million high‐accuracy models released in the AlphaFold Database are expected to guide protein science in the coming decades. Partitioning these AlphaFold models into domains and assigning them to an evolutionary hierarchy provide an efficient way to gain functional insights into proteins. However, classifying such a large number of predicted structures challenges the infrastructure of current structure classifications, including our Evolutionary Classification of protein Domains (ECOD). Better computational tools are urgently needed to parse and classify domains from AlphaFold models automatically. Here we present a Domain Parser for AlphaFold Models (DPAM) that can automatically recognize globular domains from these models based on inter‐residue distances in 3D structures, predicted aligned errors, and ECOD domains found by sequence (HHsuite) and structural (Dali) similarity searches. Based on a benchmark of 18,759 AlphaFold models, we demonstrate that DPAM can recognize 98.8% of domains and assign correct boundaries for 87.5%, significantly outperforming structure‐based domain parsers and homology‐based domain assignment using ECOD domains found by HHsuite or Dali. Application of DPAM to the massive AlphaFold models will enable efficient classification of domains, providing evolutionary contexts and facilitating functional studies.
Keywords: domain classification, domain parser, protein domains, structural predictions
1. INTRODUCTION
Annotating proteins with their constituent domains is a fundamental step toward understanding their evolution and function. Protein domains are conserved regions conveying evolutionary fitness through function (Buljan & Bateman, 2009). Partitioning a protein sequence into domains and classifying each domain into an evolutionary hierarchy is essential in the functional annotation. The predicted function can be used to interpret high‐volume data from large‐scale studies. Homologous relationships to known domains help to generate experimentally testable hypotheses about the function of poorly characterized proteins, and accurate domain boundaries are essential for designing gene constructs for experimental studies (Buljan & Bateman, 2009; Medvedev et al., 2019; Sonnhammer et al., 1998; Tunyasuvunakool et al., 2021). To date, databases for protein domain classification have fallen into two groups: sequence‐based classifications that identify domains based on sequences, such as Pfam and CDD (Apweiler et al., 2001; Tunyasuvunakool et al., 2021), and structure‐based classifications that are primarily based on experimentally determined spatial structures, such as SCOP (Andreeva et al., 2020) and CATH (Sillitoe et al., 2021). Structure classifications show advantages in identifying remote homologs and delineating domain boundaries but have been constrained to the small fraction of proteins with experimental 3D structures.
Our Evolutionary Classification Of protein Domains (ECOD) is a hierarchical classification of protein domains tailored to identifying and classifying distant evolutionary relationships (Cheng et al., 2014). Domains sharing homology deduced from sequence and profile searches or revealing structural similarity coupled with functional evidence are grouped into ECOD H‐groups. The current ECOD release (v287) consists of over 966,000 domains in 3715 H‐groups derived from nearly 642,000 polypeptide chains from over 187,000 structural depositions in PDB (Berman et al., 2000; Burley et al., 2018). ECOD has been accepted as a standard by the field: (1) every PDB entry is linked to the ECOD classification to provide the evolutionary context; (2) ECOD is incorporated into well‐established tools, such as HHsuite (Soding et al., 2005) and RUPEE (Ayoub & Lee, 2019), as a search database; (3) ECOD serves as the source of homologous domains for target classification in multiple rounds of Critical Assessment of techniques in protein Structure Prediction (CASP), a community‐wide experiment for structure predictors to test their methods against target sequences whose structures are not yet public.
The latest round of CASP (Kinch et al., 2021; Kinch, Schaeffer, et al., 2021), CASP14, revealed a breakthrough in the structure prediction field. AlphaFold (AF), developed by DeepMind, could predict 3D structures of proteins from their sequences with accuracies approaching those of experimental methods (Jumper et al., 2021; Tunyasuvunakool et al., 2021). DeepMind has been using AF to model proteins of biomedical importance. In partnership with European Molecular Biology Laboratory, they released 3D structures for over 200 million proteins from the AlphaFold protein structure DataBase (AFDB) (Varadi et al., 2022). This breakthrough transforms structural biology, where computation becomes a key component in solving 3D structures of the most challenging and important protein complexes (Fontana et al., 2022; Mace et al., 2022) and designing small molecule drugs to target specific structures (Thornton et al., 2021; Tong et al., 2021).
The breakthrough in structure prediction is expected to chart protein science in the near future by speeding up the discovery and characterization of proteins with novel and important functions. To gain insights from these predicted structures, it is essential to partition them into domains, evaluate their quality, and classify them by their evolutionary relationships. However, the massive number of AF models represents a challenge for structure classifications, including ECOD, which are currently designed to classify experimental structures. ECOD is frequently updated to include newly released experimental structures. These updates are done through a combination of automatic assignment with human expert curation. The current ECOD automated domain assignment pipeline starts from BLAST searches against ECOD domains and previously classified PDB chains to identify homologs with high sequence similarity (Schaeffer et al., 2018). Subsequently, distant sequence hits identified by HHsuite against a set of domain profiles are used to assign regions that cannot be assigned by BLAST. Then, the structural domain parser, PDP (Alexandrov & Shindyalov, 2003), is used to make minor alterations to domain boundaries. Finally, after the automatic determination of non‐domain regions, unassigned domains (5%–10% cases) are subject to manual curation.
Incorporating a large number of AF models into structure classifications raises several challenges. First, AF models contain a significant fraction of regions unsuitable for globular domain classification, including disordered segments, single transmembrane helices, protein sorting peptides, linkers between globular domains, and coiled coils. We refer to these regions as “non‐domain regions” for simplicity. Structure similarity in such regions frequently arises from convergent evolution. Therefore, annotating the non‐domain regions is an essential task. Second, structure classifications often rely on manual curation to confirm structure similarity, which cannot be scaled up to hundreds of millions of AF models. Thus, better computational tools are necessary to recognize globular domains and facilitate automatic domain classification.
Here, we present a domain parser to recognize single globular domains from AF protein models. Our Domain Parser for AlphaFold Models (DPAM) combines several types of evidence, including the residue‐residue distances, the Predicted Aligned Errors (PAE) associated with each AF model, and candidate homologous ECOD domains detected by HHsuite (Steinegger et al., 2019) and Dali (Holm, 2019). Our benchmark based on previously classified proteins in ECOD shows that this domain parser can recognize 98.8% of these domains, and the boundaries of 87.5% of these domains agree with the ECOD definitions. Such performance is around two times better than the previous structure domain parsers, PDP (Alexandrov & Shindyalov, 2003) and PUU (Holm & Sander, 1994). Once an AF model is partitioned into single domains, assigning the domains to an evolutionary hierarchy becomes simplified. Therefore, we expect this tool to be broadly helpful for researchers interested in analyzing AF models.
2. DEVELOPING BENCHMARK AND REFERENCE SETS FOR PARSING DOMAINS
We needed a set of correctly parsed domains to benchmark our domain parser. We derived such a dataset from the overlap between AF models and proteins whose experimental structures had been previously classified in ECOD. In June 2022, AFDB contained 992,000 models that covered proteins from model organisms and human pathogens and reviewed entries from Uniprot. We obtained the 3D coordinates and the PAE plots for these models. We extracted the sequences of all ECOD domains (v285) (http://prodata.swmed.edu/ecod/complete/distribution). We searched for homologous ECOD domains to each AF model by DIAMOND (Buchfink et al., 2021) and found ECOD hits for 585,000 AF models (e‐value < 0.0001). 87,000 AF models were partially classified by ECOD (i.e., showing ≥95% sequence identity to ECOD domains) because 3D structures of some domains in these proteins were solved in experimental structures.
After removing redundancy by mmseqs2 (Steinegger & Soding, 2017) (identity ≥50%, coverage > 80%) and mapping these 87,000 proteins to ECOD, we obtained 18,759 AF models that were used as the benchmark set. 10,545 (56%) of these AF models were fully classified in ECOD, and the rest were partially classified because the experimental structure did not cover the entire protein. 6776 (36%) AF models in this benchmark contain multiple previously classified ECOD domains. This dataset was used to test the performance of our method.
We obtained the PDB70 database (PDB chains filtered by 70% sequence identity, date 220,313) for HHsuite from http://wwwuser.gwdg.de/compbiol/data/hhsuite/databases/hhsuite_dbs/. We parsed the PDB70 database to get the 92,111 representative PDB chains and found 126,416 ECOD domains annotated from these PDB chains. We further removed redundancy in these ECOD domains by mmseqs2 (identity ≥ 70%, coverage > 80%), and a total of 63,065 representative ECOD domains were selected as a result. These ECOD domains were included in a Dali search database called the ECOD70. ECOD70 was used as the reference sets for parsing domains by homology to these domains.
3. GATHER DATA FOR HOMOLOGY‐BASED DOMAIN PARSING
For each AF model in the benchmark set, we identified its sequence hits by HHsuite search against the PDB70 database. The vast majority of PDB entries were classified in ECOD. Based on these classifications, we partitioned each PDB70 hit into ECOD domains. In addition, we identified the structural hits for each AF model by Dali search against the ECOD70 database. Although Dali remains the best tool to find structural similarities (Holm, 2019), in most cases, it aligns an ECOD domain to only one segment in a query structure, even if the query contains multiple copies of this ECOD domain. Thus, Dali cannot detect duplicated domains in proteins. To alleviate this problem, we developed an iterative Dali alignment procedure (Figure 1e) for ECOD hits found by a traditional Dali search run. In iterations, the segment of a query aligned to an ECOD domain in a previous round was excluded, and the remaining structure was used to perform a Dali search until no similarity was found between the remaining portion of the query and the ECOD domain.
FIGURE 1.
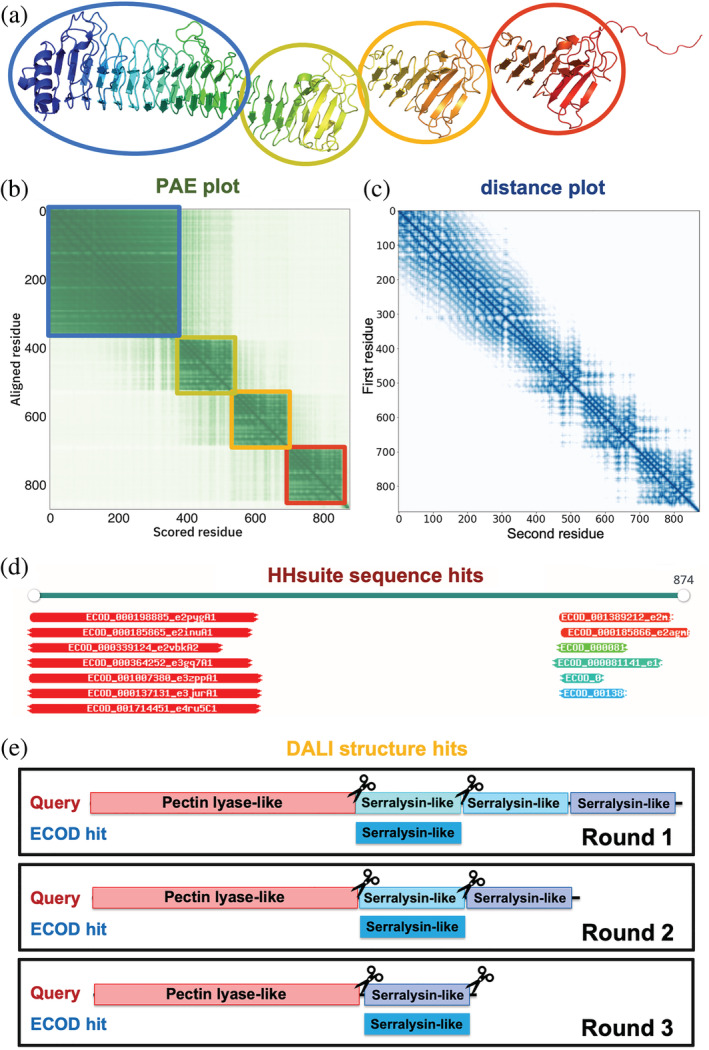
Evidence to parse an AF model into globular domains. (a) An example AF model (UniProt accession: Q9ZFH0). (b) PAE plot for the same model. As the PAE value increases, the color changes from dark green to light green. Residues in the blue, yellow, orange, and red circles in (a) correspond to the blue, yellow, orange, and red squares in (b) with lower PAE values inside. (c) Minimal inter‐residue distance plot for the same model. The color changes from dark blue to light blue as distance increases. (d) Similar sequences detected by HHsuite. (e) Similar structures detected by Dali. We aligned an ECOD hit to a query in an iterative fashion to allow the detection of duplicated domains in the query
To avoid simplifying the task to parse domains by finding a highly similar (frequently identical) ECOD domain, we detected each protein's closely related ECOD domains by BLAST (Camacho et al., 2009). We removed these close homologs from the HHsuite, and Dali hits, respectively. We then identified “acceptable HHsuite hits” using two criteria: (1) the aligned residues covered 40% of the ECOD domains, and (2) the HHsuite probability was at least 50%. These “acceptable HHsuite hits” were used for domain parsing. Similarly, we defined “acceptable Dali hits” as those satisfying any of the following criteria: (1) the top Dali hit in this region is from the same ECOD H‐group as the current hit; (2) the Dali z‐score between the query and this hit divided by the Dali z‐score when aligning this hit to itself is higher than 0.25; (3) the fraction of aligned residues in the hit is more than 50%; (4) the Dali z‐score between query and this hit is better than the 25% quantile of Dali z‐scores for comparisons of domains within the same ECOD H‐group; (5) the Dali z‐score between query and this hit is better than the 25% quantile of Dali z‐scores for comparisons between the hit domain and other domains from the same ECOD H‐group as the hit; (6) the fraction of aligned residues in the hit domain is higher than the 25% quantile of such fractions for comparisons between the hit domain and other domains from the same ECOD H‐group as the hit; (7) the same hit is also detected by HHsuite. The “acceptable HHsuite hits” and “acceptable Dali hits” are not necessarily homologous to domains in the query, but we considered them sufficiently confident to assist domain parsing.
4. GATHER FEATURES TO PARSE AF MODELS INTO DOMAINS
Each AF model is accompanied by a PAE plot (Figure 1b) that specifies the estimated errors in inter‐residue distances. PAEs reflect predicted flexibility between residue pairs. If a flexible linker connects two domains (Figure 1a), residue pairs within the same domain are expected to show low PAEs, while pairs from different domains obtain high PAEs. Thus, as AF developers also noted in AFDB, the PAE plots suggest domain boundaries (Figure 1b). Additionally, the PAE plots can be deployed to detect intrinsically disordered regions: disordered residues exhibit high PAE values except for those nearby in sequence. However, a PAE plot is insufficient to identify evolutionary units because two ECOD domains might be closely packed against each other, show low cross‐domain PAEs, and appear as a single domain in a PAE plot. Thus, we exploited additional features to parse domains, including the inter‐residue distances in AF models (Figure 1c) and similar ECOD domains found by sequence (HHsuite, Figure 1d) and structure (Dali, Figure 1e) searches. Although these criteria were designed to parse AF models into ECOD domains, they could be generalized to other classifications if HHsuite and Dali were used against other domain databases.
Based on our benchmark of 18,759 AF models, we used the above features to calculate the probabilities for a pair of residues to be in the same domain. We binned the residue pairs by their PAEs (Figure 2a ) and their distances (Figure 2b ) in the 3D structure, respectively. We counted the number of residue pairs () in the same domains and the number of pairs () in different domains in each bin, and a residue pair in this bin will receive a “same domain” probability of . The probabilities derived from PAEs and distances are denoted as and , respectively. Based on this benchmark, if two residues show PAE values of less than 8 Å, the probability for them to be in the same domain is at least 50%. Similarly, if two residues are less than 35 Å, the probability for them to be in the same domain is at least 50%.
FIGURE 2.
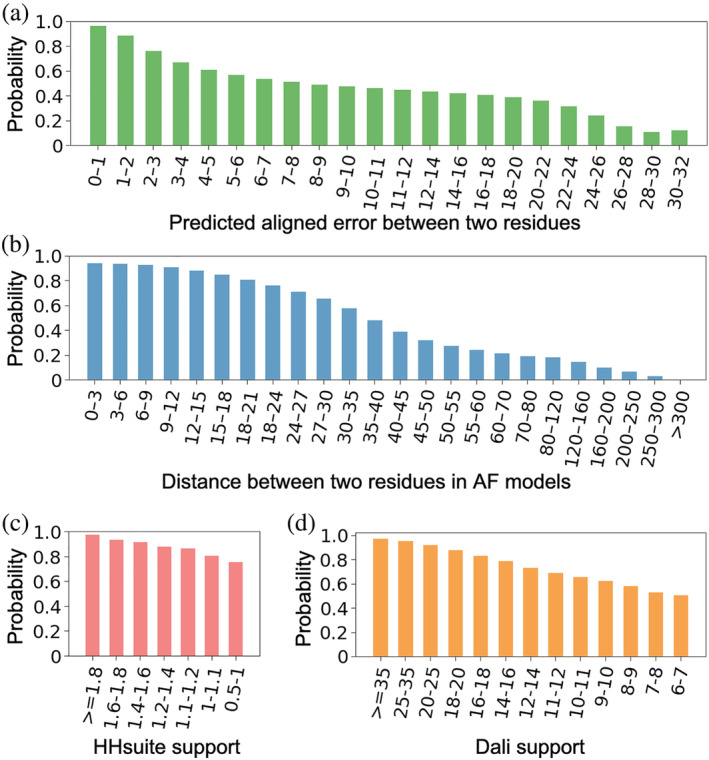
The probabilities for a residue pair to be in the same domain are derived from four parameters: (a) PAE, (b) inter‐residue distance, (c) HHsuite support, and (d) Dali support. The values for these parameters were binned, and probability was calculated as the fraction of residue pairs to be in the same domain in each bin based on our benchmark
Being aligned to the same ECOD domain by sequence or structure comparison tools provides additional support for two residues to be in the same domain. We expect the strength of such support to depend on whether the detected domain is a confident homolog. We used the well‐established confidence measurements, HHsuite probabilities (, range between 0 and 1) and Dali z‐scores (), respectively, to evaluate the confidence of HHsuite and Dali hits. For a pair of residues, we identified all “acceptable HHsuite hits” to which both residues were aligned, and we termed them supporting hits. We integrated the confidence of these supporting hits using the following formula to obtain the HHsuite support for a residue pair:
We added to the maximal HHsuite probability, where is the total number of supporting HHsuite hits. This term allows a pair of residues to receive better support from HHsuite if they are simultaneously aligned to multiple HHsuite hits. Similarly, we identified all “acceptable Dali hits” for each pair of residues and obtained Dali support by the following formula:
We binned the residue pairs by their HHsuite support values (Figure 2c ) and Dali support values (Figure 2d ), respectively. Similar to our treatment of PAEs and inter‐residue distances, a residue pair in a bin will receive the “same domain” probability of , where and are the numbers of residue pairs from the same and different domains in this bin, respectively. The probabilities derived from HHsuite and Dali hits are denoted as and , respectively. From our benchmark, and () are always larger than 0.5 because even less confident ECOD hits still primarily map to single domains in a query protein. Therefore, we assigned of 0.5 for residue pairs that were never aligned to the same HHsuite hit with of at least 0.5. Similarly, we assigned of 0.5 for residue pairs that were never aligned to the same hit with of at least 6.
We calculated the weighted geometric mean of , , , and to get the combined probabilities, , using the following formulas:
The weights for different components were optimized by the ability for to distinguish residue pairs of the same domains from residue pairs of different domains. We scanned the values of , , , and from 0 to 1 with a step size of 0.1, and quantified the performance of in ranking residue pairs from the same domains above those from different domains using the area under the “receiver operating characteristic (ROC)” curves (AUC). ROCs for individual components, and their optimized combination, are shown in Figure S1, and the AUCs are shown in Table 1. Supports from HHsuite and Dali hits contribute more than inter‐residue distances and PAEs to the optimized , but the latter two are expected to be helpful in the absence of confident sequence and structure hits.
TABLE 1.
Performance of different components and their combination
|
|
|
|
|
AUC | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0.753 | ||||
| 0 | 1 | 0 | 0 | 0.790 | ||||
| 0 | 0 | 1 | 0 | 0.765 | ||||
| 0 | 0 | 0 | 1 | 0.803 | ||||
| 0.3 | 0.7 | 0 | 0 | 0.798 | ||||
| 0 | 0 | 0.5 | 0.5 | 0.873 | ||||
| 0.1 | 0.1 | 0.4 | 0.4 | 0.899 |
5. IDENTIFY DISORDERED REGIONS AND FLEXIBLE INTER‐DOMAIN LINKERS
We hypothesize that disordered regions can be characterized as segments showing high PAE values against the rest of a protein. We tested this hypothesis using a subset of the benchmark AF models (7881 models) whose 3D structures had been entirely determined (BLAST identity ≥95%, coverage ≥90%) in experimental structures from PDB. We derived the sets of ordered residues as consistently observed in experimental structures. In contrast, residues that were always missing in the experimental structures despite being included in the experimental constructs were considered disordered. Most proteins from this benchmark are ordered proteins with disordered segments. We tested the following procedure for identifying the disordered regions in these proteins using AF models.
For each target residue, we identified other residues that are at least (tested values: 5, 10, 15, 20, 25, 30; optimized value: 20) residues away from the target residue in sequence but show PAE less than (tested values: 4, 6, 8, 10, 12; optimized value: 6) to the target residue. Despite being distant from the target residue, such residues are rigid in 3D structures relative to the target residue, and we thus term them PAE neighbors of the target. We considered a segment of (tested values: 5,10; optimized value: 5) residues to be disordered if the total number of PAE neighbors for residues in this segment is no more than (tested values: 5, 10, 15, 20, 25; optimized value: 10). We performed a grid search for the parameters used in this method, i.e., , , , and , as indicated in the parenthesis after each parameter. We calculated the precision and recall for identifying disordered residues using each combination of parameters. The optimal parameters were selected to maximize the harmonic mean of precision and recall, that is, F‐score. The chosen parameters show a precision of 0.80 and a recall of 0.64 on our benchmark set, that is, the subset of 7881 models discussed above.
In addition to disordered regions, globular domains in proteins are frequently linked by long helices. The above procedure was extended to identify flexible helical linkers or coiled coils between domains. These helical linkers could be long, and it is important to evaluate if residues in the linker show high PAE values to residues from other secondary structure elements. Therefore, we first defined secondary structure elements in AF models aided by DSSP (Kabsch & Sander, 1983): three or more consecutive residues annotated as “B” or “E” by DSSP are considered as a beta‐strand. Six or more successive residues annotated as “G”, “H”, or “I” by DSSP are regarded as an alpha helix. We modified the above procedure by excluding the PAE neighbors from the same secondary structure elements. Finally, HHsuite and Dali hits could be used to indicate globular domains. We identified residues aligned to “acceptable HHsuite hits” or “acceptable Dali hits” in ECOD, and we dubbed them “candidate intra‐domain residues”. Inter‐domain linkers should not contain a high fraction of “candidate intra‐domain residues”, and thus we further modified the above procedure by requiring this fraction to be no more than 40%.
6. CLUSTER RESIDUES INTO DOMAINS BY COMBINED PROBABILITIES
A visual summary of the principles behind DPAM is presented in Figure 3: inter‐residue distances, PAE values, and homology‐based evidence are combined to cluster 5‐residue segments into domains. We partitioned residues in a protein into non‐overlapping and consecutive segments of 5 residues. We chose 5‐residue segments to balance the performance and speed, and we observed a slight performance improvement (<1%) if we decided to cluster single residues. We excluded this segment if three or more residues were from disordered regions or flexible helical domain linkers. For all remaining segments, we computed the average for every pair of segments. We then clustered segments showing relatively high into the same domain using the following procedure.
FIGURE 3.
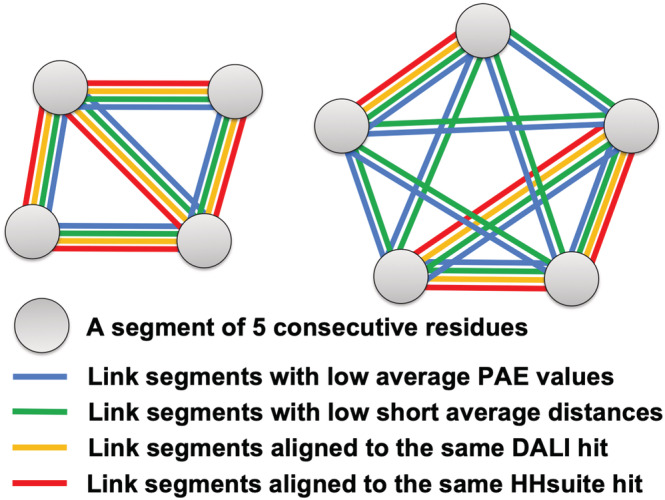
Illustration of the DPAM method. Eight 5‐residue segments of a protein are clustered into two domains based on a consensus of PAEs, inter‐residue distances, similar domains found by HHsuite, and similar domains found by Dali
We sorted segment pairs by , and only considered those pairs with greater than , a parameter to be optimized. The top‐ranking segment pairs were clustered into one candidate domain. Starting from the second pair, we iterated over the existing candidate domains to identify those containing segments from this pair. We handled three possible scenarios. First, if neither segment in the current pair was previously included in a candidate domain, we created a new candidate domain. Second, if only one segment in this pair was present in a previously defined candidate domain, we examined if the other segment should be merged into that candidate domain by comparing the average for segments within the candidate domain () and the average between the new and existing segments in the candidate domain (). We merged the new segment into the candidate domain if , where was another parameter to be optimized. Third, if the two segments in this pair were present in two previously defined candidate domains, we examined if these two candidate domains should be merged. We computed the average for segments within the first domain as , average for segments within the second domain as , and the average for pairs of segments from two different domains as . We merged the two candidate domains if or was satisfied; was the same parameter as used in the previous scenario.
We optimized the two parameters, and , in the above procedure to maximize the percentage of correctly predicted domains (overlapping residues >75% of all residues in the ECOD defined domains). Since is the minimal to classify two segments into the same domain, we expect its value to be slightly above 0.5. Since is the ratio between average intra‐domain and average inter‐domain when merging two candidate domains, we expect its value to be slightly above 1. We performed a grid search to find the optimal and values, and the results are shown in Figure S2. The following cutoffs, 0.54 for and 1.07 for , resulted in the best performance and were chosen for the current version of DPAM.
The above clustering procedure resulted in an initial set of domains, and these domains were further refined. First, we observed that short ( 10 residues) inserted segments might be excluded from a domain if they are considered disordered or loosely packed against the domain, and we merged such short segments into the domains. Afterward, if a domain contains multiple discontinuous segments, we excluded segments containing less than 15 () residues. Finally, short domains of less than 20 () residues were removed. The parameters used in this domain refinement process, that is, , , and , were optimized to maximize the fraction of corrected predicted domains (overlapping residues >75% of all residues in ECOD defined domains).
7. PERFORMANCE EVALUATION
Since DPAM integrates structure‐based and homology‐based evidence, we compared its performance against these two types of methods. The structure domain parsers, PDP and PUU, were applied to AF models in our benchmark set. In addition, among the “acceptable HHsuite hits” (closely related ECOD domains detectable by BLAST were removed), we identified a set of non‐overlapping best hits. We first ranked these hits by HHsuite probability. Starting from the top‐ranking hit, we included a hit to this set if the majority (>75%) of the query residues it mapped to were not covered by previous hits. The query segments aligned to these “best HHsuite hits” were regarded as HHsuite‐based domains. Similarly, we identified a set of “best Dali hits” according to Dali z‐scores and detected domains based on Dali for each query AF model. Thus, we obtained domains by five methods for each AF model, including DPAM, PDP, PUU, HHsuite, and Dali.
We compared the parsed domains by each method against the domains defined by ECOD. Out of the 18,759 AF models in our benchmark set, ECOD currently annotated 28,348 domains. We removed 166 domains that significantly overlap (>25% residues in the domain) with the disordered regions or flexible domain linkers we detected and kept the remaining 28,182 as reference domains. If over 75% of residues in a reference domain were included in the predicted domains by a method, we considered this domain to be covered by that method. The fraction of domains covered by different methods is shown as the blue bars in Figure 4a. Both PDP (96.5%) and DPAM (98.8%) domains covered most of the ECOD domains; however, the high coverage of ECOD domains by PDP is because PDP included a much higher fraction of residues in its domains (92.3%) than DPAM (Figure 4b). However, a significant fraction (6%) of residues in PDP domains belong to disordered regions or flexible linkers in AF models, and they should not be included in the globular domains.
FIGURE 4.
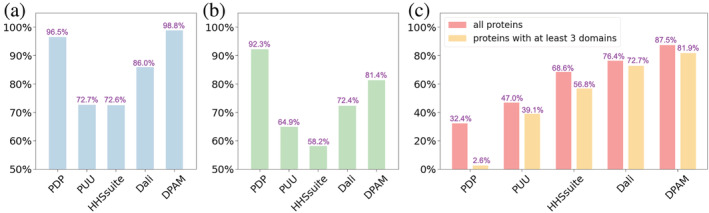
Performance evaluation of DPAM against existing structure‐based domain parsers (PDP and PUU) and assignment based on similar ECOD domains found by sequence (HHS: HHsuite) and structure similarity searches (Dali). (a) The fraction of ECOD domains covered in domains annotated by different methods. (b) The fraction of residues covered in domains annotated by different methods. (c) The fraction of domains whose boundaries were correctly predicted by different methods
We further analyzed the accuracy of different methods in delineating domain boundaries. We considered a predicted domain to have accurate boundaries if it satisfies the following criteria: (1) the fraction of overlapping residues is higher than 75% of all residues for both the reference domain and the predicted domain, or (2) the numbers of non‐overlapping residues in both the reference domain and the predicted domain are no more than 10. The fraction accurately delineated domains by different methods is shown in Figure 4c. DPAM outperforms other methods and shows accurate domain boundaries for 87.5% of domains. Even the remaining 12.5% of DPAM domains still mostly overlap with reference ECOD domains by more than 50% of residues (Figure S3). The second‐best method is Dali. Indeed, domain assignment by structure similarity to known domains is efficient as long as a confident template can be detected. However, domain parsing by structural evidence is helpful when a confident template cannot be detected. DPAM integrates both homology and structural evidence, and thus it could be particularly useful for domains that cannot be easily assigned by homology.
To further evaluate the performance of DPAM, we manually studied the results for AF models containing a large number of domains (≥5). The performance of DPAM on these proteins is slightly worse (81.9%), but it is still higher than other methods (Figure 4c, orange bars). Several correctly parsed models are shown in Figure 5a–e. These examples suggest that DPAM can correctly parse multi‐domain proteins, even when domains are tightly packed against each other or proteins containing tandem repeat domains. Our manual study also revealed scenarios where DPAM tends to make mistakes. The first is when a domain is not compact and thus lacks long‐range contacts, such as the elongated beta‐sheet in blue circles in Figure 5f. The second is when domains are small or poorly modeled, such as the zinc fingers in Figure 5g. Zinc fingers are small domains whose folding relies on zinc ions. Due to the lack of zinc in AF models, zinc fingers tend to be poorly modeled and prone to errors in domain parsing.
FIGURE 5.
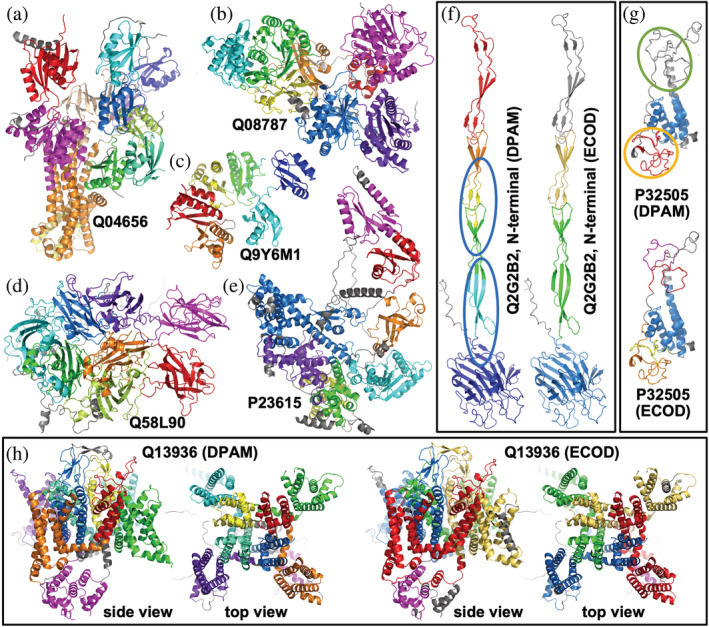
Examples of parsed domains in AF models by DPAM. Different domains in a protein are colored from blue (or purple, N‐terminal) through green, yellow, to red (or magenta, C‐terminal). Non‐domain regions are colored in gray. (a–e) cases where DPAM domain definitions agree with ECOD definitions. (f) a case where DPAM domain definitions are not all accurate, and domains that are incorrectly split are in blue circles. (g) a case where DPAM missed some poorly modeled zinc fingers (in a green circle) and combined multiple consecutive zinc fingers (in orange circles). (H) a case where the boundaries of DPAM domains differ from ECOD domain boundaries, but the DPAM domains appear more meaningful
However, in many cases, although DPAM's domain boundaries differ remarkably from the ECOD definition, a close inspection revealed that DPAM's domain definitions are equally or more accurate. One example is shown in Figure 5h, the voltage‐dependent L‐type calcium channel subunit alpha‐1C (CAC1C). CAC1C contains 24 transmembrane helices (TMHs) and adopts a 4‐fold pseudo‐symmetry. These TMHs were parsed into four domains by ECOD (Figure 5h right), and each domain with 6 TMHs corresponds to one asymmetric unit. Each of the four domains utilizes two THMs to form the central channel for calcium to go through. Because these two central TMHs are used for oligomerization between the 4‐domains, they do not pack tightly against the other four peripheral TMHs in each domain. Therefore, DPAM considered the two central TMHs to form a separate domain from the four peripheral TMHs, a more reasonable decision from the structural perspective but less meaningful from the evolutionary standpoint. CAC1C contains another cytoplasmic domain (magenta in Figure 5h) that was both classified in ECOD and recognized by DPAM. However, DPAM assigned a more reasonable boundary (Figure 5h left) for this domain, while the ECOD domain missed two helices (gray in Figure 5h right).
8. CONCLUSION
We developed a domain parser for AF models that combines predicted aligned errors, inter‐residue distances in the 3D structures, and similar domains found by sequence and structural similarities. DPAM significantly outperforms existing structure‐based domain parsers and homology‐based domain assignments. Although DPAM was developed based on ECOD, it can be easily extended to work with other structure classifications. We expect this tool to simplify and accelerate the classification of AF models into their evolutionary context and allow the scientific community to benefit the most from these valuable structural data. We have applied the DPAM to classify domains in a series of model organisms, and the results, together with our scripts, are released through GitHub at https://github.com/CongLabCode/DPAM.
AUTHOR CONTRIBUTIONS
Jing Zhang: Conceptualization (lead); data curation (lead); methodology (lead); writing – original draft (lead); writing – review and editing (equal). Richard Dustin Schaeffer: Data curation (lead); methodology (lead); software (equal); writing – review and editing (supporting). Jesse Durham: Data curation (equal); methodology (supporting); software (equal); writing – original draft (supporting); writing – review and editing (supporting). Qian Cong: Funding acquisition (equal); project administration (equal); supervision (lead); writing – original draft (equal); writing – review and editing (equal). Nick Grishin: Funding acquisition (lead); methodology (supporting); project administration (lead); supervision (lead); writing – review and editing (equal).
FUNDING INFORMATION
QC is a Southwestern Medical Foundation endowed scholar. A training grant RP210041 from Cancer Prevention and Research Institute of Texas supports JZ. This research is also supported by grant I‐2095‐20220331 to QC and I‐1505 to NVG from Welch Foundation. This research is also funded by NSF 2224128 (DBI) and NIH GM127390 to NVG.
Supporting information
Figure S1. Performance (receiver operating characteristic, ROC, curves) of different features and their combinations in distinguishing intra‐domain residue pairs from residue pairs of different domains. Combinations of features are named by the weights of different components, including inter‐residue distances, PAEs, HHsuite, and Dali. These weights were optimized to maximize area under ROC curves.
Figure S2. Optimization of parameters used to cluster 5‐residue segments into domains. The values in the cells are the fractions of accurately predicted domains (overlapping residues with reference ECOD domains >75% of domain length) using the parameters labeled by the axes. The cells with higher values are colored in darker green.
Figure S3. Performance of different methods to delineate domain boundaries. The percentage of domains at different error rates are shown as bar plots. We computed the overlap between a predicted domain and the reference ECOD domain, and the error rate is defined as the average of two values: (1) the number of non‐overlapping residues in the predicted domain divided by the number of residues in the predicted domain, and (2) the number of non‐overlapping residues in the reference ECOD domain divided by the number of residues in the ECOD domain.
Zhang J, Schaeffer RD, Durham J, Cong Q, Grishin NV. DPAM: A domain parser for AlphaFold models. Protein Science. 2023;32(2):e4548. 10.1002/pro.4548
Jing Zhang and R. Dustin Schaeffer have contributed equally to this study.
Review Editor: Nir Ben‐Tal
Funding information Cancer Prevention and Research Institute of Texas, Grant/Award Number: RP210041; National Institute of General Medical Sciences, Grant/Award Number: GM127390; National Science Foundation, Grant/Award Number: 2224128; Welch Foundation, Grant/Award Numbers: I‐1505, I‐2095‐20220331
Contributor Information
Qian Cong, Email: qian.cong@utsouthwestern.edu.
Nick V. Grishin, Email: grishin@chop.swmed.edu.
DATA AVAILABILITY STATEMENT
Code has been made available through GitHub at http://github.com/CongLabCode/DPAM. Benchmark sets have been made available at the ECOD website (http://prodata.swmed.edu/ecod)
REFERENCES
- Alexandrov N, Shindyalov I. PDP: protein domain parser. Bioinformatics. 2003;19(3):429–30. [DOI] [PubMed] [Google Scholar]
- Andreeva A, Kulesha E, Gough J, Murzin AG. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020;48(D1):D376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29(1):37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub R, Lee Y. RUPEE: a fast and accurate purely geometric protein structure search. PLoS One. 2019;14(3):e0213712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree‐of‐life scale using DIAMOND. Nat Methods. 2021;18(4):366–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M, Bateman A. The evolution of protein domain families. Biochem Soc Trans. 2009;37(Pt 4):751–5. [DOI] [PubMed] [Google Scholar]
- Burley SK, Berman HM, Christie C, Duarte JM, Feng Z, Westbrook J, et al. RCSB protein data Bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018;27(1):316–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, et al. ECOD: an evolutionary classification of protein domains. PLoS Comput Biol. 2014;10(12):e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana P, Dong, Y, Pi X , Tong AB, Hecksel CW, Wang L , et al. Structure of cytoplasmic ring of nuclear pore complex by integrative cryo‐EM and AlphaFold. Science. 2022;376(6598):eabm9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. Benchmarking fold detection by DaliLite v.5. Bioinformatics. 2019;35(24):5326–7. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Parser for protein folding units. Proteins. 1994;19(3):256–68. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12):2577–637. [DOI] [PubMed] [Google Scholar]
- Kinch LN, Schaeffer RD, Kryshtafovych A, Grishin NV. Target classification in the 14th round of the critical assessment of protein structure prediction (CASP14). Proteins. 2021;89(12):1618–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinch LN, Pei J, Schaeffer RD, Grishin NV . Topology evaluation of models for difficult targets in the 14th round of the critical assessment of protein structure prediction. Proteins. 2021;89:1673‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace K, Vadakkepat AK, Redzej A , Lukoyanova N, Oomen C, Braun N , et al. Cryo‐EM structure of a type IV secretion system. Nature. 2022;607:191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev KE, Kinch LN, Schaeffer RD, Grishin NV. Functional analysis of Rossmann‐like domains reveals convergent evolution of topology and reaction pathways. PLoS Comput Biol. 2019;15(12):e1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer RD, Liao Y, Grishin NV. Searching ECOD for homologous domains by sequence and structure. Curr Protoc Bioinformatics. 2018;61(1):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I, Bordin N, Dawson N, Waman VP, Ashford P, Scholes HM, et al. CATH: increased structural coverage of functional space. Nucleic Acids Res. 2021;49(D1):D266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server issue):W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R . Pfam: multiple sequence alignments and HMM‐profiles of protein domains. Nucleic Acids Res. 1998;26(1):320–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger SJ, Söding J. HH‐suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics. 2019;20(1):473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M, Soding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35(11):1026–8. [DOI] [PubMed] [Google Scholar]
- Thornton JM, Laskowski RA, Borkakoti N. AlphaFold heralds a data‐driven revolution in biology and medicine. Nat Med. 2021;27(10):1666–9. [DOI] [PubMed] [Google Scholar]
- Tong AB, Burch JD, McKay D, Bustamante C, Crackower MA, Wu H. Could AlphaFold revolutionize chemical therapeutics? Nat Struct Mol Biol. 2021;28(10):771–2. [DOI] [PubMed] [Google Scholar]
- Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596(7873):590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50(D1):D439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Performance (receiver operating characteristic, ROC, curves) of different features and their combinations in distinguishing intra‐domain residue pairs from residue pairs of different domains. Combinations of features are named by the weights of different components, including inter‐residue distances, PAEs, HHsuite, and Dali. These weights were optimized to maximize area under ROC curves.
Figure S2. Optimization of parameters used to cluster 5‐residue segments into domains. The values in the cells are the fractions of accurately predicted domains (overlapping residues with reference ECOD domains >75% of domain length) using the parameters labeled by the axes. The cells with higher values are colored in darker green.
Figure S3. Performance of different methods to delineate domain boundaries. The percentage of domains at different error rates are shown as bar plots. We computed the overlap between a predicted domain and the reference ECOD domain, and the error rate is defined as the average of two values: (1) the number of non‐overlapping residues in the predicted domain divided by the number of residues in the predicted domain, and (2) the number of non‐overlapping residues in the reference ECOD domain divided by the number of residues in the ECOD domain.
Data Availability Statement
Code has been made available through GitHub at http://github.com/CongLabCode/DPAM. Benchmark sets have been made available at the ECOD website (http://prodata.swmed.edu/ecod)