

Abstract
Skeletal muscle relies upon regeneration to maintain homeostasis and repair injury. This process involves the recruitment of the tissue’s resident stem cell, the muscle progenitor cell, and a subsequent proliferative response by newly generated myoblasts, which must then align and fuse to generate new muscle fibers. During regeneration, cells rely on environmental input for direction. Extracellular matrix (ECM) represents a crucial component of a cell’s microenvironment that aids in guiding muscle regeneration. We hypothesized that ECM extracted from skeletal muscle would provide muscle progenitor cells and myoblasts with an ideal substrate for growth and differentiation ex vivo. To test this hypothesis, we developed a method to extract ECM from the large thigh muscles of adult rats and present it to cells as a surface coating. Myogenic cells cultured on ECM extract experienced enhanced proliferation and differentiation relative to standard growth surfaces. As the methodology can be applied to any size muscle, these results demonstrate that bioactive ECM can be readily obtained from skeletal muscle and used to develop biomaterials that enhance muscle regeneration. Furthermore, the model system demonstrated here can be applied to the study of interactions between the ECM of a particular tissue and a cell population of interest.
Keywords: Extracellular matrix, Muscle, Progenitor cell, Cell culture, Cell proliferation
1. Introduction
Extracellular matrix (ECM) is a term used to represent the complex network of biomolecules occupying the space between cells, tissues, and organs. The contribution of ECM to the structural and mechanical integrity of tissues and organs is well recognized. Additional roles for ECM including the regulation of cellular processes such as proliferation, migration, and differentiation have also been described, and the appreciation of the contribution of ECM to the control of these processes continues to grow. ECM can exert control over cells in several ways. It can provide cells with signals directly through the binding of several classes of membrane proteins to ECM [1]. ECM can also serve as a reservoir for growth factors that become embedded within the matrix and available to local cells [2]. Finally, the previous two types of interactions between cells and ECM can serve as the context in which additional cellular and soluble signals are received and processed, allowing ECM to also have a more indirect or moderating influence on cellular behavior. While many components of ECM are shared between tissues and organs, each has a unique composition tailored to its specific physiologic and mechanical requirements and to the particular types of cells that reside within. It follows that the interaction between unique combinations of cells and ECM molecules within each tissue and organ plays a crucial role in its development and maintenance, as well as its regeneration and repair following injury.
Skeletal muscle tissue relies heavily upon ECM for organization, structural support, and mechanical function [3]. ECM of the epimysium, perimysium, and endomysium, in addition to the basement membrane, establishes the hierarchial organization of skeletal muscle by enveloping the entire muscle, muscle fascicles, and muscle fibers, respectively [4]. For skeletal muscle to function properly, interaction between myofibers and ECM molecules within the basement membrane is crucial. The importance of ECM to proper skeletal muscle function is illustrated by Duchenne’s muscular dystrophy and other congenital forms of muscular dystrophy, which result from mutations in genes encoding the molecular components involved in coupling the cytoskeleton to the ECM [5].
In addition to its physical and mechanical roles, skeletal muscle ECM is required for the cellular maintenance of tissue homeostasis. Skeletal muscle tissue is maintained and repaired through regeneration. When skeletal muscle becomes damaged through usage or injury, a regenerative response is initiated to repair the tissue and restore or enhance muscle function. The resident stem/progenitor cell within skeletal muscle, known as the satellite cell or muscle progenitor cell (MPC), initiates this regenerative response [6,7]. MPCs reside within the basement membrane at the periphery of myofibers and are recruited to proliferate in response to the need for muscle regeneration [8–10]. Proliferation of MPCs generates myoblasts that undergo several rounds of division before fusing to form myofibers, which comprise the basic cellular unit of skeletal muscle. During skeletal muscle regeneration, ECM molecules play a key role in the proliferation of MPCs and myoblasts as well as the subsequent stages of differentiation required to form new myofibers [11–17]. The study of skeletal muscle ECM regulation of MPC and myoblast behavior presents a significant challenge. In vivo, it is difficult to isolate the effects of ECM from those of other environmental components. For ex vivo studies, it is possible to study the role of one or more purified ECM proteins, to use commercially available products to approximate tissue ECM, or to use cultured cells to produce ECM [18–23]. However, in all cases, it is unlikely that the material used represents the specific combination of ECM molecules that exists within skeletal muscle tissue.
To address these challenges, we have developed a method to extract ECM from skeletal muscle tissue. Skeletal muscle ECM extract can be applied as a surface coating, and MPCs or myoblasts can then be seeded onto ECM extract-coated surfaces to study the influence of skeletal muscle ECM on the regenerative capacity of myogenic cells. We hypothesized that two aspects of skeletal muscle regeneration, (1) the proliferation of MPCs and myoblasts and (2) their differentiation into myotubes, would be significantly enhanced by the presence of a skeletal muscle ECM-derived substrate. The goal of this study was to develop a model system whereby skeletal muscle ECM-derived coatings could be utilized to test the interaction of skeletal muscle matrix and MPCs and myoblasts, and to investigate the effect of this microenvironment on several aspects of progenitor cell behavior.
2. Materials and methods
2.1. Animals
Tissue and primary cells were obtained from adult Fisher 344/Brown Norway F1 rats. All animal procedures were conducted under a protocol approved by the Wake Forest University Animal Care and Use Committee.
2.2. Extraction of skeletal muscle ECM
To obtain skeletal muscle ECM, tissue was harvested from the quadriceps and hamstring muscle groups of 4–7 month-old rats. The tissue was frozen at −80 °C and then thinly sliced (<500 μm) using a razor blade at −20 °C. Tissue slices were decellularized through exposure to a series of solutions at 4 °C with continuous agitation at a ratio of 1 L solution to 40 g of starting material. The number of solution changes performed for each step is given in parentheses. First, the tissue was washed in ultrapure water for 2 days (3 changes on day 1; 2 changes on day 2). Next, tissue was exposed to 0.05% trypsin with EDTA for 1 h. Trypsin was neutralized with DMEM + 10% FBS overnight. The decellularization process continued with 5 days of exposure to 1% Triton X-100 (1 change per day) and was completed with 2 days of rinsing in ultrapure water (1 change per day) and 1 day of rinsing in PBS.
To extract ECM from the decellularized tissue, slices were cut into small pieces and washed with ultrapure water for 1 h. The solution was then drained, and the tissue pieces were frozen for 1 day at −80 °C prior to lyophilization. The tissue pieces were lyophilized (Freeze-Dry Systems, Labconco, Kansas City, MO) and processed in a micro-grinder for 15 min while frozen under liquid nitrogen. The powdered product was then dissolved in 2 m urea with shaking for 3 days at 4 °C. Insoluble material was removed via centrifugation for 20 min at 6000 rpm in an ultra centrifuge. The supernatant was filtered through a 40 μm mesh into dialysis tubing. The soluble ECM was dialyzed against ultrapure water for 2–3 days. The solution was frozen in 50 mL conical tubes at −80 °C overnight and then lyophilized until dry to obtain the final ECM extract. Yield was measured by weighing the starting material as wet tissue and the final lyophilized product in pre-weighed 50 mL conical tubes.
2.3. Coating with ECM
For use as a coating material, ECM extract was solubilized in a minimal volume of 0.1% acetic acid. Protein concentration was determined by the Lowry method and adjusted to the desired level by dilution. Coating solution was applied to the substrate surface for 48 h in a humidified 37 °C incubator. For consistency across experiments carried out on different-sized surfaces, 0.33 mL of coating solution was used per 1 cm2 surface area. When used as a control, rat tail type I collagen (BD Biosciences, San Jose, CA) was also diluted in 0.1% acetic acid and applied under the same conditions as the ECM solution. All uncoated surfaces were covered in 0.1% acetic acid during the coating period as a control. The coating solution was removed and the surface was washed twice with PBS prior to cell seeding. For treatment of coated surfaces with collagenase, 0.1% collagenase type III (BD Biosciences) in HBSS was applied for 60–90 min in a 37 °C incubator prior to an additional PBS wash and subsequent cell seeding.
2.4. Contact angle measurement
The static, advanced contact angle of a drop of deionized water on coated and uncoated surfaces was measured using a CAM 100 contact angle meter and software (KSV Instruments, Helsinki, Finland) to measure the angle formed by the surface of interest and the tangent to the drop at the surface. Uncoated surfaces were exposed to the coating solution solvent (0.1% acetic acid) during the coating process.
2.5. Primary cell isolation and culture
Rat MPCs were obtained from quadriceps and gastrocnemius muscles using previously described myofiber explant culture methodology [24–26]. Rat MPC growth medium consisted of DMEM + 10% FBS + 1% penicillin/streptomycin (P/S). Human MPCs were similarly obtained from a small skeletal muscle biopsy and expanded in SkGM2 (Lonza, Walkersville, MD). Differentiation medium consisted of DMEM/F12 + 2% horse serum + 1% P/S.
2.6. Myoblast culture
C2C12 murine myoblasts were propagated in DMEM + 10% FBS + 1% P/S [27]. Differentiation medium consisted of DMEM/F12 + 2% horse serum + 1% P/S.
2.7. Cell proliferation assay
Cell numbers were measured using MTS assay reagents according to the manufacturer’s instructions (Promega, Madison, WI). The MTS assay utilizes a colorometric reaction to measure cellular dehydrogenase activity as an indicator of the number of live, metabolically active cells present. The conditions used for each cell type were optimized to allow 2–4 population doublings without allowing the cells to reach confluence over the course of the experiment as over-confluence can skew assay results. Assays were carried out in 96-well plates, and plates were read using an ELX-500 UV plate reader (Bio-Tek, Winooski, VT).
2.8. Western blot analysis
Standard Western blotting procedures were employed. Briefly, cell lysates were harvested in the presence of protease and phosphatase inhibitors. Protein content was quantified by the Bradford method. Equal amounts of protein from each sample were loaded into the wells of 10% SDS-polyacrylamide gels and resolved by electrophoresis. Protein was transferred to a PVDF membrane. Membranes were blocked with 5% BSA in TBS + 0.05% Tween + 0.025% sodium azide (blocking solution) for 2 h at room temperature. Primary antibody was applied overnight in blocking solution at 4 °C. Membranes were washed and secondary antibody was applied for 35–40 min at room temperature. Membranes were washed and imaged. Images were obtained with the LAS 300 imaging system, and MultiGauge v3.0 was used to quantify the signals by densitometry (both FujiFilm Life Science, Stamford, CT). A biotinylated protein ladder was used for molecular weight measurement (Cell Signaling Tech., Danvers, MA, #7727 with #7075 for detection).
2.9. Antibodies
All antibodies used are listed as antigen (vendor: clone or product #): phospho-Erk1/2 (Cell Signaling Tech., #9101); Erk1/2 (Cell Signaling Tech., #9102); MyoD (Santa Cruz Biotechnology, Santa Cruz, CA: 5.8A); myogenin (Santa Cruz, F5D), myosin heavy chain (University of Iowa Hybridoma facility, Iowa City, IA: MF20); α-tubulin (Santa Cruz, B-5-1-2); HRP-conjugated anti-mouse IgG (Cell Signaling Tech., #7076); HRP-conjugated anti-rabbit IgG (Cell Signaling Tech., #7074).
2.10. Collagen assay
Collagen content as a percentage of total protein content was determined using the Sircol collagen assay kit according to the manufacturer’s instructions (Biocolor Ltd., Newtownabbey, United Kingdom).
2.11. Myotube formation assay
Standard immunofluorescence staining procedures were employed. Briefly, cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton-X 100 for 5 min. Cells were blocked with 10% goat serum for 30 min. Primary antibody (anti-MyHC, MF20) was applied in 3% goat serum for 1 h. Secondary antibody (goat anti-mouse Alexa Fluor 594; Invitrogen, Carlsbad, CA) was applied in 3% goat serum for 45 min. Cells were mounted with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA) and imaged on an Axio Imager (Carl Zeiss MicroImaging, Thornwood, NY). All steps were done at room temperature with 3 PBS washes between steps when necessary.
To quantify the number of myotubes per field, images from 6 random fields of each surface treatment group (ECM or uncoated) were obtained and the number of MyHC-positive myotubes was counted. Additionally, the number of nuclei for each myotube counted was recorded. The average number of myotubes as well as the average number and percentage of myotubes comprised of 1, >1, or >2 nuclei could then be determined for each surface treatment. For the ECM treatment group, 3 different extracts were used and 6 fields from each extract were analyzed.
2.12. Silver staining
For silver staining of ECM proteins, 20 μg of protein from three independent ECM extractions was loaded into the wells of an 8% SDS-polyacrylamide gel and resolved by electrophoresis. The gel was silver stained using Silver Stain Plus reagents according to manufacturer’s instructions (Bio-Rad, Hercules, CA). High range and low range Silver Stain SDS-PAGE Standards were used for molecular weight standards (Bio-Rad).
2.13. Statistical analysis
Data are presented as mean ± SEM. To determine statistical significance between experimental groups, Student’s t-tests were employed. The threshold for significance was set at p < 0.05.
3. Results
3.1. Extraction of skeletal muscle ECM
Skeletal muscle tissue was processed into thin slices (<500 μm) prior to decellularization. By processing the tissue into thin sections, surface area was increased and removal of cellular material made more efficient. Decellularization was confirmed with H&E and DAPI staining (Fig. 1A–D) as evidenced by the lack of cell bodies and punctate nuclei, respectively. ECM was then extracted and freeze dried as described.
Fig. 1.
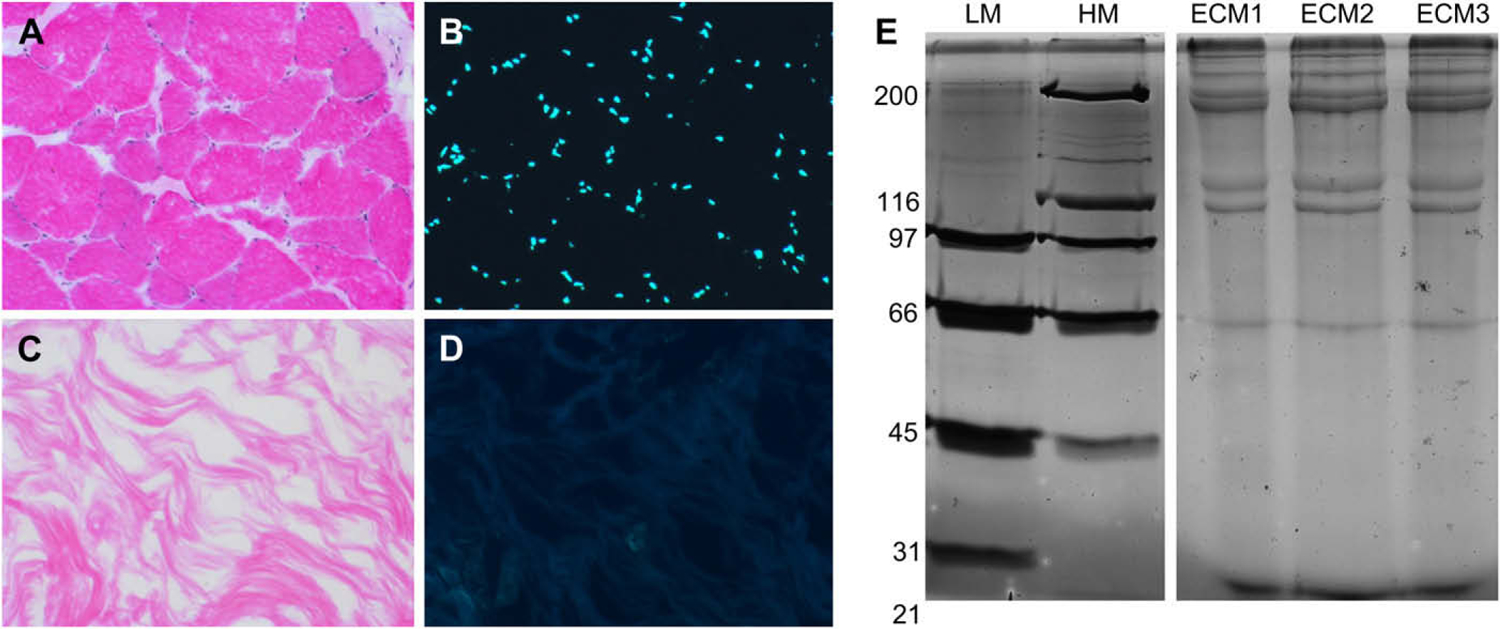
The extraction of ECM protein from skeletal muscle tissue is selective and consistent. Skeletal muscle tissue was stained with H & E (A, C) or DAPI (B, D). (A, B) Prior to decellularization, cellular muscle fibers and abundant nuclei are visible. (C, D) Following decellularization, soluble cellular material and nuclei are absent, while ECM remains. (E) ECM was extracted from the skeletal muscle of three groups (2–4 animals) of rats. ECM proteins were resolved on an 8% polyacrylamide gel and visualized by silver staining. Each of the three extracts (ECM1–3) contained several high molecular weight (>100 kDa) proteins and exhibited nearly identical banding patterns. Note also the presence of abundant low molecular weight proteins/peptides. LM and HM = high and low molecular weight standards, respectively. Numbers indicate molecular weight of standards in kDa.
To confirm the consistency of the extraction method, ECM extract was resolved on an SDS-PAGE gel and visualized by silver stain. Silver staining demonstrated the presence of several high molecular weight proteins and the consistency of the banding pattern between three independent extracts of skeletal muscle ECM from 3 to 5 animals each (Fig. 1E). The collagen content of the ECM extract was found to be 72 ±9%. This finding is consistent with our expectations for skeletal muscle ECM, the abundance of high molecular weight proteins in the polyacrylamide gel, and the extraction method used, which preferentially yields insoluble ECM molecules.
To verify that ECM extract could be used as a coating material, it was applied to tissue culture treated surfaces at a concentration of 0.2 mg/mL. Coated surfaces were compared to uncoated and collagen I-coated surfaces by measuring the contact angle of deionized water on each surface. The contact angle of ECM extract-coated surfaces was 55.9 ± 2.7°. This was significantly less than the contact angle of an uncoated surface (68.1 ± 2.1) and similar to the contact angle of a collagen I-coated surface (56.1 ± 3.3). The reduction in contact angle following the application of an ECM extract coating indicates that a coating is deposited on the surface that renders it more hydrophilic. Together, these results demonstrate that dense skeletal muscle tissue can be effectively decellularized, its ECM can be consistently extracted to yield both collagen and non-collagen proteins, and the ECM extract can be applied as a surface coating.
3.2. Proliferation of myogenic cells on muscle ECM coating
To demonstrate the biologic activity of skeletal muscle ECM extract, myogenic cells were cultured on tissue culture surfaces coated with ECM extract for comparison with culture on uncoated and collagen I-coated surfaces. When murine C2C12 myoblasts were cultured on tissue culture surfaces coated with increasing concentrations of skeletal muscle ECM extract, the level of proliferation increased (Fig. 2A and Supplementary Fig. 1). Significant increases in proliferation were detected as coating concentration increased to 0.2 mg/mL. Above this concentration, proliferation was no longer significantly enhanced. At every coating concentration tested, the levels of C2C12 cell proliferation observed on skeletal muscle ECM extract-coated surfaces were significantly greater than those observed for cells grown on either uncoated tissue culture plastic or collagen I-coated surfaces. When coated surfaces were treated with collagenase prior to the addition of cells, the proliferation-enhancing effect of the ECM extract coatings was significantly reduced, indicating an important but not exclusive role for collagen molecules in mediating the proliferation enhancement of skeletal muscle ECM extract.
Fig. 2.
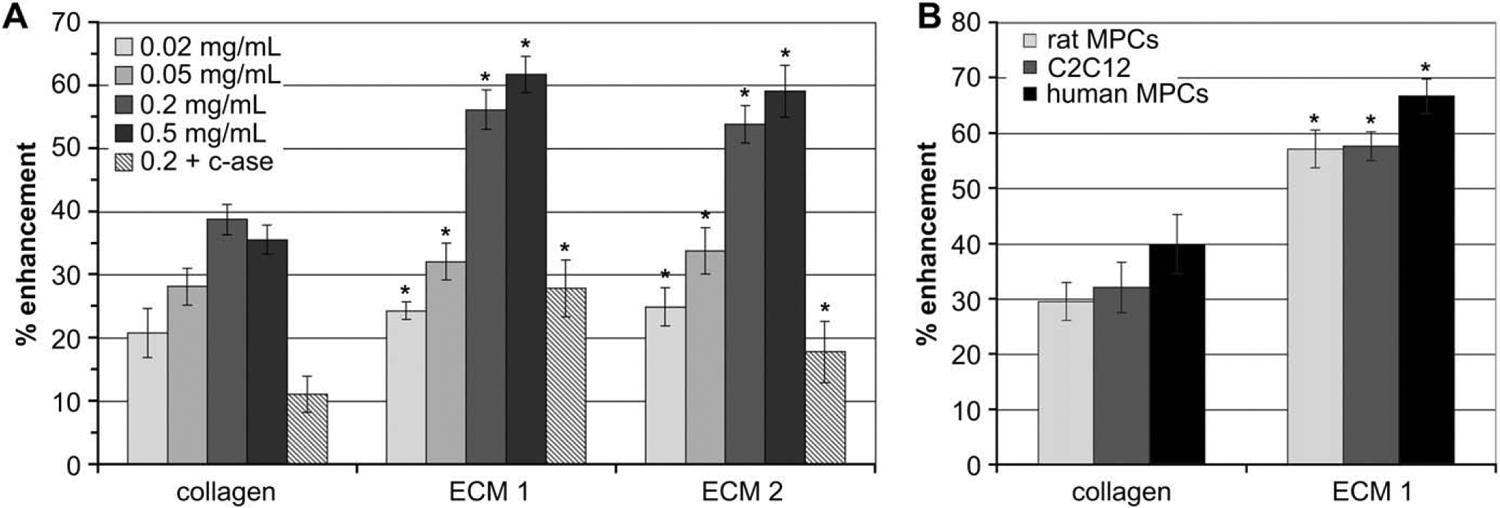
ECM extract enhances the proliferation of myoblasts and primary MPCs in a concentration dependent manner and is sensitive to collagenase. (A) Increasing concentrations of two skeletal muscle ECM extracts were used to coat tissue culture surfaces. Some surfaces were treated with type III collagenase (c-ase) for 60 min. C2C12 myoblasts were seeded onto the surfaces and allowed to proliferate for 48 h. Note the consistency in the results from two independent ECM extracts. (B) Primary rat MPCs, human MPCs, or C2C12 myoblasts were cultured on surfaces coated with 0.2 mg/mL skeletal muscle ECM extract, collagen type I, or on uncoated wells and allowed to proliferate for 54 (human) or 72 h (rat). In all cases, proliferation was quantified by MTS assay and normalized to growth on an uncoated surface (basline). * indicates a statistically different (p < 0.05) result compared to collagen-coated wells at the same concentration and for the same cell type. All results are statistically greater than growth on an uncoated surface. Data are presented as the average of 8 replicates ± SEM.
To further characterize the proliferation of cells grown on ECM extract-coated and uncoated surfaces, C2C12 cell lysates obtained on days −1, +1, and +3 (relative to the induction of differentiation on day 0) were assayed for the expression and phosphorylation of extracellular signal-regulated kinases 1 and 2 (Erk1/2). Erk1/2 are members of the mitogen activated protein kinase (MAPK) family, which become activated through a cascade of phosphorylation initiated at the cell surface [28]. Erk1/2 signaling is known to play a role in the proliferation of skeletal muscle MPCs and myoblasts and has also been implicated in myogenic differentiation [29–31]. Western analysis revealed that higher levels of phospho-Erk1/2 were present in cells cultured in the presence of muscle ECM on days +1 and +3, as the cells continued to grow and differentiate.
To verify that the effect of skeletal muscle ECM extract on cellular proliferation extended beyond the C2C12 myoblast line, the proliferation of primary MPCs was also measured. MPCs are believed to represent a primitive cell within the myogenic lineage, and their proliferation and commitment to differentiation would be required to generate myoblasts similar to C2C12 cells [10]. When primary MPCs obtained from rat or human skeletal muscle tissue were cultured on skeletal muscle ECM extract-coated surfaces, their proliferation was also significantly enhanced over that observed on uncoated and collagen I coated surfaces (Fig. 2B and Supplementary Fig. 1). Together these results demonstrate that the proliferation of the progenitor cell population (MPCs) as well as the main proliferative cell population (myoblasts) within skeletal muscle tissue is significantly enhanced by providing the cells with a substrate for growth derived from skeletal muscle ECM.
These results also indicate that the major component of this material is collagen; however, the proliferation-enhancing effect of skeletal muscle ECM extract is significantly greater than that of a collagen I coating. Furthermore, collagenase treatment does not completely abolish the effect, even when carried out for 90 min (data not shown). Thus, when ECM extract is used to coat surfaces prior to cell seeding, it deposits both collagen and non-collagen molecules that provide myogenic cells with an environment that enhances their proliferation ex vivo.
3.3. Myogenic differentiation on muscle ECM coating
To determine if growth on a skeletal muscle ECM extract-coated surface influences the differentiation of myoblasts, C2C12 cells were cultured on ECM extract-coated and uncoated surfaces for 7 days (days −3 to +3). For the first 3 days (days −3 to −1), cells were grown in proliferation medium in parallel to the experiments described above. After reaching 80–90% confluency, cells were transferred to a low serum medium known to facilitate differentiation and the formation of myotubes in confluent and near confluent cultures of C2C12 cells. The switch to differentiation medium marked day 0, with the days that followed referred to as + days. To monitor the differentiation of the cells grown on ECM extract-coated and uncoated surfaces, cell lysates obtained on days −1, +1, and +3 were assayed for several progressive indicators of myogenic differentiation, including MyoD, myogenin, and myosin heavy chain (MyHC). MyoD and myogenin are members of a family of transcription factors involved in myogenic differentiation, with MyoD expression preceding myogenin expression [32–35]. The final stage of myogenic differentiation that is achieved in two-dimensional culture is the fusion of cells into multinucleated myotubes expressing MyHC.
Western analysis revealed that the expression of phospho-Erk1/2 was enhanced and the progression of myogenic transcription factor expression was accelerated in cells cultured in the presence of muscle ECM (Fig. 3A). On day +3, MyoD expression was greater in myoblasts cultured on uncoated surfaces; however, the expression of myogenin, a transcription factor expressed during later stages of myogenic differentiation, was greater in cells cultured on skeletal muscle ECM extract-coated surfaces. A more rapid progression of transcription factor expression would be expected to result in a more rapid generation of myotubes. The detection of greater levels of MyHC expression in cells cultured on ECM extract-coated surface suggested that this was indeed the case. In addition to Western blot analysis, myotube formation was assayed by immunofluorescent microscopy for the expression of MyHC. On day +3, cultures were stained for MyHC and counterstained with DAPI to compare the number of myotubes per field and the number of nuclei per myotube between cells grown on ECM and those grown on uncoated dishes. As myotubes continue to mature, they gain nuclei and grow in size. The total number of nuclei contained within all myotubes of a field was not significantly different between cells grown on ECM-coated and uncoated surfaces; however, their distribution was different. While cells grown on uncoated surfaces displayed more small myotubes per field, cells cultured on ECM formed fewer, larger myotubes with more nuclei/myotube. This result is indicative of advanced differentiation into more mature myotubes by cells cultured on ECM-coated surfaces. Together, these results demonstrate that the myogenic differentiation of myoblasts grown on skeletal muscle ECM extract-coated surfaces proceeds faster and yields more mature myotubes with more myosin heavy chain than cells grown on an uncoated surface.
Fig. 3.
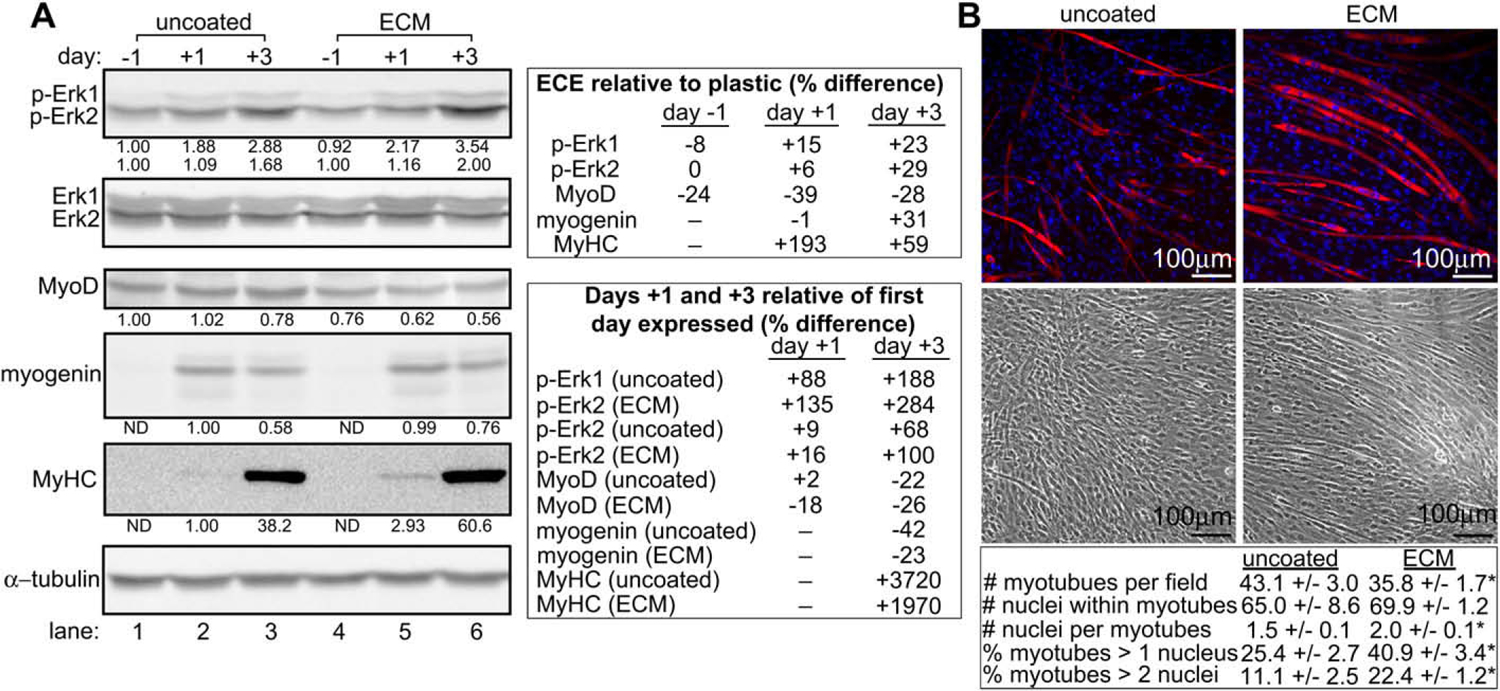
Cells grown on muscle ECM undergo enhanced Erk1/2 phosphorylation and myogenic differentiation over the course of 7 days. (A) Lysates from C2C12 cells grown on uncoated or skeletal muscle ECM extract-coated dishes were assayed for expression of phosphorylated Erk1/2 (p-Erk1/2) and markers of myogenic differentiation by Western analysis. Day numbers indicate the day of culture relative to the addition of myogenic differentiation medium on day 0. For densitometry in p-Erk1/2 blots, Erk1/2 values were normalized to α-tubulin. p-Erk1/2 values were then normalized to the adjusted Erk1/2 values, and the p-Erk1/2 values obtained from cells grown on uncoated dishes on day −1 were set to 1.00. For densitometry in remaining blots, values were normalized to α-tubulin. Values obtained from cells grown on uncoated dishes on the first day of their expression were set to 1.00. All bands are of the expected molecular weight. A representative experiment is shown. (B) C2C12 cells grown on uncoated or ECM-coated surfaces were assayed for the formation of myotubes by immunofluorescence microscopy. The number of myotubes per field and nuclei per myotube were compared between cells grown on uncoated and ECM-coated surfaces. * indicates a statistically different (p < 0.05) result compared to uncoated wells. Data are presented as average ± SEM.
4. Discussion
In the work presented here, skeletal muscle was used as a model tissue to test the hypothesis that the ECM of a particular tissue would enhance the regenerative mechanisms utilized by that tissue’s resident stem/progenitor cell population. We chose to use skeletal muscle because it is a tissue with a well-defined progenitor cell population that relies heavily upon regeneration for both repair and maintenance. Furthermore, there is great interest in engineering skeletal muscle ex vivo for the replacement and/or repair of damaged or diseased tissue, resulting in a need for biomaterials that might better facilitate this endeavor. Naturally-derived, tissue-specific ECM is an attractive biomaterial for use in tissue engineering as it would seem to be well-suited to support the regeneration of the tissue from which it is derived. In pursuing the use of such biomaterials, it would be beneficial to have simple model systems in which hypotheses regarding the interplay between ECM and stem/progenitor cells can be tested. Extracting ECM down to a powdered form that can be solubilized and used to coat substrates prior to the seeding of stem/progenitor cells provides a simple system in which cell–ECM interactions can be observed. In addition to its utility in studying ECM as a biomaterial for potential use in tissue engineering applications, this type of methodology can be used to compare the ECM from the same tissue source in different states. For example, ECM can be extracted from diseased and healthy or young and old tissues and its impact on the behavior of a given cell type can be compared.
Skeletal muscle is a dense tissue with abundant cellular material, making it difficult to decellularize. However, acellular skeletal muscle matrices have been previously developed and employed in the repair of abdominal wall and diaphragm defects [36–38]. These matrices were generated from the same thin muscles for which they were intended to replace using a protocol that relies on repeated water, sodium deoxycholate, and DNase treatment to facilitate decellularization. When seeded with myoblasts, the constructs proved adequate to repair both types of defects. Furthermore, the tissue generated by the constructs continued to mature over time frames as long as 9 months, thereby demonstrating the utility of myoblast-seeded skeletal muscle ECM in tissue engineering [37]. Another way in which acellular skeletal muscle has been used is in the creation of grafts that promote and guide nerve regeneration [39,40]. Again, relatively small and thin muscles such as the gracilis, abdominal muscles, and segments of soleus, adductor magnus, and biceps were used to generate the scaffolds [41–44]. In most of these studies, the tissue was repeatedly snap frozen in liquid nitrogen to lyse the water-containing cells. This simple method of decellularization produces a conduit with architecture similar to that of peripheral nerves. The use of detergent-based methods to facilitate complete removal of cellular material from these scaffolds has shown additional benefits [45,46]. These grafts have been shown to be effective at bridging nerve defects. They are especially effective when seeded with Schwann cells, which can condition the muscle ECM scaffolding to better promote neurogenesis [43,44]. While the aforementioned studies demonstrate the utility of skeletal muscle ECM through its use as an acellular scaffold, our work is the first to extract skeletal muscle ECM down to a form that facilitates the investigation of cell–ECM interactions in a simple, ex vivo model system.
Here, we set out to extract ECM from skeletal muscle present in the large and thick upper leg muscles of adult rats to study its effect on myogenic cells ex vivo and independent from other environmental factors. To facilitate the decellularization of thick skeletal muscle, the tissue was thinly sliced, thereby allowing complete decellularization without consequence to the final product. This methodology can easily be applied to other solid tissues. Our results demonstrate that the material extracted from skeletal muscle contains several high molecular weight proteins, most of which are collagen molecules. While a thorough characterization of all proteins present within each isolate of skeletal muscle ECM was beyond the scope of this work, collagen content analysis, silver staining of SDS-PAGE gels, and the consistency of the biologic effects observed demonstrate that our ECM extraction method yielded a consistent preparation. Additionally, the silver staining banding pattern is similar to that obtained with a lyophilized form of urinary bladder matrix [47]. The extraction method is not without weaknesses. While our enzymatic and detergent-based decellularization method effectively decellularized the skeletal muscle tissue slices, this effect came at the expense of the loss of some factors that normally exist within the ECM. For example, GAGs are lost during decellularization protocols such as that employed here. As expected, when we assayed our skeletal muscle ECM extract for the presence of GAGs, they were below the limits of detection (data not shown). Thus, while the ECM extract used in these studies had a desirable impact on the proliferation and differentiation of myogenic cells, it is possible that these effects could be further enhanced with the development of a method that preserves skeletal muscle ECM in a more native state.
Despite these weaknesses, our results demonstrate that the skeletal muscle ECM extract obtained using our methodology can have profound effect on the ex vivo proliferation and differentiation of myogenic cells. The extract can be used to coat tissue culture surfaces using the same techniques commonly used to apply coatings of single purified ECM proteins, such as collagen, laminin, or fibronectin as substrates for cell culture. True to our hypothesis, when MPCs and myoblasts were cultured on skeletal muscle ECM-coated surfaces, the number of cells produced was significantly greater than that of an uncoated or collagen I coated surface. Furthermore, culture on skeletal muscle ECM extract-coated surfaces also affected the differentiation of myoblasts into myotubes. Cells cultured on ECM extract differentiated faster and generated more mature myotubes in comparison to those cultured on an uncoated tissue culture treated surface. Similar results demonstrating that surfaces coated with ECM molecules accelerate and enhance the proliferation and differentiation of myogenic cells, including C2C12 myoblasts, have been obtained by others [48,49]. Additionally, it has been shown that coating scaffolds composed of Poly-l-Lactic Acid (PLLA), a synthetic material commonly used in tissue engineering, with single ECM molecules like fibronectin and laminin or more complex mixtures of ECM molecules found in commercially available gel products enhances myogenesis from satellite cells/MPCs and skeletal muscle myoblasts [50]. Our results suggest that skeletal muscle ECM extract could provide an additional option for testing cell–biomaterial interactions or even for the engineering skeletal muscle tissue on decellularized tissue scaffolds.
Perhaps most importantly, the work presented here demonstrates the utility of a relatively simple model system for studying the influence of tissue-specific ECM on a cell population of interest. Importantly, this experimental system allows the possibility for ECM to be isolated as the independent variable, something that is nearly impossible to do in in vivo experiments where the effects of additional environmental variables cannot be effectively separated from the unique effects of ECM. While such a system cannot provide a perfect representation of a tissue’s native ECM and cannot recreate the same complex spatial distribution of ECM that exists in vivo, it provides a convenient and effective way to study cell–ECM interactions and investigate hypotheses that might otherwise go untested due to the technical difficulty associated with doing so.
5. Conclusions
We have demonstrated that ECM molecules extracted from skeletal muscle positively influence the proliferation and differentiation of the MPCs and myoblasts responsible for the regenerative maintenance of skeletal muscle tissue. These data suggest that the inclusion of skeletal muscle specific ECM in tissue engineering strategies for the replacement and/or repair of skeletal muscle might provide beneficial effects. The relatively simple and easily modified experimental system employed in this study can be used in the future to gain insight into how ECM obtained from skeletal muscle tissue in different states, such as diseased vs. healthy or young vs. old, differs in content and in its effect on the regenerative capacity of myogenic cells.
Supplementary Material
Acknowledgements
We would like to acknowledge Kristen Carnagey, Ph.D. for her technical assistance, and Chad Markert, Ph.D. and Martin Childers, M.D. for providing C2C12 cells and protocols. This work is partially supported by the Wake Forest Claude D. Pepper Older Americans Independence Center (P30 AG021332).
Footnotes
Appendix. Supplementary material
Skeletal muscle ECM extract enhances the proliferation of myoblasts and human primary MPCs. C2C12 myoblasts and human MPCs were seeded onto uncoated, collagen I-coated, or skeletal muscle ECM-coated surfaces and maintained in growth medium for 48 and 54 h, respectively. Phase-contrast images were then obtained from the areas of lowest and highest cell density for each experimental group. Note that myoblast and MPC density appears to be greater and the cells appear to be more organized when cultured on ECM-coated surfaces relative to both uncoated and collagen-coated surfaces. Supplementary data associated with this article can be found in the online version at doi:10.1016/j.biomaterials.2008.12.069.
References
- [1].Mayer U. Integrins: redundant or important players in skeletal muscle? J Biol Chem 2003;278(17):14587–90. [DOI] [PubMed] [Google Scholar]
- [2].Baeg GH, Perrimon N. Functional binding of secreted molecules to heparan sulfate proteoglycans in Drosophila. Curr Opin Cell Biol 2000;12(5):575–80. [DOI] [PubMed] [Google Scholar]
- [3].Clark KA, McElhinny AS, Beckerle MC, Gregorio CC. Striated muscle cytoarchitecture: an intricate web of form and function. Annu Rev Cell Dev Biol 2002;18:637–706. [DOI] [PubMed] [Google Scholar]
- [4].Sanes JR. The basement membrane/basal lamina of skeletal muscle. J Biol Chem 2003;278(15):12601–4. [DOI] [PubMed] [Google Scholar]
- [5].Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve 2000;23(10):1456–71. [DOI] [PubMed] [Google Scholar]
- [6].Gamble HJ, Fenton J, Allsopp G. Electron microscope observations on human fetal striated muscle. J Anat 1978;126(Pt 3):567–89. [PMC free article] [PubMed] [Google Scholar]
- [7].Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 1961;9:493–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Campion DR. The muscle satellite cell: a review. Int Rev Cytol 1984;87:225–51. [DOI] [PubMed] [Google Scholar]
- [9].Grounds MD, White JD, Rosenthal N, Bogoyevitch MA. The role of stem cells in skeletal and cardiac muscle repair. J Histochem Cytochem 2002;50(5): 589–610. [DOI] [PubMed] [Google Scholar]
- [10].Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004;84(1):209–38. [DOI] [PubMed] [Google Scholar]
- [11].Blaschuk KL, Guerin C, Holland PC. Myoblast alpha v beta3 integrin levels are controlled by transcriptional regulation of expression of the beta3 subunit and down-regulation of beta3 subunit expression is required for skeletal muscle cell differentiation. Dev Biol 1997;184(2):266–77. [DOI] [PubMed] [Google Scholar]
- [12].Boettiger D, Enomoto-Iwamoto M, Yoon HY, Hofer U, Menko AS, Chiquet-Ehrismann R. Regulation of integrin alpha 5 beta 1 affinity during myogenic differentiation. Dev Biol 1995;169(1):261–72. [DOI] [PubMed] [Google Scholar]
- [13].Buck CA, Horwitz AF. Cell surface receptors for extracellular matrix molecules. Annu Rev Cell Biol 1987;3:179–205. [DOI] [PubMed] [Google Scholar]
- [14].Eklund L, Piuhola J, Komulainen J, Sormunen R, Ongvarrasopone C, Fassler R, et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc Natl Acad Sci U S A 2001;98(3):1194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Menko AS, Boettiger D. Occupation of the extracellular matrix receptor, integrin, is a control point for myogenic differentiation. Cell 1987;51(1):51–7. [DOI] [PubMed] [Google Scholar]
- [16].Nandan D, Clarke EP, Ball EH, Sanwal BD. Ethyl-3,4-dihydroxybenzoate inhibits myoblast differentiation: evidence for an essential role of collagen. J Cell Biol 1990;110(5):1673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Osses N, Brandan E. ECM is required for skeletal muscle differentiation independently of muscle regulatory factor expression. Am J Physiol Cell Physiol 2002;282(2):C383–94. [DOI] [PubMed] [Google Scholar]
- [18].Beach RL, Burton WV, Hendricks WJ, Festoff BW. Extracellular matrix synthesis by skeletal muscle in culture: proteins and effect of enzyme degradation. J Biol Chem 1982;257(19):11437–42. [PubMed] [Google Scholar]
- [19].Foster RF, Thompson JM, Kaufman SJ. A laminin substrate promotes myogenesis in rat skeletal muscle cultures: analysis of replication and development using antidesmin and anti-BrdUrd monoclonal antibodies. Dev Biol 1987;122(1):11–20. [DOI] [PubMed] [Google Scholar]
- [20].Hauschka SD, Konigsberg IR. The influence of collagen on the development of muscle clones. Proc Natl Acad Sci U S A 1966;55(1):119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li AA, MacDonald NC, Chang PL. Effect of growth factors and extracellular matrix materials on the proliferation and differentiation of microencapsulated myoblasts. J Biomater Sci Polym Ed 2003;14(6):533–49. [DOI] [PubMed] [Google Scholar]
- [22].Lyles JM, Amin W, Weill CL. Matrigel enhances myotube development in a serum-free defined medium. Int J Dev Neurosci 1992;10(1):59–73. [DOI] [PubMed] [Google Scholar]
- [23].Maley MA, Davies MJ, Grounds MD. Extracellular matrix, growth factors, genetics: their influence on cell proliferation and myotube formation in primary cultures of adult mouse skeletal muscle. Exp Cell Res 1995;219(1):169–79. [DOI] [PubMed] [Google Scholar]
- [24].Bekoff A, Betz W. Properties of isolated adult rat muscle fibres maintained in tissue culture. J Physiol 1977;271(2):537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bischoff R. Proliferation of muscle satellite cells on intact myofibers in culture. Dev Biol 1986;115(1):129–39. [DOI] [PubMed] [Google Scholar]
- [26].Bischoff R. Analysis of muscle regeneration using single myofibers in culture. Med Sci Sports Exerc 1989;21(5 Suppl.):S164–72. [PubMed] [Google Scholar]
- [27].Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977;270(5639):725–7. [DOI] [PubMed] [Google Scholar]
- [28].Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 1997;9(2):180–6. [DOI] [PubMed] [Google Scholar]
- [29].Campbell JS, Wenderoth MP, Hauschka SD, Krebs EG. Differential activation of mitogen-activated protein kinase in response to basic fibroblast growth factor in skeletal muscle cells. Proc Natl Acad Sci U S A 1995;92(3):870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gredinger E, Gerber AN, Tamir Y, Tapscott SJ, Bengal E. Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J Biol Chem 1998;273(17):10436–44. [DOI] [PubMed] [Google Scholar]
- [31].Jones NC, Fedorov YV, Rosenthal RS, Olwin BB. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol 2001;186(1):104–15. [DOI] [PubMed] [Google Scholar]
- [32].Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol 2005;16(4–5):585–95. [DOI] [PubMed] [Google Scholar]
- [33].Fuchtbauer EM, Westphal H. MyoD and myogenin are coexpressed in regenerating skeletal muscle of the mouse. Dev Dyn 1992;193(1):34–9. [DOI] [PubMed] [Google Scholar]
- [34].Smith CK 2nd, Janney MJ, Allen RE. Temporal expression of myogenic regulatory genes during activation, proliferation, and differentiation of rat skeletal muscle satellite cells. J Cell Physiol 1994;159(2):379–85. [DOI] [PubMed] [Google Scholar]
- [35].Yablonka-Reuveni Z, Rivera AJ. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev Biol 1994;164(2):588–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Conconi MT, Bellini S, Teoli D, de Coppi P, Ribatti D, Nico B, et al. In vitro and in vivo evaluation of acellular diaphragmatic matrices seeded with muscle precursors cells and coated with VEGF silica gels to repair muscle defect of the diaphragm. J Biomed Mater Res A 2008. [DOI] [PubMed] [Google Scholar]
- [37].De Coppi P, Bellini S, Conconi MT, Sabatti M, Simonato E, Gamba PG, et al. Myoblast-acellular skeletal muscle matrix constructs guarantee a long-term repair of experimental full-thickness abdominal wall defects. Tissue Eng 2006;12(7):1929–36. [DOI] [PubMed] [Google Scholar]
- [38].Gamba PG, Conconi MT, Lo Piccolo R, Zara G, Spinazzi R, Parnigotto PP. Experimental abdominal wall defect repaired with acellular matrix. Pediatr Surg Int 2002;18(5–6):327–31. [DOI] [PubMed] [Google Scholar]
- [39].Fawcett JW, Keynes RJ. Muscle basal lamina: a new graft material for peripheral nerve repair. J Neurosurg 1986;65(3):354–63. [DOI] [PubMed] [Google Scholar]
- [40].Ide C. Nerve regeneration through the basal lamina scaffold of the skeletal muscle. Neurosci Res 1984;1:379–91. [Google Scholar]
- [41].Fansa H, Keilhoff G. Comparison of different biogenic matrices seeded with cultured Schwann cells for bridging peripheral nerve defects. Neurol Res 2004;26(2):167–73. [DOI] [PubMed] [Google Scholar]
- [42].Fansa H, Keilhoff G, Wolf G, Schneider W, Gold BG. Tissue engineering of peripheral nerves: a comparison of venous and acellular muscle grafts with cultured Schwann cells. Plast Reconstr Surg 2001;107(2):495–6. [DOI] [PubMed] [Google Scholar]
- [43].Feneley MR, Fawcett JW, Keynes RJ. The role of Schwann cells in the regeneration of peripheral nerve axons through muscle basal lamina grafts. Exp Neurol 1991;114(3):275–85. [DOI] [PubMed] [Google Scholar]
- [44].Keilhoff G, Pratsch F, Wolf G, Fansa H. Bridging extra large defects of peripheral nerves: possibilities and limitations of alternative biological grafts from acellular muscle and Schwann cells. Tissue Eng 2005;11(7–8):1004–14. [DOI] [PubMed] [Google Scholar]
- [45].Mligiliche N, Kitada M, Ide C. Grafting of detergent-denatured skeletal muscles provides effective conduits for extension of regenerating axons in the rat sciatic nerve. Arch Histol Cytol 2001;64(1):29–36. [DOI] [PubMed] [Google Scholar]
- [46].Mligiliche N, Tabata Y, Endoh K, Ide C. Peripheral nerve regeneration through a long detergent-denatured muscle autografts in rabbits. Neuroreport 2001;12(8):1719–22. [DOI] [PubMed] [Google Scholar]
- [47].Freytes DO, Martin J, Velankar SS, Lee AS, Badylak SF. Preparation and rheo-logical characterization of a gel form of the porcine urinary bladder matrix. Biomaterials 2008;29(11):1630–7. [DOI] [PubMed] [Google Scholar]
- [48].Lan MA, Gersbach CA, Michael KE, Keselowsky BG, Garcia AJ. Myoblast proliferation and differentiation on fibronectin-coated self assembled monolayers presenting different surface chemistries. Biomaterials 2005; 26(22):4523–31. [DOI] [PubMed] [Google Scholar]
- [49].Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM. Enhanced myogenic differentiation by extracellular matrix is regulated at the early stages of myogenesis. In Vitro Cell Dev Biol Anim 2003;39(3–4):163–9. [DOI] [PubMed] [Google Scholar]
- [50].Cronin EM, Thurmond FA, Bassel-Duby R, Williams RS, Wright WE, Nelson KD, et al. Protein-coated poly(l-lactic acid) fibers provide a substrate for differentiation of human skeletal muscle cells. J Biomed Mater Res A 2004;69(3):373–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.