

Abstract
The danger signals that activate the NLRP1 inflammasome have not been established. Here, we report that the oxidized, but not the reduced, form of thioredoxin-1 (TRX1) binds to NLRP1 and represses inflammasome activation. We discovered that oxidized TRX1 associates with the NACHT-LRR region of NLRP1 in an ATP-dependent process, forming a stable complex that restrains inflammasome activation. Consistent with these findings, patient-derived and ATPase-inactivating mutations in the NACHT-LRR region that cause hyperactive inflammasome formation interfere with TRX1 binding. Overall, this work strongly suggests that reductive stress, the cellular perturbation that will eliminate oxidized TRX1 and abrogate the TRX1-NLRP1 interaction, is a danger signal that activates the NLRP1 inflammasome.
One Sentence Summary:
The oxidized form of thioredoxin-1 binds to and restrains NLRP1 inflammasome activation.
INTRODUCTION
The human NLRP1 (nucleotide-binding domain leucine-rich repeat pyrin domain-containing 1) protein has an N-terminal (NT) pyrin domain (PYD), followed by a disordered region and nucleotide-binding (NACHT), leucine-rich repeat (LRR), function-to-find (FIIND), and caspase activation and recruitment (CARD) domains (Fig. 1A). CARD8 is the only other human protein with a FIIND, but it lacks the NT structured domains (Fig. 1A). Both NLRP1 and CARD8 undergo autoproteolysis within their FIINDs, generating autoinhibitory NT and inflammatory C-terminal (CT) fragments that remain associated (1-3). The proteasome-mediated degradation of the NT fragments releases the CT fragments from autoinhibition (4, 5). Initially, each freed CT fragment is restrained in a ternary complex with one copy of the full-length (FL) sensor protein (NLRP1 or CARD8) and one copy of either dipeptidyl peptidase 8 or 9 (DPP8/9) (6, 7). However, if enough CT fragments are released or if the ternary complex is disrupted by DPP8/9-binding ligands, the CT fragments oligomerize into caspase-1-activating structures called inflammasomes and trigger pyroptosis.
Fig. 1. The NACHT-LRR region of NLRP1 associates with TRX1.
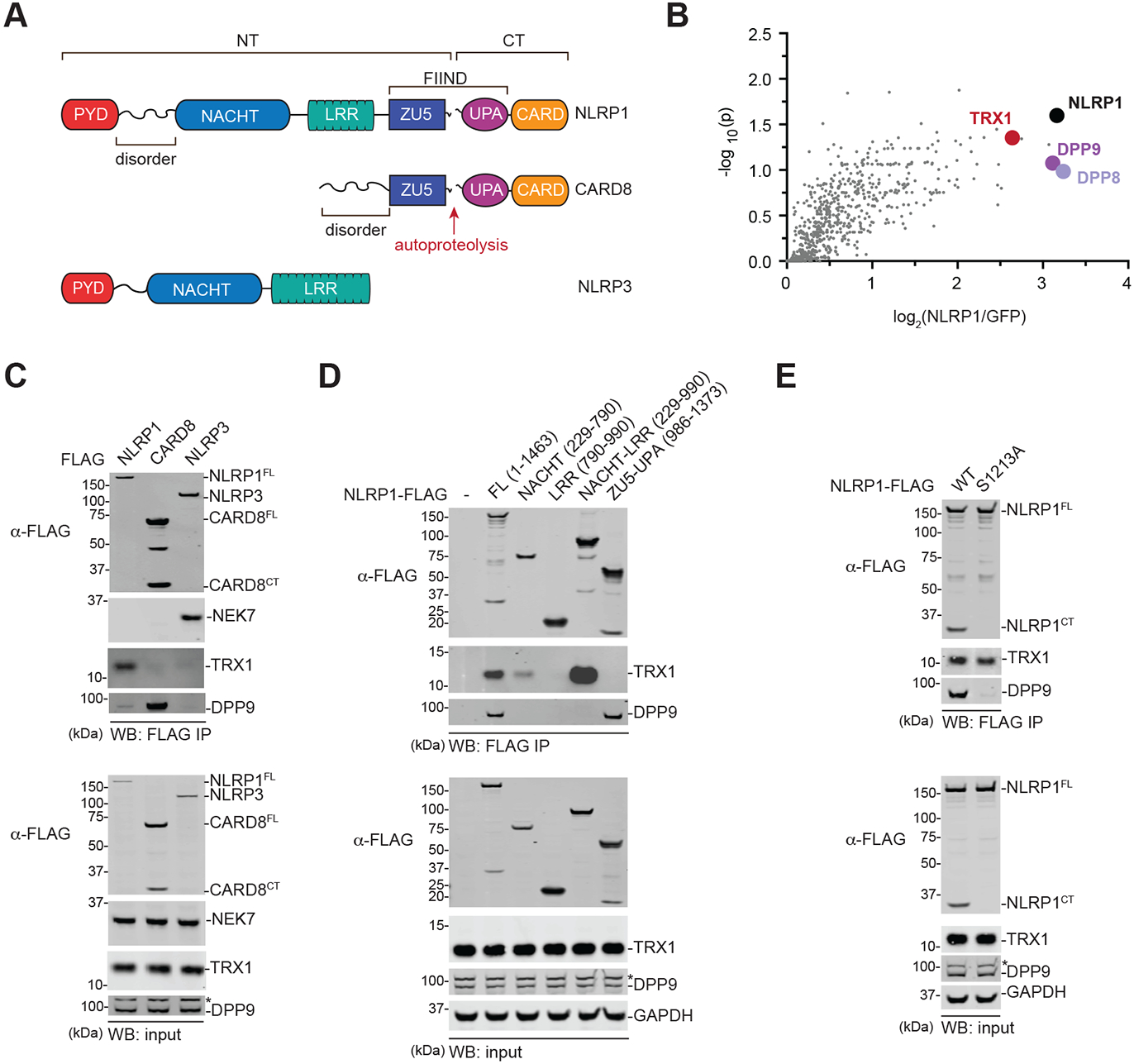
(A) Domain organization of the human NLRP1, CARD8, and NLRP3 proteins. The FIINDs of NLRP1 and CARD8 undergo autoproteolysis between the ZU5 and UPA subdomains. NT, N-terminus. CT, C-terminus. (B) HEK 293T cells were transfected with GFP- or NLRP1-FLAG, subjected to anti-FLAG IP, and analyzed by quantitative mass spectrometry. The volcano plot depicts proteins enriched in the NLRP1-FLAG IP. (C-E) HEK 293T cells were transiently transfected with the indicated FLAG-tagged constructs, subjected to anti-FLAG IP, and analyzed by immunoblotting. In D, numbers indicate amino acid residues. Asterisks (*) denote background bands.
At least three seemingly unrelated agents – enteroviral 3C proteases (8, 9), long double-stranded RNA (dsRNA) (10), and DPP8/9 inhibitors (11-16) – activate the human NLRP1 inflammasome. 3C proteases cleave within the disordered linker between the PYD and NACHT domains, creating an unstable neo-N-terminus that is degraded by the N-end rule proteasome pathway. dsRNA directly interacts with the NACHT-LRR region, triggering ATP hydrolysis and presumably NT degradation, via unknown mechanisms. DPP8/9 inhibitors, including Val-boroPro (VbP), both accelerate the proteasome-mediated degradation of the NT fragment through a poorly characterized pathway and destabilize the NLRP1FL-NLRP1CT-DPP8/9 repressive ternary complex (6, 7, 14, 16, 17). Of these three triggers, only DPP8/9 inhibitors also activate the rat and mouse NLRP1 inflammasomes (15) and the human CARD8 inflammasome (rodents do not express CARD8) (13, 18). As such, only DPP8/9 inhibitors, and not dsRNA and 3C proteases, are likely related to the primordial function of the NLRP1 inflammasome (19). Interestingly, we recently discovered that XP-containing peptides (X is any amino acid), which are the endogenous substrates of DPP8/9, mimic VbP and are thus naturally occurring NLRP1/CARD8-activating danger signals (20-22).
We have hypothesized that the NT domains of all NLRP1 proteins evolved to detect at least one as yet unknown danger signal, perhaps related in some way to DPP8/9 (19). Here, we report that the oxidoreductase thioredoxin-1 (TRX1) regulates NLRP1NT degradation. TRX1 exists in either an oxidized (disulfide) or a reduced (dithiol) state, the relative levels of which are dependent on the intracellular redox potential (23). We found that the oxidized, but not the reduced, form of TRX1 binds to the NACHT-LRR region of the NLRP1NT, which stabilizes this region and restrains inflammasome activation. Moreover, we discovered that mutations in the NACHT-LRR region that cause NLRP1 hyperactivation, including those in patients with autoinflammatory disorders and those that disrupt ATPase activity (24-26), impair oxidized TRX1 binding. Overall, these results show that the NLRP1NT fragment detects the disappearance of the oxidized form of thioredoxin, indicating that a low intracellular redox potential – or reductive stress – is an NLRP1 inflammasome-activating danger signal.
RESULTS
The NLRP1 NACHT-LRR region binds to oxidized TRX1
The NLRP3 inflammasome sensor protein is structurally similar to the NLRP1NT fragment (Fig. 1A). Notably, the mitotic kinase NEK7 directly interacts with NLRP3’s NACHT-LRR region to license inflammasome activation (27-29). We reasoned that an endogenous protein might similarly bind to the NT domains of NLRP1 and regulate inflammasome formation. We therefore performed an immunoprecipitation-mass spectrometry (IP-MS) experiment in HEK 293T cells with FLAG-tagged NLRP1 or FLAG-tagged GFP to identify potential NLRP1 binding partners (Fig. 1B, Table S1, Data file S1). This experiment not only identified DPP8 and DPP9 as NLRP1-binding proteins, as expected, but also revealed that thioredoxin-1 (TRX1) potentially interacted with NLRP1 as well (Fig. 1B). We confirmed that TRX1 indeed co-immunoprecipitated human NLRP1, but not CARD8 or NLRP3, by immunoblotting (Fig. 1C). Moreover, we found that TRX1 also interacted with all functional rat and mouse NLRP1 alleles (Fig. S1A,B), showing that TRX1 binding, like DPP8/9 binding, is an evolutionarily conserved function of NLRP1.
We next expressed various truncated forms of FLAG-tagged NLRP1 to identify the region that interacts with TRX1. We found that the isolated NACHT domain (we initially defined the NACHT as residues 229-790, but the AlphaFold prediction suggests that residues 730-790 are likely part of a linker region and the start of the LRR (30)) retained some affinity for TRX1, but the isolated LRR and FIIND domains did not (Fig. 1D). However, the entire NACHT-LRR region bound more strongly to TRX1 than the isolated NACHT, indicating that these two domains together mediate this protein-protein interaction. DPP9 associates with the NLRP1 FIIND (6, 7, 14), and, as expected, only the full-length NLRP1 protein and isolated FIIND bound to DPP9. Thus, TRX1 and DPP9 bind to distinct regions of NLRP1. Consistent with these distinct binding regions, autoproteolysis-deficient NLRP1 (S1213A), which has dramatically impaired binding to DPP9, retained binding to TRX1 (Fig. 1E).
TRX1 is a ubiquitous, highly conserved oxidoreductase that maintains redox balance in the cytosol and protects proteins from oxidative damage and aggregation (31, 32). TRX1 uses a cysteine (Cys32) within a conserved CGPC motif (residues 32-35) to react with disulfide bonds in oxidized proteins. The second cysteine (Cys35) then resolves the disulfide bond between the target protein and TRX1, thereby reducing the target protein and oxidizing TRX1. The disulfide bond in oxidized TRX1 is then reduced by thioredoxin reductase-1 (TXNRD1), which uses NADPH as an electron donor. To determine if Cys32 and/or Cys35 were critical for binding to NLRP1, we expressed FLAG-tagged WT, C32S, C35S, or C32S/C35S TRX1 together with untagged NLRP1 before performing an anti-FLAG IP. We found that both Cys32 and Cys35 were required for binding (Fig. 2A, Fig. S2A). It should be noted that NLRP1 and TRX1 could, in theory, form a stable intermolecular disulfide bond. However, this likely does not occur, as the C35S mutation, which would trap such a bond, dramatically weakened the NLRP1-TRX1 interaction.
Fig. 2. Only oxidized TRX1 binds NLRP1.
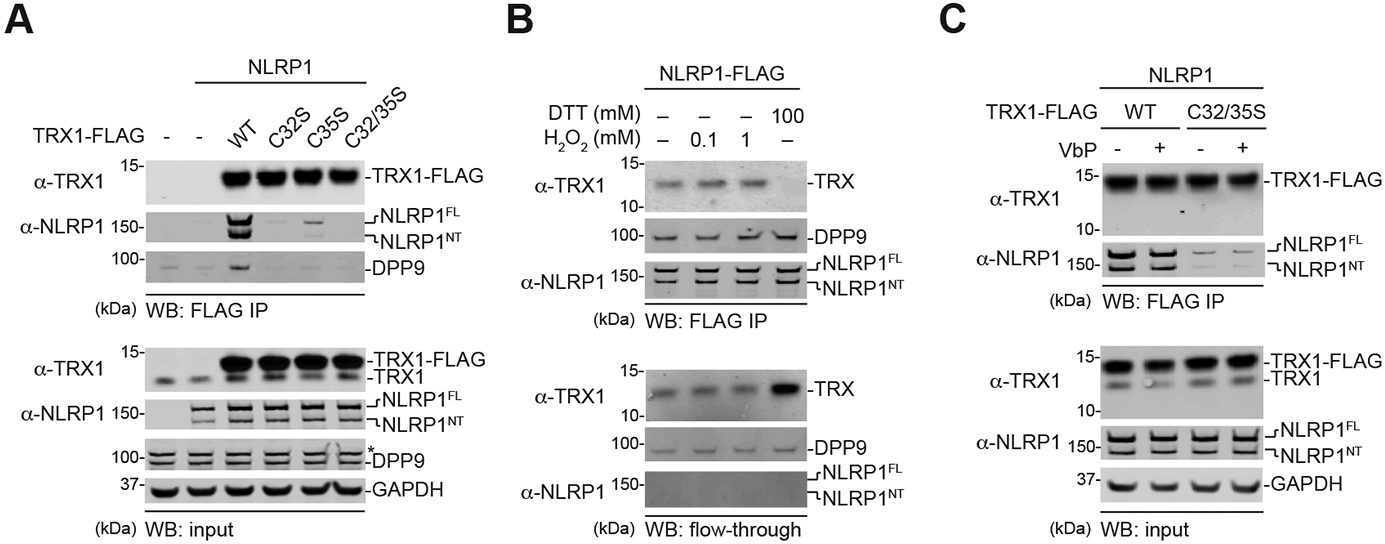
(A-C) HEK 293T cells were transfected with the indicated constructs before lysates were harvested, subjected to anti-FLAG IP, and immunoblotted. Anti-FLAG beads were treated with the indicated concentrations of (anti)oxidants in B and VbP (10 μM) in C. The flow-through in B is the unbound fraction after (anti)oxidant treatment. Note that cells were transfected with untagged NLRP1 in A and C. An asterisk (*) denotes a background band.
Instead, we hypothesized that NLRP1 was exclusively binding to either the reduced or the oxidized form of TRX1. To determine which state of TRX1 was bound to NLRP1, we immobilized FLAG-tagged NLRP1 protein from HEK 293T lysates on anti-FLAG beads, and then treated these beads with the oxidizing agent hydrogen peroxide (H2O2) or the reducing agent dithiothreitol (DTT) (Fig. 2B, Fig. S2B). We found that H2O2 slightly strengthened the TRX1 interaction, and conversely that DTT abolished the interaction. Similarly, we found that reduced glutathione (GSH) dramatically weakened the interaction as well (Fig. S2B). Thus, only oxidized TRX1 (i.e., the disulfide form of TRX1) binds to NLRP1. Notably, neither H2O2 nor DTT affected the NLRP1-DPP9 interaction (Fig. 2B, Fig S2C). Moreover, the DPP8/9 inhibitor VbP weakened the NLRP1-DPP9 interaction in this assay (Fig. S2C), as expected, but had no direct impact on the NLRP1-TRX1 interaction (Fig. 2C, Fig. S2C). The dsRNA analog poly(I:C), which reportedly binds to the NACHT-LRR region of NLRP1 (10), did not affect NLRP1’s interaction with either DPP9 or TRX1 (Fig. S2C).
TRX1 restrains NLRP1 activation
We next wanted to determine if TRX1 binding either restrained or potentiated NLRP1 inflammasome formation. The free NLRP1CT oligomerizes with the adapter protein ASC (apoptosis-associated speck-like protein containing a CARD) to form foci called specks before recruiting caspase-1, and VbP induces the formation of visible ASC specks in the cytosol of HEK 293T cells ectopically expressing NLRP1 and GFP-tagged ASC (14, 33). Although TRX1 deficiency imposes a fitness cost on cell lines (Fig. S3A), we managed to generate TRX1-deficient HEK 293T cells using sgRNAs targeting TXN1 (the gene encoding TRX1) (Fig. S3B). We then transiently transfected control (Cas9 expressing) or TRX1-deficient HEK 293T cells with constructs encoding NLRP1 and GFP-tagged ASC, treated cells with DMSO or VbP, and assessed speck formation by fluorescence microscopy (Fig. S3C,D). We found that TRX1-deficient cells had significantly higher numbers of basal and VbP-induced specks compared to control cells. These data indicated that TRX1 binding restrains the NLRP1 inflammasome.
We next sought to evaluate the impact of TRX1 protein depletion on NLRP1 inflammasome activation in more physiologically relevant systems. Notably, VbP induces NLRP1-dependent pyroptosis in human immortalized N/TERT-1 keratinocytes (14). We therefore electroporated CRISPR/Cas9-based ribonucleoprotein particles (RNPs) targeting TXN1 and known NLRP1 inflammasome genes into N/TERT-1 keratinocytes, treated these cells with DMSO or VbP, and monitored for signs of pyroptosis, including lactate dehydrogenase (LDH) release, IL-1β/−18 cleavage and release, and GSDMD cleavage (Fig. 3A-D, Fig. S3E-I). As expected, NLRP1−/−, CASP1−/−, PYCARD−/−, and GSDMD−/− N/TERT-1 keratinocytes were all resistant to VbP (Fig. S3G-I). In contrast, TRX1-deficient N/TERT-1 keratinocytes (we only achieved a transient ~80-90% reduction in TRX1 protein levels for 2-3 passages before expression was restored, likely due to the essentiality of TRX1) were significantly more sensitive to VbP, which induced more rapid and intense LDH and IL-1β/−18 release in these cells relative to controls (Fig. 3A-D). In addition, VbP induced significantly more ASC oligomerization and cytosolic accumulation of cleaved IL-1β in TRX1-deficient GSDMD−/− keratinocytes (GSDMD−/− cells were used to prevent cell lysis) (Fig. S3J). As expected, we found that TRX1-deficient NLRP1−/− keratinocytes were completely resistant to VbP (Fig. 3E,F), confirming that NLRP1 indeed mediated this increased pyroptosis. It should be noted TRX1-deficient cells did not undergo spontaneous pyroptosis, indicating that loss of TRX1 alone is not sufficient to activate the NLRP1 inflammasome. In addition, TRX1 protein loss did not substantially shift the half-maximal effective concentration (EC50) of VbP, but rather increased the intensity of pyroptosis that VbP induced at each dose (Fig. S3K). We speculate that the DPP8/9 ternary complexes effectively sequester the CT fragments released due to the loss the of TRX1 binding in the absence of VbP, and that a certain concentration of VbP is required to sufficiently destabilize these complexes. In contrast to TRX1 deficiency, we found that overexpression of WT TRX1, and not the C32S/C35S TRX1 mutant, slightly, but significantly, reduced VbP-induced IL-1β release (Fig. S3L-N). Overall, these data show that TRX1 binding restrains NLRP1 inflammasome activation in N/TERT-1 keratinocytes.
Fig. 3. Thioredoxin restrains NLRP1 activation.

(A-D) N/TERT-1 keratinocytes were electroporated with control or TXN1 RNPs before being treated with VbP (10 μM) for 6 h or 16 h. Supernatants were analyzed for levels of LDH (A), IL-18 (B), and IL-1β (C) release, and both lysates and supernatants were subjected to immunoblotting analyses (D). (E,F) WT and NLRP1−/− N/TERT-1 keratinocytes were electroporated with control or TXN1 RNPs and treated with VbP (10 μM, 8 h). Supernatants were assessed for IL-1β release (E), and lysates and supernatants were evaluated by immunoblotting (F). Data in A-C and E are means ± SEM of three biological replicates. (G) Lysates from BMDMs from the indicated mouse strains were evaluated by immunoblotting. (H,I) The indicated mice were treated with vehicle or VbP (1 mg/kg, i.p.). The levels or serum cytokines were evaluated after 6 h by ELISA (H) or Luminex (I). Data in H are means ± SEM. Data in I are the column sum normalization of each cytokine (all data in Fig. S4D) depicted using Morpheus Software (Broad Institute). n = 8 and 11 for vehicle-treated WT and Txn1−/− mice, respectively, and n = 10 and 12 for VbP-treated WT and Txn1−/− mice, respectively. *p < 0.05, **p < 0.01,***p < 0.001 by two-sided Student’s t-test.
VbP activates the NLRP1A and B inflammasomes and thereby elevates the levels of many serum cytokines in C57BL/6J mice (12, 15). As TRX1 associates with mNLRP1A/B (Fig. S1B), we next wanted to evaluate the impact of TRX1 protein loss on the induction of these cytokines. Txn1 knockout is embryonic lethal in mice (34), and we thus generated myeloid lineage-specific Txn1-null mice by crossing mice with a floxed Txn1 allele with mice harboring Cre recombinase expression under the myeloid lineage-specific Lyz2 promoter. We found that bone marrow-derived macrophages (BMDMs) from Txn1tm1.1Ees/J:B6.129P2-Lyz2tm1(cre)lfo/J homozygous mice (referred to hereafter as Txn1−/−) had a ~93% loss of TRX1 protein by immunoblot (Fig. 3G). These mice were overtly normal, having similar weights (Fig. S4A), peripheral blood cell counts (Fig. S4B), blood chemistry (Fig. S4C), and baseline serum cytokine levels (Fig. 3H,I, Fig. S4D) as control mice. Consistent with our data in cell culture, we found that VbP induced significantly higher levels of many cytokines, including G-CSF, IL-1β, and IL-18, in the serum of Txn1−/− mice (Fig. 3H,I, S4D). Thus, TRX1 restrains the mouse NLRP1 inflammasome in vivo. It should be noted that TRX1 loss did not impact the transcription of any pyroptotic genes, including NLRP1, GSDMD, CASP1, IL1B, or IL18, in either N/TERT-1 human keratinocytes or mouse BMDMs (Fig. S5A,B, Table S2, Data files S2,S3), consistent with the effect being due to loss of the direct protein-protein interaction and not a global rewiring of the inflammasome pathway.
Hyperactive NACHT-LRR mutations impair TRX1 binding
Patient-derived mutations in the PYD, NACHT-LRR, and FIIND domains of NLRP1 cause autoinflammatory disorders (Fig. 4A, top) (24-26, 35). Mutations in the PYD destabilize the domain’s structure (24), which likely increases NT degradation by protein quality control pathways (36, 37). The P1214R mutation in the FIIND abrogates DPP9 binding and thereby destabilizes the ternary complex checkpoint (6, 14). How the mutations in the NACHT-LRR cause inflammasome hyperactivation has not yet been determined. NACHT domains belong to the signal transduction ATPases with numerous domains (STAND) superfamily of AAA+ ATPases (38). These domains use two conserved sequences called Walker A and Walker B motifs to bind and hydrolyze ATP, respectively (39-41). Intriguingly, mutation of the Walker A site in NLRP3 inactivates the protein (39), but conversely mutation of the Walker A site in mouse NLRP1B generates a hyperactive protein (42, 43). The mechanistic basis for this striking difference is also not yet understood. We confirmed that ectopic expression of the patient-derived PYD (A54T, A66V, and M77T), NACHT-LRR (R726W, T755N, and ΔF787-R843), and FIIND (P1214R) mutations, as well as Walker A (K340R) and B (D413A and E414A) mutations, caused spontaneous ASC speck formation in the HEK 293T GFP-ASC assay (Fig. S6A-F). Moreover, we expressed a subset of these mutants (M77T, K340R, R726W, and P1214) in NLRP1−/− N/TERT-1 keratinocytes, and found that they induced spontaneous pyroptosis (Fig. 4B-D). In addition, we discovered that mutation of both Walker A and B sites together (K340R/D413R/E414A) caused more spontaneous pyroptosis than mutation of either site alone (Fig. 4B-D). Interestingly, we also observed that VbP still stimulated more inflammasome activation in cells expressing PYD and NACHT-LRR mutants, but not the P1214R mutant or the isolated NLRP1CT (Fig. 4B-D, Fig. S6E). These results indicate that DPP8/9 still represses at least some inflammasome formation caused by mutations in the NLRP1NT.
Fig. 4. Hyperactive NLRP1 NACHT-LRR mutations weaken TRX1 binding.

(A) Diagram of NLRP1 mutations. Patient-derived mutations are shown above the protein. ATPase- and FIIND-inactivating mutations are shown below the protein. (B-D) NLRP1−/− N/TERT-1 keratinocytes were complemented with the indicated NLRP1 mutant behind a tetracycline-inducible promotor. Cells were stimulated with doxycycline (1 μg/mL) in the presence or absence of VbP (10 μM, 24 h) before analysis of inflammasome activation by LDH release (B), IL-1β ELISA (C), or immunoblotting (D). (E,F) HEK 293T cells were transiently transfected with the indicated FLAG-tagged NLRP1 constructs before lysates were harvested, subjected to anti-FLAG IP, and immunoblotted. Data in B and C are means ± SEM of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by two-sided Student’s t-test. Asterisks (*) denote background bands.
We next wanted to evaluate the impact of these mutations on binding to TRX1 and DPP9. We found that the patient-derived (R726W, T755N, and ΔF787-R843), Walker A (K340R), and Walker B (E414A) mutations in the NACHT-LRR partially disrupted the NLRP1-TRX1 interaction (Fig. 4E, Fig. S6G). The Walker B D413A mutation even more severely interfered with this interaction, and mutation of both the Walker A and B sites (K340R/D413A/E414A) completely abrogated it altogether (Fig. 4F). Notably, these mutations did not impact binding to DPP9 (Fig. 4E). Interestingly, a Q593P mutation in the NACHT-LRR region of mouse NLRP1A causes neutrophilic inflammatory disease in mice (35). We found that expression of this mutant similarly causes spontaneous inflammasome activation in NLRP1−/− N/TERT-1 keratinocytes (Fig. S6H,I) and weakens binding to TRX1 (Fig. S6J). Conversely, we observed that the S1213A and P1214R mutations in the FIIND did not affect binding to TRX1, but impaired binding to DPP9 (Fig. 4E, Fig S6G). In contrast and as expected, neither patient-derived mutations in the PYD nor removal of the PYD entirely affected binding to either TRX1 or DPP9 (Fig. S6K). Overall, these data show that hyperactive NACHT-LRR and FIIND mutations have compromised binding to TRX1 and DPP9, respectively.
TRX1 binding stabilizes the NACHT-LRR region
As mentioned above, patient-derived PYD mutations are thought to compromise the folding of the domain, thereby accelerating NT degradation and leading to inflammasome activation (24, 36, 37). To confirm that these PYD mutations impact domain stability, we transiently expressed isolated WT, A66V, and M77T PYD domains in HEK 293T cells. We then harvested cell lysates, separated the soluble and insoluble fractions by centrifugation, and analyzed these fractions by immunoblotting (Fig. 5A). Consistent with compromised folding, we found that the mutant PYD domains pelleted into the insoluble fraction much more than the WT PYD. Moreover, a cellular thermal shift assay (CETSA) of these lysates revealed that A66V and M77T PYD were substantially less stable than the WT PYD (Fig. 5B). We hypothesized that the NACHT-LRR mutations might similarly affect domain stability and thereby lead to inflammasome formation. Indeed, we found that the isolated Walker A, Walker B, and patient-derived R726W and ΔF787-R843 mutant NACHT-LRR regions were less soluble (Fig. 5C) and thermally stable (Fig. 5D) than the WT region in similar fractionation and CETSA analyses, respectively.
Fig. 5. TRX1 stabilizes the NACHT-LRR region.
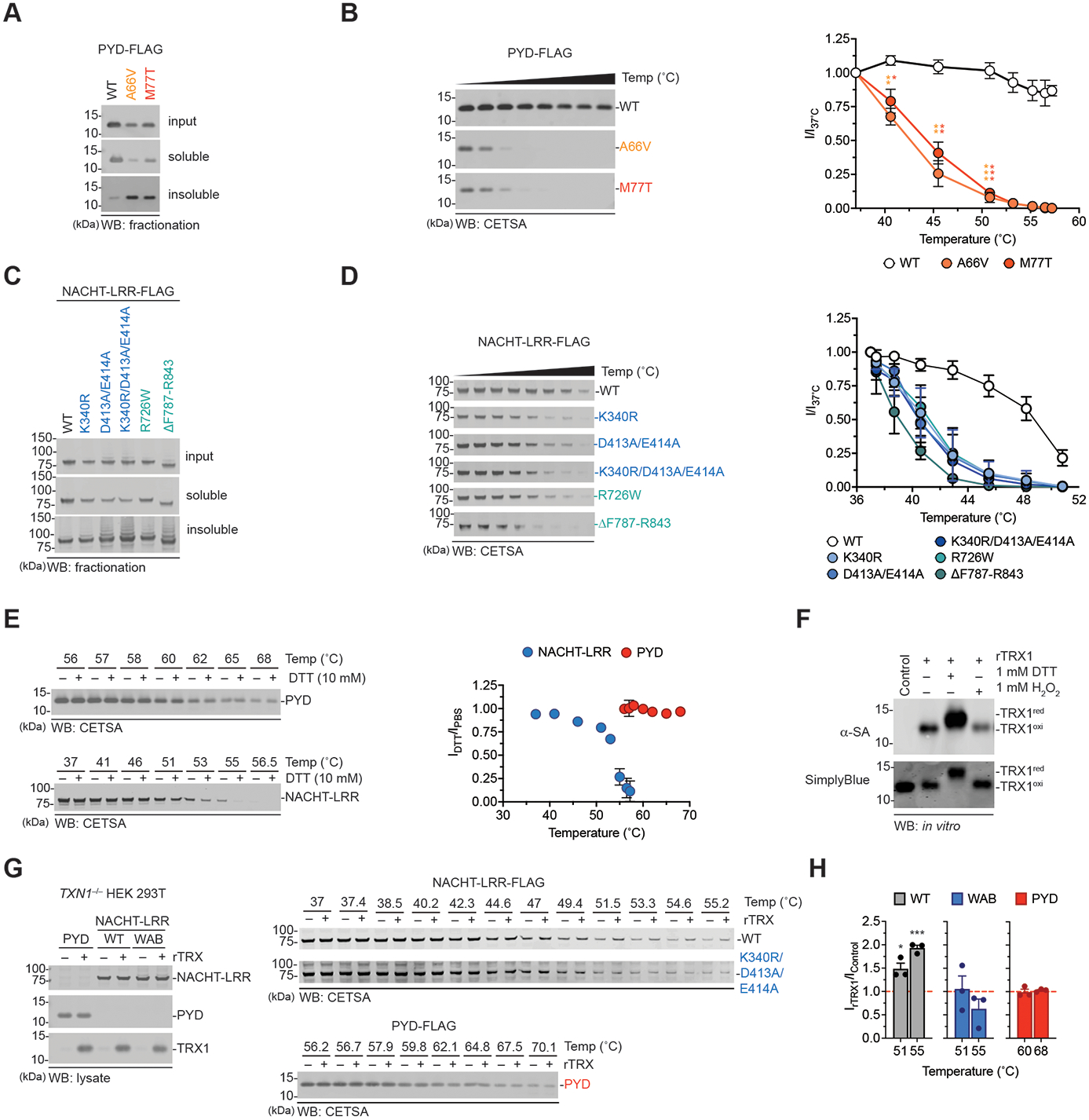
(A-E) HEK 293T cells were transfected with constructs encoding isolated FLAG-tagged WT or mutant PYD and NACHT-LRR domains as indicated. Lysates were harvested, fractionated by centrifugation (A,C) or subjected to CETSA analysis (B,D, and E) and probed by immunoblotting. In E, CETSA was performed in the presence or absence of 10 mM DTT. The graphs in B and D are the quantifications of immunoblot densitometry. The graph in E is the ratio of the immunoblot densitometry in the presence of DTT over PBS alone. (F) Recombinant TRX1 was incubated with DTT or H2O2 (1 mM) for 30 min at 37°C before the addition of iodoacetamide-biotin (20 mM, 2 h, 37°C) and immunoblotting analysis. (G) Constructs encoding isolated FLAG-tagged PYD, NACHT-LRR (WT or K340R/D413A/E414A) were transiently transfected into TXN1-deficient HEK 293T cells. Lysates were subjected to CETSA analysis in the presence or absence of recombinant oxidized TRX1 protein (1.58 μM). (H) Gel band intensities from G were standardized relative to lane 1 (no rTRX1, 37°C). The graph depicts the ratio of immunoblot density in the presence or absence of rTRX1 (IrTRX1/IControl) at 51°C and 55°C for the NACHT-LRRs and 60°C and 68°C for the PYD constructs (the hashed red line depicts no change in stabilization). Graphs in B, D, E, and H are means ± SEM of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001 by a two-sided Student’s t-test.
Based on these results, we reasoned that oxidized TRX1 binding might stabilize the fold of the NACHT-LRR region, thereby repressing NT degradation and inflammasome activation. To explore this premise, we next expressed the isolated WT PYD and NACHT-LRR domains in HEK 293T cells, harvested lysates, and performed CETSA analyses in the presence or absence of DTT (Fig. 5E). Consistent with our hypothesis, we found that DTT only compromised the stability of the NACHT-LRR region (Fig. 5E). We expressed the isolated WT PYD, WT NACHT-LRR, or Walker A/B triple mutant (K340R/D413A/E414A) NACHT-LRR domains in TXN1 deficient HEK 293T cells, harvested lysates, and performed analogous CETSA experiments in the presence or absence of recombinant oxidized TRX1 (rTRX1) (Fig. 5F-H). Intriguingly, we found that rTRX1 significantly stabilized, albeit only slightly, the WT NACHT-LRR region, but not the PYD or Walker A/B triple mutant NACHT-LRR region. Collectively, these data indicate that oxidized TRX1 specifically stabilizes NLRP1’s NACHT-LRR region.
ATP hydrolysis is required to form the inactive NLRP1-TRX1 complex
Our results with the Walker A and B motif mutations strongly indicated that ATP binding and hydrolysis is required for NLRP1 to adopt an inactive, TRX1-bound state. If correct, we reasoned that incubating the NLRP1-TRX1 complex on beads in the absence of nucleotides would eventually cause ADP to dissociate and the complex to separate. To explore this idea, we expressed a FLAG-tagged isolated NACHT-LRR region in HEK 293T cells, immobilized the NACHT-LRR-TRX1 complex on anti-FLAG beads, washed the beads once with PBS, and incubated these beads with shaking at various temperatures. Although the complex was stable at room temperature, we found that TRX1 was eluted from the beads after several hours at 37°C (Fig. 6A). We next co-incubated the immobilized NLRP1-TRX1 complexes with 1 mM of ATP, ADP, or the non-hydrolyzable ATP analog adenosine 5’-[γ-thio]triphosphate (ATPγS) at 37°C (Fig. 6B). All three nucleotides, and most significantly ATP, substantially slowed TRX1 dissociation. We found that high (≥ 100 μM), non-physiologically-relevant concentrations of ADP were needed to stabilize the interaction, but that only 10 μM ATP or ATPγS was sufficient (Fig. 6C). Unlike ATPγS, we found that the non-hydrolyzable ATP analogs adenylyl-imidodiphosphate (AMP-PNP), β, γ-methyleneadenosine 5'-triphosphate (AMP-PCP), α,β-methyleneadenosine 5’-triphosphate (AMP-CPP) did not prevent TRX1 elution (Fig. 6D). As expected, AMP, adenosine 3’,5’-cyclic monophosphate (cAMP), GTP, GDP, and CTP did not affect the interaction (Fig. 6E).
Fig. 6. ATP binding and hydrolysis is coupled to TRX1 binding.
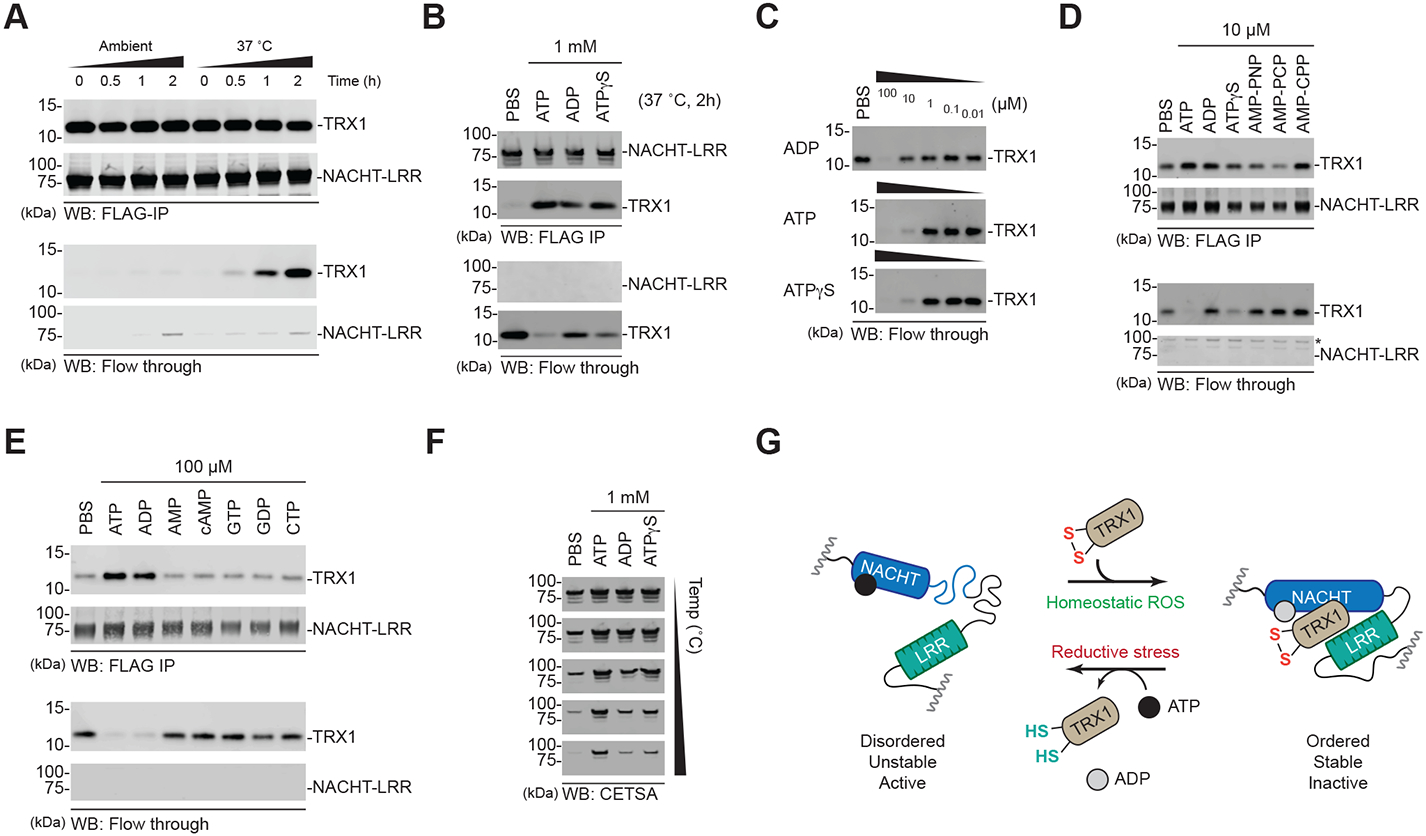
(A-E) HEK 293T cells were transfected with constructs encoding the isolated FLAG-tagged NLRP1 NACHT-LRR region. Protein was immobilized on anti-FLAG beads, washed with PBS, and incubated for the indicated times under the specified conditions. Dissociated (flow through) and complexed (FLAG-IP) TRX1 levels were assessed by immunoblotting. (F) Eluates from B were subjected to CETSA analysis. (G) Proposed model for the nucleotide-dependent states of NLRP1’s NACHT-LRR region.
We next assessed the stability of the complexes eluted from beads after 2 h shaking with the various nucleotides at 37ΔC (Fig. 6B, E). CETSA analysis revealed that complexes incubated with ATP were more stable than those incubated with either ADP or ATPγS (Fig. 6F). Notably, the relative stabilities of these complexes perfectly corresponded to the amount of TRX1 still associated with the NACHT-LRR region (Fig. 6B). Together, these data indicate that, during normal homeostasis, ATP-bound NACHT-LRR domains associate with oxidized TRX1, and that this interaction at least partially stabilizes the region. ATP hydrolysis into ADP then induces the formation of a stable, “inactive” conformer (Fig. 6G). ADP and/or TRX1 will eventually dissociate, thereby regenerating the ATP-bound, TRX1-free protein. During reductive stress, TRX1 does not interact with the NACHT-LRR domains, leaving it in the disordered, “active” conformation.
Common antioxidants do not potentiate inflammasome activation
Although only ~5-10% of TRX1 is oxidized in the cytosol of unstressed human cells (23), TRX1 is among the most abundant proteins in mammalian cells and therefore a sufficient level of oxidized TRX1 should normally exist to repress the much less abundant NLRP1 protein (44). Indeed, our genetic knockout and mutation data above shows that TRX1 restrains the NLRP1 inflammasome in cells. We reasoned that lowering the intracellular redox potential (i.e., inducing reductive stress) should decrease the fraction of oxidized TRX1, dissociate TRX1 from NLRP1, destabilize the NACHT-LRR region, and potentiate inflammasome activation. Unfortunately, however, we are not aware of any published agent that lowers the intracellular redox potential and decreases the fraction of oxidized TRX1. Regardless, we next wanted to assess the impact of a panel of oxidants and antioxidants, including DTT, N-acetyl-cysteine (NAC), H2O2, and antimycin, on NLRP1 inflammasome activation (Fig. S7A). Although several of these agents were toxic on their own at high doses, none impacted basal- or VbP-induced pyroptosis in human NLRP1-dependent N/TERT-1 keratinocytes (Fig. S7B,C), mouse NLRP1B-dependent RAW 264.7 cells (Fig. S7D-F), or human CARD8-dependent MV4;11 cells (Fig. S7G). Consistent with our results, DTT did not substantially reduce a fluorescent biosensor for TRX1 redox (TrxRFP1) in HEK 293T cells below its basal oxidation state (45). Thus, our data strongly indicate reductive stress will potentiate NLRP1 activation, but, as described below, additional research is needed to identify and characterize reagents that induce reductive stress and alter the TRX1 redox couple in cells.
DISCUSSION
Here, we have established that oxidized, but not reduced, TRX1 directly binds to the NACHT-LRR domains of NLRP1 and represses inflammasome activation. Although TRX1 transiently interacts with many intracellular proteins in the process of reducing disulfide bonds, it is only known to stably interact with a handful of proteins, including apoptosis signal-regulating kinase 1 (ASK1) and thioredoxin interacting protein (TXNIP) (46, 47). Interestingly, the reduced form of TRX1 binds to ASK1 and TXNIP, but the oxidized form binds to NLRP1. Based on this data, we now propose a model that reductive stress, or a profound lack of ROS, eliminates the oxidized form of TRX1, causing it to dissociate from NLRP1 and thereby destabilizing the folding of the NACHT-LRR region (Fig. 7). We speculate that the core 20S proteasome, which degrades disordered and misfolded proteins independent of ubiquitin conjugation (48, 49), then rapidly destroys the TRX1-free, disordered NLRP1NT and liberates the NLRP1CT from autoinhibition. Consistent with this idea, we recently reported that the 20S proteasome controls the activation of the related CARD8 inflammasome independent of ubiquitination (50). The details of the NLRP1 degradation process, and whether it involves ubiquitination, requires further investigation.
Fig. 7. Proposed mechanism for NLRP1 activation.
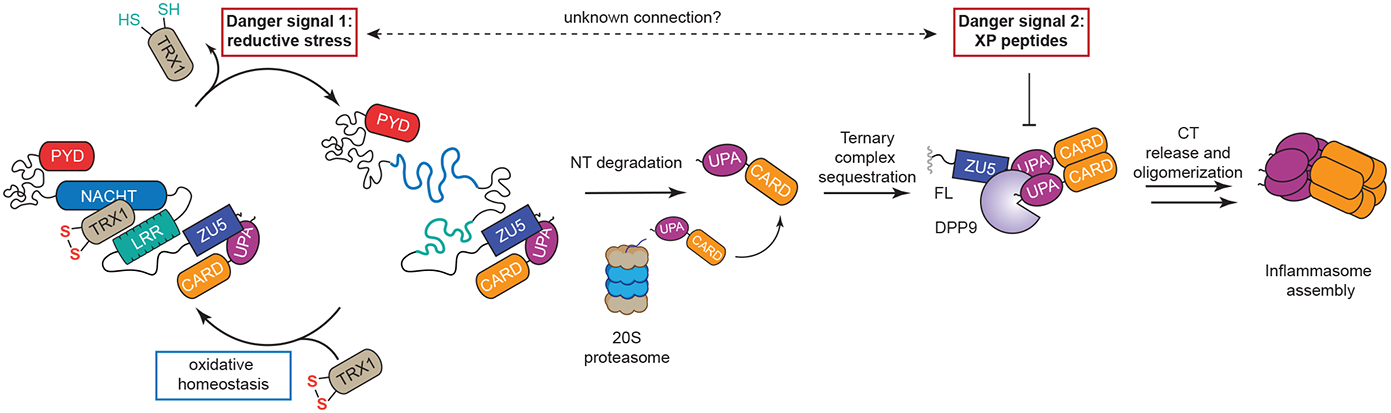
During homeostasis, oxidized TRX1 is complexed with NLRP1, stabilizing it in an inactive conformation. In the presence of reductive stress (“Danger Signal 1”), the levels of oxidized TRX1 are lowered, causing it to dissociate from NLRP1. The TRX1-free form of NLRP1 is more unstable, leading to accelerated degradation by the proteasome. The CT fragments released by this mechanism are initially sequestered in a ternary complex with DPP9 and NLRP1FL. The accumulation of XP-containing peptides (“Danger Signal 2”) disrupts this ternary complex and cause inflammasome activation. It remains unclear if these two danger signals are in some way related.
We also discovered that the ATPase activity of the NACHT domain is required to form the repressive complex with TRX1. Briefly, we found that NLRP1 likely occupies either an ATP-bound, TRX1-free active state or an ADP-bound, TRX1-bound inactive state (Fig. 6G), and that ATPase-inactivating mutations block the formation of the ADP-bound inactive state and thereby cause spontaneous inflammasome assembly (42, 43). Intriguingly, we found that patient-derived mutations in the NACHT-LRR domains, like ATPase-inactivating mutations, interfere with TRX1 binding and cause inflammasome hyperactivation. How these patient-derived mutations abrogate TRX1 binding remains unclear. Notably, these mutations occur near the boundary between the NACHT and LRR domains (Fig. S8), a region known to act as a hinge between the ATP and ADP bound conformational states in the analogous NACHT-LRR regions of NLRP3, NAIP, and NLRC4 (29, 51). As such, we speculate that the patient-derived mutations in the NLRP1 NACHT-LRR restrict the ability of this region to adopt the ADP-bound inactive form. However, it is alternatively possible that these mutations directly abrogate TRX1 binding or destabilize the intrinsic ability of the NACHT-LRR to fold. Future structural studies will be required to distinguish between these plausible mechanisms.
Regardless of these mechanistic details, our work now reveals that reductive stress is likely an NLRP1 inflammasome-activating danger signal. Unfortunately, however, no agents or perturbations, including commonly used antioxidants, are known to induce reductive stress. Thus, additional research is needed to identify and characterize reductive stress inducers. We recently posted a preprint that strongly suggests certain ferroptosis-inhibiting small molecules, including JSH-23, appear to induce reductive stress and potentiate both NLRP1 and CARD8 activation (22). However, it remains unknown precisely how these molecules induce reductive stress, and what, if any, natural events (e.g., pathogen infection or metabolic dysfunction) lead to this perturbed cell state. Lastly, CARD8 lacks the NACHT-LRR region and does not bind TRX1, but also appears to sense reductive stress (22). Thus, it will be important to determine how CARD8 monitors the intracellular redox environment.
In summary, we have now identified that NLRP1 detects at least two key danger signals: reductive stress and XP-containing peptides (Fig. 7). It remains unclear if these signals are in some way related or are entirely distinct. Interestingly, genetic loss of TRX1 does not cause inflammasome activation, likely because the DPP8/9 ternary complex restrains the CT fragments freed by this mechanism. We speculate that reductive stress (as opposed to genetic TRX1 loss) may in some way also cause DPP8/9 inhibition, and that the ternary complex acts as a “checkpoint” to confirm that reductive stress is indeed occurring. Alternatively, DPP8/9 inhibition might be a consequence of a distinct perturbation, and that reductive stress and DPP8/9 inhibition act as “AND” gates for inflammasome activation. Future studies are needed to investigate the relationship between these two key danger signals. Nevertheless, our study here now reveals a new fundamental mechanism that links the intracellular redox environment to the activation of the innate immune system.
Materials and Methods
Study design
The aim of this study was to identify cellular factors that control the activation of the NLRP1 inflammasome. We used an immunoprecipitation-mass spectrometry assay to identify that TRX1 binds to NLRP1. We then used a variety of genetic perturbations to show that TRX1 binding restrains inflammasome activation. In addition, we used several biochemical approaches to show that only the oxidized form of TRX1 binds to NLRP1, and that TRX1 binding stabilizes the folding of the N-terminal region of NLRP1.
Plasmids, antibodies, and reagents
NLRP1 and CARD8 transcript variant 1 DNA obtained commercially from Origene (pCMV6-entry clones RC216481 and RC230245, respectively). NLRP3, TXN1, Nlrp1a, and Nlrp1b2 DNA was purchased from Genscript (OHu26815D, OHu15866D, OMu19634, and OMu00866D, respectively). DNA encoding Nlrp1b1 was cloned directly from RAW 264.7 macrophages. DNA encoding the mouse Nlrp1b3 and Nlrp1b5 were obtained from R. Vance and J. Mogridge. DNA encoding the rat Nlrp1 alleles 1-5 were cloned from Sprague Dawley, CDF, Zucker, Copenhagen, and Lewis rodents, respectively. Expression constructs were first amplified by polymerase chain reaction (PCR) and subcloned into pDONR221, then shuttled into modified pLEX307 vectors originating from pLEX307 (Addgene #41392) using Gateway cloning technology (ThermoFisher Scientific). For doxycycline induction experiments, constructs originating in pDONR221 vectors were shuttled into the pInducer20 vector (Addgene #44012), also using Gateway cloning. TXN1 CRISPR sgRNAs were cloned into the pLentiGuide expression system (Addgene #52963) for HEK 293T gene ablation. Viral transductions were performed with the psPAX2 lentiviral packaging vector (Addgene #11260) and VSV-G envelope expression plasmid, pMD2.G (Addgene #12259). Unless otherwise indicated, all plasmid transfections were performed with FuGene HD (Promega) following manufacturer’s recommendations. For sgRNA:Cas9 Ribonucleoprotein (RNP) complex gene modifications, the Alt-R CRISPR-Cas9 system was used (Integrated DNA Technologies, IDT). Pre-designed crRNA for NLRP1, CASP1, PYCARD, GSDMD, and TXN1 and with tracrRNA and Alt-R S.p. Cas9 Nuclease V3 recombinant protein (IDT) was delivered to cells using the Neon transfection system (ThermoFisher Scientific). Where necessary, knockout cell lines were prepared following successive rounds of electroporation until protein expression was no longer observed by Western blot. Point mutations were prepared using QuikChange II site-directed mutagenesis (Agilent), following manufacturer’s suggested protocols.
Antibodies used in this study were: anti-FLAG antibody (F1804, Millipore SiGMa), anti-NEK7 (3057S, Cell Signaling Technology), anti-V5 (V8012, Sigma-Aldrich), anti-HA (2367S, Cell Signaling Technology), anti-TRX1 (ab86255, Abcam; 2429S, Cell Signaling Technology), anti-DPP9 (ab42080, Abcam), anti-GAPDH (2118S, Cell Signaling Technology), anti-NLRP1 (AF6788, R&D systems), anti-CARD8 (ab194585, Abcam), anti-ASC (AF3806, R&D systems), anti-CASP1 (2225S, Cell Signaling Technology; ab179515, Abcam), anti-GSDMD (NBP2-33422, Novus), anti-mGSDMD (ab209845, Abcam) anti-IL-1β (AF201NA, R&D systems), anti-IL-18 (AF2548, R&D systems), IRDye 800CQ streptavidin (926-32230, LI-COR).
Experiments involving Poly(I:C) were performed using the long synthetic analog of dsRNA, Poly(I:C) HMW (InvivoGen). Disuccinimidyl suberate (DSS), Iodoacetyl-PEG2-Biotin, Adenosine-5’-triphsophate (ATP), and Adenosine-5’-diphosphate (ADP) were purchased from ThermoFisher Scientific. VX-765 was obtained from APExBio; Guanosine 5’-triphosphate, Guanosine 5’-diphosphate, Cytidine 5’-triphosphate, α,β-Methyleneadenosine 5’-triphosphate (AMP-CPP), β,γ,-Methyleneadenosine 5’-triphosphate (AMP-PCP), Antimycin A from Streptomyces sp., N-acetyl cysteine (NAC), 1,4-dithiothreitol, hydrogen peroxide, Diamide, MitoTEMPO, and Adenosine 5’-[γ-thio]thiotriphosphate (ATPγS) were purchased from EMD Millipore. Adenylyl-imidodiphosphate (AMP-PNP) was purchased from ROCHE. Tert-butyl hydrogen peroxide was obtained from ACROS Organics; Oxidized and reduced L-Glutathione, and Adenosine-5’-monophosphate (AMP) were obtained from Alfa Aesar; Adenosine 3’,5’-cyclic monophosphate (cAMP) was purchased from TCI. Val-boroPro, Doxycycline, and Vitamin K3 were obtained from Cayman Chemical Company; HOECHST 33342, trihydrochloride, trihydrate was obtained from Invitrogen. Recombinant TRX1 was purchased from SiGMa (T0910).
Cell culture
HEK293T and RAW264.7 were purchased from ATCC and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with L-glutamine and supplemented with 10% fetal bovine serum (FBS) and 1X penicillin/streptomycin (100 U/mL penicillin and 100 μg/mL streptomycin, Gibco). MV4;11 cells were purchased from the Liebnez Institute DSMZ’s German collection of microorganisms and cell cultures GmbH and cultured in Roswell Park Memorial Institute (RPMI) medium 1640 with L-glutamine supplemented with 10% FBS and 1X penicillin/streptomycin. N/TERT-1 cells, a gift from James Rheinwald (52), were cultured in Keratinocyte serum free medium (KSFM, Gibco) supplemented with 1X penicillin /streptomycin (Gibco), bovine pituitary extract (20 μg/ml, Gibco), and recombinant epidermal growth factor (EGF, 2 ng/ml, Gibco).
All cells were grown at 37°C in a 5% CO2 atmosphere incubator. Cell lines were tested for mycoplasma using the MycoAlert™ Mycoplasma Detection Kit (Lonza).
Bone marrow derived macrophage preparation
Mouse femurs were resected from mice euthanized by CO2 asphyxiation followed by cervical dislocation. Whole femur samples were mechanically cleaned of soft tissue before grinding with a ceramic mortar and pestle in the presence of PBS supplemented with 2.5% FBS. Bone and tissue debris were removed by filtration through a 70 μm cell strainer and the cellular fraction of the filtrate condensed by centrifugation. The resulting pellet was resuspended in 1X RBC lysis buffer (BioLegend) followed by centrifugation to reform a cell pellet. The final cell population was then taken up in DMEM supplemented with 15% L929 cell medium, transferred to untreated tissue culture plates, and left for 6-8 days to differentiate before collecting macrophages and plating for the indicated experiments using Opti-MEM (Gibco).
Genetically modified cell lines
TXN1-deficient HEK 293T cells were prepared by transient transfection of HEK 293T cells stably expressing Cas9 with sgRNA for TXN1 packaged in lentiGuide-Puro (Addgene #52963) using the following oligo sequences (5’-ACGTGATATTCCTTGAAGTA-3’; 5’-TAGTTGACTTCTCAGCCACG-3’, 5’-GGTGAAGCAGATCGAGAGCA-3’). 48 h following transfection, cells were selected with 1 μg/mL puromycin for 5 days and subjected to single cell cloning protocols to obtain a population enriched for the gene modification.
CASP1−/−, NLRP1−/−, PYCARD−/−, and GSDMD−/− N/TERT-1 keratinocyte cells were prepared by delivery of sgRNA:Cas9 ribonucleoprotein complexes containing an Alt-R CRISPR-Cas9 sgRNA and recombinant Cas9 (IDT) using the Neon Transfection System (ThermoFisher Scientific) following the manufacturer’s recommendations with modifications to optimize for N/TERT-1 cells. Briefly, sgRNA complexes were prepared by annealing predesigned Alt-R CRISPR-Cas9 crRNAs (see Table S3) with Alt-R CRISPR-Cas9 tracrRNA to 44 μM and annealing by heating to 95°C for 5 min followed by gradual cooling to ambient temperature over 30 min. To form the RNP complexes, sgRNA samples were combined with recombinant Alt-R S.p. Cas9 Nuclease V3 enzyme and incubated for 20 min at ambient temperature. RNP complexes were then introduced to 5x105 wild-type N/TERT-1 keratinocyte cells with Alt-R Cas9 Electroporation Enhancer (IDT) and electroporated with 3 cycles of 1700 V using a pulse width of 20 ms. Cells were then transferred to warm supplemented KSFM without antibiotics to recover for 72 h, expanded, and evaluated by immunoblot for protein expression levels. Where protein was detected by immunoblot, cells were subjected to further rounds of RNP electroporation and revaluated until protein expression could no longer be detected.
As stable TXN1−/− N/TERT-1 cells could not be generated using this, or other methods, to prepare TRX1-deficient N/TERT-1 keratinocytes for experiments, 5x105 cells were electroporated using the same protocol as above, however, RNP complexes contained sgRNAs for both TXN1-AB and TXN1-AD CRISPR-Cas9 crRNA sequences (in our hands, TXN1-AC did not result in suppression of TRX1 protein levels by immunoblot analysis) and cells were electroporated as individual replicates prior to each experiment.
Western Blotting
Cell pellets harvested from tissue culture plates were resuspended in 1X PBS and unless otherwise indicated, mechanically lysed by sonication before protein standardization by DC assay (BIO-RAD). Standardized lysates were then mixed with an equal volume of Western blot sample buffer (2X protein loading buffer (LI-COR) and 100 mM DTT or 2X NuPAGE sample reducing agent (ThermoFisher Scientific) in molecular biology grade water (Corning)) prior to heat-denaturation at 97°C for 10 min on a heating block.
For supernatants samples, protein pellets were collected by precipitation with cold acetone and overnight chilling (−20°C) followed by centrifugation at 3000 g for 30 min at 4°C and decanting. Dried pellets were then suspended in 1X PBS and combined 1:1 with 2X sample loading buffer before heating to 97°C for 10 min.
All protein samples were separated by SDS-PAGE, transferred to nitrocellulose membranes using the Trans-Blot Turbo transfer system (BIO-RAD) before blocking and incubating with 1° antibodies for the indicated protein overnight at 4°C with shaking. The following day, membrane samples were washed and incubated with fluorophore-tagged 2° antibodies for 1 h at ambient temperature with shaking, washed again, and imaged on a LI-COR Odyssey imager. Image files were saved as .TIFF format and image handling was performed in Photoshop and Illustrator software (Adobe Creative Suite).
FLAG Immunoprecipitations
Cells were transfected with the given FLAG-tagged construct as defined in the cell culture section of methods. Collected cell pellet samples were lysed by sonication and clarified by centrifugation at 1000 g for 10 min at 4°C. Aliquots from the clarified lysate samples were combined with ANTI-FLAG M2 affinity beads (Millipore SiGMa) in Pierce Micro-Spin columns (ThermoFisher Scientific) and the remaining lysate was used to prepare input samples for Western analysis. The Micro-Spin samples were incubated for 2 h at ambient temperature or 4°C overnight, eluted to obtain flow through samples, and washed with PBS before the immunoprecipitated proteins were obtained by 3X-FLAG peptide incubation and sample preparation for Western blot analysis.
TMT labeling for IP-MS/MS analysis
3x106 HEK 293T cells were seeded in a 10 cm tissue culture dish and transfected the following day with an expression plasmid for the given FLAG-tagged protein construct. The cells were harvested 48 h post transfection and lysed in 1 mL of 0.5% NP-40 in PBS on ice for 30 min. Lysate samples were placed in a tabletop centrifuge for rotation at 2000 g for 10 min at 4°C. The soluble fraction was separated, and protein content normalized (DCA protein assay, BIO-RAD). Protein samples were globally reduced by application of 10 mM TCEP (tris(2-carboxyethyl) phosphine) for 1h at 56°C with shaking, followed by alkylation of free thiols with 20 mM iodoacetamide for 30 min at ambient temperature, protected from light. Protein samples were then precipitated from solution by adding −20°C acetone to six sample volumes and incubated overnight at −20°C. Precipitated protein was isolated by sample rotation at 8000 g for 10 min at 4°C and decanting the liquid phase. The pellet samples were resuspended in in 100 μL 50 mM triethylammonium bicarbonate (TEAB) followed by total protein quantification by DCA assay prior to digestion with 2.5 mg trypsin per 100 mg sample at 37°C overnight.
TMTsixplex sample reagents (ThermoFisher Scientific) were equilibrated to ambient temperature and 0.8 mg of label (126, 127 = GFP-FLAG; 128, 129 = NLRP1-FLAG; 130, 131 = unused sample) diluted into 41 μL pure acetonitrile before use. 41 μL of each TMT label reagent were added to each protein sample and incubated at ambient temperature for 1 h. 8 μL of a 5% hydroxylamine solution was added to quench each sample and incubated at ambient temperature for 15 min. Samples were then equally combined and purified by a High pH Reversed-Phase Peptide Fractionation Kit (Pierce) and dried with a Genevac EZ-2 evaporator to yield peptide pellets submitted for tandem LC-MS/MS/MS analysis.
TMT LC-MS/MS/MS
Mass spectrometry data was collected on an Orbitrap Fusion Lumos mass spectrometer coupled to an Easy-nLC 1200 (ThermoFisher Scientific). Peptides were separated over a 200 min gradient of 0-50% acetonitrile in water with 0.1 % formic acid at a flow of 300 nL/min on a 50 cm PepMap RSLC C18 column (2 mm, 100 Å, 75 μm x 50 cm). The full MS spectrum was acquired in the Orbitrap at a resolution of 120,000. The 10 most intense MS1 ions were selected for MS2 analysis. The isolation width was set at 0.7 m/z and isolated precursors were fragmented by CID (35% CE). Following acquisition of each MS2 spectrum, a synchronous precursor selection (SPS) MS3 scan was collected on the top 10 most intense ions in the MS2 spectrum. The isolation width was set at 1.2 m/z and isolated precursors were garmented using HCD.
Proteomics analysis
MS raw files were converted to MGF and processed using Proteome Discoverer version 2.2.0.388 (Thermo Scientific) by searching against the Uniprot human database supplemented with common contaminant protein sequences and quantified according to SPS MS3 reporter ions. Identification was based on CID spectra using SequestHT. Search criteria included: 20 ppm mass tolerance for MS spectra, 0.5 Da fragment mass tolerance for MS/MS spectra, a maximum of two allowed missed cleavages, static carbamidomethylation (+57.021 Da) of cysteine and TMTsixplex (+229.163 Da) of lysine and of peptide N-terminus, dynamic modifications of methionine oxidation (+15.995 Da), N-terminal protein acetylation (+42.011 Da), asparagine or glutamine deamidation (+0.984 Da), and serine, tyrosine, or tryptophan phosphorylation (+79.966 Da) and a false discovery rate of < 0.01.
DSS cross-linking
GSDMD−/− N/TERT-1 keratinocytes were given either control or TXN1 RNPs as described in the methods above and left to recover for 72 h before the indicated treatment conditions. Cells were harvested and lysed with 0.5% NP-40 in PBS for 30 min on ice. The lysates were clarified by centrifugation at 1,000 g at 4°C for 10 min, reserving clarified lysate for input Western Blot samples. The remaining lysates were fractionated at 20,000 g at 4°C for 20 min, the soluble portion removed, and the insoluble fraction resuspended with CHAPS buffer (50 mM HEPES pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, and 0.1% wt/vol CHAPS) before the addition of 2 mM disuccinimidyl suberate (DSS). The samples were incubated at 37°C with shaking on an orbital platform for 45 min before the samples were prepared for immunoblot analysis.
LDH cell death assay
Supernatant samples were collected directly from experiments and analyzed by CyQUANT LDH cytotoxicity assay (ThermoFisher Scientific) following the manufacturer’s recommendations in black 384-well microplates. The samples were analyzed on a Cytation 5 multimodal plate reader (BioTek) and exported to spreadsheet software for statistical analysis. The data was visualized using GraphPad PRISM 9.3.1. Unless otherwise indicated, data represent the mean value ± SEM for 3 biological replicates.
ELISA assays
Supernatant samples were collected and pre-diluted 4- to 20-fold before quantification of cytokines by ELISA assays targeting IL-1β (R&D systems) or IL-18 (Abcam) following the manufacturer’s recommendations. For mouse serum G-CSF analysis, serum samples isolated from blood were diluted by 10- to 20-fold before measurement by ELISA (R&D systems), following the manufacture’s recommendations. The samples were analyzed on a Cytation 5 multimodal plate reader (BioTek) or an Infinite M1000 Pro plate reader (Tecan) and the data exported to spreadsheet software for statistical analysis. Data visualization was performed on GraphPad PRISM 9.3.1. Unless otherwise indicated, data represent the mean value ± SEM for at least 3 biological replicates.
Microscopy
0.75-2x105 HEK 293T cells were seeded per well in 12-well tissue culture plates (Corning) in the appropriate media prior to transfection of the given plasmids for 24-48 h. Unless otherwise indicated, cells were stimulated with VbP (10 μM) for 6 h. Cell nuclei were stained with Hoechst 33342 dye (0.8 μM) prior to imaging on a Zeiss Axio Observer.Z1 inverted wide-field microscope using 40×/0.95NA air objective. For each replicate well, 8-12 field positions were imaged for bright-field, DAPI, TRITC, and FITC channels and the raw data saved to .czi format for downstream analysis in ImageJ/FIJI using custom macros to calculate the total number of GFP-ASC specks and individual cell nuclei before data visualization in GraphPad PRISM. ASC foci quantitation was calculated as the ratio of the number of GFP-ASC foci over the total number of nuclei within the field and expressed as a percentage. Data represent the mean value from the average of all field positions imaged within the biological replicate. Representative images from each condition were produced within ImageJ/FIJI.
Mouse cytokine stimulation studies
8–10-week-old, age-matched wild-type C57BL/6J (The Jackson Laboratory) or Txn1−/− mice were treated with either vehicle (PBS) or 1 mg/kg VbP via intraperitoneal injection for 6 h. Whole blood samples were collected in Microtainer tubes containing a serum separator (BD) via cardiac puncture following euthanasia by CO2 asphyxiation and centrifuged to obtain serum samples. This animal protocol was reviewed and approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee (IACUC). No animals were excluded from analysis. The experiments were not randomized, and the investigators were not blinded. G-CSF levels were determined by ELISA (R&D Systems) following the manufacturer’s recommendations. For Luminex cytokine panel profiling, serum samples were pre-diluted 5-fold and analyzed in two technical replicates using the 36-Plex Mouse ProcartaPlex™ Panel 1A (ThermoFisher Scientific) following the manufacturer’s recommendations. Data were acquired on a Luminex FLEXMAP 3D system. Undetected analytes were assigned a value of zero. Analytes measured below the limit of detection were estimated as the lowest value within the given standards available.
Necropsy and histopathology
Mice were euthanized with CO2. Following gross examination of all organs were fixed in 10% neutral buffered formalin, followed by decalcification of bone in a formic acid solution (Surgipath Decalcifier I, Leica Biosystems). Tissues were then processed in ethanol and xylene and embedded in paraffin in a Leica ASP6025 tissue processor. Paraffin blocks were sectioned at 5 μm, stained with hematoxylin and eosin (H&E), and examined by a board-certified veterinary pathologist. The following tissues were processed and examined: heart, thymus, lungs, liver, gallbladder, kidneys, pancreas, stomach, duodenum, jejunum, ileum, cecum, colon, lymph nodes (submandibular, mesenteric), salivary glands, skin (trunk and head), urinary bladder, uterus, cervix, vagina, ovaries, oviducts, adrenal glands, spleen, thyroid gland, esophagus, trachea, spinal cord, vertebrae, sternum, femur, tibia, stifle join, skeletal muscle, nerves, skull, nasal cavity, oral cavity, teeth, ears, eyes, pituitary gland, brain.
Hematology
For hematology, blood was collected into tubes containing EDTA. Automated analysis was performed on an IDEXX Procyte DX hematology analyzer and the following parameters were determined: white blood cell count, red blood cell count, hemoglobin concentration, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, red blood cell distribution width standard deviation and coefficient of variance, reticulocyte relative and absolute counts, platelet count, platelet distribution width, mean platelet volume, and relative and absolute counts of neutrophils, lymphocytes, monocytes, eosinophils, and basophils.
Serum chemistry
For serum chemistry, blood was collected into tubes containing a serum separator, the tubes were centrifuged, and the serum was obtained for analysis. Serum chemistry was performed on a Beckman Coulter AU680 analyzer, and the concentration of the following analytes was determined: alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, creatine kinase, gamma-glutamyl transpeptidase, albumin, total protein, globulin, total bilirubin, blood urea nitrogen, creatinine, cholesterol, triglycerides, glucose, calcium, phosphorus, chloride, potassium, and sodium. Na/K ratio, albumin/globulin ratio were also calculated.
RNASeq gene expression analysis
Following culture conditions reported above, BMDMS from WT or Txn1−/− C57BL/6J mice and WT or TXN1 RNP treated N/TERT-1 keratinocytes were harvested by and purified for total RNA content by RNeasy (Qiagen) following the manufacturer’s recommendations. RNA samples were analyzed by Nanodrop (ThermoFisher Scientific) for quality before RNASeq quality control, library preparation, and sequencing, performed (GENEWIZ, LLC./AZENTA US, Inc).
Library preparation, polyA selection, and sequencing were performed as follows. Briefly, RNA samples were quantified using a Qubit 2.0 fluorometer (ThermoFisher Scientific) and RNA integrity assessed with a 4200 TapeStation (Agilent Technologies). Samples were then normalized using the ERCC RNA Spike-In Mix kit (ThermoFisher Scientific) following the manufacturer’s recommendations. The RNA sequencing library was prepared using the NEBNext Ultra II RNA Library Prep Kit for Illumina (New England Biolabs) following the manufacturer’s recommendations. Sequencing libraries were multiplexed and clustered onto a flowcell and loaded onto the Illumina HiSeq 4000 instrument according to manufacturer’s instructions and sequenced using a 2x150 bp Paired End configuration. Image analysis and base calling were conducted by the Illumina Control Software. Raw sequencing data (.bcl files) were converted to fastq and de-multiplexed using Illumina bcl2fastq 2.20 software. One mismatch was allowed for index sequence identification.
For data analysis, sequence reads were trimmed using Trimmomatic v.0.36 and mapped to the Mus musculus GRCm38 or Homo sapiens GRCh38 reference genomes (ENSEMBL) using the STAR aligner v.2.5.2b. Unique gene hit counts were summarized and reported using the gene_id feature in the annotation file. Only unique reads within exon regions were counted. Gene hit count tables were used for downstream differential expression analysis. DESeq2 was used for comparative gene expression analysis between sample groups. The Wald test was used to generate p-values and log2FC. Genes with an adjusted p-value < 0.05 and absolute log2FC > 1 were defined as differentially expressed genes for each comparison. Gene ontology analysis was performed on the statistically significant set of genes using GeneSCF v.1.1-p2 software.
TRX1 complex dissociation studies
HEK 293T cells were transiently transfected with the given FLAG-tagged NLRP1 NACHT-LRR construct before lysate samples were immunoprecipitated onto anti-FLAG M2 affinity gel beads (Millipore SiGMa). Beads samples were then washed 3X with PBS prior to incubation for the indicated time and temperature, and with the given additive in PBS. Elution from these incubations were used for flow through immunoblot samples. The beads were washed again 3X with PBS and immunoprecipitated protein recovered following incubation with 3X-FLAG peptide (Millipore SiGMa) prior to Western blot analysis.
CETSA assays
Protein samples for CETSA analysis were aliquoted into PCR strips in the presence of the indicated conditions, placed in a thermocycler (Eppendorf), and heated to the indicated temperatures for 30 min, cooled to 4°C. The insoluble fraction was then separated by centrifugation at 13, 500 g for 20 min at 4°C. This soluble fraction was then used to prepare Western blot samples.
Reduced disulfide labeling experiment
Recombinant human TRX1 (ThermoFisher) was taken up to 100 μL total volume to a concentration of 400 nM in DAB buffer (6 M urea, 200 mM Tris-HCl, 10 mM EFTA, 0.5% SDS, pH 8.5) before the introduction of either 1 mM DTT or H2O2 for 15 min at 37°C with shaking. To this was added 100 μL of 20 mM Iodoacetamide-biotin in DAB buffer following by an incubation at 37°C for 2 h with shaking. The mixture was then spun at 17000 g to pellet the insoluble fraction and the supernatant was transferred to a 5 mL centrifuge tube, diluted with 3 volumes of H2O, then 5 volumes of cold acetone, and incubated at −20°C for 2 h. To collect the proteinaceous material, the sample was placed in a centrifuge to spin at 4500 g for 10 min and decanted, air dried, and resuspended in PBS before standard Western blot sample preparation and immunoblot or total protein content by SimplyBlue SafeStain (Invitrogen) analysis following the manufacturers recommendations.
Mitochondrial ROS analysis
3x104 CASP1−/− N/TERT-1 cells were seeded per well of a black, clear bottom 96-well microplate and cultured at 37°C overnight. The following day, cellular ROS was evaluated for oxidants/antioxidants using the DCFDA cellular ROS assay (Abcam) following manufacturer’s recommendations. Briefly, culture media was aspirated, and the cells washed with 1X dilution buffer (DB) before 45 min incubation with the DCFDA reagent at 37°C protected from light. Cells were further washed with DB, aspirated again, followed by the introduction of the indicated compounds as a 150 μL addition in PBS. The microplate was then immediately placed within a Cytation 5 multi-modal plate reader (BioTek) and the fluorescence (ex/em: 485/535 nm) was read over a kinetic time course. The data was exported to spreadsheet software for statistical analysis and visualized using GraphPad PRISM 9.1.2. Unless otherwise indicated, data represent the mean ± SEM for 6 replicates.
Cell viability measurements
2x103 cells/well were plated in white 384-well clear-bottom tissue culture microplates (Corning) using an EL406 Microplate Washer/Dispenser (BioTek) to 25 μL final volume with the appropriate cell culture medium. The indicated compounds were introduced analytically using a CyBio pintool and the plates left to incubate at 37°C prior to the addition of the CellTiter-Glo reagent (Promega, G7573) according to the manufacturer’s recommendations. Assay plates were placed on an orbital sharker for 2 min and incubated at 25°C for 10 min before luminescence was read on a Cytation 5 multimodal plate reader (BioTek). The data was exported to spreadsheet software for analyses and visualized with GraphPad PRISM software (v 9.2.0).
Supplementary Material
Acknowledgments:
We thank members the Laboratory of Comparative Pathology, Molecular Cytology, and the Microchemistry & Proteomics MSKCC core facilities for technical assistance. We also thank the past and present members of the Bachovchin lab for helpful discussions.
Funding:
This work was supported by the Pew Charitable Trusts (D.A.B. is a Pew-Stewart Scholar in Cancer Research), the NIH (R01 AI137168, R01 AI163170, and R01 CA266478 to D.A.B.; T32 GM007739-Hsu and F30 CA243444 to A.R.G.; T32 GM136640-Tan to C.D.W.; the MSKCC Core Grant P30 CA008748), Mr. William H. and Mrs. Alice Goodwin, the Commonwealth Foundation for Cancer Research, The Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center (D.A.B.), the Emerson Collective (D.A.B.), the Marie-Josée Kravitz Women in Science Endeavor (WISE) fellowship (SDR), and the Anna Fuller Trust.
Footnotes
Competing interests: MSK has filed a patent application on behalf of D.A.B. and Q.W. for inventions relating to the induction of reductive stress. The remaining authors declare no competing interests.
Data and materials availability: The RNASeq data summarized in Data files S1 (N-TERT-1 cells) and S2 (C57BL/6 BMDMs) is available in GEO under accession number GSE215036. All other data are available in the main text or the supplementary materials.
References and Notes:
- 1.D'Osualdo A et al. , CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS One 6, e27396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finger JN et al. , Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem 287, 25030–25037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frew BC, Joag VR, Mogridge J, Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8, e1002659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandstrom A et al. , Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chui AJ et al. , N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingsworth LR et al. , DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 592, 778–783 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang M et al. , Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 592, 773–777 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson KS et al. , Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 370, (2020). [DOI] [PubMed] [Google Scholar]
- 9.Tsu BV et al. , Diverse viral proteases activate the NLRP1 inflammasome. bioRxiv, 2020.2010.2016.343426 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V, Human NLRP1 is a sensor for double-stranded RNA. Science, (2020). [DOI] [PubMed] [Google Scholar]
- 11.Okondo MC et al. , DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol 13, 46–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okondo MC et al. , Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem Biol 25, 262–267 e265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson DC et al. , DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med 24, 1151–1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong FL et al. , Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem 293, 18864–18878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gai K et al. , DPP8/9 inhibitors are universal activators of functional NLRP1 alleles. Cell Death Dis. 10, 587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griswold AR et al. , DPP9's Enzymatic Activity and Not Its Binding to CARD8 Inhibits Inflammasome Activation. ACS Chem. Biol 14, 2424–2429 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chui AJ et al. , Activation of the CARD8 Inflammasome Requires a Disordered Region. Cell Rep 33, 108264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharif H et al. , Dipeptidyl peptidase 9 sets a threshold for CARD8 inflammasome formation by sequestering its active C-terminal fragment. Immunity, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachovchin DA, NLRP1: a jack of all trades, or a master of one? Mol. Cell 81, 423–425 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Rao SD et al. , M24B aminopeptidase inhibitors selectively activate the CARD8 inflammasome. Nat. Chem. Biol, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orth-He EL et al. , Cytosolic peptide accumulation activates the NLRP1 and CARD8 inflammasomes. bioRxiv, 2022.2003.2022.485298 (2022). [Google Scholar]
- 22.Wang Q et al. , The NLRP1 and CARD8 inflammasomes detect reductive stress. bioRxiv, 2022.2003.2022.485209 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson WH et al. , Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem 278, 33408–33415 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Zhong FL et al. , Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 167, 187–202 e117 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Drutman SB et al. , Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc. Natl. Acad. Sci. U. S. A 116, 19055–19063 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grandemange S et al. , A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis 76, 1191–1198 (2017). [DOI] [PubMed] [Google Scholar]
- 27.He Y, Zeng MY, Yang D, Motro B, Nunez G, NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H et al. , NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol 17, 250–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharif H et al. , Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jumper J et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu J, Holmgren A, The thioredoxin antioxidant system. Free Radic. Biol. Med 66, 75–87 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Cheng Z, Zhang J, Ballou DP, Williams CH Jr., Reactivity of thioredoxin as a protein thiol-disulfide oxidoreductase. Chem. Rev 111, 5768–5783 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ball DP et al. , Caspase-1 interdomain linker cleavage is required for pyroptosis. Life Sci Alliance 3, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsui M et al. , Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol 178, 179–185 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Masters SL et al. , NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37, 1009–1023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell PS, Sandstrom A, Vance RE, The NLRP1 inflammasome: new mechanistic insights and unresolved mysteries. Curr. Opin. Immunol 60, 37–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taabazuing CY, Griswold AR, Bachovchin DA, The NLRP1 and CARD8 inflammasomes. Immunol. Rev, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snider J, Thibault G, Houry WA, The AAA+ superfamily of functionally diverse proteins. Genome Biol. 9, 216 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duncan JA et al. , Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. U. S. A 104, 8041–8046 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coll RC et al. , MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol 15, 556–559 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Hu Z et al. , Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Liao KC, Mogridge J, Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect. Immun 81, 570–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chavarria-Smith J, Mitchell PS, Ho AM, Daugherty MD, Vance RE, Functional and Evolutionary Analyses Identify Proteolysis as a General Mechanism for NLRP1 Inflammasome Activation. PLoS Pathog. 12, e1006052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, von Mering C, Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15, 3163–3168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan Y, Makar M, Wang MX, Ai HW, Monitoring thioredoxin redox with a genetically encoded red fluorescent biosensor. Nat. Chem. Biol 13, 1045–1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kosek D et al. , Biophysical and structural characterization of the thioredoxin-binding domain of protein kinase ASK1 and its interaction with reduced thioredoxin. J. Biol. Chem 289, 24463–24474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshihara E et al. , Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol 4, 514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu CW, Corboy MJ, DeMartino GN, Thomas PJ, Endoproteolytic activity of the proteasome. Science 299, 408–411 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baugh JM, Viktorova EG, Pilipenko EV, Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol 386, 814–827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsiao JC et al. , A ubiquitin-independent proteasome pathway controls activation of the CARD8 inflammasome. J. Biol. Chem, 102032 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tenthorey JL et al. , The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 358, 888–893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dickson MA et al. , Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol 20, 1436–1447 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.