

SUMMARY
As opposed to de novo mutation, β-lactam resistance in S. pneumoniae is often conferred via homologous recombination during horizontal gene transfer. We hypothesize that β-lactam resistance in pathogenic streptococci is restricted to naturally competent species via intra-/interspecies recombination due to in vivo fitness trade-offs of de novo penicillin-binding protein (PBP) mutations. We show that de novo mutant populations have abrogated invasive disease capacity and are difficult to evolve in vivo. Conversely, serially transformed recombinant strains efficiently integrate resistant oral streptococcal DNA, gain penicillin resistance and tolerance, and retain virulence in mice. Large-scale changes in pbp2X, pbp2B, and non-PBP-related genes occur in recombinant isolates. Our results indicate that horizontal transfer of β-lactam resistance engenders initially favorable or minimal cost changes in vivo compared with de novo mutation(s), underscoring the importance of recombination in the emergence of β-lactam resistance and suggesting why some pathogenic streptococci lacking innate competence remain universally susceptible.
Graphical abstract
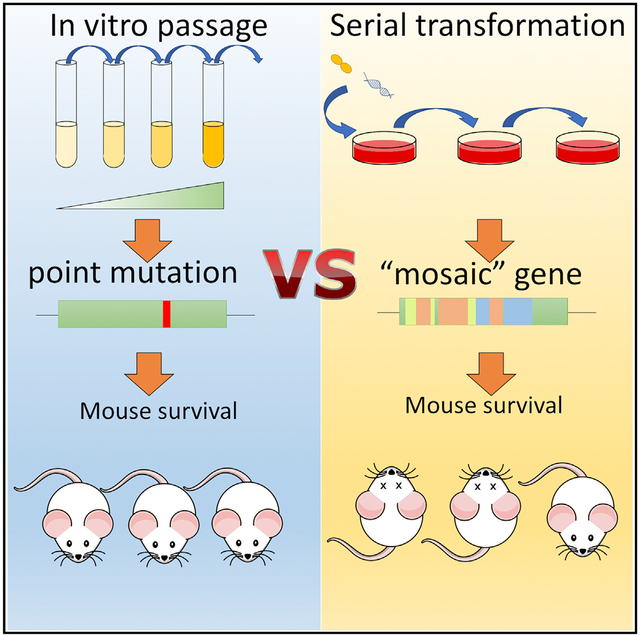
In brief
Nishimoto et al. show that S. pneumoniae can initially acquire resistance to β-lactam antibiotics via interspecies recombination without a virulence loss in vivo, in contrast to resistance acquired by de novo mutation. These findings provide insight on how fitness trade-offs constrain β-lactam resistance acquisition in the pneumococcus and other pathogenic streptococci.
INTRODUCTION
Streptococcus pneumoniae remains a major human pathogen that causes pneumonia, otitis media, bacteremia, and meningitis in children and adults. Globally, an estimated 300,000 deaths in children were attributed to pneumococcal infection in 2015 despite the introduction and use of pneumococcal-conjugate vaccines.1 Additionally, pneumococcal resistance to antibiotics in the United States has not appreciably decreased over the past few years, with resistance estimates hovering between 2% and 2.4% for penicillin over the past 5 years.2–5 In 2019 alone, drug-resistant S. pneumoniae caused an estimated 900,000 infections and 3,600 deaths; it has been designated a “serious threat” by the Centers for Disease Control and Prevention.6 Moreover, β-lactam antibiotics are among the most used drugs to treat pneumococcal infection, and while there is ongoing debate about the clinical impact of β-lactam-resistant S. pneumoniae on mortality and illness severity,7,8 it remains important to understand the mechanisms by which resistance arises.
S. pneumoniae is a naturally competent pathogen that readily uptakes genomic material from neighboring pneumococcal cells and closely related streptococcal species.9–11 Consequently, the pneumococcus can gain resistance via alterations in the genes encoding drug targets, such as the penicillin-binding proteins (PBPs).10,12,13 Previous work has identified recombination events, as opposed to spontaneous single-nucleotide polymorphisms (SNPs), as the major driver of rapid resistance acquisition.14 Indeed, commensal streptococcal species resistant to penicillin and other β-lactams have been identified as reservoirs with which S. pneumoniae can readily recombine and incorporate new genetic material.15,16 However, genomic recombination in response to drug-selective pressure can often result in virulence and fitness defects in exchange for drug resistance, making for a delicate balance between resistance and in vivo fitness.17,18 Therefore, understanding the consequences of these resistance-acquiring recombination events at the host-pathogen interface and the determinants driving this selection are essential to understanding antibiotic resistance.
Importantly, the intrinsic ability to exchange genetic material via natural competency is not shared by all other pathogenic streptococci. Streptococcus pyogenes and Streptococcus agalactiae, for example, are both causes of potentially life-threatening invasive disease, but neither is considered to be innately competent.19,20 Interestingly, both species of streptococci are also considered to be universally susceptible to β-lactams, which are the primary drugs of choice during infection caused by these organisms.21–23 Cases of resistance to β-lactams in clinical isolates of either species occur very rarely compared with S. pneumoniae,22,24–26 and resistant isolates would presumably require de novo mutation, not horizontal gene transfer, for stable changes in β-lactam susceptibility. Thus, an intriguing question persists as to whether the high frequency of β-lactam resistance in the pneumococcus is a consequence of its proclivity toward recombination, in contrast to de novo mutational pathways restricted to the typically non-resistant pathogenic streptococcal species.
Here, we propose that steep fitness trade-offs associated with de novo mutational pathways to β-lactam resistance restrict their emergence in streptococcal populations and that horizontal transfer of resistance genes via interspecies recombination may be favored due to reduced fitness trade-offs. Using β-lactam-resistant recombinant pneumococcal strains, we observed enhanced virulence in vivo with initial recombinant strains compared with antibiotic-evolved, resistant pneumococcal strains possessing de novo point mutations in PBPs. Although in vitro characterization revealed only modest differences in adherence to mammalian cells, invasive potential, macrophage phagocytosis, and affinity for PBPs between strains, recombinant strains demonstrating initial small increases in minimum inhibitory concentration (MIC) displayed increased virulence compared with wild-type (WT) parental strains in murine models. Moreover, in vitro-evolved strains containing de novo mutations conferring slight MIC increases to penicillin resulted in complete attenuation of virulence in vivo. Genome sequencing of recombinant strains revealed large regions of recombination, including a conserved pattern of recombination around the pbp2X locus in all recombinant strains tested. These findings indicate that horizontal gene transfer, as opposed to de novo mutation, is readily achievable and confers decreased susceptibility to β-lactams without a significant in vivo cost to S. pneumoniae. Overall, these data suggest that high-level β-lactam resistance may be restricted to streptococcal species that can readily undergo horizontal gene transfer due to the fitness consequences of the initial de novo mutations in response to antibiotic pressure.
RESULTS
De novo mutations in response to penicillin result in attenuation of virulence
We initially modeled the evolutionary trajectories of S. pneumoniae to increasing concentrations of penicillin during serial in vitro passaging. Passaging was undertaken at various concentrations of penicillin with the highest permissive concentration allowing growth used to seed the following passage (Figure 1A). This continuously applied pressure was undertaken for 32 passages representing approximately 115 generations of antibiotic selection, after which a 3× shift in MIC was observed in liquid culture. Despite continuous selection, only relatively small (3× or less) increases in the penicillin MIC (Figure 1B; Table S1) among the eight in vitro-evolved populations (T4EP1 through T4EP8) were observed, making these strains well below established clinical breakpoints for resistance. Mutational mapping of the evolved isolates against their penicillin-free passaged population counterparts revealed similar conserved (100% of the sequenced population) point mutations arising as a result of antibiotic selective pressure (Table 1). Despite the similar fixed mutational changes in T4EP1 through T4EP4, the predominant mutations in each population remained unique to each respective population at every passage tested (days 7, 14, 21, and 32), though these mutations did not grow to fixation (Figure S1). This suggests that these populations were evolutionarily distinct. Decreased susceptibility for T4EP1, T4EP2, T4EP3, and T4EP4 most likely stemmed from a single Y586S amino acid change in PBP2X, which was identified in all populations. Other changes included single-point mutations in the cell wall biosynthesis component murE, the capsular polysaccharide biosynthesis gene cps4E, a gene encoding a DHH subfamily 1 protein, and an ATP-binding cassette transporter. SNPs in these genes commonly recurred across all four penicillin-evolved populations, and many of the mutant genes have been linked either as resistance determinants or virulence factors in S. pneumoniae.27–29
Figure 1. De novo mutations in response to penicillin result in attenuation of virulence.
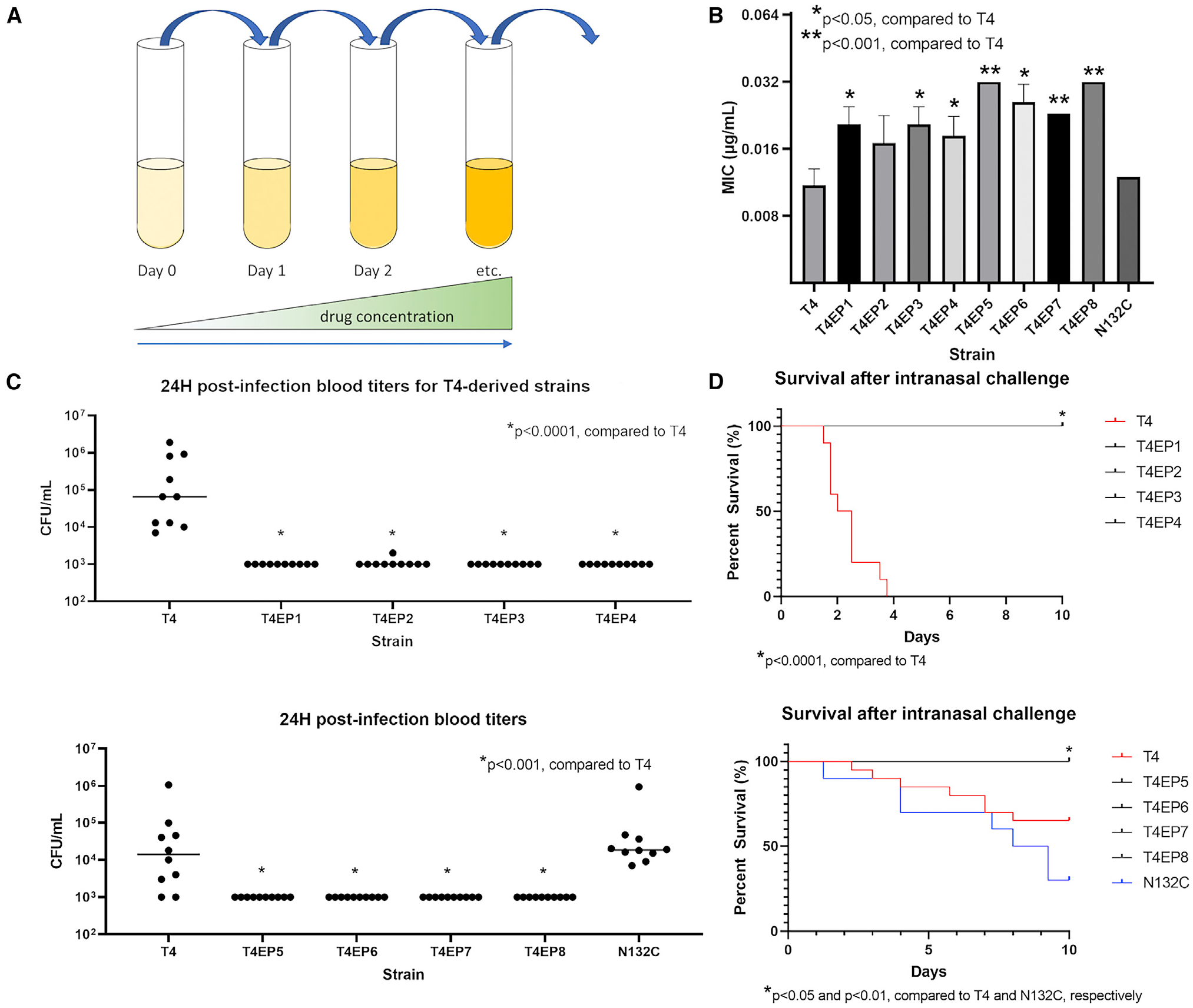
(A) Penicillin resistance was generated via de novo mutation in T4-derived pneumococcal cells by continuous passaging of planktonic cells in liquid culture containing gradually increasing subinhibitory concentrations of penicillin.
(B) Penicillin MICs of WT pneumococcal strain T4 and its derivative experimentally evolved pneumococcal populations. Data are represented as mean MIC ± SD (n = 3).
(C) Blood titers at 24 h post-infection with WT T4 or a derivative strain.
(D) Mouse survival followed 10 days after intranasal challenge with experimentally evolved versus WT pneumococci. Seven-week-old BALB/c mice were intranasally infected with 106 CFUs of T4 or an experimentally evolved derivative population (n = 10 mice/groups). p values of 0.05 or less as determined by Mann-Whitney U test were considered significant.
Table 1.
Mutations in the penicillin-resistant in vitro-evolved populations of S. pneumoniae
| Population | Position | Mutationa | Gene | Encoded protein description |
|---|---|---|---|---|
| T4EP1 | 308,636 | Y586S (TAT→TCT) | SP_0336 | PBP2X |
| 324,228 | P80L (CCT→CTT) | SP_0350 | Cps4E | |
| 1,280,284 | F218L (TTT→CTT) | SP_1358 | ABC transporter, ATP-binding/permease protein | |
| 1,442,712 | A430S (GCG→TCG) | SP_1530 | MurE | |
| 2,125,225 | A586E (GCG→GAG) | SP_2205 | DHH subfamily 1 protein | |
| T4EP2 | 308,636 | Y586S (TAT→TCT) | SP_0336 | PBP2X |
| 324,228 | P80L (CCT→CTT) | SP_0350 | Cps4E | |
| 1,280,284 | F218L (TTT→CTT) | SP_1358 | ABC transporter, ATP-binding/permease protein | |
| 1,442,712 | A430S (GCG→TCG) | SP_1530 | MurE | |
| 2,125,225 | A586E (GCG→GAG) | SP_2205 | DHH subfamily 1 protein | |
| T4EP3 | 308,636 | Y586S (TAT→TCT) | SP_0336 | PBP2X |
| 324,228 | P80L (CCT→CTT) | SP_0350 | Cps4E | |
| 1,442,712 | A430S (GCG→TCG) | SP_1530 | MurE | |
| 2,125,225 | A586E (GCG→GAG) | SP_2205 | DHH subfamily 1 protein | |
| T4EP4 | 308,636 | Y586S (TAT→TCT) | SP_0336 | PBP2X |
| 324,228 | P80L (CCT→CTT) | SP_0350 | Cps4E | |
| 1,207,068 | Δ1 bp | SP_1274 | licD2 protein | |
| 1,280,284 | F218L (TTT→CTT) | SP_1358 | ABC transporter, ATP-binding/permease protein | |
| 1,442,712 | A430S (GCG→TCG) | SP_1530 | MurE | |
| 2,125,225 | A586E (GCG→GAG) | SP_2205 | DHH subfamily 1 protein | |
| T4EP5 | 136,365 | Δ1 bp | SP_0137 | ABC transporter ATP-binding protein |
| 308,635 | Y586H (TAT→CAT) | SP_0336 | PBP2X | |
| 945,790 | P509P (CCA→CCG) | SP_1003 | hypothetical protein | |
| 1,494,178 | V407V (GTT→GTC) | SP_1588 | pyridine nucleotide-disulfide oxidoreductase | |
| 2,048,117 | Δ1 bp | SP_2136 | choline-binding protein A | |
| 2,150,991 | L159L (CTA→TTA) | SP_2231 | copper ABC transporter permease | |
| T4EP6 | 308,636 | Y586C (TAT→TGT) | SP_0336 | PBP2X |
| 1,207,173 | G40E (GGA→GAA) | SP_1274 | licD2 protein | |
| T4EP7 | 103,388 | Δ1 bp | SP_0101 | MFS transporter |
| 308,458 | M527V (ATG→GTG) | SP_0336 | PBP2X | |
| 1,674,302 | +3 bp (+CCC) | SP_RS08765 | glycosyltransferase family 2 protein | |
| T4EP8 | 308,636 | Y586C (TAT→TGT) | SP_0336 | PBP2X |
| 1,207,823 | E257* (GAG→TAG) | SP_1274 | phosphorylcholine transferase LicD | |
| 1,635,159 | A507V (GCT→GTT) | SP_1732 | serine/threonine protein kinase pknB | |
| 1,674,302 | +1 bp (+C) | SP_RS08765 | glycosyltransferase family 2 protein |
Cps4E, capsular polysaccharide biosynthesis protein; MurE, UDP-N-acetylmuramoylalanyl-D-glutamate–2, 6-diaminopimelate ligase; PBP2X, penicillin-binding protein 2X.
Mutations listed were present in at least 50% of the sequenced population and were absent from either the parent strain or its drug-free passaged counterpart.
To eliminate the possibility of cross-contamination between passaged cultures as an alternative explanation for the similar penicillin-resistance-related SNPS observed between populations, we used T4 strains barcoded with unique identifier sequences as the progenitors of the evolved populations T4EP5, T4EP6, T4EP7, and T4EP8. As with the other passaged strains, each barcoded population passaged in penicillin possessed a predominant mutation in PBP2X (Table 1). Of the 8 passaged populations, 7 of them altered the Y586 residue of PBP2X, which is conserved in Gram-positive cocci and known to confer resistance.30 Notably among the unique mutations that were present in the individual lineages, a nonsense mutation arose in the phosphorylcholine metabolism gene licD2 in T4EP6. licD2− mutants have been shown to be resistant to penicillin-induced lysis in late logarithmic phases.31 Interestingly, T4EP4 also contained a single base-pair deletion in licD2 resulting in a frameshift, possibly suggesting that licD2 mutation may play a role in pneumococcal resilience to penicillin.
Despite modest alterations in MIC and genetic changes associated with them, we hypothesized that these first step mutations toward resistance would negatively impact the invasive capacity of the pneumococcus. To compare in vivo fitness and other phenotypic traits between β-lactam-evolved strains, we challenged 7-week-old BALB/c mice intranasally with either the parental strain or each of the independently experimentally evolved penicillin-resistant populations and subsequently monitored survival levels of bacteremia following challenge. We observed that all the in vitro experimentally evolved isolates were highly attenuated in vivo, exhibiting significantly lower blood titers compared with the TIGR4 (T4, serotype 4) parent isolate and complete survival 10 days post-infection (Figures 1C and 1D). These data indicate that even minor de novo mutational steppingstones toward β-lactam resistance can be constrained by severe in vivo fitness consequences.
Fitness consequences of interspecies recombination conferring β-lactam resistance
We next sought to ascertain whether the fitness trade-offs observed in the experimentally evolved isolates would hold true during interspecies recombination as previous work has identified recombination events rather than spontaneous SNPs as the major driver of rapid β-lactam resistance acquisition.14 Many of these recombination events are thought to occur between the pneumococcus and other streptococcal species, with recombination between closely related oral streptococcal species being a primary reservoir for β-lactam resistance in the pneumococci.
To test this, recombinant β-lactam-resistant pneumococcal strains were generated via transformation of T4 with genomic DNA (gDNA) from clinical isolates of viridans group streptococci (VGS) with varying degrees of β-lactam resistance (Table S1). Second- and third-round recombinants, so named because they are the result of successive transformations of the previous “round” of successful transformants, were subsequently created by repeating transformation an additional one or two times, respectively, in the presence of VGS gDNA, with colony selection on penicillin-containing blood agar plates (Figure 2A). Although the first-round recombinant pneumococcal strain, T4R1P, possessed a penicillin MIC similar to the in vitro-evolved strains, the second- and especially the third-round recombinants showed a capacity for much larger increases in penicillin MIC, possessing MICs almost 50 times greater than the T4 MIC (Figure 2B). The initial recombinant strain, T4R1P, exhibited marked increased virulence in vivo with high 24 h blood titers present in mice and significantly increased mortality compared with that of T4 (Figures 2C and 2D), suggesting the initial rounds of recombination conferring the initial steps toward resistance did not impair invasive disease capacity. However, second- and third-round recombinants behaved similarly to in vitro-evolved strains, with little to no detectable presence in the blood after 24 h and complete mouse survival 10 days post-infection. We also tested in vivo the recombinant pneumococcal strain T4trDAW7, which was generated from T4 transformed with gDNA taken from DAW7, a β-lactam-resistant pneumococcal clinical isolate. The recombinant isolate possessed a relatively high penicillin MIC and was highly virulent in vivo (Figures 2B–2D). Interestingly, T4trDAW7 exhibited a significantly higher mortality than either the parent strain T4 or the donor isolate DAW7, suggesting that this initial recombination confers significant fitness advantages in vivo over either strain individually. Based on our observations, early recombinants do not possess attenuated traits and may even enhance virulence in vivo, though attenuation was not observed until S. pneumoniae underwent subsequent recombination events via horizontal gene transfer. When resistant pneumococcal DNA was utilized as a source of gDNA, virulence was likewise maintained. Furthermore, the levels of attenuation observed in the later-recombinant pneumococcal strains were comparable to the decreased virulence seen in mice challenged with the experimentally evolved isolates. These data suggest that early recombinants can possess decreased susceptibility to β-lactam agents without significant impairment of in vivo fitness and may provide initial fitness advantages during invasive disease. These data further indicate that acquisition of β-lactam resistance from multiple recombination events requires a balance between antimicrobial susceptibility and fitness in vivo.
Figure 2. Fitness consequences of interspecies recombination conferring β-lactam resistance.
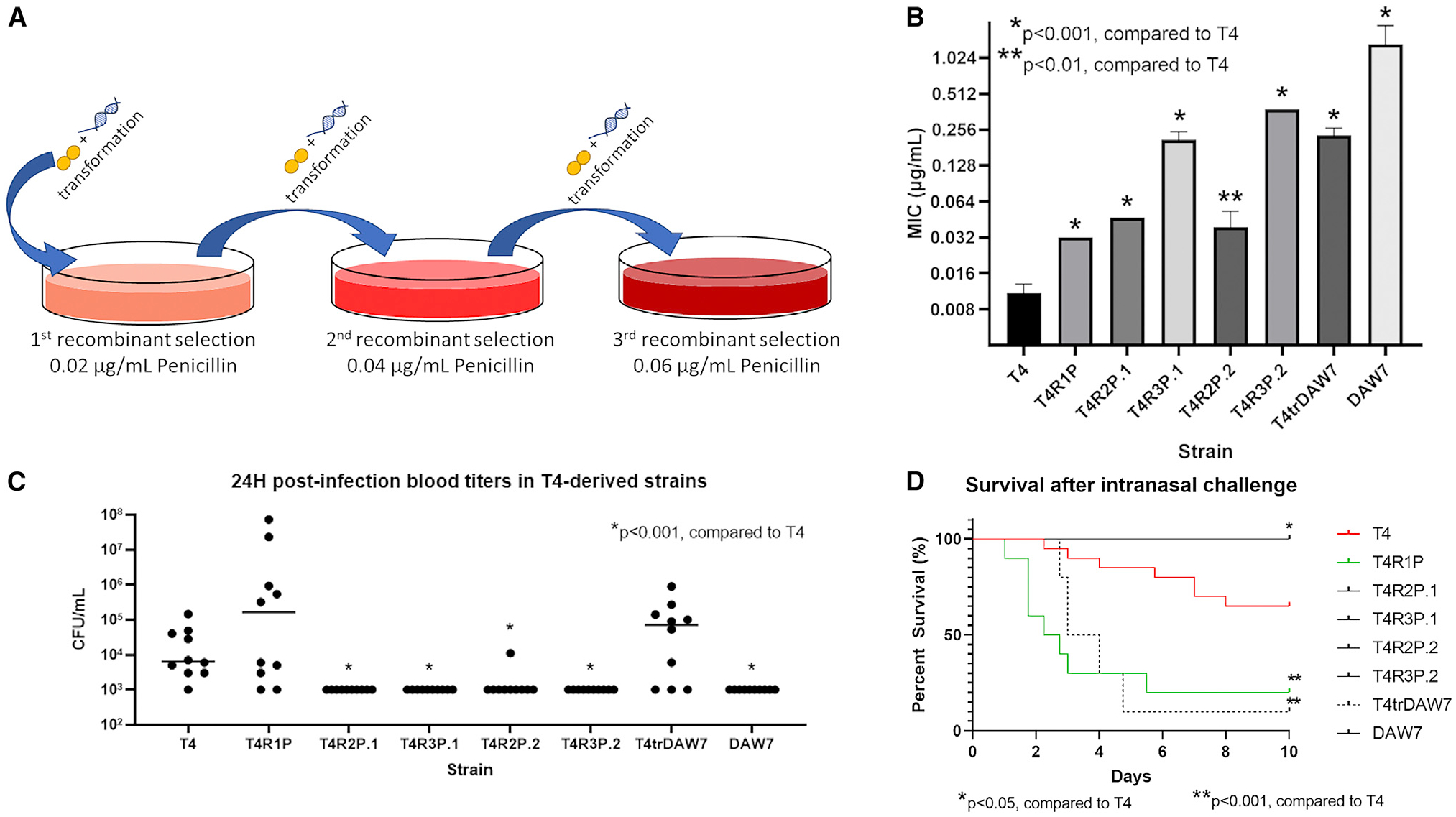
(A) Penicillin resistance was developed via serial transformation of T4 cells with 5 μg gDNA from either penicillin-resistant VGS strains or the pneumococcal strain DAW7. Colonies were screened on penicillin-containing blood agar plates and used for subsequent re-transformation with gDNA.
(B) Penicillin MICs of WT pneumococcal strain T4 and its derivative recombinant pneumococcal populations. Data are represented as mean MIC ± SD.
(C) Blood titers at 24 h post-infection.
(D) Mouse survival after intranasal challenge with recombinant versus WT. Seven-week-old BALB/c mice were intranasally infected with 106 CFUs of T4 or recombinant strains and followed for 10 days (n = 10 mice/group). p values of 0.05 or less as determined by Mann-Whitney U test were considered significant.
Bacterial burden in different host niches supports virulence differences across recombinant strains in vivo, but differences in markers of inflammation lack significance
To further characterize the effects of early recombination in pneumococcal pathogenesis, we examined the virulence of first-round recombinants and the less penicillin-susceptible third-round recombinants in different host niches, including blood, lungs, and nasal passages. At 24 h post-infection, mice infected with T4R1P had relatively equivalent bacterial burden in the three niches compared with mice infected with T4 (Figures S2A–S2C). In contrast, mice infected with T4-derived third-round recombinants had significantly lower bacterial burden in the blood and nasal passages (p < 0.05). Compared with the initial virulence experiment, these results recapitulated that mutations from initial recombination did not diminish S. pneumoniae’s ability to colonize and invade the host. Mutations resulting from subsequent transformations, which consequently had further decreased antibiotic susceptibility, may decrease the bacteria’s virulence.
Furthermore, using sectioned lung tissues collected at 24 h post-infection, we examined the signs of inflammatory response and extent of lung injuries from infection with the recombinant mutants and their respective WT strains, including vasodilation of blood vessels (veins, arteries, arterioles), neutrophil migration from the bloodstream into the pulmonary air space, vascular injuries, leukocytes in lungs, collapsed alveoli, and pulmonary edema. Cumulative pathologic scoring of the right lobar sections from the five mice in each group revealed slightly higher severity of interstitial inflammation, alveolar involvement, and the presence of alveolar neutrophils in T4R1P (25, 36, and 40, respectively) and T4R3P.1 (32, 28, and 34, respectively) compared with T4 (12, 21, and 20, respectively) (Figures S2D–S2H). Thus, although bacterial burden of the third-round recombinant in the host lungs was significantly lower, there appeared to be modest, but increased, histologic signs of damage and invasion with both recombinant strains compared with T4. Lastly, heat-killed versions of T4 and T4R1P lacked any evidence of infection or inflammation, suggesting that dead cells and cell fragments were not responsible for damage in the lung.
During acute inflammation of lung tissues caused by pneumococcal infection, neutrophils that infiltrate the lungs and migrate into the pulmonary airspace induced the expression of regulatory inflammatory cytokines, such as granulocyte macrophage colony-stimulating factor (GM-CSF), interleukin 1β (IL-1β), IL-6, IL-10, IL-12 (p40), and tumor necrosis factor α (TNF-α).32 There were few significant differences in the expression of cytokines responsible for the acute inflammatory phase collected and analyzed from mice infected with first- or third-round recombinants compared with their parental strains (Figure S3). Indeed, only the expression of IL-6 and TNF-α in the serum of T4R3P.1-infected mice, compared with that of T4-infected mice, was significantly decreased (p = 0.0079). Consistent with the pulmonary histopathologic findings, most differences in cytokines induced by recombinants, compared with their parent strains, were slight. Overall, the degree of inflammation and inflammatory markers, as a result of infection with recombinant strains, closely resembled that of the parent strains, indicating that the recombinants’ abilities to cause inflammation and tissue injury was not attenuated.
Inter- and intraspecies recombinants exhibit similar transformation efficiencies
The gDNA of 20 clinical VGS strains were isolated for use as donor DNA for generating recombinant β-lactam-resistant S. pneumoniae. Of these, the gDNA from 10 VGS strains yielded successful transformants when co-incubated with WT T4 or a T4 recombinant. The gDNA from a known resistant pneumococcal isolate was also transformed into the parent strains as a comparator. The transformation efficiencies for the interspecies recombinants varied between approximately 10−6 and 10−8 per 5 μg gDNA, which was comparable to the transformation efficiencies of generating the intraspecies recombinant T4trDAW7 (Figure S4). Subsequent transformations using first-round transformants also did not alter transformation efficiency when generating second- and third-round recombinants. Overall, our results suggest that for VGS strains whose DNA was readily taken up by S. pneumoniae, the transformation efficiency was similar to that of intraspecies recombination under the same conditions.
Recombinant pneumococcal isolates show large regional genomic changes
When examining the genome sequence of recombinant pneumococcal isolates against that of the parent strains, we identified several large SNP-rich regions, likely indicating sections of the genome where recombination with the exogenous VGS gDNA took place (Figure 3; Tables S4 and S5). For example, in the first recombinant series, T4R1P, a segment of roughly 4 kb containing multiple genes, including the gene encoding the β-lactam target PBP2X, was densely populated with SNPs. Presumably, this region underwent recombination with the VGS gDNA from isolate SV5, in which antibiotic pressure selected for recombinant pbp2X. As one might expect, recombinant pbp2X was conserved upon successive transformations screened on penicillin, and additional SNPs were accumulated after each transformation. Indeed, changes in the ~300,000 bp region containing the pbp2X locus were prominent and conserved in every recombinant isolate sequenced (Figure 3). The third series recombinants, T4R3P.1 and T4R3P.2, were both derived from T4R1P and possessed recombinant pbp2X. Also, both had integrated recombinant changes in pbp2B, even though the donor DNA used to generate the recombinants was sourced from two different VGS isolates.
Figure 3. Recombinant pneumococcal isolates show large regional genomic changes.
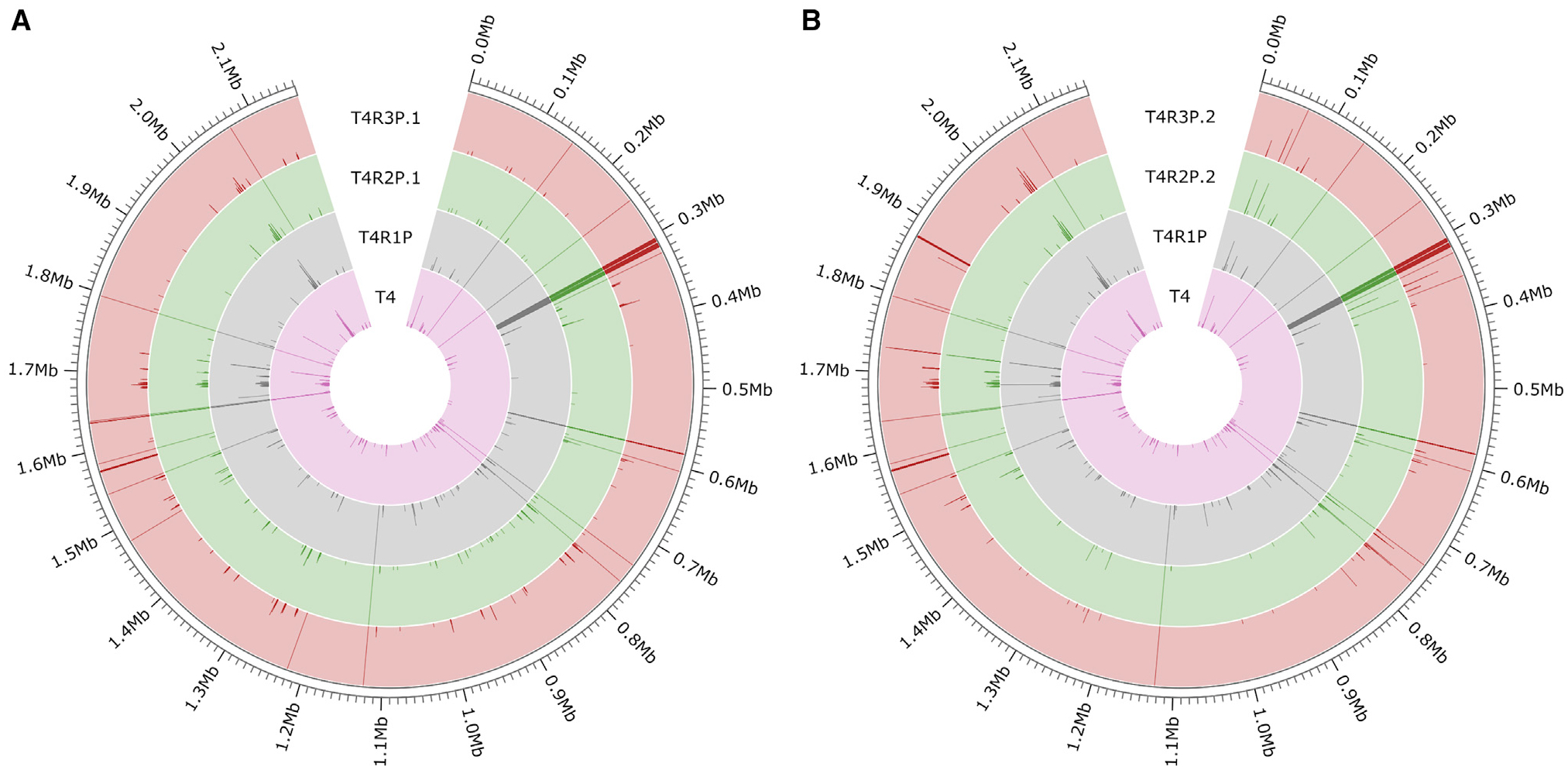
Maps of genomic changes present in the T4 parental strain (A) and the recombinant series T4R1P, T4R2P.1, and T4R3P.1 (B). The T4 and recombinant series T4R1P, T4R2P.2, and T4R3P.2 were compared with the reference strain TIGR4 (NCBI: NC_003028.3). Strains are denoted by the colored rings. SNPs are illustrated by dark lines, with frequency denoted by length of the line across the width of the ring (i.e., SNPs present in 100% of the sequenced population are depicted as lines spanning the width of each respective ring). Position along the pneumococcal genome is circumscribed on the outer edge of each figure.
Additionally, we observed variable changes in non-PBP regions of the genome for each recombinant. Both isolated polymorphisms and SNP-heavy regions of recombination were present in all recombinant series. Notably, T4R1P and all its derivative strains possessed a recombinant murM gene, which is involved in the production of pneumococcal branched-stem peptides and PBP-mediated β-lactam resistance.33 Also, within this series of recombinant isolates was a 5 kb recombinatorial region encompassing, among others, genes encoding LuxS, which helps regulate virulence and fitness genes during biofilm formation; ClpB, a heat-shock and stress-response protein; and DexB, a highly conserved glucosidase known to flank one side of the pneumococcal capsular polysaccharide locus. No other PBP-associated genes were localized in this region, suggesting that one or more of these changes is advantageous for the pneumococcal cell with regards to β-lactam exposure.
In vitro phenotypes of recombinant versus experimentally evolved pneumococcal strains do not significantly differ
To further assess the phenotypes of the resistant recombinant strains, compared with the in vitro-evolved pneumococcal isolates, we incubated strains with A549 human alveolar basal epithelial cells to compare strain capacity for invasion and adhesion (Figures S5A and S5B). Although we observed some alterations in the ability of the recombinant strains to adhere to cells, the results were inconsistent between the tested strains and the overall discernible patterns being observed. Nonetheless, these data might, in part, account for the differences in attenuation seen in vivo between the second- and third-round recombinant series compared with the first-round recombinant series. For example, the second- and third-round recombinants, T4R2P.1, T4R3P.1, T4R2P.2, and T4R3P.2, showed elevated adhesion to alveolar epithelial cells compared with T4 (Figure S5A). Although target cell adhesion may assist with invasion and colonization, it did not explain (and was somewhat counterintuitive to) the attenuation we observed with these strains in vivo, in which we expected enhanced adhesion to facilitate virulence in T4R1P. Quantification of pneumococcal cell invasion of A549 cells revealed only significantly higher invasive potential with the third-round recombinant T3R3P.2, suggesting that these observed differences in invasion were not primarily responsible for fitness differences in vivo (Figure S5B). Additionally, the penicillin-evolved strain T4EP1 showed no significant differences in either adhesion or invasion of A549 alveolar epithelial cells compared with T4.
We next assessed whether macrophage phagocytosis differed across recombinant strains, their parents, and experimentally evolved isolates. For this, we co-incubated J774A.1 murine macrophages with respective pneumococcal strains at an approximate multiplicity of infection (MOI) of 50. Unfortunately, not all strains differed from their respective parent, as measured by the proportion of intracellular S. pneumoniae cells present inside lysed macrophages versus total pneumococcal cells present (Figure S5C). The measure of intracellular S. pneumoniae may indicate a significant alteration in the ability of the pneumococcal cells to survive intracellularly after phagocytosis and/or the ability of macrophages to ingest the invading pneumococcus. However, we saw no strong trends in recombination versus de novo mutation, lending to consistent changes in killing by macrophages.
Lastly, to determine the alterations in PBP-binding affinity of the recombinant strains and the experimentally evolved isolates, we added BOCILLIN FL, a fluorescent penicillin derivative, to actively dividing bacteria during the mid-logarithmic phase and then purified and quantified the whole-cell protein. A standard concentration of 10 μg was loaded and run via gel electrophoresis, which allowed for the visualization of fluorescent PBP bands; band intensity correlated with PBP abundance and BOCILLIN FL-binding affinity. Quantification of band intensity revealed no large changes in PBP binding between any strain and its parent (Figure S6). Overall, many recombinant strains showed very slight decreases in band intensity for many, if not all, of the six visualized PBPs, perhaps suggesting a slight decrease in the PBPs’ affinity to BOCILLIN FL compared with that of the parent strain. However, our data indicated that the differences in BOCILLIN FL PBP-binding affinities across recombinant strains was also modest and therefore probably not wholly responsible for differences in virulence and fitness seen between recombinant strains.
Interspecies recombination can promote antibiotic tolerance
Antibiotic tolerance, whereby there are minimal shifts in MIC but markedly reduced kill kinetics, has been recognized in the pneumococcus for decades34,35 and has been associated with antibiotic resistance and poorer treatment outcomes.36–39 Due to their subtle increases in MICs to penicillin, we next sought to investigate whether recombinant isolates displayed tolerance phenotypes by examining their kill kinetics in the presence of antibiotics. The Tupelo strain, a prototypical tolerant pneumococcal strain, served as a reference strain and positive control for tolerance experiments.40 Upon exposure to lethal concentrations of penicillin (10× MIC), both the in vitro-evolved populations and the T4-derived first- and second-round recombinants displayed a lack of tolerance to penicillin, with ≤ 1% colony-forming units (CFUs) remaining within 2 h of penicillin exposure (Figures 4A–4D). Third-round recombinants exhibited a more gradual decline in bacterial CFUs than did the susceptible WT (Figure 4E). The kill kinetics of the third-round, penicillin-screened recombinants also more closely resembled those of the control Tupelo strain, indicating that these recombinants had acquired tolerance to otherwise lethal concentrations of the antibiotics. Thus, in our hands, the repeated uptake and recombination of penicillin-resistant gDNA into S. pneumoniae resulted in not only measurable increases in penicillin MIC but also tolerance acquisition after multiple exposures. A single transformation of penicillin-resistant pneumococcal gDNA into T4, as in the recombinant T4trDAW7 strain, was also not sufficient to confer tolerance to penicillin, suggesting that resistance, rather than tolerance, is more easily attained under penicillin-selective pressure during intraspecies horizontal gene transfer.
Figure 4. Interspecies recombination can promote antibiotic tolerance.
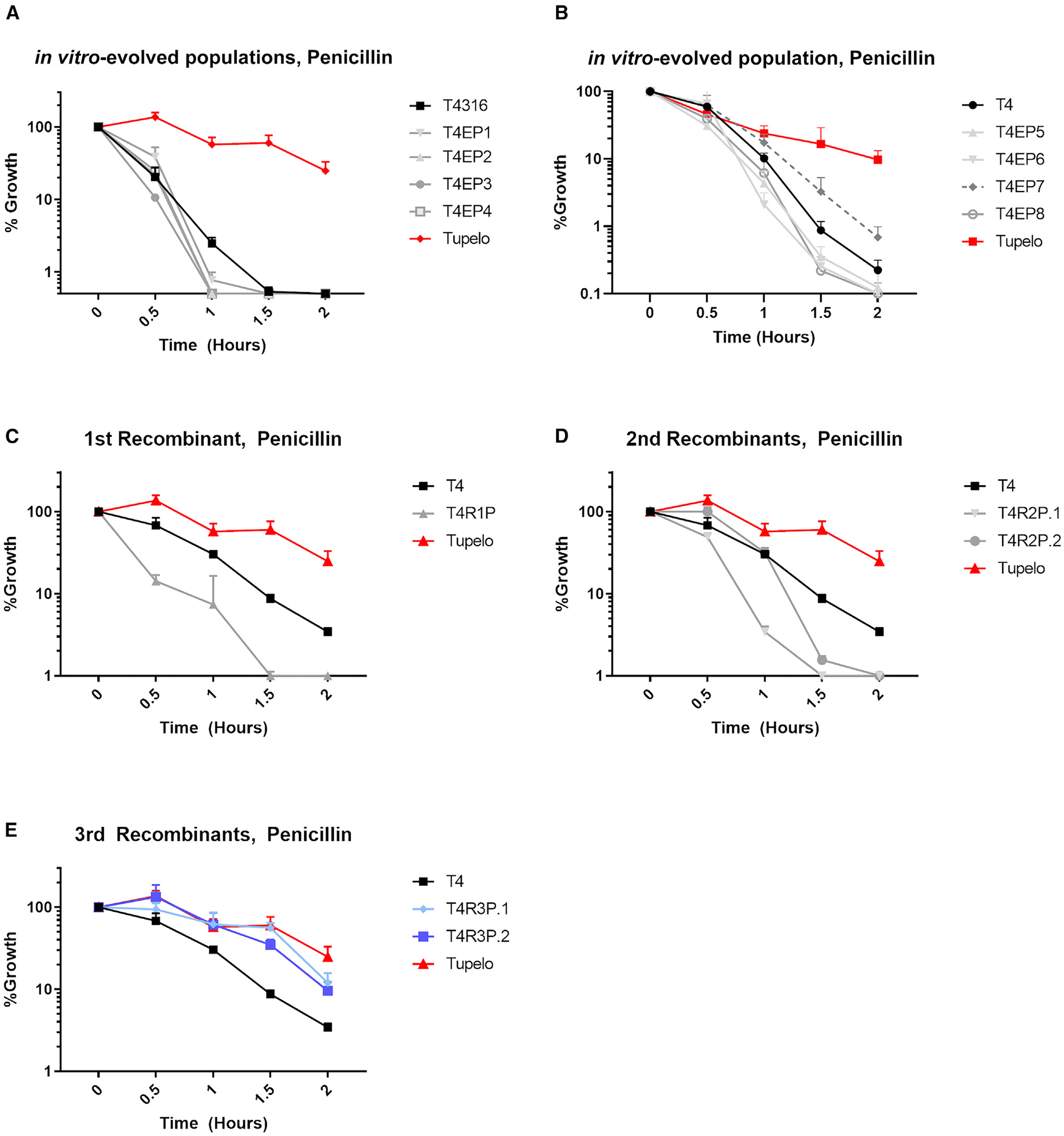
Half-hour bacterial titers were taken after exposure to 10× MIC of penicillin for T4-derived in vitro-evolved strains (A and B), T4-derived first-round recombinant strains (C), T4-derived second-round recombinant strains (D), and T4-derived third-round recombinant strains (E). Bacterial growth is represented as a percentage of the bacterial CFU/mL taken at time = 0, prior to the addition of antibiotic. The prototypical multidrug-tolerant S. pneumoniae strain known as Tupelo was included as a reference for tolerance phenotype. Data are represented as mean CFU counts ± SD (n = 3).
It should also be noted that among the VGS strains used for transformation and horizontal gene transfer, all except for SV4 showed attenuated killing by 10× MIC of penicillin, indicating that these donor strains were also penicillin tolerant (Figure 5). Recombination of the non-tolerant T4R2P.2 with gDNA from the non-tolerant SV4 generated the penicillin-tolerant T4R3P.2. It is unknown whether this emergence of tolerance after recombination between non-tolerant strains is an unmasked phenotype from previous recombination events with the tolerant SV5 and/or SV8. Conversely, it is also possible that tolerance arose de novo in T4R3P.2 after piecemeal transfer of various genetic components contributing to a penicillin-tolerant phenotype. These data underscore the potential for rapid emergence of tolerance to cell-wall-active antibiotics via intraspecies recombination events that could contribute to antibiotic treatment failure.
Figure 5. VGS donor strains were predominantly penicillin tolerant.
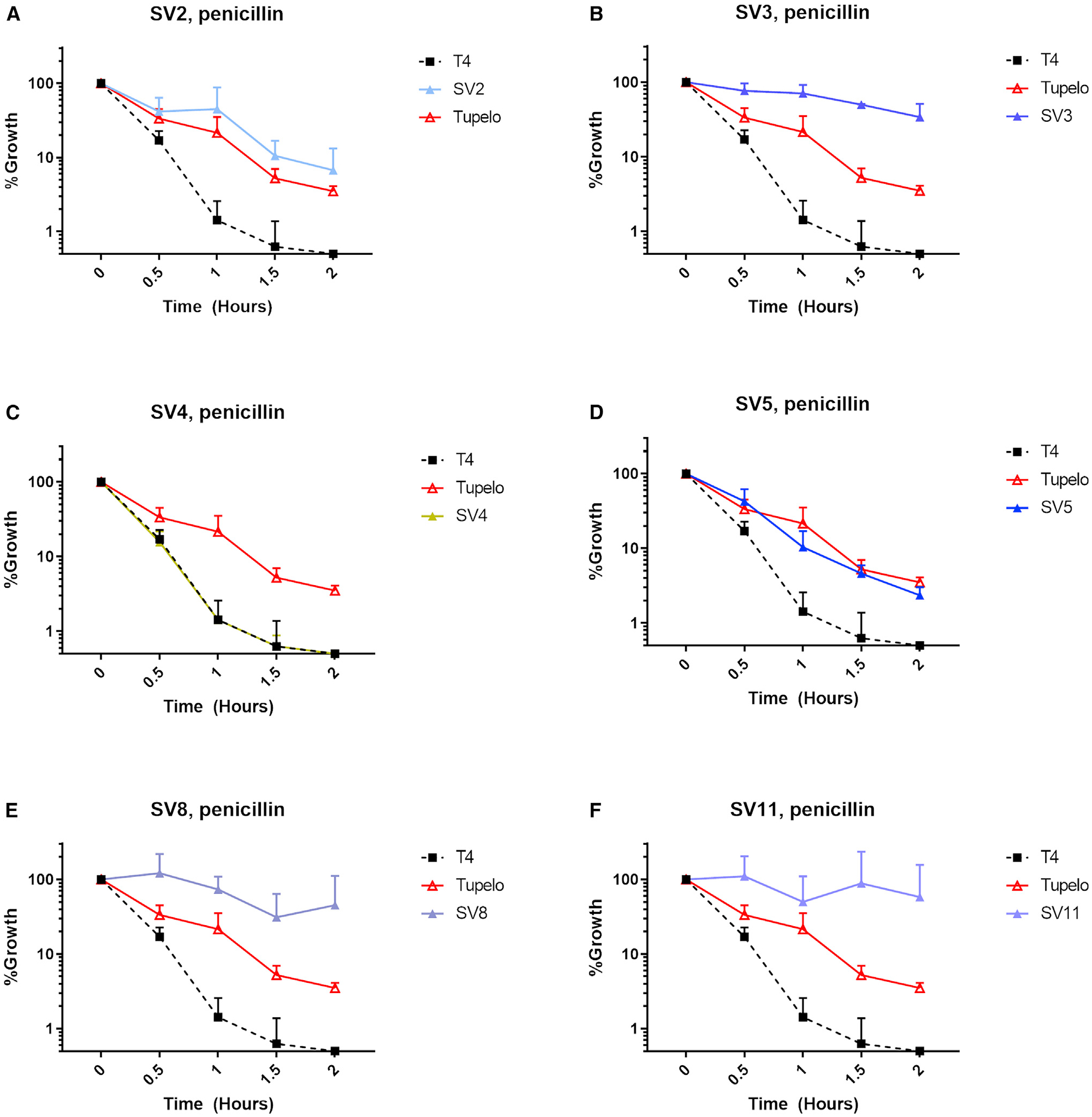
Bacterial titers were measured 30 min after exposure to 10× MIC of penicillin for the WT T4 strain and VGS strains SV2 (A), SV3 (B), SV4 (C), SV5 (D), SV8 (E), and SV11 (F). Bacterial growth is represented as a percentage of the bacterial CFU/mL taken at time = 0, prior to the addition of an antibiotic. The prototypical multidrug-tolerant S. pneumoniae strain known as Tupelo was included as a reference for tolerance phenotype. Data are represented as mean CFU counts ± SD (n = 3).
De novo PBP mutations occur infrequently and do not persist in vivo under penicillin selective pressure
Because our data suggest that de novo mutation comes with a high in vivo fitness trade-off, we hypothesized that de novo PBP mutations occur infrequently or, alternatively, are quickly outcompeted in vivo. Therefore, we next sought to determine the frequency and persistence of de novo-mutation-containing pneumococcal strains in vivo during antibiotic treatment. To test this, we passaged the T4 parental strain through 15 successive mice treated with penicillin in three independent lineages. Briefly, mice were intranasally inoculated with pneumococci and treated 8 h later with intraperitoneal injections of penicillin. 24 h post-infection, mice were euthanized, and pneumococci was recovered from the lungs of infected mice and used as the inoculum for the next successive mouse infection. Penicillin doses were slowly increased up to 40 mg/kg to allow for resistance development. As expected, mutations were observed in several PBP genes, including pbp1A, pbp2A, pbp2B, and pbp2X, in the later passages of each lineage (Figure 6). However, each PBP mutation occurred in <10% of the sequenced pneumococcal population among which it was detected, and none of these mutations were retained in the following passages of their respective lineages. Overall, SNPs in PBP genes were observed in only 8 passaged populations: generation 15 of lineage 1; generations 6, 8, 12, and 13 of lineage 2; and generations 7, 8, and 15 of lineage 3. Taken together, these data suggest that even under antibiotic stress, de novo PBP mutations occur relatively infrequently and are quickly outcompeted in vivo. This further supports the notion that de novo resistance mutations to the β-lactams are disadvantageous in vivo.
Figure 6. De novo PBP mutations occur infrequently and do not persist in vivo under penicillin selective pressure.
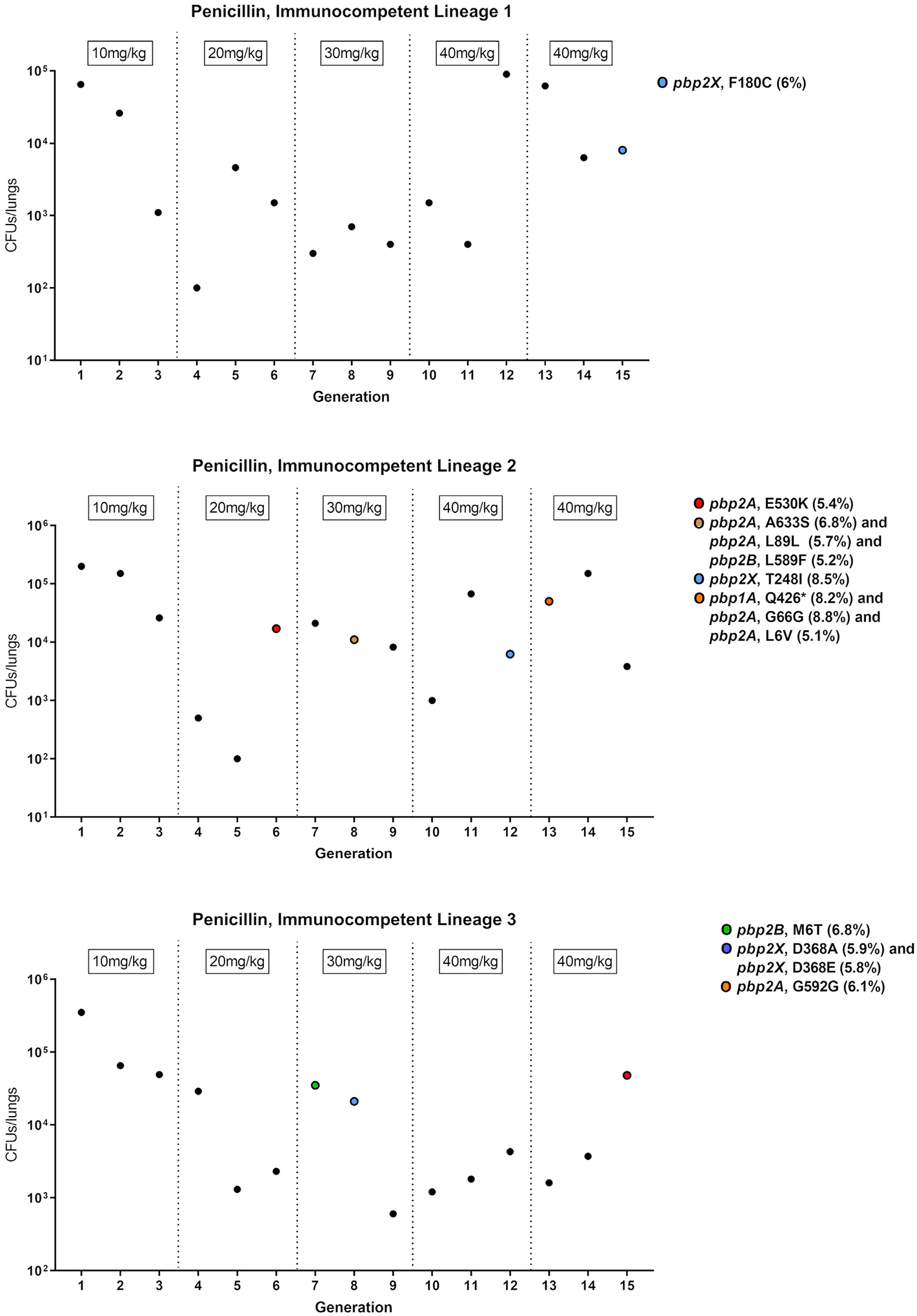
Data points represent the total CFU recovered from the lungs of infected mice for three separate lineages of passaged pneumococcal strains. The corresponding penicillin dose given to each mouse is labeled above the data points and incrementally increased with successive generations. Whole-genome sequencing confirmed the presence of PBP mutations. Colored points represent pneumococcal populations containing one or more mutations in PBP genes. SNPs and SNP frequencies for the respective PBP genes are listed in the legend.
DISCUSSION
Phenotypic and genotypic distinction between recombination and de novo mutation in resistance acquisition
Gaining an understanding of why some pathogens become antibiotic resistant while others remain sensitive despite decades of antibiotic exposure is a fundamental question in microbiology. While de novo mutation is one route to resistance, oftentimes resistance rapidly spreads via horizontal gene transfer, raising the question to whether one route may be more advantageous for a given antibiotic-pathogen combination. To address this question, we generated both in vitro-evolved and recombinant pneumococcal isolates with altered susceptibilities to β-lactam antibiotics. The fitness of the isolates was variable in murine models of intranasal infection, where bacterial blood and lung tissue titers, nasal colonization, and overall lethality tended to be inversely related to serial recombination and increased MIC. The in vitro experimentally evolved pneumococcal isolates demonstrated in vivo attenuation for all isolates tested. However, first-round recombinants (including T4trDAW7) exhibited in vivo fitness similar to, if not greater than, the WT parent strain despite elevated MICs to the β-lactams. In fact, despite roughly comparable bacterial titers in vivo, the survival of the first-round recombinant strains, T4R1P and T4trDAW7, was significantly decreased compared with T4. This suggests that initial genetic changes in both strains enhanced their overall virulence compared with the parental strain. These data further underscore that β-lactam resistance in S. pneumoniae is tightly constrained by in vivo fitness determinants.
We identified a few significant changes to the virulence phenotypes in the recombinant isolates compared with WT. Furthermore, many in vitro differences did not appear to translate in vivo. For example, the second- and third-round recombinants (T4R2P.1, T4R2P.2, T4R3P.1, and T4R3P.2) all exhibited increased adhesion to A549 cells in vitro, which suggests possible advantages to colonization. However, nasal colonization titers of T4R3P.1 were lower than those of T4 in mice 24 h post-infection. Similarly, the scoring of lung histology (i.e., comparisons of the resistant recombinant pneumococcal strains) suggested a subjective increase in inflammation with recombinant S. pneumoniae strains, compared with WT, despite the lack of significance in inflammatory cytokine profiling of the lungs and blood. Consistent with the alterations in PBP2X found in all resistant pneumococcal recombinants, we found a slight decrease in BOCILLIN FL-binding affinity for PBP2X in all recombinants compared with the parental isolate. Our results appear to indicate that changes to any one virulence trait investigated here were relatively minor and that the cumulative effect of these deficits, and potentially others not identified, was the overall attenuation seen in vivo with some of the recombinant strains.
Although the evolution of antibiotic resistance can often be accompanied by a trade-off in fitness, growth, and/or virulence, the underlying cost associated with resistance development is highly variable and dependent on the genetic mechanisms driving resistance.18,41 Altered β-lactam susceptibility has been associated with differences in virulence and transmission in pneumococcal isolates.42,43 Specifically, resistant pbp alleles are acquired at the expense of fitness determinants in competitive nasal colonization models.17 Our findings demonstrated that resistant strains evolve via different mechanisms (i.e., horizonal transfer versus de novo mutation) and can have strikingly dissimilar in vivo fitness despite exhibiting similar susceptibilities. The attenuation to invasion without significant change in colonization of the de novo mutant strains in vivo indicates that resistance acquired by horizontal transfer may be preferable from an evolutionary standpoint. Mechanistically, horizontal transfer may allow a diverse exchange of genetic material among isolates, which, under drug pressure, might engender selection for low-cost resistance traits or accompanying compensatory genetic changes. In contrast, we observed in vivo that de novo PBP mutations occur with low frequency and are not retained even under penicillin selective pressure, suggesting that pneumococci with de novo PBP mutations are disadvantaged in vivo.
In all cases of β-lactam-induced resistance within the isolates, we identified changes in PBP2X, and in many cases, we saw alterations in PBP2B, especially in later-round recombinants. Mutations in pbp1A and pbp2X can act as compensatory mutations that may restore fitness to S. pneumoniae, stabilizing resistance mutations by allowing their dissemination in the face of competition with WT strains.44 We found more than 200 amino acid changes in PBP2X in many of the recombinant series, which contrasts with the experimentally evolved, penicillin-resistant T4EP populations, which possessed only single amino acid substitutions in the central transpeptidase domain of PBP2X. Despite the relative lack of PBP changes, all in vitro-evolved isolates were greatly attenuated in vivo. In contrast, the recombinant pneumococcal isolates showed attenuated in vivo characteristics only after larger increases in MIC in the later transformations, unsurprisingly when many of the isolates showed large changes in PBP2B. Thus, it appears that the changes to PBP2X in the first-round recombinant isolates had a net-neutral or net-minimal cost to fitness in vivo, with possible compensatory changes conferred by substitutions within PBP2X or from other alterations in the recombinant genome. Several large recombination regions in resistant isolates also involved genes not previously associated with β-lactam resistance. For example, T4R1P possessed genomic recombinations involving luxA, dexB, and clpB, which are not directly involved in β-lactam resistance. These alterations, namely the changes that occurred in the tolerant third-round recombinant strains, may be of interest for investigating determinants of tolerance and resistance. However, the broad scale of change in many of the genes poses a significant hurdle to identifying the contributions of individual mutations.
Relationship between recombination, innate competency, and adaptive β-lactam resistance across streptococcal species
Here, we showed that S. pneumoniae’s innate competence provides for greater genetic diversification and adaption than does de novo mutation. This advantage has important implications for not only pneumococcus but also non-pneumococcal Streptococcus spp. In part, this flexibility afforded by genetic recombination via horizontal gene transfer may explain the lack of β-lactam resistance observed in non-competent Streptococcus spp., such as S. pyogenes (Table S3).21 Despite the presence of orthologous genes that are required for transformation, S. pyogenes generally does not undergo genetic transformation even after induction of competency genes.20 As such, a reliance on de novo mutation without accompanying plasticity to allow compensatory changes could explain the constraints in PBP variability seen in S. pyogenes, for example.45 This could also potentially explain the low rates of resistance seen in group B streptococcus, which is not thought to be naturally competent.19 In contrast, the genomic plasticity of S. pneumoniae allows greater diversity, selection, and adaptation in low-fitness environments,46,47 such as in the presence of antimicrobial agents. Based on this and our current data, we propose that genetic variability via recombination grants a considerable advantage not only in developing antibiotic resistance but also in simultaneously adapting to the host’s response.
Limitations of the study
Commensal streptococcal species have long been known to recombine effectively with S. pneumoniae to confer resistance. However, the cross-species specificity and efficiency of this recombination, while prominently studied in vitro, lacks in vivo data, especially during acute infection, in part due to the technical challenges involved. Additionally, while we have demonstrated the retention and attenuation of virulence in vivo in the first recombinant strain and later recombinants, respectively, the mechanism and precise genetic changes responsible for these differences are yet to be identified. Our work characterized slight changes in damage and inflammation in the lung based on histological scoring among the first recombinant and third recombinant strains, for example, but further investigation of the differences in genomic recombination would be needed to clarify the specific contributors behind the observed differences in mortality seen in our mouse models.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jason Rosch (jason.rosch@stjude.org).
Materials availability
Streptococcal isolates and strains used in this study are readily available upon request from the lead contact.
Data and code availability
Whole genome sequence data have been deposited at the NCBI SRA database and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Streptococcus pneumoniae: serotype 4 | Laboratory strain, gifted by | N/A |
| strain: TIGR4 | Dr. Elaine Tuomanen | |
| Streptococcus pneumoniae: serotype 4 | Laboratory strain, gifted by | N/A |
| strain: TIGR4 | Dr. Timothy van Opijnen | |
| Streptococcus pneumoniae: serotype 23 strain: DAW7 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV2 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV3 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV4 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV5 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV8 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV11 | Clinical isolate, this paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R1P.1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R2P.1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R2P.2 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R3P.1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R3P.2 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP2 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP3 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP4 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP5 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP6 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP7 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP8 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N132P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N232P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N332P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N432P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4trDAW7 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| BOCILLIN™ FL Penicillin, Sodium Salt | Invitrogen | Cat# B13233 |
| Pfizerpen® penicillin G potassium | Pfizer, requested from St. Jude Pharmacy | CAS: 113-98-4 |
| Neomycin | Sigma | Cat# N6386; CAS: 1405-10-3 |
| Defibrinated Sheep’s Blood | Lampire | Cat# 103-100-3 |
| Triton X-100 | Sigma | Cat# X100; CAS: 9036-19-5 |
| Complete, EDTA-free protease inhibitor cocktail tablets | Roche | Cat# 11-836-170 |
| NuPAGE™ LDS Sample Buffer (4X) | ThermoFisher Scientific | Cat# NP0007 |
| TNF-α | ThermoFisher Scientific | Cat# 554618 |
| F12K | ThermoFisher Scientific | Cat# 21127022 |
| Fetal Bovine Serum | Atlanta Biologicals | Cat# S11150 |
| Gentamicin sulfate | Sigma | Cat# G1914 |
| Trypsin-EDTA | ThermoFisher Scientific | Cat# 15400054 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher Scientific | Cat# 15140122 |
| DMEM | ATCC | Cat# 30-2002 |
| BSA | Sigma | Cat# A9418 |
| Proteinase K | Sigma | Cat# P4850 |
| Phenol:Chloroform:isoamyl alcohol | Sigma | Cat# 77617 |
| Chloroform:isoamyl alcohol | Sigma | Cat# C0549 |
| Penicillin E-test strips | Liofilchem | Cat# 921030 |
| p-chloro-phenylalanine | Cayman | Cat# 26168 |
| ExTaq HS Polymerase | Takara | Cat# RR006B |
| erythromycin | Sigma | Cat# E5389 |
| Critical commercial assays | ||
| Milliplex® MAP Kit | EMD Millipore | Cat# MCYTOMAG-70K |
| Nextera XT DNA Library Prep Kit | Illumina | Cat# FC-131-1096 |
| Deposited data | ||
| Pneumococcal genome sequence data | This paper | SRA: PRJNA737504, BioSample: SAMN29540204 |
| Experimental models: Cell lines | ||
| Human: A549 lung epithelial cells | ATCC | CCL-185 |
| Mouse: J774A.1 macrophages | ATCC | TIB-67 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/c | The Jackson Laboratory | RRID: IMSR_JAX:000651 |
| Oligonucleotides | ||
| Primers for barcoded pneumococcal strains, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| Trim Galore version 0.6.4 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| breseq version 0.35.5 | Deatherage et al.48 | https://github.com/barricklab/breseq/releases |
| bowtie2 version 2.3.5 | Langmead et al.49 | https://github.com/BenLangmead/bowtie2/releases |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Female wildtype BALB/c mice were purchased from The Jackson Laboratory and housed in groups of five under a 12-h light and dark cycle with regular husbandry handled by in-house staff of the St. Jude Animal Resource Center. All mice were approximately seven weeks old at the outset of all experiments, and mice were randomly assigned to experimental groups. All experiments, protocols, and procedures involving mice were performed with approval of and in accordance with the NIH Guide for the Care and Use of Laboratory Animals and guidelines of the St. Jude Institutional Animal Care and Use Committee.
METHOD DETAILS
Strains and growth conditions
Streptococcus pneumoniae from frozen glycerol stocks was inoculated onto tryptic soy agar (Millipore Sigma, Billerica, MA) plates containing 20 μg/mL neomycin and 3% defibrinated sheep blood and then incubated overnight at 37°C with 5% CO2. Because S. pneumoniae is naturally resistant to neomycin, it was added to prevent contamination of other bacterial species. Liquid cultures in semi-defined media C + Y or Todd-Hewitt Broth and 2% yeast extract were inoculated with the bacteria grown on the blood agar plate.50
MIC determination
Bacterial strains were inoculated into C + Y from overnight growth on agar plates and incubated at 37°C with 5% CO2 until reaching mid-logarithmic phase at OD620 ~0.4. Then, the bacterial culture (100 μL) was plated on blood agar plates supplemented with neomycin before E-test strips were added to the center of the plates. Plates were incubated overnight at 37°C with 5% CO2. MIC was determined, in terms of μg/mL, where the symmetrical inhibition ellipse edge intersected the E-test strips on the plate. MIC determination was performed in triplicate for each strain.
Generation and transformation of recombinant pneumococcal strains
T4 (from the Rosch laboratory) was grown in C + Y media to OD620 ~0.07 to 0.1. 5 μg gDNA from penicillin-resistant S. pneumoniae or S. viridans strains and competence stimulating peptide 2 was added to T4 cultures to induce and enhance competence.51 Specifically, T4 was transformed with gDNA from SV5 to generate T4R1P. T4R1P was transformed with gDNA from SV3 and SV8 to generate T4R2P.1 and T4R2P.2, respectively. T4R2P.1 was transformed with gDNA from SV11, and T4R2P.2 was transformed with gDNA from SV4 to generate strains T4R3P.1 and T4R3P.2, respectively. Transformation with genomic DNA of the Tn-seq library mutants served as the positive control, as a measure of relative competence during each experiment. The Tn-seq library mutants represented a group that had transposon insertions in nonessential genes in S. pneumoniae. The bacteria were incubated at 37°C with 5% CO2 for 3 h, and the transformants were selected on blood agar plates supplemented with concentrations of penicillin sufficient to prevent growth of the parent strain (0.02 μg/mL, 0.04 μg/mL, and 0.06 μg/mL for the first-, second-, and third-round recombinants, respectively), 3% sheep blood, and 20 μg/mL neomycin after overnight incubation at 37°C and 5% CO2. Transformation efficiency was calculated as the ratio of the number of transformant colonies (CFUs/mL) selected on penicillin-containing plates to the total number of bacteria on neomycin-containing plates.
Generation of barcoded isolates
Barcoded strains were generated through insertion of a 6-nucleotide index sequence unique to that isolate into the neutral CEP locus.52 To generate the barcoded strains without the addition of an antibiotic resistant cassette, a modified PhunSweet cassette was inserted into the CEP site before addition of the barcode (Figure S7).53 To reduce the potential for false positive colonies with kanamycin selection, the Phunsweet cassette was modified by replacing the kanamycin resistance kan with an erythromycin resistance erm, generating PhunSweetErm. The PhunSweet cassette (minus kan) was amplified from TIGR4Δcps:PhunSweet gDNA using primers PhunSweet_F and PhunSweet-kan_R to generate fragment PhunSweet-kan (Table S2).54 The erythromycin resistance cassette erm was amplified from TIGR4ΔspxB:Erm gDNA using primers Erm_F and Erm_R to generate fragment Erm. PhunSweet-kan and Erm PCR products were spliced together using PhunSweet_F and Erm_R primers (PCR3) using SOE PCR.55 To insert PhunSweetErm into the CEP locus, 1 kb fragments upstream and downstream of the CEP insertion site were amplified from TIGR4 gDNA using primers CEP_Up_F/CEP_Up_R and CEP_Down_F/CEP_Down_R, respectively. Of note, in order to generate a tool for other strain backgrounds, more of the transposase between treR (Sp_1885) and amiF (Sp_1887) was excluded than in previous publications in order to increase the homology shared in other strains (D39, BHN97).52,56 These amplicons were spliced with the PhunSweetErm cassette with primers CEP_Up_F and CEP_Down_R to construct CEPΩPhunSweetErm. For generation of TIGR4 CEPΩPhunSweetErm, TIGR4 was grown in 10 mL C + Y until OD620~0.08. 1 mL of culture was incubated with 3 μL of 1 mg/mL CSP2 and the CEPΩPhunSweetErm amplicon for three hours at 37°C, 5% CO2.51 Transformants were selected on plates containing 1 μg/mL erythromycin. Correct insertion of the PhunSweetErm was confirmed through lack of growth on counter-selection plates (15 mM chlorinated-phenylalanine, 10% sucrose). Next, to generate the barcoded isolates, 1 kb fragments upstream and downstream of the CEP insertion site were amplified from TIGR4 gDNA using primers CEP_Up_F/CEP_Index#_R and CEP_Index#_F/CEP_Down_R, respectively, with paired index numbers. These primers were designed such that the unique 6 nucleotide index in each primer pair sat between the binding sequence and the overhang sequence such that a double stranded index would be inserted at the CEP site upon SOE PCR, generating CEPΩIndex#. Barcoded isolates were obtained through transformation of TIGR4 as above using the CEPΩIndex# amplicon as donor DNA and selection on plates containing 15 mM chlorinated-phenylalanine and 10% sucrose. Transformants were confirmed through lack of growth on counter-selection plates (1 μg/mL erythromycin). Correct index sequence was confirmed through Sanger sequencing of the CEP site. All PCR products were amplified using exTaq polymerase (TAKARA) following the recommended guidelines. gDNA was extracted using the aqueous/organic extraction protocol, as described previously.53
In vitro experimental evolution
T4 was used as parental strain in antibiotic-evolution experiments. For strains T4EP1, T4EP2, T4EP3, and T4EP4, the T4 parent strain used to generate each of these populations were from the van Opijnen laboratory. T4 was streaked on blood agar plates and a different individual colony was selected each time to start four, independent cultures of S. pneumoniae. Cultures were continuously passaged in liquid media containing gradually increasing subinhibitory concentrations of penicillin. Briefly, every day populations were inoculated in fresh media at an initial optical density at 600 (OD600) of 0.1 and incubated at 37°C in 5% CO2. After approximately three doubling times, planktonic cells were collected by centrifugation and used as inoculum for next passages. Starting at 0.50xMIC, every five passages the penicillin concentration was increased up to an additional 0.5xMIC and held steady for an additional five passaged before the next increment (MIC based on wild-type T4). Four additional replicate populations were serially passaged in media without antibiotic as controls to identify background mutations unrelated to antibiotic stress. Whole-genome sequencing was performed for each population after 32 passages (approximately 115 generations). To ensure completely independent populations and to eliminate the possibility of cross-contamination between cultures, four additional strains of barcoded T4 (from the Rosch laboratory) possessing unique identifier sequences inserted within the CEP locus were passaged in the same manner. All populations were serially passaged with and without penicillin to identify background mutations.
In vivo virulence determination
All mouse experiments were performed with approval of and in accordance with guidelines of the St. Jude Institutional Animal Care and Use Committee. To examine the bacteria’s virulence, the WT T4 and the derivative, transformant mutants were grown in C + Y to logarithmic phase OD620 ~0.8 and frozen as glycerol stocks at −80°C. Prior to infection, the glycerol stocks were thawed, washed, resuspended in 1× PBS, and enumerated to confirm the correct CFUs infection dose. Seven-week-old female BALB/c mice from Jackson Laboratory were anesthetized with 2.5% isoflurane and infected with the WT and the mutants via low-volume intranasal instillation of 30 μL containing 107 CFUs. After the infection, mice were monitored for survival and disease progression, including lethargy, conjunctivitis, meningitis, pneumonia, and bacteremia. At 24-h post-infection, blood samples were collected via tail-bleeding (5 μL of collected blood was diluted into 1× PBS containing heparin), and lungs and nasal passage samples were collected via dissection. Mice surviving through the study period and moribund mice that were too sick to continue in the study were euthanized using 3 L/min CO2 followed by cervical dislocation. Bacterial burden was enumerated by serially diluting into 1× PBS and plating onto blood agar, and bacterial titers were compared using the nonparametric Mann-Whitney U test. Mouse survival curves were analyzed using the Mantel-Cox log rank test.
Lung harvest and histology
From each infected mouse, the left lung was collected for enumeration of bacterial burden, and the right lung was sent to the Veterinary Pathology Core at St. Jude Children’s Research Hospital for evaluation of inflammation severity. Briefly, right lobes were perfused and fixed with 10% formalin. Sections were stained with hematoxylin-eosin for evaluation under light microscopy. A scoring system was developed to quantify and compare the histopathology of lungs infected by the recombinants with that of their respective WT parental strains. Criteria for the scoring system included vascular injury, leukocytes in lungs, collapsed alveoli, and pulmonary edema. The scoring system was graded on severity and extent of injuries, ranging from 0 = within normal limits, 1 = minimal (rare or inconspicuous lesions), 2 = mild (multifocal or small, focal, or widely separated, but conspicuous lesions), 3 = moderate (multifocal, prominent lesions), 4 = marked (extensive to coalescing lesions or areas of inflammation with some loss of structure), or 5 = severe (diffuse lesion with effacement of normal structure). Cytokines were also collected and analyzed for inflammatory signals, including GM-CSF, IL-1β, IL-6, IL-10, IL-12 (p40), and TNF-α, by using the Milliplex MAP Kit (EMD Millipore Catalog# MCYTOMAG-70K, Burlington, MA). Briefly, serum or homogenized lung tissue supernatant was diluted and assayed against a standard curve using magnetic beads coated with cytokine-specific antibodies. Streptavidin-phycoerythrin was incubated with samples for detection and median fluorescence intensity (MFI) was read via Luminex 200. The MFI output was normalized based on dilution and standard curve and converted to cytokine concentration in each sample.
In vivo nasal colonization determination
To enumerate bacterial burden in the nasopharynx of the infected mice at 24 h post-challenge, the nasal passage composed of nasal cavity, nasopharynx, paranasal sinus was isolated, mashed on a cell strainer, and washed with 750 μL 1× PBS. The mixture was then serially diluted and spotted on blood agar plates prior to incubation at 37°C supplemented with 5% CO2 overnight.
In vivo passaging
Seven-week-old BALB/c mice were intranasally infected with 100 μL of 106 CFU of TIGR4. Eight hours post-infection, infected mice were injected intraperitoneally with 100 μL of penicillin, beginning at 10 mg/kg. Mice were euthanized 24 h post-infection and mouse lungs were excised for processing. Briefly, mouse lungs were homogenized and resuspended in 1 mL of PBS and the resulting suspension was centrifuged for 5 min at 300× g to separate lung tissue and pneumococci. The supernatant was serially diluted and plated onto 3% blood agar plates containing 20 μg/mL neomycin for colony counting. The remaining supernatant was spread across five additional blood agar plates containing neomycin to allow for pneumococcal outgrowth. Agar plates were incubated at 37°C overnight and pneumococci was harvested from outgrowth plates into approximately 15 mL of C + Y media. The resulting culture was aliquoted into 1 mL frozen stocks to be used in the following passage, while the remainder was centrifuged at 6000×g for 5 min to pellet cells for genomic DNA processing. Penicillin doses were increased every 3 “generations” or passages up to a maximum concentration of 40 mg/kg.
BOCILLIN FL–penicillin-binding protein affinity assay
From overnight cultures on blood agar plates, the bacterial strains were inoculated into C + Y and grown to OD620 ~0.4. Then 1 mL of each bacterial culture was pelleted, washed with 100 μL 1× PBS, and resuspended in 100 μL 1× PBS containing 0.5 μg BOCILLIN FL penicillin. After a 30-min incubation at 37°C, the bacteria were pelleted at 6,500 ×g for 10 min and washed with 100 μL 1× PBS to remove unbound BOCILLIN FL penicillin. Each bacterial pellet was resuspended in 100 μL 1× PBS, Triton X-100 (0.1%), and 1× complete miniprotease inhibitor tablet before lysis at 37°C for 30 min and freezing overnight at −20°C. BCA assay was performed to determine the concentration of proteins in each pellet. The pellets were boiled for 3 min at 100°C and then 10 μg protein per sample was resuspended with 1× LDS sample buffer without reducing agent and loaded onto an SDS-PAGE gel (4%–12%). PBP bands were visualized using ChemiDoc with Pro-Q Emerald 488 settings and quantified using Image Lab software.
Adhesion and invasion assays
A modified version of the adhesion and invasion protocol developed by Orihuela and colleagues was followed.57 Briefly, A549 pulmonary epithelial cells were cultivated in 24-well tissue culture plates until having 90%–95% confluent monolayers with approximately 4 × 105 cells/mL. After being activated at 37°C with TNF-α (10 ng/mL) in F12K media supplemented with 10% fetal bovine serum and 20 μg/mL gentamicin, cells were washed twice with 1× PBS and then infected with 1 mL of ~107 bacteria at mid-logarithmic phase OD620 ~0.4, which yields the MOI of ~25:1. Once bacteria and cells were incubated for 30 min at 37°C, the supernatant was serially diluted and plated onto blood agar to determine the number of nonadherent bacteria. Cells with adhered bacteria were washed twice with 1× PBS and detached with 100 μL 0.1% Trypsin-EDTA in 1× PBS for 5 min at 37°C. Cells were resuspended in 900 μL 1× PBS. To determine the number of adherent bacteria, the cell suspension was serially diluted and plated onto blood agar. Ratios of adherent bacteria to total bacteria were calculated for each strain. Total bacteria number was calculated by adding adherent bacteria and nonadherent bacteria. Each bacterial strain was represented in three biological replicates in independent experiments.
For the invasion assay, A549 cells were seeded, activated, and infected, as in the adhesion assay, except for the following differences. After bacterial infection, cells were incubated at 37°C with 5% CO2 for 2 h and washed twice with 1× PBS. To eliminate extra-cellular bacteria, cells were incubated with 1 mL infection media, which contained F12K media, 1:100 Pen/Strep solution, and 200 μL gentamicin), for 1 h at 37°C and 5% CO2 before washing 3 times with 1× PBS. Then 100 μL 0.1% Trypsin-EDTA in 1× PBS was added to each well to detach the cells, and the suspension was incubated for 5 min at 37°C. Cells were lysed by incubating with 200 μL 0.025% ice-cold Triton X-100 in water for 8 min at 37°C. Finally, to determine the number of intracellular bacteria, cells were resuspended by pipetting, and 200 μL of the suspension was spread onto blood agar plates. Ratios of intracellular bacteria to total number of bacteria were compared with those of TIGR4. The total number of bacteria was equal to the sum of intracellular bacteria and extra-cellular bacteria, which were determined by plating the supernatant after a 3-h incubation post-infection with bacteria. Each bacterial strain was assayed in three biological replicates conducted in independent experiments.
Macrophage phagocytosis assay
J774A.1 murine macrophages were subcultured in DMEM (Dulbecco’s Modified Eagle Medium) supplemented with fetal bovine serum (10% vol/vol), 50 mg/mL streptomycin, and 50 units/mL penicillin. Once reaching 70%–80% confluent monolayers in 24-well plates, the cells were washed 3 times with 1× PBS and resuspended in DMEM containing 3% BSA without antibiotics. Cells were activated with fresh murine serum prior to infection with 100 μL bacteria at OD620 ~0.1, which corresponds to an MOI of 50 bacteria to 1 macrophage). Cells were incubated with bacteria at 37°C for 30 min or 1 h and then washed followed by incubation for 1 min with DMEM containing 100 μg/mL gentamicin. The washing and incubation process was repeated three times. Finally, the macrophages were lysed using 0.025% Triton X-100, serially diluted, and plated onto blood agar plates to enumerate viable engulfed, intracellular bacteria.
Time-kill assay
The WT and derivative, transformant, mutant strains were grown in ThyB medium to early-logarithmic phase OD620 ~0.2. The bacteria were exposed to 10× MIC of either penicillin or cefepime. Bacterial CFUs were titrated and plated onto neomycin-containing blood agar plates prior to antibiotic exposure and then titrated every half-hour for 2 h post-exposure.
Genomic DNA extraction
Genomic DNA was extracted from the WT and the derivative, transformant strains via the aqueous/organic extraction method using phenol chloroform. Briefly, the bacteria were grown in C + Y to late logarithmic phase at OD620 ~0.6 and then pelleted by centrifugation at 6000 ×g for 10 min, followed by lysis in 1 mL 1× PBS containing 50 μL 10% deoxycholate, 50 μL 10% SDS, and 10 μL Proteinase K (10 mg/mL). The suspension was incubated until clear (~5 min) at 37°C, mixed with 500 μL phenol:chloroform:isoamyl alcohol, transferred to phase-lock tubes, and centrifuged at maximum speed for 5 min to separate the aqueous and organic phases. Then 500 μL chloroform:isoamyl alcohol was added to and mixed with the aqueous phase and centrifuged (13,000 ×g for 5 min). To precipitate the gDNA, the aqueous phase was transferred to an Eppendorf tube containing 100% ethanol. Precipitated DNA was washed with chilled 70% ethanol, pelleted, dried, and resuspended in nuclease-free water.
Whole-genome sequencing and analysis
Extracted gDNA samples from the recombinants were sequenced in the Hartwell Center at St. Jude. Sequence libraries were barcoded using the Nextera kit, and run on the Illumina HiSeq2000 platform as previously described.58 The resulting paired-end reads were cleaned by Trim Galore version 0.6.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The polymorphism mode of breseq pipeline version 0.35.548 with bowtie2 version 2.3.549 was used to map the trimmed reads and identify genomic variants resulting from recombination events in the S. pneumoniae TIGR4 reference genome (NC_003028.3). Variants that were at a frequency of 5% or more were included. All samples had an average mapped read depth of at least 100×.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were performed using Graphpad Prism 9 software. Results were considered to have reached statistical significance with p values of 0.05 or less. Significance is indicated on respective figures as p < 0.05, p < 0.01, p < 0.001, and p < 0.0001. Statistical significance of titered bacterial counts in the blood, lungs and nasal passages of mice (n = 5) was performed using Mann-Whitney U test. Determination of significance between derivative strains and the parent pneumococcal strain was also performed using Mann-Whitney U test in invasion, adhesion, macrophage phagocytosis and cytokine profiling experiments. A log rank (Mantel-Cox) test was used in determining significance differences in survival curves between infecting strains.
Supplementary Material
Highlights.
Horizontal gene transfer preserves virulence during β-lactam resistance acquisition
De novo resistance mutations in PBPs come at a high fitness cost
Antibiotic tolerance can also be gained via interspecies recombination
Genetic recombination drives initial resistance gain with little fitness trade-offs
ACKNOWLEDGMENTS
This research was supported by NIH grant (5U01AI124302-05) and the American Lebanese Syrian Associated Charities (ALSAC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111835.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Wahl B, O’Brien KL, Greenbaum A, Majumder A, Liu L, Chu Y, Lukšić I, Nair H, McAllister DA, Campbell H, et al. (2018). Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: global, regional, and national estimates for 2000–15. Lancet Global Health 6, e744–e757. 10.1016/S2214-109X(18)30247-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention (2015). Active bacterial core surveillance (ABCs) report, emerging infections program network, Streptococcus pneumoniae https://www.cdc.gov/abcs/reports-findings/survreports/spneu15.pdf.
- 3.Centers for Disease Control and Prevention (2016). Active bacterial core surveillance (ABCs) report, emerging infections program network, Streptococcus pneumoniae https://www.cdc.gov/abcs/reports-findings/survreports/spneu16.pdf.
- 4.Centers for Disease Control and Prevention (2017). Active bacterial core surveillance (ABCs) report, emerging infections program network, Streptococcus pneumoniae https://www.cdc.gov/abcs/reports-findings/survreports/spneu17.html.
- 5.Centers for Disease Control and Prevention (2018). Active bacterial core surveillance (ABCs) report, emerging infections program network, Streptococcus pneumoniae https://www.cdc.gov/abcs/reports-findings/survreports/spneu18.pdf.
- 6.Centers for Disease Control and Prevention (2019). Antibiotic resistance threats in the United States https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf.
- 7.Tleyjeh IM, Tlaygeh HM, Hejal R, Montori VM, and Baddour LM (2006). The impact of penicillin resistance on short-term mortality in hospitalized adults with pneumococcal pneumonia: a systematic review and meta-analysis. Clin. Infect. Dis 42, 788–797. 10.1086/500140. [DOI] [PubMed] [Google Scholar]
- 8.Metlay JP (2004). Antibacterial drug resistance: implications for the treatment of patients with community-acquired pneumonia. Infect. Dis. Clin 18, 777–790. 10.1016/j.idc.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Bracco RM, Krauss MR, Roe AS, and Macleod CM (1957). Transformation reactions between Pneumococcus and three strains of Streptococci. J. Exp. Med 106, 247–259. 10.1084/jem.106.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laible G, Spratt BG, and Hakenbeck R (1991). Interspecies recombinational events during the evolution of altered PBP 2x genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Mol. Microbiol 5, 1993–2002. 10.1111/j.1365-2958.1991.tb00821.x. [DOI] [PubMed] [Google Scholar]
- 11.Dowson CG, Hutchison A, Brannigan JA, George RC, Hansman D, Liñares J, Tomasz A, Smith JM, and Spratt BG (1989). Horizontal transfer of penicillin-binding protein genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 86, 8842–8846. 10.1073/pnas.86.22.8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hakenbeck R, König A, Kern I, van der Linden M, Keck W, Billot-Klein D, Legrand R, Schoot B, and Gutmann L (1998). Acquisition of five high-Mr penicillin-binding protein variants during transfer of high-level beta-lactam resistance from Streptococcus mitis to Streptococcus pneumoniae. J. Bacteriol 180, 1831–1840. 10.1128/JB.180.7.1831-1840.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibold C, Henrichsen J, König A, Martin C, Chalkley L, and Hakenbeck R (1994). Mosaic pbpX genes of major clones of penicillin-resistant Streptococcus pneumoniae have evolved from pbpX genes of a penicillin-sensitive Streptococcus oralis. Mol. Microbiol 12, 1013–1023. 10.1111/j.1365-2958.1994.tb01089.x. [DOI] [PubMed] [Google Scholar]
- 14.Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, et al. (2011). Rapid pneumococcal evolution in response to clinical interventions. Science 331, 430–434. 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen A, Valdórsson O, Frimodt-Møller N, Hollingshead S, and Kilian M (2015). Commensal streptococci serve as a reservoir for beta-lactam resistance genes in Streptococcus pneumoniae. Antimicrob. Agents Chemother 59, 3529–3540. 10.1128/AAC.00429-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakayama A, and Takao A (2003). Beta-lactam resistance in Streptococcus mitis isolated from saliva of healthy subjects. J. Infect. Chemother 9, 321–327. 10.1007/s10156-003-0286-y. [DOI] [PubMed] [Google Scholar]
- 17.Trzcinski K, Thompson CM, Gilbey AM, Dowson CG, and Lipsitch M (2006). Incremental increase in fitness cost with increased beta-lactam resistance in pneumococci evaluated by competition in an infant rat nasal colonization model. J. Infect. Dis 193, 1296–1303. 10.1086/501367. [DOI] [PubMed] [Google Scholar]
- 18.Vogwill T, and MacLean RC (2015). The genetic basis of the fitness costs of antimicrobial resistance: a meta-analysis approach. Evol. Appl 8, 284–295. 10.1111/eva.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glaser P, Rusniok C, Buchrieser C, Chevalier F, Frangeul L, Msadek T, Zouine M, Couvé E, Lalioui L, Poyart C, et al. (2002). Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol. Microbiol 45, 1499–1513. 10.1046/j.1365-2958.2002.03126.x. [DOI] [PubMed] [Google Scholar]
- 20.Mashburn-Warren L, Morrison DA, and Federle MJ (2012). The cryptic competence pathway in Streptococcus pyogenes is controlled by a peptide pheromone. J. Bacteriol 194, 4589–4600. 10.1128/JB.00830-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horn DL, Zabriskie JB, Austrian R, Cleary PP, Ferretti JJ, Fischetti VA, Gotschlich E, Kaplan EL, McCarty M, Opal SM, et al. (1998). Why have group A streptococci remained susceptible to penicillin? Report on a symposium. Clin. Infect. Dis 26, 1341–1345. 10.1086/516375. [DOI] [PubMed] [Google Scholar]
- 22.Oppegaard O, Skrede S, Mylvaganam H, and Kittang BR (2020). Emerging threat of antimicrobial resistance in beta-hemolytic streptococci. Front. Microbiol 11, 797. 10.3389/fmicb.2020.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuchat A (1999). Group B streptococcus. Lancet 353, 51–56. 10.1016/S0140-6736(98)07128-1. [DOI] [PubMed] [Google Scholar]
- 24.Genovese C, D’Angeli F, Di Salvatore V, Tempera G, and Nicolosi D (2020). Streptococcus agalactiae in pregnant women: serotype and antimicrobial susceptibility patterns over five years in Eastern Sicily (Italy). Eur. J. Clin. Microbiol. Infect. Dis 39, 2387–2396. 10.1007/s10096-020-03992-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fluegge K, Supper S, Siedler A, and Berner R (2004). Antibiotic susceptibility in neonatal invasive isolates of Streptococcus agalactiae in a 2-year nationwide surveillance study in Germany. Antimicrob. Agents Chemother 48, 4444–4446. 10.1128/AAC.48.11.4444-4446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim L, McGee L, Tomczyk S, and Beall B (2016). Biological and epidemiological features of antibiotic-resistant Streptococcus pneumoniae in pre- and post-conjugate vaccine eras: a United States perspective. Clin. Microbiol. Rev 29, 525–552. 10.1128/CMR.00058-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cron LE, Stol K, Burghout P, van Selm S, Simonetti ER, Bootsma HJ, and Hermans PWM (2011). Two DHH subfamily 1 proteins contribute to pneumococcal virulence and confer protection against pneumococcal disease. Infect. Immun 79, 3697–3710. 10.1128/IAI.01383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hava DL, and Camilli A (2002). Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol. Microbiol 45, 1389–1406. [PMC free article] [PubMed] [Google Scholar]
- 29.Todorova K, Maurer P, Rieger M, Becker T, Bui NK, Gray J, Vollmer W, and Hakenbeck R (2015). Transfer of penicillin resistance from Streptococcus oralis to Streptococcus pneumoniae identifies murE as resistance determinant. Mol. Microbiol 97, 866–880. 10.1111/mmi.13070. [DOI] [PubMed] [Google Scholar]
- 30.Mouz N, Gordon E, Di Guilmi AM, Petit I, Pétillot Y, Dupont Y, Hakenbeck R, Vernet T, and Dideberg O (1998). Identification of a structural determinant for resistance to beta-lactam antibiotics in Gram-positive bacteria. Proc. Natl. Acad. Sci. USA 95, 13403–13406. 10.1073/pnas.95.23.13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang JR, Idanpaan-Heikkila I, Fischer W, and Tuomanen EI (1999). Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol. Microbiol 31, 1477–1488. 10.1046/j.1365-2958.1999.01291.x. [DOI] [PubMed] [Google Scholar]
- 32.Abraham E (2003). Neutrophils and acute lung injury. Crit. Care Med 31, S195–S199. 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 33.Filipe SR, and Tomasz A (2000). Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc. Natl. Acad. Sci. USA 97, 4891–4896. 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Handwerger S, and Tomasz A (1985). Antibiotic tolerance among clinical isolates of bacteria. Annu. Rev. Pharmacol. Toxicol 25, 349–380. 10.1146/annurev.pa.25.040185.002025. [DOI] [PubMed] [Google Scholar]
- 35.Henriques Normark B, Novak R, Ortqvist A, Källenius G, Tuomanen E, and Normark S (2001). Clinical isolates of Streptococcus pneumoniae that exhibit tolerance of vancomycin. Clin. Infect. Dis 32, 552–558. 10.1086/318697. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Gefen O, Ronin I, Bar-Meir M, and Balaban NQ (2020). Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 367, 200–204. 10.1126/science.aay3041. [DOI] [PubMed] [Google Scholar]
- 37.Windels EM, Michiels JE, Fauvart M, Wenseleers T, Van den Bergh B, and Michiels J (2019). Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J 13, 1239–1251. 10.1038/s41396-019-0344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher RA, Gollan B, and Helaine S (2017). Persistent bacterial infections and persister cells. Nat. Rev. Microbiol 15, 453–464. 10.1038/nrmicro.2017.42. [DOI] [PubMed] [Google Scholar]
- 39.Fauvart M, De Groote VN, and Michiels J (2011). Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol 60, 699–709. 10.1099/jmm.0.030932-0. [DOI] [PubMed] [Google Scholar]
- 40.McCullers JA, English BK, and Novak R (2000). Isolation and characterization of vancomycin-tolerant Streptococcus pneumoniae from the cerebrospinal fluid of a patient who developed recrudescent meningitis. J. Infect. Dis 181, 369–373. 10.1086/315216. [DOI] [PubMed] [Google Scholar]
- 41.Andersson DI, and Levin BR (1999). The biological cost of antibiotic resistance. Curr. Opin. Microbiol 2, 489–493. 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 42.Azoulay-Dupuis E, Rieux V, Muffat-Joly M, Bédos JP, Vallée E, Rivier C, Isturiz R, Carbon C, and Moine P (2000). Relationship between capsular type, penicillin susceptibility, and virulence of human Streptococcus pneumoniae isolates in mice. Antimicrob. Agents Chemother 44, 1575–1577. 10.1128/Aac.44.6.1575-1577.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Domenech de Cellès M, Opatowski L, Salomon J, Varon E, Carbon C, Boëlle PY, and Guillemot D (2011). Intrinsic epidemicity of Streptococcus pneumoniae depends on strain serotype and antibiotic susceptibility pattern. Antimicrob. Agents Chemother 55, 5255–5261. 10.1128/Aac.00249-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albarracín Orio AG, Piñas GE, Cortes PR, Cian MB, and Echenique J (2011). Compensatory evolution of pbp mutations restores the fitness cost imposed by beta-lactam resistance in Streptococcus pneumoniae. PLoS Pathog 7, e1002000, ARTN e1002000. 10.1371/journal.ppat.1002000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayes A, Lacey JA, Morris JM, Davies MR, and Tong SYC (2020). Restricted sequence variation in Streptococcus pyogenes penicillin binding proteins. mSphere 5, e00090–20. 10.1128/mSphere.00090-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanage WP, Fraser C, Tang J, Connor TR, and Corander J (2009). Hyper-recombination, diversity, and antibiotic resistance in pneumococcus. Science 324, 1454–1457. 10.1126/science.1171908. [DOI] [PubMed] [Google Scholar]
- 47.Muzzi A, and Donati C (2011). Population genetics and evolution of the pan-genome of Streptococcus pneumoniae. Int. J. Med. Microbiol 301, 619–622. 10.1016/j.ijmm.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Deatherage DE, and Barrick JE (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol 1151, 165–188. 10.1007/9781-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lacks S, and Hotchkiss RD (1960). A study of the genetic material determining an enzyme in Pneumococcus. Biochim. Biophys. Acta 39, 508–518. 10.1016/0006-3002(60)90205-5. [DOI] [PubMed] [Google Scholar]
- 51.Pozzi G, Masala L, Iannelli F, Manganelli R, Havarstein LS, Piccoli L, Simon D, and Morrison DA (1996). Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: two allelic variants of the peptide pheromone. J. Bacteriol 178, 6087–6090. 10.1128/jb.178.20.6087-6090.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guiral S, Hénard V, Laaberki MH, Granadel C, Prudhomme M, Martin B, and Claverys JP (2006). Construction and evaluation of a chromosomal expression platform (CEP) for ectopic, maltose-driven gene expression in Streptococcus pneumoniae. Microbiology (Read.) 152, 343–349. 10.1099/mic.0.28433-0. [DOI] [PubMed] [Google Scholar]
- 53.Echlin H, and Rosch JW (2020). Advancing genetic tools in Streptococcus pneumoniae. Genes 11, 965. 10.3390/genes11090965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Echlin H, Frank MW, Iverson A, Chang TC, Johnson MDL, Rock CO, and Rosch JW (2016). Pyruvate oxidase as a critical link between metabolism and capsule biosynthesis in Streptococcus pneumoniae. PLoS Pathog 12, e1005951. 10.1371/journal.ppat.1005951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horton RM, Cai ZL, Ho SN, and Pease LR (1990). Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8, 528–535. [PubMed] [Google Scholar]
- 56.Sorg RA, Kuipers OP, and Veening JW (2015). Gene expression platform for synthetic biology in the human pathogen Streptococcus pneumoniae. ACS Synth. Biol 4, 228–239. 10.1021/sb500229s. [DOI] [PubMed] [Google Scholar]
- 57.Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala’Aldeen DAA, and Tuomanen EI (2009). Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest 119, 1638–1646. 10.1172/JCI36759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burcham ZM, Schmidt CJ, Pechal JL, Brooks CP, Rosch JW, Benbow ME, and Jordan HR (2019). Detection of critical antibiotic resistance genes through routine microbiome surveillance. PLoS One 14, e0213280. 10.1371/journal.pone.0213280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequence data have been deposited at the NCBI SRA database and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Streptococcus pneumoniae: serotype 4 | Laboratory strain, gifted by | N/A |
| strain: TIGR4 | Dr. Elaine Tuomanen | |
| Streptococcus pneumoniae: serotype 4 | Laboratory strain, gifted by | N/A |
| strain: TIGR4 | Dr. Timothy van Opijnen | |
| Streptococcus pneumoniae: serotype 23 strain: DAW7 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV2 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV3 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV4 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV5 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV8 | Clinical isolate, this paper | N/A |
| Group viridans streptococcus: SV11 | Clinical isolate, this paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R1P.1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R2P.1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R2P.2 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R3P.1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4R3P.2 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP1 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP2 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP3 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP4 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP5 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP6 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP7 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4EP8 | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N132P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N232P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N332P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: N432P | This paper | N/A |
| Streptococcus pneumoniae: serotype 4 strain: T4trDAW7 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| BOCILLIN™ FL Penicillin, Sodium Salt | Invitrogen | Cat# B13233 |
| Pfizerpen® penicillin G potassium | Pfizer, requested from St. Jude Pharmacy | CAS: 113-98-4 |
| Neomycin | Sigma | Cat# N6386; CAS: 1405-10-3 |
| Defibrinated Sheep’s Blood | Lampire | Cat# 103-100-3 |
| Triton X-100 | Sigma | Cat# X100; CAS: 9036-19-5 |
| Complete, EDTA-free protease inhibitor cocktail tablets | Roche | Cat# 11-836-170 |
| NuPAGE™ LDS Sample Buffer (4X) | ThermoFisher Scientific | Cat# NP0007 |
| TNF-α | ThermoFisher Scientific | Cat# 554618 |
| F12K | ThermoFisher Scientific | Cat# 21127022 |
| Fetal Bovine Serum | Atlanta Biologicals | Cat# S11150 |
| Gentamicin sulfate | Sigma | Cat# G1914 |
| Trypsin-EDTA | ThermoFisher Scientific | Cat# 15400054 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher Scientific | Cat# 15140122 |
| DMEM | ATCC | Cat# 30-2002 |
| BSA | Sigma | Cat# A9418 |
| Proteinase K | Sigma | Cat# P4850 |
| Phenol:Chloroform:isoamyl alcohol | Sigma | Cat# 77617 |
| Chloroform:isoamyl alcohol | Sigma | Cat# C0549 |
| Penicillin E-test strips | Liofilchem | Cat# 921030 |
| p-chloro-phenylalanine | Cayman | Cat# 26168 |
| ExTaq HS Polymerase | Takara | Cat# RR006B |
| erythromycin | Sigma | Cat# E5389 |
| Critical commercial assays | ||
| Milliplex® MAP Kit | EMD Millipore | Cat# MCYTOMAG-70K |
| Nextera XT DNA Library Prep Kit | Illumina | Cat# FC-131-1096 |
| Deposited data | ||
| Pneumococcal genome sequence data | This paper | SRA: PRJNA737504, BioSample: SAMN29540204 |
| Experimental models: Cell lines | ||
| Human: A549 lung epithelial cells | ATCC | CCL-185 |
| Mouse: J774A.1 macrophages | ATCC | TIB-67 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/c | The Jackson Laboratory | RRID: IMSR_JAX:000651 |
| Oligonucleotides | ||
| Primers for barcoded pneumococcal strains, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| Trim Galore version 0.6.4 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| breseq version 0.35.5 | Deatherage et al.48 | https://github.com/barricklab/breseq/releases |
| bowtie2 version 2.3.5 | Langmead et al.49 | https://github.com/BenLangmead/bowtie2/releases |