

Summary
Human mitochondrial genome is transcribed bidirectionally, generating long complementary RNAs that can form double-stranded RNAs (mt-dsRNAs). When released to the cytosol, these mt-dsRNAs can activate antiviral signaling. Here, we present a detailed protocol for the analysis of mt-dsRNA expression. The protocol provides three approaches that can complement one another in examining mt-dsRNAs. While the described protocol is optimized for human cells, this approach can be adapted for use in other animal cell lines and tissue samples.
For complete details on the use and execution of this protocol, please refer to Kim et al. (2022).1
Subject areas: Biotechnology and bioengineering, Cell biology, Immunology, Molecular biology, Protein biochemistry, Protein expression and purification
Graphical abstract
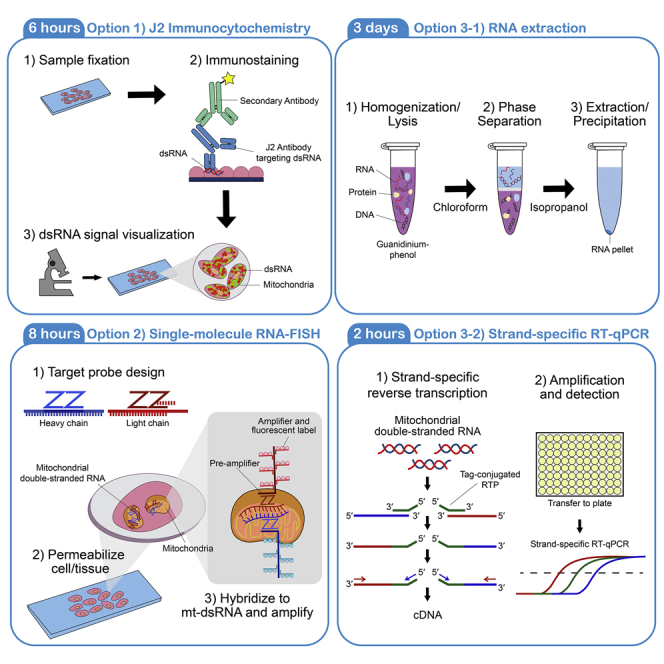
Highlights
-
•
Analysis of mitochondrial double-stranded RNA (mt-dsRNA) expression
-
•
Immunocytochemistry using anti-dsRNA J2 antibody
-
•
Direct mt-dsRNA quantification via fluorescent in situ RNA hybridization
-
•
Strand-specific RT-qPCR for semi-quantitative analysis of mt-dsRNAs
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Human mitochondrial genome is transcribed bidirectionally, generating long complementary RNAs that can form double-stranded RNAs (mt-dsRNAs). When released to the cytosol, these mt-dsRNAs can activate antiviral signaling. Here, we present a detailed protocol for the analysis of mt-dsRNA expression. The protocol provides three approaches that can complement one another in examining mt-dsRNAs. While the described protocol is optimized for human cells, this approach can be adapted for use in other animal cell lines and tissue samples.
Before you begin
The protocol below describes specific steps for mt-dsRNA analysis in human chondrosarcoma cell line SW1353, human salivary gland acinar cell line NS-SV-AC, and human embryonic kidney cell line HEK-293T. However, we have also applied a similar protocol in several human and mouse cell lines as well as in human and mouse tissue samples.
Institutional permissions
All research conducted in this study was approved by the Institutional Review Board (IRB-21-4-92) of Korea Advanced Institute of Science and Technology (KAIST). Anyone who wishes to replicate this protocol must acquire permissions from the relevant institutions.
Cell preparation
Timing: 1 week (for step 1)
CRITICAL: All steps in this part must be performed in a biosafety cabinet. Buffers and experimental materials, including microtubes and microtips, must be autoclaved in advance. All materials must be sterilized with 70% ethanol and stored in a biosafety cabinet.
-
1.Thaw cell stock in a 60-mm dish.
-
a.Pre-warm the cell culture medium in 37°C water bath for at least 15 min.
-
b.Take a frozen cell stock vial (2 × 106 cells/vial) out of a liquid nitrogen tank and gently swirl it in the water bath until only a thin layer of ice remains.CRITICAL: Cells may not adhere well to the surface if the cell stock was kept at 20°C–25°C for a few minutes after thawing.Note: Use personal protective equipment (PPE) such as a lab coat, gloves, and safety goggles whenever handling liquid nitrogen.CRITICAL: Avoid water from entering the cryogenic vial.
-
c.Gently pipette up and down the cells with 1 mL of pre-warmed cell culture medium and transfer them into a new 15 mL conical tube.
-
d.Add 8 mL of the pre-warmed medium into the 15 mL conical tube.
-
e.Centrifuge the tube at 380 × g for 3 min at 20°C–25°C.
-
f.Aspirate the supernatant carefully to keep the pellet intact.
-
g.Gently pipette up and down the cell pellet with 1 mL of fresh medium and transfer the medium into the 60-mm dish.
-
h.Shake the dish until the medium is evenly distributed.
-
i.Incubate the dish in a humid 37°C incubator with 5% CO2 for 18–24 h.
-
j.Replace the medium with fresh medium.Note: Cells should be passaged every 2–3 days at least twice before using them for experiments.
-
a.
-
2.Maintain cells in a 100-mm dish.
-
a.Pre-warm the cell culture medium and phosphate-buffered saline (PBS) in a 37°C water bath for at least 15 min.
-
b.Aspirate the medium and wash the cells with 5 mL of PBS.
-
c.Add 1 mL of 0.05% (w/v) Trypsin-EDTA and shake the dish until Trypsin-EDTA is evenly distributed.
-
d.Aspirate Trypsin-EDTA.
-
e.Incubate the dish in a humid 37°C with 5% CO2 incubator for 3–5 min.Note: Tap the dish to confirm whether cells are lifted. If cells are still attached to the surface, incubate them longer.
-
f.Pipette up and down cells with 4 mL of medium using a pipette aid.
-
g.Transfer 0.5 mL of suspended cells to a new dish.
-
h.Add 7.5 mL of medium to the new dish with cells.
-
i.Incubate cells in a humid 37°C incubator with 5% CO2.
-
a.
Probe preparation
-
3.Prepare a target probe for RNA FISH.
-
a.Search the target RNA at https://acdbio.com/catalog-probes.
-
b.If the target RNA is not included in the catalog list, design the custom probe at https://acdbio.com/target-probes-made-order.
-
a.
Note: When designing the probe, make sure to use different channels for heavy and light strands. If channel 1 (C1) is chosen as the probe targeting the heavy strand mtRNAs, a different channel (channel 2 (C2) or channel 3 (C3)) must be used for the complementary light strand mtRNAs in order to detect both of them simultaneously.
Note: The following method is a modified version of the ACD RNAscope manual. For the original manual, refer to the ACD RNAscope manual (cat# 320293).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| J2 monoclonal antibody, mouse, IgG2a, Kappa chain, 100× | SCICONS | Cat# 10010200; RRID: AB_2651015 |
| Donkey mouse IgG (H+L) secondary antibody, Alex Fluor 555, 1,000× | Invitrogen | Cat# A-31570; RRID: AB_2536180 |
| Chemicals, peptides, and recombinant proteins | ||
| TRIzol reagent | Invitrogen | Cat# 10296028 |
| Recombinant DNase I (RNase-free) | TaKaRa | Cat# 2270 |
| SuperScript IV Reverse Transcriptase | Invitrogen | Cat# 18090200 |
| Paraformaldehyde | Sigma | Cat# P6148; CAS: 30525-89-4 |
| Triton X-100, molecular biology grade | Promega | Cat# H5141 |
| Bovine serum albumin (BSA) | RMBIO | Cat# BSA-BSH |
| 4′,6-Diamidino-2-phenylindole, dihydrochloride (DAPI) | Invitrogen | Cat# D1306; CAS: 28718-90-3 |
| MitoGreen | PromoKine | Cat# PK-CA707-70054 |
| Chloroform | Sigma | Cat# 496189 |
| Acid-phenol:chloroform, pH 4.5 | Invitrogen | Cat# AM9722 |
| 3 M Sodium acetate, pH 5.5 | Invitrogen | Cat# AM9740 |
| GlycoBlue Coprecipitant (15 mg/mL) | Invitrogen | Cat# AM9516 |
| Recombinant RNase Inhibitor (40 U/μL) | TaKaRa | Cat# 2313A |
| SensiFAST SYBR Lo-Rox Kit | Bioline | Cat# BIO-94020 |
| dNTP mmixture (2.5 mM) | TaKaRa | Cat# 4030 |
| Dithiothreitol (DTT) | VWR Chemicals | Cat# 0281 |
| Gelatin, type A | MP Biomedicals | Cat# 901771 |
| Ethanol, absolute | Alfa Aesar | Cat# A9951 |
| Isopropanol | Merck | Cat# 8.18766 |
| Critical commercial assays | ||
| RNAscope Multiplex Fluorescent V2 Assay | ACD | N/A |
| Deposited data | ||
| Unprocessed microscopy data deposited to Mendeley Data | This paper | https://doi.org/10.17632/3r7r9r2n6t.2; https://data.mendeley.com/drafts/3r7r9r2n6t |
| Experimental models: Cell lines | ||
| SW1353 | ATCC | Cat# HTB-94; RRID: CVCL_0543 |
| HEK293T | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| NS-SV-AC | RRID: CVCL_UD37 | |
| Oligonucleotides | ||
| See Table S1 for the List of tag for strand-specific RT-qPCR | This paper | https://doi.org/10.17632/3r7r9r2n6t.2; https://data.mendeley.com/drafts/3r7r9r2n6t |
| See Table S2 for the List of primers for strand-specific reverse transcription | This paper | https://doi.org/10.17632/3r7r9r2n6t.2; https://data.mendeley.com/drafts/3r7r9r2n6t |
| See Table S3 for the List of primers for strand-specific qPCR | This paper | https://doi.org/10.17632/3r7r9r2n6t.2; https://data.mendeley.com/drafts/3r7r9r2n6t |
| Software and algorithms | ||
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Illustrator | Adobe | N/A |
| Other | ||
| Dulbecco’s modified eagle medium (DMEM; High-glucose, with 4 mM L-glutamine, 100 mg/L sodium pyruvate, and 3,700 mg/L sodium bicarbonate) | Welgene | Cat# LM001-05 |
| Phosphate buffered saline (PBS) Tablets | TaKaRa | Cat# T9181 |
| Qualified fetal bovine serum (FBS) | Gibco | Cat# 26140079 |
| Trypsin-EDTA | Welgene | Cat# LS015-01 |
| G-Tube Flat Top Microcentrifuge Tube, Natural | Bio Plas | Cat# 4030 |
| 1.7 mL microcentrifuge tube | Axygen | Cat# MCT-175-C |
| AriaMx Real-time PCR system | Agilent | N/A |
| QuantStudio 1 Real-time PCR system | Thermo Scientific | N/A |
| HybEZ II Oven System | ACDBio | N/A |
| Eppendorf BioPhotometer D30 | Eppendorf | N/A |
| Confocal dish | SPL | Cat# 101350 |
| 8-chamber slide | Thermo Scientific | Cat# 154534PK |
| 60-mm dish | SPL | Cat# 20060 |
| 100-mm dish | SPL | Cat# 20100 |
| 6-well plate | SPL | Cat# 30006 |
| 0.22 μm filter | Merck | Cat# SLGP033RB |
| 15 mL conical tube | SPL | Cat# 50015 |
| MicroAmp 96-well plate | Thermo Scientific | Cat# N8010560 |
| MicroAmp optical adhesive film | Thermo Scientific | Cat# 4311971 |
Materials and equipment
Culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Dulbecco’s Modified Eagle’s Medium – High glucose | N/A | 500 mL |
| Qualified Fetal Bovine Serum | 10% | 50 mL |
| Total | N/A | 550 mL |
Store at 4°C. The solution can be used for up to one month.
Alternatives: Different composition of culture media is required for different cell lines.
4% Paraformaldehyde
| Reagent | Final concentration | Amount |
|---|---|---|
| Paraformaldehyde | 4% | 2 g |
| PBS | N/A | To 50 mL |
| Total | 4% | 50 mL |
Store at 4°C. The solution is stable for up to 1 month. Protect the solution from light.
0.1% Gelatin
| Reagent | Final concentration | Amount |
|---|---|---|
| Gelatin | 0.1% | 0.1 g |
| ddH2O | N/A | To 100 mL |
| Total | 0.1% | 550 mL |
Autoclave and filter the solution with a 0.22 μm filter in a biosafety cabinet. Store the solution at 4°C for up to 3 months.
Secondary antibody mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| Secondary antibody | 0.2 μL | |
| MitoGreen | 500 μM | – |
| DAPI | 450 nM | – |
| 1% BSA in PBST | N/A | 200 μL |
Use a freshly prepared solution. Protect the solution from light.
RNAscope single-molecule RNA-FISH wash buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 50× wash buffer | 1× | 30 mL |
| ddH2O | N/A | 1,470 mL |
| Total | 0.1% | 1.5 L |
Store at 20°C–25°C for up to one month. Prepare the solution in advance because the buffer contains detergent and easily forms bubbles. The solution can be poured down the sink after use.
Step-by-step method details
Option 1: J2 immunocytochemistry
This option describes how to visualize endogenous dsRNAs in cultured cells.
-
1.
Coat a confocal dish with 1 mL of 0.1% gelatin and incubate in a humid 37°C incubator with 5% CO2 for 18 h.
Alternatives: Instead of a confocal dish, coverslips can be used after flame sterilization.
-
2.
Aspirate the gelatin and wash the dish with the cell culture medium.
-
3.
Seed 1 × 105 cells on the confocal dish in 1 mL of the cell culture medium (70%–80% cell density) and incubate in a 37°C incubator with 5% CO2 for 18 h.
-
4.
Wash cells once with PBS pre-warmed to 37°C.
-
5.
Add 1 mL of 4% paraformaldehyde and incubate cells for 10 min at 20°C–25°C.
-
6.
Rinse cells with PBS twice.
-
7.
Wash cells with PBS twice for 5 min.
-
8.
Permeabilize cells in 1 mL of 0.1% Triton X-100 in PBS for 10 min at 20°C–25°C.
Note: Before diluting the 0.1% Triton X-100, prepare 10% Triton X-100 in PBS in advance. It is difficult to make 0.1% Triton X-100 directly from Triton X-100 because it is very viscous.
-
9.
Rinse cells twice with PBST.
Note: To make PBST, add 1 mL of Tween-20 to 1 L of PBS. Make PBST one day prior to the experiment in order to prevent excess bubble formation.
-
10.
Add 1 mL of 1% bovine serum albumin (BSA) in PBST and incubate cells for 1 h at 20°C–25°C.
Note: 1% BSA solution must be cleaned with a 0.22 μm filter before use to remove precipitants, which may increase background noise. Filtered BSA solution can be stored at 4°C for up to a month.
-
11.
Remove the solution.
-
12.
Add 200 μL of 1:100 diluted J2 antibody in 1% BSA and incubate for 2 h at 20°C–25°C. Troubleshooting 1.
Note: The antibody solution can be stored at 4°C for up to a month and reused at most three times.
-
13.
Rinse cells with PBST twice.
-
14.
Wash cells with PBST three times, each time for 10 min, at 20°C–25°C.
-
15.
Add 200 μL of secondary antibody mixture and incubate for 45 min at 20°C–25°C.
-
16.
Rinse cells with PBST twice.
-
17.
Wash cells with PBST three times for 10 min each at 20°C–25°C.
-
18.
Detach the cover glass from the confocal dish (Methods video S1).
-
19.
Put 10–20 μL of mounting solution on a new microscope slide.
-
20.
Place the cover glass on the microscope slide with the mounting solution.
Note: Avoid bubbles forming between the cover glass and the microscope slide as they result in high autofluorescence. To remove bubbles, press lightly on the area with bubbles to release air from that area (Methods video S2).
A video demonstration to separate the cover glass from the confocal dish.
-
21.
Seal the edges of the cover glass with clear nail polish.
-
22.
Air-dry for 5 min at 20°C–25°C.
-
23.
Store the sample in a light-protective box and keep it at 4°C for 18 h.
-
24.
Observe the cells and capture images using a confocal microscope with a 63× objective. Troubleshooting 2. Troubleshooting 3.
Alternatives: 40× and 100× objectives can be used for imaging.
A video demonstration to place the cover glass on the microscope slide.
Option 2: Single-molecule RNA-FISH using RNAscope
This option describes the process of visualizing heavy and light strands of mt-dsRNAs independently at a single cell level.
Note: The protocol is optimized for adherent cells. When suspension cells and formalin-fixed, paraffin-embedded (FFPE) tissues are used for mt-dsRNA visualization, users must test and optimize the conditions appropriately. Troubleshooting 4.
-
25.
Seed 2 × 104 cells on an 8-chamber slide in 200 μL of the cell culture medium (70%–80% cell density) and incubate in a humid 37°C incubator with 5% CO2 for 18 h.
-
26.
Remove the culture medium and wash cells with cold (4°C) PBS once.
-
27.
Remove PBS, add 200 μL of 4% paraformaldehyde per each well, and incubate for 10 min at 20°C–25°C to fix the cells.
-
28.
Remove the paraformaldehyde and rinse the cells with PBS twice.
-
29.
Carefully remove the frame from the 8-chamber slide (Figure 1).
Note: Lift the frame slowly from the end. The slide can break if you lift the frame directly up with too much force.
-
30.
Add 80 mL of 50% ethanol in a staining jar and submerge the slide for 5 min at 20°C–25°C.
-
31.
Replace the solution with 80 mL of 70% ethanol and submerge the slide for 5 min at 20°C–25°C.
-
32.
Replace the solution with 80 mL of 100% ethanol and submerge the slide for 5 min at 20°C–25°C.
-
33.
Replace the ethanol solution with fresh 100% ethanol and submerge the slide for 10 min at 20°C–25°C.
Pause point: The slides can be stored in 100% ethanol at −20°C for up to 6 months.
-
34.
Submerge the slide in 70% ethanol for 2 min at 20°C–25°C.
-
35.
Replace the solution with 80 mL of 50% ethanol and submerge the slide for 2 min at 20°C–25°C.
-
36.
Replace the solution with PBS and submerge the slide for 10 min at 20°C–25°C.
Note: The dehydration and rehydration process helps prevent damage to the cell morphology during the RNA-FISH protocol.
-
37.
Draw a boundary around each well with a hydrophobic barrier pen and dry the barrier for about 30 s.
-
38.
Rinse slides with PBS.
-
39.
Prepare diluted protease III.
Diluted protease III
| Reagent | Amount |
|---|---|
| Protease III | 4 μL |
| PBS | 56 μL |
Figure 1.

Handling the 8-chamber slide
Picture of the method to remove the frame without breaking the slide glass.
For multiple samples, multiply each volume by the number of samples. The solution should be prepared immediately before use.
-
40.
Drop 60 μL of diluted protease III on each well and incubate for 10 min at 20°C–25°C.
-
41.
Warm up the humidity control tray (PN310012) of the HybEZ II hybridization system with wet paper towels at 40°C (Figure 2).
Note: During incubation, warm probes for 10 min at 40°C in the hybridization system.
Alternatives: A dry bath or other ovens can be used instead of the hybridization system to pre-warm probes and other reagents.
-
42.
Wash the slide glass twice with PBS.
-
43.
Prepare a probe mixture. Vortex for 5 s and spin down.
Probe mixture
| Reagent | Amount |
|---|---|
| Probe C1 | 78.4 μL |
| Probe C2 | 1.6 μL |
For multiple samples, multiply each volume by the number of samples.
Note: The probes can be stored at 4°C for up to 6 months.
-
44.
Remove excess liquid from the slide glass (Figure 3).
Note: If you shake off the liquid, cells may fall off.
-
45.
Place the slide glass in the HybEZ slide rack.
-
46.
Drop 80 μL of the mixed probe on each well.
-
47.
Close the tray and incubate it in the humid HybEZ II hybridization system at 40°C for 2 h.
-
48.
Remove the tray from the hybridization system.
-
49.
Wash the slides in the RNAscope single-molecule RNA-FISH wash buffer (wash buffer) for 2 min at 20°C–25°C (Figure 4). Repeat the wash step once more.
Note: To save the use of wash buffer, prepare an empty 10 μL microtip rack. Pour 50 mL of wash buffer into the rack. Separate the slide glass from the tray and immerse it in the wash buffer.
Note: For detection and signal enhancement, four amplification (Amp) reagents are used in order. The last Amp (Amp 4-FL) has several alternatives to determine the final fluorescence per channel as summarized below. In our protocol, Amp 4-FL Alt C is used because we used MitoGreen dye with 488 nm excitation wavelength to visualize mitochondria.
| Probe channel ID | Amp 4-FL Alt A | Amp 4-FL Alt B | Amp 4-FL Alt C |
|---|---|---|---|
| C1 | Alexa 488 | Atto 550 | Atto 550 |
| C2 | Atto 550 | Alexa 488 | Atto 647 |
-
50.
During incubation, warm Amp 1-FL for 5 min at 40°C in the hybridization system.
-
51.
Remove excess liquid from the slide.
-
52.
Put the slide back into the slide rack.
-
53.
Add 2 drops (around 60 μL) of the Amp 1-FL to each well.
-
54.
Close the tray and incubate it in the hybridization system at 40°C for 30 min.
Note: During incubation, warm Amp 2-FL for 5 min at 40°C in the hybridization system.
-
55.
Remove the tray from the hybridization system and submerge the slides in the wash buffer for 2 min at 20°C–25°C. Repeat the wash step once more.
-
56.
Remove excess liquid from the slide.
-
57.
Put the slide back into the slide rack.
-
58.
Add 2 drops of the Amp 2-FL to each well.
-
59.
Close the tray and incubate it in the hybridization system at 40°C for 15 min.
Note: During incubation, warm Amp 3-FL for 5 min at 40°C in the hybridization system.
-
60.
Remove the tray from the hybridization oven and submerge the slides in the wash buffer for 2 min at 20°C–25°C. Repeat the wash step once more.
-
61.
Remove excess liquid from the slide.
-
62.
Put the slide back into the slide rack.
-
63.
Add 2 drops of the Amp 3-FL to each well.
-
64.
Close the tray and incubate it in the hybridization system at 40°C for 30 min.
Note: During incubation, warm Amp 4-FL-Alt C for 5 min at 40°C in the hybridization system.
-
65.
Remove the tray from the hybridization system and submerge the slides in the wash buffer for 2 min at 20°C–25°C. Repeat the wash step once more.
-
66.
Remove excess liquid from the slide.
-
67.
Put the slide back into the slide rack.
-
68.
Add 2 drops of the Amp 4-FL-Alt C to each well.
-
69.
Close the tray and incubate it in the hybridization system at 40°C for 15 min.
-
70.
Remove the tray and submerge the slides in the wash buffer for 2 min at 20°C–25°C.
-
71.
Repeat the wash step once more.
-
72.
Add 80 μL of the mitochondria staining mixture on each well.
Mitochondria staining mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| MitoGreen | 1 μM | |
| DAPI | 450 nM | |
| PBST | 80 μL |
For multiple samples, multiply each volume by the number of samples.
-
73.
Incubate for 15 min at 20°C–25°C.
-
74.
Wash the slide twice for 2 min with PBST in a 10 μL pipette tip tray.
-
75.
Add a drop (around 10 μL) of mounting solution on each well and place the cover glass on top, avoiding bubbles.
-
76.
Seal the edges with clear nail polish.
-
77.
Air-dry for 5 min at 20°C–25°C.
-
78.
Store the sample in a light-protective box and keep it at 4°C for 18 h.
-
79.
Observe the cells and capture images using a confocal microscope with a 63× objective. Troubleshooting 2. Troubleshooting 3.
Figure 2.

Setup of the humidity control tray in the HybEZ II hybridization system
Creating a gasket-sealed, temperature-controlled humidifying chamber necessary for optimized single-molecule RNA-FISH assay using the HybEZ II hybridization system.
Figure 3.

Removing the excess liquid using a 3M paper
Figure 4.

Washing the slide glass while minimizing the use of the wash buffer
Option 3: RNA extraction and strand-specific RT-qPCR
This option covers the steps of extracting RNA from cultured cells and quantitatively analyzing the expression of mtRNAs from both heavy and light strands.
Option 3-1: RNA extraction
Note: It is essential to remove all RNases and fine dust before starting the experiment by wiping the bench with 10% bleach (sodium hypochlorite) followed by 70% EtOH. RNases, especially those in the RNase A family, contain multiple cysteine residues that form intramolecular disulfide bonds.2 In particular, RNase A acts as an endonuclease by specifically cleaving the P-O5′ bond of RNA after pyrimidine residues.3 Bleach eliminates the universally present RNase A by inducing the cleavage of disulfide bonds.4 Commercial systems, such as RNaseZap, can be used instead.
-
80.Seed cells in an appropriate dish.
-
a.After trypsinization, transfer 3 × 105 cells in 1 mL of the culture medium to a 6-well plate.
-
b.Incubate cells in a humid 37°C incubator with 5% CO2.
-
a.
-
81.
Aspirate the culture medium.
-
82.
Add 1 mL/well of TRIzol directly onto cells grown in a 6-well plate.
-
83.
Suspend the solution by pipetting up and down until all cells are dissolved in TRIzol and transfer the solution into a 1.7 mL microcentrifuge tube.
-
84.
Add 200 μL of chloroform and vortex the solution thoroughly for 30 s.
-
85.
Centrifuge the sample at 13,523 × g for 15 min at 4°C.
-
86.
Carefully transfer the supernatant to a 1.5 mL G-Tube.
Note: It is better to use a 200 μL rather than 1,000 μL tip to carefully and thoroughly remove the aqueous layer.
-
87.
Add 600 μL of an isopropanol mixture solution.
Isopropanol mixture
| Reagent | Final ratio | Amount |
|---|---|---|
| Sodium acetate (pH 5.5) | 10% | 50 μL |
| Isopropanol | 110% | 550 μL |
| GlycoBlue | 0.5 μL |
For multiple samples, multiply each volume by the number of samples.
-
88.
Vortex for 5 s and incubate the sample at −20°C for 18 h.
-
89.
Centrifuge the sample at 21,130 × g for at least 1 h at 4°C. Troubleshooting 5.
-
90.
Remove the supernatant and wash the pellet with 1 mL of 75% ethanol at 13,523 × g for 5 min at 4°C.
-
91.
Remove the supernatant and repeat the wash step.
-
92.
Air-dry for 15 min at 20°C–25°C.
Note: While air-drying, protect the RNA pellet by covering the microtubes with plastic wrap.
-
93.
Add 42 μL of RNase-free water to the sample and dissolve the pellet thoroughly by pipetting.
-
94.
Add 8 μL of a DNase I mixture solution.
DNase I mixture
| Reagent | Volume |
|---|---|
| 10× DNase I buffer | 5 μL |
| RNase Inhibitor | 1 μL |
| DNase I | 2 μL |
For multiple samples, multiply each volume by the number of samples.
-
95.
Mix the solution by tapping and spin down the sample.
-
96.
Incubate the sample in a 37°C heat block for 30 min.
-
97.
Add 150 μL of RNase-free water and 200 μL of acid-phenol.
-
98.
Vortex vigorously for 30 s.
-
99.
Centrifuge the sample at 13,523 × g for 5 min at 20°C–25°C.
Note: Close the microtube lid tightly to prevent leakage. White crystals of acid-phenol may form inside the centrifuge, which needs to be cleaned for safety.
-
100.
Carefully transfer 200 μL of supernatant to a fresh G-tube.
-
101.
Add 1 mL of 100% ethanol, 20 μL of sodium acetate (pH 5.5), and 0.5 μL of GlycoBlue.
-
102.
Vortex for 5 s and incubate the sample at −80°C for 18 h.
-
103.
Centrifuge the sample at 21,130 × g for at least 1 h at 4°C. Troubleshooting 5.
-
104.
Remove the supernatant and wash the RNA pellet with 75% ethanol at 13,523 × g for 5 min at 4°C.
-
105.
Remove the supernatant and repeat the wash step.
-
106.
Air-dry for 15 min at 20°C–25°C.
-
107.
Dissolve the pellet in 30 μL of RNase-free water. Troubleshooting 6.
Note: After dissolving the RNA pellet, measure the concentration and analyze RNA quality. The ratio of 28S:18S rRNA observed by gel electrophoresis should be greater than 2.0 (Figure 5). The ratio of A260/A280 and A260/A230 should be 1.8–2.0 and higher than 2.0, respectively, when analyzed by a spectrophotometer.
Figure 5.
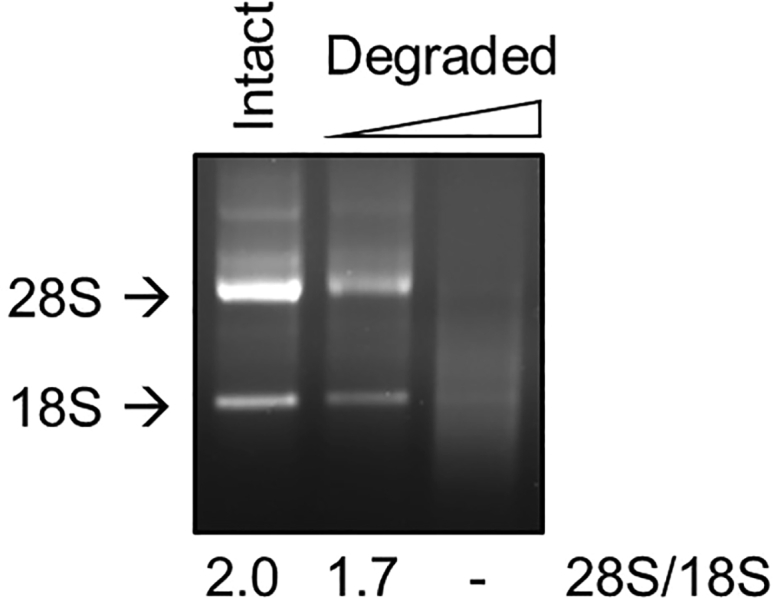
Analyzing RNA quality using gel electrophoresis
1 μg of intact and degraded total RNAs extracted from HEK-293T cells were run on a 1.5% agarose gel. The 28S and 18S ribosomal RNA (rRNA) bands were detected by EtBr staining and the intensity ratio (28S rRNA/18S rRNA) was determined, which was 2.0 for the intact RNA sample. In the case of partially degraded sample, the rRNA ratio was 1.7, and for the completely degraded sample, we could not calculate the ratio.
Option 3-2: Strand-specific RT-qPCR
Figure 6.

Comparison of GAPDH Cq values for three different RTP tags on the same amount of RNAs extracted from HEK-293T cells
Each Cq value is the average of three biological replicates.
-
108.Prepare 4 μM RTP stock.
-
a.To make an RTP stock, mix 100 μM of primer stocks to make a total concentration of 4 μM.
-
a.
Note: For example, if you have four 100 μM primers, add 1 μL of each primer and 96 μL of triple distilled water (TDW).
-
109.
Make a strand-specific RT pre-mixture solution in a PCR tube.
Strand-specific RT pre-mixture
| Reagent | Amount |
|---|---|
| RNA | 500 ng - 2 μg |
| 4 μM RTP | 0.5 μL |
| 2.5 mM dNTP | 4 μL |
| TDW | To 13 μL |
-
110.
Heat the pre-mixture solution at 65°C for 5 min and cool the sample at 4°C.
-
111.
Add 7 μL of a strand-specific RT second mixture solution. Troubleshooting 7.
Strand-specific RT second mixture
| Reagent | Amount |
|---|---|
| 5× SSIV buffer | 4 μL |
| 100 mM DTT | 1 μL |
| Recombinant RNase inhibitor | 1 μL |
| SuperScript IV Reverse Transcriptase (200 U/μL) | 1 μL |
For multiple samples, multiply each volume by the number of samples.
Alternatives: For reverse transcription, other reverse transcriptases such as RevertAid (Thermo Fisher Scientific) can be used.
-
112.
Incubate the sample in a PCR machine with the following procedure.
Strand-specific RT reaction condition
| Steps | Temperature | Time |
|---|---|---|
| Reaction | 52.5°C | 10 min |
| Inactivation | 80°C | 10 min |
| Hold | 4°C | Forever |
-
113.
To prepare a 3 μM of qPCR primer mix, add 3 μL of the target (sense or antisense) and tag-only qPCR primer stock (100 μM) to 94 μL of TDW.
-
114.
Prepare a strand-specific qPCR pre-mixture.
Strand-specific qPCR pre-mixture
| Reagent | Amount |
|---|---|
| 2× SYBR reagent | 7.5 μL |
| TDW | 4.5 μL |
| 3 μM qPCR primer mix | 2 μL |
For multiple samples, multiply each volume by the number of samples.
Note: For accurate analysis, a technical triplicate is highly recommended.
-
115.
Add 14 μL of the strand-specific qPCR pre-mixture in each well of a 96-well PCR plate.
-
116.
Carefully add 1 μL of cDNA and seal the plate with an optical adhesive film.
Note: Be careful not to generate bubbles as they can cause inaccurate fluorescent readings. To remove bubbles, spin down the plate for 30 s using a centrifuge.
-
117.
Centrifuge the plate for 5 s.
-
118.
Run qPCR using the following procedure. Troubleshooting 8.
qPCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 3 min | 1 |
| Denaturation | 95°C | 5 s | 40 cycles |
| Annealing | 60°C | 10 s | |
| Melt Curve Stage 1 | 95°C | 15 s | 1 |
| Melt Curve Stage 2 | 60°C | 1 min | 1 |
| Melt Curve Stage 3 | 95°C | 1 s | 1 |
Expected outcomes
Representative images of dsRNAs visualized through J2 immunocytochemistry (ICC) are shown in Figure 7. Mitochondria were stained with MitoGreen, and dsRNAs were shown in red (Figure 7A). These images were obtained using a LSM780 confocal microscope with 15% laser and 11.6 s frame time setting. Considering that the range of signal intensity was from 0 to 85 for all three colors, we defined the true signal as fluorescence intensities greater than 20 (Figure 7B). Typically, we expect more than 50% of cells to exhibit strong signals. In this experiment, we transfected NS-SV-AC cells with 20 μg/mL of polyinosinic:polycytidylic acid (poly(I:C)) for 14 h and examined the expression of mt-dsRNAs. Consistent with a recent study, we observed increased expression of dsRNAs by poly(I:C) stimulation.5 The overlapping signals between dsRNAs and mitochondria indicate a potential increase in mt-dsRNA expression.
Figure 7.

The expression of dsRNAs and analysis of the dsRNA signal
(A) The expression of dsRNAs (visualized by J2 antibody, red) and mitochondria (MitoGreen, green) in NS-SV-AC cells upon poly(I:C) transfection. The white and yellow scale bars indicate 50 and 20 μm, respectively.
(B) Analysis of the dsRNA signal and background noise. The light yellow line across the signal represents the fluorescence intensity per pixel.
Although the J2 antibody detects long dsRNAs including mt-dsRNAs, it can also recognize other cellular dsRNAs, such as those from inverse repeats of Alus, and even poly(I:C) RNAs. Therefore, the increased red fluorescent signal from Figure 7A may not reflect the increased expression of mt-dsRNAs. Through single-molecule RNA-fluorescent in situ hybridization (RNA-FISH), we could further analyze the expression of mt-dsRNAs by directly visualizing both heavy and light strands of mtRNAs. Representative images from the RNA-FISH experiment are shown in Figure 8. As a representative mt-dsRNA, we examined ND5 mRNA (ND5 Heavy) and its complementary non-coding RNA (ND5 Light) in poly(I:C) transfected NS-SV-AC cells. The mitochondrial localization of these RNAs can be confirmed by co-staining the cells with MitoGreen. For imaging, we used 2% laser and 15.5 s frame time setting. Clearly, we could confirm the increased expression of ND5 mt-dsRNA by poly(I:C) stimulation as both ND5 Heavy and ND5 Light RNA fluorescent signals were enhanced. In particular, our analysis using the Image J program showed that the overlapping yellow signal was increased from 25% to 35% of the green signal in response to poly(I:C) transfection.
Figure 8.

Analyzing mt-dsRNAs via RNA-FISH
The expression of ND5 mtRNAs (red and green for heavy and light strand mtRNAs, respectively) and MitoGreen (cyan) in NS-SV-AC cells upon poly(I:C) transfection, visualized through RNA-FISH. The white and yellow scale bars indicate 50 and 20 μm, respectively.
Single-molecule RNA-FISH is a powerful technique that allows the quantification of transcript expression without a normalization control. However, it requires separate probes and imaging for individual mt-dsRNAs. For the analysis of multiple mt-dsRNAs in response to poly(I:C) stimulation, we applied strand-specific RT-qPCR. Our data showed that the levels of all analyzed mt-dsRNAs were significantly increased upon poly(I:C) transfection (Figure 9).
Figure 9.

Analyzing mt-dsRNA expression through strand-specific RT-qPCR
Strand-specific RT-qPCR analysis showing mt-dsRNA expression level in cells transfected with 20 μg/mL of poly(I:C) for 14 h. The graph represents three independent experiments with error bars denoting means ± SEM. The statistical significances were calculated using one-tailed Student's t-tests, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Quantification and statistical analysis
Statistical analyses were performed using Microsoft Excel. Data are presented as means ± standard error of the mean (SEM). P-values were determined using a one-tailed Student’s t test. (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Limitations
Currently, J2 ICC is broadly used in analyzing mt-dsRNA expression.6,7,8 However, one limitation is that J2 antibody does not specifically detect mt-dsRNAs and can capture other cellular dsRNAs including short interspersed nuclear elements (SINE) and endogenous retroviruses (ERVs).9,10,11 Previous studies that employed high-throughput sequencing on RNAs captured by the J2 antibody showed that the major RNA antigen of J2 antibody is SINE elements.11,12 By analyzing signals that colocalize with mitochondria, J2 ICC can still offer reliable analysis of the mt-dsRNA expression. Yet, the exact identity of dsRNA visualized by J2 ICC should be further confirmed via additional experiments such as single-molecule RNA-FISH.
RNA-FISH can specifically detect both heavy and light strands of mtRNAs. By combining the signals from two separate strands, we can easily evaluate whether mt-dsRNA is formed at a single-molecule resolution. In addition, the mt-dsRNA location can be evaluated by staining other cell organelles such as mitochondria. However, setting up a proprietary platform and designing RNA probes are relatively costly, which makes it challenging to analyze all mt-dsRNAs. Furthermore, RNAscope probes may show different target efficiency because each probe relies on sequence-specific interaction with the target mtRNA, which may result in underestimated co-localization signals. Lastly, as mitochondria from other animals have different genomes, new probes are required to analyze mt-dsRNAs from different species.
RT-qPCR is one of the most conventional methods in the semi-quantitative analysis of RNA expression.13 Strand-specific RT-qPCR is accessible and economical in analyzing all strands of mt-dsRNAs. However, one caveat of the strand-specific RT-qPCR is that it relies on a housekeeping gene such as GAPDH as a normalization control. Considering that the expression of housekeeping genes can be altered between two conditions, it is highly recommended to use additional normalization controls (such as β-Actin) or complement the data with additional analysis such as RNA-FISH, which does not require a normalization control. Another critical note regarding the strand-specific RT-qPCR is that the RT and qPCR efficiency may vary depending on the RTP tags. In our experiment, we tested SP6 and CMV promoters and GST sequences as candidate RTP tags. Notably, they resulted in very different Cq values even when the same amount of RNA was used to amplify the same target gene (Figure 6). One factor is the GC content as the SP6 tag with the lowest GC content (33.3%, Tm 45.2°C) resulted in the highest Cq values. Still, CMV sequences showed better RT and PCR amplification efficiencies than GST sequences despite both RTP tags sharing the same GC content of 66.67% (Tm 63.7°C). A possible explanation is the location of the GC sequences because CMV tag contains most of its GC sequences in the 3′ end (Table S1). Therefore, among the three tags we tested, CMV sequence was the most suitable one for the strand-specific RT-qPCR. However, as the list of our candidate RTP tags was not exhaustive, we recommend that users must confirm the RT and PCR efficiencies of the RTP tag of choice in advance.
Troubleshooting
Problem 1
The RNA fluorescent signal is too weak (step 12).
Potential solution
-
•
Improve the staining efficiency by preparing a fresh antibody mix.
-
•
Increase the antibody concentration.
Problem 2
The background signal is too high (steps 24 and 79).
Potential solution
-
•
Increase the number of wash steps.
-
•
Avoid bubbles between the cover glass and the microscope slide.
-
•
Use freshly made gelatin.
Problem 3
The nuclear morphology is damaged (steps 24 and 79).
Cells are lost during the protocol (steps 24 and 79).
Potential solution
-
•
Use cells that have been incubated for at least 18 h after seeding.
-
•
Increase cell density.
-
•
Prevent cell damage due to hydraulic pressure by adding the solution along the wall of the well rather than directly onto the cells.
Problem 4
Application of RNA-FISH in suspension cells and formalin-fixed, paraffin-embedded (FFPE) tissue.
Potential solution
-
•
For RNA-FISH in suspension cells, cells must be air-dried to attach to the cover glass.
-
•
For FFPE tissue RNA-FISH, retrieval steps are required to expose the antigen epitope before the protease treatment (step 40). For antigen retrieval, slides with FFPE tissue pieces are incubated for 15 min in hot (around 99°C) target retrieval reagent from ACDBio.
Problem 5
The RNA pellet is too small, and the RNA concentration is low (steps 89 and 103).
Potential solution
-
•
Centrifuge the sample for a longer time (up to 3 h) during ethanol precipitation. Tapping the wall of the microtube and providing additional GlycoBlue (0.5–1 μL) may be needed in the middle of centrifugation.
-
•
Use larger dishes to harvest more cells.
Problem 6
Low RNA quality (step 107).
Potential solution
-
•
Before the RNA extraction, wipe the bench with freshly made 10% bleach and 70% ethanol.
-
•
Use clean equipment wiped with 10% bleach.
-
•
Always close the lid on microtip racks except when in use and avoid microtips from touching elsewhere.
-
•
Vortex the solution with max speed to thoroughly mix two phases for a longer time (up to 1 min) (steps 84 and 98).
-
•
Avoid touching the layer during the TRIzol-chloroform and Acid-phenol extraction steps (steps 86 and 100).
Problem 7
Compromised efficiency of reverse transcription (step 111).
Potential solution
-
•
Check the RNA concentration. Using too much RNA may reduce the reverse transcription efficiency. The maximum amount of RNA for reverse transcription is 5 μg.
-
•
Tap the microtubes to mix the solution and spin down to collect all the samples to the bottom of the tube.
-
•
High GC content of RTP tags tends to result in higher RT and PCR amplification efficiencies.
-
•
Concentrate GC sequences toward the 3′ end of tags for better amplification efficiencies.
Problem 8
A large difference between technical replicates of qPCR (step 118).
Potential solution
-
•
Make sure to put the same volume of reagents in each well.
-
•
Centrifuge the qPCR plate for 30 s to collect all reagents to the bottom of the tube.
-
•
Run a self-diagnosis of the qPCR machine to check for errors such as failed heating of the thermal block.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yoosik Kim (ysyoosik@kaist.ac.kr).
Materials availability
No materials were generated in this study.
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2022R1C1C1008850) funded by the Korean government’s Ministry of Science and ICT. S.K. was supported by NRF-2020R1A6A3A13076492.
Author contributions
Conceptualization, S.K., Y.K.; Methodology, S.K., Y.K.; Investigation, S.K., J.Y.; Writing – original draft, S.K.; Writing – review & editing, J.Y., K.L., Y.K.; Funding acquisition, S.K., Y.K.; Supervision, Y.K.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.102007.
Contributor Information
Sujin Kim, Email: kuki430@kaist.ac.kr.
Yoosik Kim, Email: ysyoosik@kaist.ac.kr.
Supplemental information
Data and code availability
-
•
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/3r7r9r2n6t.2.
-
•
This manuscript does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Kim S., Lee K., Choi Y.S., Ku J., Kim H., Kharbash R., Yoon J., Lee Y.S., Kim J.H., Lee Y.J., Kim Y. Mitochondrial double-stranded RNAs govern the stress response in chondrocytes to promote osteoarthritis development. Cell Rep. 2022;40:111178. doi: 10.1016/j.celrep.2022.111178. [DOI] [PubMed] [Google Scholar]
- 2.Klink T.A., Woycechowsky K.J., Taylor K.M., Raines R.T. Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A. Eur. J. Biochem. 2000;267:566–572. doi: 10.1046/j.1432-1327.2000.01037.x. [DOI] [PubMed] [Google Scholar]
- 3.Messmore J.M., Fuchs D.N., Raines R.T. Ribonuclease a: revealing structure-function relationships with semisynthesis. J. Am. Chem. Soc. 1995;117:8057–8060. doi: 10.1021/ja00136a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karimi M., Crossett B., Cordwell S.J., Pattison D.I., Davies M.J. Characterization of disulfide (cystine) oxidation by HOCl in a model peptide: evidence for oxygen addition, disulfide bond cleavage and adduct formation with thiols. Free Radic. Biol. Med. 2020;154:62–74. doi: 10.1016/j.freeradbiomed.2020.04.023. [DOI] [PubMed] [Google Scholar]
- 5.Yoon J., Lee M., Ali A.A., Oh Y.R., Choi Y.S., Kim S., Lee N., Jang S.G., Park S., Chung J.-H., et al. Mitochondrial double-stranded RNAs as a pivotal mediator in the pathogenesis of Sjӧgren’s syndrome. Mol. Ther. Nucleic Acids. 2022;30:257–269. doi: 10.1016/j.omtn.2022.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnaiz E., Miar A., Dias Junior A.G., Prasad N., Schulze U., Waithe D., Nathan J.A., Rehwinkel J., Harris A.L. Hypoxia regulates endogenous double-stranded RNA production via reduced mitochondrial DNA transcription. Front. Oncol. 2021;11:779739. doi: 10.3389/fonc.2021.779739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhir A., Dhir S., Borowski L.S., Jimenez L., Teitell M., Rötig A., Crow Y.J., Rice G.I., Duffy D., Tamby C., et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018;560:238–242. doi: 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J.H., Shim Y.R., Seo W., Kim M.H., Choi W.M., Kim H.H., Kim Y.E., Yang K., Ryu T., Jeong J.M., et al. Mitochondrial double-stranded RNA in exosome promotes interleukin-17 production through toll-like receptor 3 in alcoholic liver injury. Hepatology. 2020;72:609–625. doi: 10.1002/hep.31041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ku Y., Park J.H., Cho R., Lee Y., Park H.M., Kim M., Hur K., Byun S.Y., Liu J., Lee Y.S., et al. Noncanonical immune response to the inhibition of DNA methylation by Staufen1 via stabilization of endogenous retrovirus RNAs. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016289118. e2016289118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee A.K., Pan D., Bao X., Hu M., Li F., Li C.Y. Endogenous retrovirus activation as a key mechanism of anti-tumor immune response in radiotherapy. Radiat. Res. 2020;193:305–317. doi: 10.1667/RADE-20-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehdipour P., Marhon S.A., Ettayebi I., Chakravarthy A., Hosseini A., Wang Y., de Castro F.A., Loo Yau H., Ishak C., Abelson S., et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature. 2020;588:169–173. doi: 10.1038/s41586-020-2844-1. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y., Park J., Kim S., Kim M., Kang M.G., Kwak C., Kang M., Kim B., Rhee H.W., Kim V.N. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol. Cell. 2018;71:1051–1063.e6. doi: 10.1016/j.molcel.2018.07.029. [DOI] [PubMed] [Google Scholar]
- 13.Wong M.L., Medrano J.F. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39:75–85. doi: 10.2144/05391RV01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video demonstration to separate the cover glass from the confocal dish.
A video demonstration to place the cover glass on the microscope slide.
Data Availability Statement
-
•
Original data have been deposited to Mendeley Data: https://doi.org/10.17632/3r7r9r2n6t.2.
-
•
This manuscript does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.