

Abstract

Kazal inhibitors hold high potential as scaffolds for therapeutic molecules, taking advantage of the easily exchangeable canonical binding loop. Different Kazal inhibitor backbones have been suggested to be therapeutically useful, but the impact of different Kazal-like scaffolds on binding properties is still largely unknown. Here, we identified trypsin-targeting human serine protease inhibitor Kazal type 1 (SPINK1) homologues in different mammalian species that cluster in two P2–P1 combinations, implying the coevolution of these residues. We generated loop exchange variants of human SPINK1 for comparison with Kazal inhibitors from related species. Using comprehensive biophysical characterization of the inhibitor–enzyme interactions, we found not only affinity but also pH resistance to be highly backbone-dependent. Differences are mostly observed in complex stability, which varies by over one order of magnitude. We provide clear evidence for high backbone dependency within the Kazal family. Hence, when designing Kazal inhibitor-based therapeutic molecules, testing different backbones after optimizing the canonical binding loop can be beneficial and may result in increased affinity, complex stability, specificity, and pH resistance.
Introduction
Canonical serine protease inhibitors are divided into 18 families which all share a common mechanism.1,2 Serine proteases are inhibited through reversible tight-binding interactions, commonly described as the “standard mechanism”.3 The 18 inhibitor families are contained within 13 different clans, indicating that this mechanism of inhibition evolved independently numerous times. Binding of a canonical inhibitor is mediated by the binding loop present in all inhibitors of this family. The inhibitor interacts with the protease in a substrate-like manner, which is followed by cleavage of the P1–P1′ scissile bond (Schechter and Berger nomenclature).4 Protease and inhibitor exist in an equilibrium between intact and cleaved reactive site peptide bonds, and after complex dissociation, the free inhibitor molecule can exist in both the intact and cleaved state.5,6 The reactive site of the inhibitor is contained within structurally similar loops whose conformation and geometry upon complex formation are essentially identical in all 18 families.3,7−9 Despite the varying supporting scaffolds of each protein family, binding was thought to be independent of the scaffold and mostly mediated by the canonical binding loop itself, with each amino acid contributing independently toward binding.10,11 Hereof, inter- and intrascaffolding additivity principles were extrapolated; however, exceptions have recently been found.12 Loop exchange variants between a Kazal and Pacifastin inhibitors showed that the Kazal inhibitor loop distorts the weak Pacifastin scaffold, while the Pacifastin binding loop was distorted by the stable Kazal inhibitor scaffold. However, the loop exchange variants were modified since the Pacifastin inhibitor possesses a completely conserved P3′ cysteine residue that participates in disulfide bonding within the inhibitor and, therefore, could not be omitted without destabilizing the overall folding of the inhibitor. Hence, as an alternative approach, we focused on loop exchange variants of a single inhibitor family. Through phylogenetic analysis of the human serine protease inhibitor Kazal type 1 (SPINK1), we identified two different P2–P1 amino acid combinations among different mammal species. It appears that either a Thr-Lys or Pro-Arg pair has evolved, which would contradict the aforementioned additivity principles, as a crosstalk between the P2 and P1 residues is implied. Therefore, we generated the four possible P2–P1 combinations (Thr-Lys, Pro-Lys, Thr-Arg, Pro-Arg) in SPINK1 and identified Kazal inhibitors of other organisms that feature binding loops with sequences identical to the mutated human SPINK1 loop. Due to the lack of productive contacts between the Kazal domain backbone and the target protease outside the canonical binding loop, identical binding behavior was expected (Figure S1). However, here we show that binding of Kazal inhibitors can be highly backbone-dependent, with the Kazal scaffold influencing complex stability as well as pH dependency of the interaction. A similar effect was previously observed for loop exchange variants of different Kunitz type inhibitors, and varying effects of the scaffold on the overall affinity were found.13 Loop exchange variants have previously been utilized to investigate interscaffolding additivity models and were considered useful only within evolutionary related scaffolds.12,14 Recently, SPINK2 was proposed as a potential scaffold for designing novel therapeutic molecules. Furthermore, already active patents describe the therapeutic use of SPINK1 and variants thereof as anticancer or anticoagulation agents.15−17 Our findings indicate that optimizing the supporting Kazal domain may be a viable approach for increasing the inhibitor’s specificity, pH resistance, complex stability, and affinity.
Materials and Methods
Protein Expression and Purification
SPINK1 variants and human trypsin isoforms were expressed and purified as previously described.18 In brief, SPINK1 was expressed in SHuffle T7 Express E. coli (New England Biolabs, Frankfurt am Main, Germany). After induction and overnight expression at 16 °C, cells were lysed by sonication and purified by immobilized metal ion affinity chromatography (IMAC). His-tags were removed by cleavage with the HRV3C protease, and proteins were purified again by IMAC. Human trypsin isoforms were expressed in E. coli BL21 (DE3) (New England Biolabs) overnight at 30 °C. Inclusion bodies were washed before solubilization and refolded in 0.9 M Gdn-HCl, 0.1 M Tris pH 8, 2 mM EDTA, 1 mM l-cysteine, and 1 mM l-cystine at 4 °C overnight. Refolded trypsin isoforms were purified by IMAC and activated with enterokinase before use. Bovine and porcine trypsin were commercially available (Sigma-Aldrich, Taufkirchen, Germany) and were thus not expressed recombinantly. Protein purity and homogeneity were verified by tricine SDS PAGE and analytical size exclusion chromatography. Calibration of the analytical size exclusion can be found in the work of Susemihl et al.19
Circular Dichroism Spectroscopy
Circular dichroism (CD) spectra of SPINK1 variants were recorded between 190 and 250 nm using a Chirascan V100 CD spectrometer (Applied Photophysics, Leatherhead, UK) in 1 mm quartz cuvettes containing 250 μg mL–1 of protein in 10 mM Tris pH 7.4 and 10 mM NaF, with adaptive sampling turned on and a step size of 0.5 nm. Three scans were accumulated and averaged before deconvolution using BeStSel.20
Activity Assays
Inhibition constants (Ki) of SPINK1 variants were determined by monitoring the conversion rate of Nα-benzoyl-l-arginine 4-nitroanilide hydrochloride (l-BAPA, Bachem, Bubendorf, Switzerland) at varying inhibitor concentrations ranging from 4.5 pM to 10 nM. The absorbance was recorded at 405 nm using a Cytation 5 microplate reader (BioTek, VT, USA). Michaelis–Menten kinetics of l-BAPA with different trypsin variants were determined using l-BAPA concentrations from 0.02 to 10 mM and fitted using eq 1.
| 1 |
For Ki determination, 100 pM of enzyme and 1 mM l-BAPA were used. Assays were carried out at 37 °C in 20 mM Hepes pH 7.4, 150 mM NaCl, 2 mM CaCl2, and 0.05% Tween20, and initial rates were determined after a 24 h lag phase at 37 °C. Data were fitted with Morrison’s quadratic equation using previously determined Km values (eq 2).
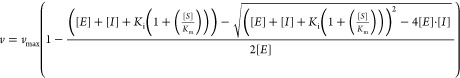 |
2 |
Reported Ki values are determined from fitting the averages of three independent experiments.
Surface Plasmon Resonance
Kinetic analyses were carried out using a BIAcore T200 instrument (Cytiva, Freiburg, Germany) at 37 °C, a flow rate of 50 μL min–1, and a data collection rate of 10 Hz. Trypsin was covalently immobilized to a CM5 sensor chip (Cytiva) using an amine coupling kit according to the manufacturer’s instructions (Cytiva). A concentration series of SPINK1 was prepared as 2-fold dilutions from 3.125 to 50 nM in HBS-P+ buffer supplemented with 2 mM CaCl2. Single cycle kinetics were recorded and double referenced. The surface was regenerated using 10 mM glycine pH 1.4. Sensorgrams were fitted to a 1:1 Langmuir model, and reported rate constants represent average and standard deviation of at least three independent experiments. Dissociation rates are expressed as half-life (t1/2, eq 3). For van’t Hoff, Eyring, and pH analyses, the temperature or buffer system was varied.
| 3 |
Phylogenetic Analyses
We used 79 sequences from mammal species homologous to human SPINK1 (Table S1). Out of the 79 sequences, 62 featured lysine as a P1 residue and 17 contained an arginine in the corresponding position. Multiple sequence alignments (MSA) were carried out using the MUSCLE algorithm.21 Phylogenies were estimated using PhyML 2.5.0+ within the MPI Bioinformatics Toolkit and visualized in iTOL v5.22−24
Structure Prediction
Apart from SPINK1 WT, for which a crystal structure was available (PDB ID: 7QE8), SPINK1 structures were predicted using AlphaFold within ChimeraX.25,26 For inhibitor–enzyme complex prediction, AlphaFold-Multimer was used.27 Multimer scores were above 90 in all cases, indicating high confidence complexes, which are consistent with available crystal structures. Amber force fields for structure relaxation were used in all cases, and the best model of each prediction was chosen for visualization and analysis. AlphaFold utilizes pLDDT (predicted local distance difference test) for ranking the model predictions.
Results
Phylogenetic Analysis of Trypsin-Binding Kazal Inhibitors
We carried out phylogenetic analyses of human SPINK1-related proteins and identified two predominant P2 and P1 residues (Figure 1A). Consistent with the lack of a dedicated S2 pocket in trypsin, the P2 residue in the different SPINK1 homologues was the least conserved residue within the canonical binding loop (Figure 1B). The P1 residues were all positively charged, containing either lysine or arginine. Subclustering according to P1 residues revealed distinct P2–P1 combinations with either Thr-Lys or Pro-Arg in the canonical binding loop (Figure 1C). Although not possessing a binding pocket for the P2 residue, trypsin can influence the orientation of the P2 residue by L104 and H63 in the case of a threonine side chain (Figure 1D). Proline, however, does not interact with the protease but assists the overall loop rigidity and aids in maintaining a favorable geometry. We proceeded to generate human SPINK1 (hSPINK1) variants containing the four possible P2–P1 combinations and identified mouse (mSPINK1) and naked mole SPINK1 (nmSPINK1) proteins as homologues containing identical canonical binding loops to the generated variants.
Figure 1.

SPINK1 sequence and structure conservation. (A) Phylogenetic analysis of trypsin-targeting SPINK1 homologues. Duplicates indicate different isoforms within the same species. SPINK1 variants featuring arginine as the P1 residue are highlighted in red. (B) Structure of the canonical binding loop (PDB ID: 7QE8) colored according to conservation using the multiple sequence alignment from (A). Conservation scores were determined using the ConSurf server.28 (C) Multiple sequence alignment of selected SPINK1 variants and consensus sequences of (A) subclustered according to P1 residues. (D) Structure of binding pockets in trypsin. The S1 pocket is highlighted in red, the oxyanion hole in blue, and L104 interacting with the P2 residue in gray.
Kinetic and Thermodynamic Profiling of SPINK1–Trypsin Interactions Reveals High Backbone Dependency
In addition to available crystal structures, missing hSPINK1 structures for the generated mutants, as well as mSPINK1 and nmSPINK1, were modeled using AlphaFold.26 All structures and models show or predict a similar Kazal-like fold (Figure 2A). Circular dichroism spectroscopy shows similar spectra for all human variants, indicating that the substitution of the P2–P1 residues did not destabilize the overall fold. mSPINK1 shows higher α-helix content compared to the human counterparts, while nmSPINK1 shows a lower α-helix content (Figure 2B). Differences between secondary structure compositions in the predicted models and determined by CD spectroscopy might be caused by the low salt content in the buffer used for CD spectroscopy measurements. Purity and homogeneity of the proteins was verified by tricine SDS-PAGE (Figure 2C) and analytical size exclusion chromatography (Figure 2D). The refolded human cationic trypsin (TRY1) was characterized using l-BAPA as model substrate (Figure 2E). The obtained Km values were used for determining Ki values of the SPINK1 variants with TRY1 at 37 °C, pH 7.4 (Figure 2F). Highest affinity binders were hSPINK1 T40P and mSPINK1. Surprisingly, nmSPINK1 showed an almost 20-fold decrease in affinity compared to that of hSPINK1 T40P, despite containing an identical binding loop. Kinetics of the interactions were characterized by surface plasmon resonance (SPR) with TRY1 as the ligand and SPINK1 variants as the analytes (Figure 2G). Binders displaying the highest affinity were again both human T40P variants as well as mSPINK1. Additionally, the higher affinity is almost exclusively caused by higher complex stability, which results in a half-life of 10 h for hSPINK1 T40P compared to 1 h for hSPINK1 WT (Figure 2H). Even though nmSPINK1 has the same binding loop as hSPINK1 T40P, the complex stability is much lower, indicating a major impact of the Kazal backbone on the overall binding properties of the inhibitor. Finally, thermodynamic analyses between all hSPINK1 variants show that the proline substitution at the P2 site results in a more favorable entropic contribution to binding, while minor enthalpy–entropy compensation is also observed (Figure 2I). T40P was omitted from the analyses due to very slow dissociation rates at lower temperatures. However, the overall impact of the proline substitution can be inferred from comparing the K41R and T40P K41R variants. Through transition state theory, we came to a similar conclusion, where the T40P substitution results in a lower enthalpic energy barrier of the transition state but a much higher entropic barrier resulting in an overall more favorable binding of proline variants (Figure 2J).
Figure 2.

Kinetic and thermodynamic characterization of SPINK1-TRY1 interactions. (A) Superposition of the crystal structure of SPINK1 WT (PDB ID: 7QE8, red) and AlphaFold models of naked mole (purple) and mouse SPINK1 (gray). (B) Far-UV circular dichroism spectra of SPINK1 variants with their secondary structure contents determined by deconvolution in BeStSel.20 (C) Reducing tricine SDS-PAGE of purified human, naked mole, and mouse SPINK1. (D) Analytical size exclusion chromatography of purified SPINK1. (E) Michaelis–Menten kinetics of TRY1 and l-BAPA at pH 7.4 and 37 °C. (F) TRY1 equilibrium activity assays at 37 °C and pH 7.4 with different SPINK1 variants and Ki values obtained with eq 2. (G) Single cycle kinetic experiments of SPINK1–TRY1 interactions using surface plasmon resonance at pH 7.4 and 37 °C. Experimental data are shown in color (as in F) and 1:1 Langmuir fits are represented in black. (H) Complex half-life (t1/2) of different SPINK1–TRY1 interactions obtained from (G) and corresponding Kd values. (I) Van’t Hoff and Eyring analyses of interactions between human SPINK1 variants and TRY1 at different temperatures. (J) Energy landscapes of selected SPINK1–TRY1 interactions.
Kinetic Profiles of SPINK1 Variants Interacting with Different Trypsin Homologues
Due to the unexpected preference of TRY1 for proline as P2 residue despite the wild-type inhibitor carrying a threonine in that position, we extended the characterization to other related proteases. Aiming to identify whether this P2 preference is a general feature for trypsin homologues or specific to human trypsin, we characterized the interaction with human anionic trypsin (TRY2), bovine, and porcine trypsin, and all SPINK1 variants (Figure S2 and Table S1).
Comparison between affinities of the different SPINK1 variants with different trypsin isoforms reveals the most prominent difference for the hSPINK1 T40P variant with TRY1 and TRY2 (Figure 3A). Conversely, bovine and porcine trypsin are barely affected by substituting the P2 residue with proline. Bovine trypsin is not affected by minor changes in the canonical binding loop within the hSPINK1 scaffold but strongly disfavors the mSPINK1 and nmSPINK1 variants despite them having identical binding loops to the human counterparts. Except for the hSPINK1 T40P K41R–porcine trypsin combination, all affinity changes are caused by alterations in complex stability rather than association rates (Figure 3B). Hence, substituting the P2 residue in hSPINK1 WT with a proline residue selectively improved binding toward human trypsin isoforms but not toward trypsin from the other species tested.
Figure 3.

Influence of mutations in the canonical binding loop and changes in inhibitor scaffold on affinity and complex stability. (A) Changes in affinity obtained from SPR experiments of different SPINK1 variants toward different trypsin variants compared to human SPINK1 WT. (B) Changes in complex stability of different SPINK1 variants toward different trypsin variants compared to human SPINK1 WT.
Free Energy Contributions of Scaffolds and P1 and P2 Residues
We showed that substitution of the P2 and P1 residues as well as the protein scaffold can influence the binding to trypsin variants (Table 1). Our data enabled us to dissect the individual free energy contributions of each feature. Using the available kinetic data, we can compare the contribution to the transition free energy of association and dissociation as well as in equilibrium (eq 4–6). The inhibitor scaffold has a greater influence on overall binding than replacing the P1 residue with lysine or arginine. Proline as P2 residue exerts the greatest influence on binding, most likely by stabilizing the overall loop geometry due to limited degrees of freedom.
| 4 |
| 5 |
| 6 |
Table 1. Impact of Binding Loop Mutations and Inhibitor Scaffold on Affinity and Kinetic Rate Constants of SPINK1 Variants Interacting with TRY1 at 37 °C.
| variant A | variant B | ka difference (log2 fold) | kd difference (log2 fold) | Kd difference (log2 fold) | ΔΔ⧧Ga(kJ mol–1) | ΔΔ⧧Gd(kJ mol–1) | ΔΔGd(kJ mol–1) |
|---|---|---|---|---|---|---|---|
| influence of point mutations within the canonical binding loop | |||||||
| WT | K41R | –0.8 | –1.3 | –0.6 | 1.4 | 2.4 | 1.0 |
| WT | T40P | –0.2 | 3.3 | 3.4 | 0.3 | –5.8 | –6.1 |
| WT | T40P K41R | –0.4 | 1.7 | 2.1 | 0.7 | –3.0 | –3.7 |
| influence of scaffold substitution | |||||||
| T40P | naked mole | 1.6 | –2.8 | –4.4 | –2.9 | 5.0 | 7.9 |
| T40P K41R | mouse | 0.3 | –0.9 | –1.2 | –0.5 | 1.6 | 2.1 |
Kazal Inhibitor Backbones Influence pH Dependency
We investigated the SPINK1–TRY1 interaction on pH sensitivity. Using SPR, we observed lower affinity for all SPINK1 variants at pH 5 (Figure 4A). For human SPINK1, Kd values increased 16-fold compared to those with the same interaction at pH 7.4, regardless of the P2–P1 variant used (Figure 4B and Figure 1H). Decreases in affinity were also prominent for the naked mole and mouse variants, which would indicate similar susceptibility to pH changes. Upon analyzing the kinetic rate constants, however, we found the largest decrease in complex stability for naked mole and mouse SPINK1. Simultaneously, naked mole and mouse SPINK1 association rates increased up to 16-fold, compensating for the lower complex stability (Figure 4B,C). Hence, we show that both association and dissociation rates are highly backbone-dependent and that different Kazal motifs can lower the pH sensitivity of the inhibitor. However, caution should be exercised when considering equilibrium binding data. Here, Kd values would suggest that naked mole and mouse SPINK1 are as susceptible to pH changes as the human counterparts, when in actuality, their interactions are the most influenced by the lower pH out of all variants.
Figure 4.

Influence of pH on binding kinetics of SPINK1–TRY1 interactions. (A) Single cycle kinetics of SPINK1–TRY1 interactions using SPR at 37 °C and pH 5. Experimental data are shown in colors, and 1:1 Langmuir fits are shown in black. (B) Change in affinity and kinetic rate constants compared to pH 7.4 reported in Figure 1H. (C) Summary of kinetic rate constants and dissociation constants obtained from (A).
Discussion
We demonstrate by various biophysical techniques that the interaction between Kazal inhibitors and different trypsin variants can be backbone-dependent despite no apparent productive contacts outside the canonical binding loop. Using phylogenetic analysis of trypsin-targeting Kazal inhibitors, homologous to human SPINK1, we identified two major P2–P1 pairs occurring naturally. Thr-Lys or Pro-Arg combinations appear to be preferred in the majority of cases. We generated the four possible P2–P1 combinations in human SPINK1 and recombinantly expressed naked mole and mouse SPINK1, which natively feature identical binding loops to the SPINK1 T40P or SPINK1 T40P K41R variants, respectively.
Unexpectedly, despite the lack of a designated S2 binding pocket in trypsin, the hSPINK1 T40P variant displayed a remarkable complex half-life of 10 h, which represents a 10-fold increase compared to that of hSPINK1 WT. Most likely, proline in the P2 position helps rigidifying the binding loop, locking it in the canonical conformation.3,9,29 In contrast to Kunitz inhibitors, replacement of the P1 Lys residue with Arg weakened the interaction, demonstrating a clear lysine preference of human trypsins for Kazal inhibitors.13,30 Although the Pro-Lys combination resulted in by far the most potent inhibitor, this combination is rarely encountered in nature. The evolutionary rationale behind the low occurrence of this variant remains to be elucidated, but unwanted cross-reactions and lower specificity in secondary functions apart from trypsin inhibition may offer one possible explanation.
Despite naked mole and mouse SPINK1 featuring identical binding loops to the human T40P and T40P K41R variants, Kd values varied up to 10-fold under physiological conditions. At lower pH values, the differences amplified up to 40-fold. In previous studies, loop exchange variants of the Kunitz inhibitors amyloid precursor protein inhibitor domain (APPI) and bovine pancreatic trypsin inhibitor (BPTI) interacting with mesotrypsin were described, and differences in affinity between both scaffolds ranged from negligible up to 10-fold depending on the specific binding loop sequence.13 Additionally, large differences in kcat values were reported, indicating an influence of the inhibitor scaffold beyond supporting the canonical binding loop. The authors concluded that the binding affinity depends mostly on the binding loop, whereas proteolysis of the inhibitor by mesotrypsin can be highly scaffold-dependent. Our results indicate that, in the case of Kazal inhibitors, the inhibitor’s scaffold can contribute significantly to the overall affinity and should not be discounted as a major contributor. Against the backdrop of recent proposals such as using SPINK2 as a scaffold for therapeutic inhibitors and several filed patents describing SPINK1 and related mutants as therapeutic molecules in coagulation and cancer therapy, our findings suggest that, while very potent inhibitors can be obtained from a single Kazal scaffold, performing a secondary scaffold screen will likely create more potent, specific, and resistant inhibitors.15−17
Furthermore, we show scaffold and loop selectivity to be protease- and species-specific, implying that care must be taken when optimizing inhibitors in different animal models. For pancreatitis and trypsin-targeting molecules specifically, transferring results obtained from animal models to clinical trials is often not possible, and outcomes are mostly unpredictable.31 Here, we offer insights into the kinetic and thermodynamic behavior of different trypsin-inhibiting Kazal inhibitors, partly explaining the lack of correlation between results from animal models and clinical trials.
In conclusion, we identified a hSPINK1 T40P variant displaying 10-fold increased potency for trypsin inhibition. We also show high scaffold dependency and illuminate the underlying kinetic and thermodynamic contributions to the different binding behaviors of scaffolds containing identical binding loops.
Conclusions
Our data show that inhibitor–protease pairs form one highly integrated unit, and a secondary scaffold screen after optimizing the canonical binding loop is advisable. Optimizing the binding loop supporting scaffold can increase affinity, specificity, pH resistance, and complex stability, resulting in higher potency inhibitors. In the context of pancreatitis, our results offer a biophysical framework for protease–inhibitor interactions and may help transfer results obtained from animal models to clinical trials.
Glossary
Abbreviations
- CD
circular dichroism
- ka
association rate
- Kd
dissociation constant
- kd
dissociation rate
- Ki
inhibition constant
- NM
naked mole
- SPINK1
serine protease inhibitor Kazal type 1
- SPR
surface plasmon resonance
- t1/2
half-life
- TRY1
human cationic trypsin
- TRY2
human anionic trypsin
- ΔG
change in free energy
- ΔH
change in enthalpy
- ΔS
change in entropy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00573.
Supplemental figures of AlphaFold models and supplemental binding assays; supplemental tables of kinetic parameters and multiple sequence alignments (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was funded by the European Research Council (ERC) Starting Grant “PredicTOOL” (637877) to M.D. and by the European Union (research project “PePPP”, Grant No. ESF/14-BM-A55-0047/16).
The authors declare no competing financial interest.
Supplementary Material
References
- Farady C. J.; Craik C. S. Mechanisms of Macromolecular Protease Inhibitors. ChemBioChem. 2010, 11 (17), 2341–2346. 10.1002/cbic.201000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krowarsch D.; Cierpicki T.; Jelen F.; Otlewski J. Canonical Protein Inhibitors of Serine Proteases. Cell. Mol. Life Sci. C 2003, 60 (11), 2427–2444. 10.1007/s00018-003-3120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski M. Jr; Kato I. Protein Inhibitors of Proteinases. Annu. Rev. Biochem. 1980, 49 (1), 593–626. 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- Schechter I.; Berger A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27 (2), 157–162. 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- Luthy J. A.; Praissman M.; Finkenstadt W. R.; Laskowski M. Detailed Mechanism of Interaction of Bovine -Trypsin with Soybean Trypsin Inhibitor (Kunitz). I. Stopped Flow Measurements. J. Biol. Chem. 1973, 248 (5), 1760–1771. 10.1016/S0021-9258(19)44256-7. [DOI] [PubMed] [Google Scholar]
- Zakharova E.; Horvath M. P.; Goldenberg D. P. Structure of a Serine Protease Poised to Resynthesize a Peptide Bond. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (27), 11034–11039. 10.1073/pnas.0902463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R. M.; Russell R. B. The Serine Protease Inhibitor Canonical Loop Conformation: Examples Found in Extracellular Hydrolases, Toxins, Cytokines and Viral Proteins11Edited by R. Huber. J. Mol. Biol. 2000, 296 (2), 325–334. 10.1006/jmbi.1999.3389. [DOI] [PubMed] [Google Scholar]
- Bode W.; Huber R. Natural Protein Proteinase Inhibitors and Their Interaction with Proteinases. EJB Rev. 1993, 43–61. 10.1007/978-3-642-78046-2_5. [DOI] [PubMed] [Google Scholar]
- Apostoluk W.; Otlewski J. Variability of the Canonical Loop Conformations in Serine Proteinases Inhibitors and Other Proteins. Proteins Struct. Funct. Genet. 1998, 32 (4), 459–474. . [DOI] [PubMed] [Google Scholar]
- Lu S. M.; Lu W.; Qasim M. A.; Anderson S.; Apostol I.; Ardelt W.; Bigler T.; Chiang Y. W.; Cook J.; James M. N. G.; Kato I.; Kelly C.; Kohr W.; Komiyama T.; Lin T. Y.; Ogawa M.; Otlewski J.; Park S. J.; Qasim S.; Ranjbar M.; Tashiro M.; Warne N.; Whatley H.; Wieczorek A.; Wieczorek M.; Wilusz T.; Wynn R.; Zhang W.; Laskowski M. Predicting the Reactivity of Proteins from Their Sequence Alone: Kazal Family of Protein Inhibitors of Serine Proteinases. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (4), 1410–1415. 10.1073/pnas.98.4.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qasim M. A.; Ganz P. J.; Saunders C. W.; Bateman K. S.; James M. N. G.; Laskowski M. Interscaffolding Additivity. Association of P1 Variants of Eglin c and of Turkey Ovomucoid Third Domain with Serine Proteinases. Biochemistry 1997, 36 (7), 1598–1607. 10.1021/bi9620870. [DOI] [PubMed] [Google Scholar]
- Boros E.; Sebák F.; Héja D.; Szakács D.; Zboray K.; Schlosser G.; Micsonai A.; Kardos J.; Bodor A.; Pál G. Directed Evolution of Canonical Loops and Their Swapping between Unrelated Serine Proteinase Inhibitors Disprove the Interscaffolding Additivity Model. J. Mol. Biol. 2019, 431 (3), 557–575. 10.1016/j.jmb.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Salameh M. A.; Soares A. S.; Navaneetham D.; Sinha D.; Walsh P. N.; Radisky E. S. Determinants of Affinity and Proteolytic Stability in Interactions of Kunitz Family Protease Inhibitors with Mesotrypsin. J. Biol. Chem. 2010, 285 (47), 36884–36896. 10.1074/jbc.M110.171348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürvanger Z.; Boros E.; Nagy Z. A.; Hegedüs R.; Megyeri M.; Dobó J.; Gál P.; Schlosser G.; Ángyán A. F.; Gáspári Z.; Perczel A.; Harmat V.; Mezö G.; Menyhárd D. K.; Pál G. Directed Evolution-Driven Increase of Structural Plasticity Is a Prerequisite for Binding the Complement Lectin Pathway Blocking MASP-Inhibitor Peptides. ACS Chem. Biol. 2022, 17 (4), 969–986. 10.1021/acschembio.2c00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte S.; Kronthaler U.; Schmidbauer S.; Weimer T.; Hofmann K.. Therapeutic Application of Kazal-Type Serine Protease Inhibitors. Patent Appl. WO/2008/098720, 2019.
- Lu X.; Block T.. Identification of Modulators of Serine Protease Inhibitor Kazal and Their Use as Anti-Cancer and Anti-Viral Agents. Patent Appl. WO/2007/030560, June 5, 2012.
- Nishimiya D.; Kawaguchi Y.; Kodama S.; Nasu H.; Yano H.; Yamaguchi A.; Tamura M.; Hashimoto R. A Protein Scaffold, Engineered SPINK2, for Generation of Inhibitors with High Affinity and Specificity against Target Proteases. Sci. Rep. 2019, 9 (1), 11436. 10.1038/s41598-019-47615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel F.; Palm G. J.; Geist N.; McDonnell T. C. R.; Susemihl A.; Girbardt B.; Mayerle J.; Lerch M. M.; Lammers M.; Delcea M. Structural and Biophysical Insights into SPINK1 Bound to Human Cationic Trypsin. International Journal of Molecular Sciences. 2022, 23, 3468. 10.3390/ijms23073468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susemihl A.; Nagel F.; Grabarczyk P.; Schmidt C. A.; Delcea M. Easy Expression and Purification of Fluorescent N-Terminal BCL11B CCHC Zinc Finger Domain. Molecules. 2021, 26, 7576. 10.3390/molecules26247576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Bulyáki É.; Kun J.; Moussong É.; Lee Y. H.; Goto Y.; Réfrégiers M.; Kardos J. BeStSel: A Web Server for Accurate Protein Secondary Structure Prediction and Fold Recognition from the Circular Dichroism Spectra. Nucleic Acids Res. 2018, 46 (W1), W315–W322. 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32 (5), 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L.; Stephens A.; Nam S. Z.; Rau D.; Kübler J.; Lozajic M.; Gabler F.; Söding J.; Lupas A. N.; Alva V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430 (15), 2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Guindon S.; Dufayard J.-F.; Lefort V.; Anisimova M.; Hordijk W.; Gascuel O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59 (3), 307–321. 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Letunic I.; Bork P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49 (W1), W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Meng E. C.; Couch G. S.; Croll T. I.; Morris J. H.; Ferrin T. E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30 (1), 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R.; O’Neill M.; Pritzel A.; Antropova N.; Senior A.; Green T.; Žídek A.; Bates R.; Blackwell S.; Yim J.; Ronneberger O.; Bodenstein S.; Zielinski M.; Bridgland A.; Potapenko A.; Cowie A.; Tunyasuvunakool K.; Jain R.; Clancy E.; Kohli P.; Jumper J.; Hassabis D. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2022, 10.1101/2021.10.04.463034. [DOI] [Google Scholar]
- Ashkenazy H.; Abadi S.; Martz E.; Chay O.; Mayrose I.; Pupko T.; Ben-Tal N. ConSurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44 (W1), W344–W350. 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulescu H.; Guo Y.; Garner T. P.; Goodwin O. Y.; Henderson G.; Laine R. A.; Macnaughtan M. A. A Kazal-Type Serine Protease Inhibitor from the Defense Gland Secretion of the Subterranean Termite Coptotermes Formosanus Shiraki. PLoS One 2015, 10 (5), e0125376. 10.1371/journal.pone.0125376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiak A.; Krokoszynska I.; Krowarsch D.; Buczek O.; Dadlez M.; Otlewski J. Inhibition of Six Serine Proteinases of the Human Coagulation System by Mutants of Bovine Pancreatic Trypsin Inhibitor*. J. Biol. Chem. 2000, 275 (43), 33346–33352. 10.1074/jbc.M006085200. [DOI] [PubMed] [Google Scholar]
- Pezzilli R. Pharmacotherapy for Acute Pancreatitis. Expert Opin. Pharmacother. 2009, 10 (18), 2999–3014. 10.1517/14656560903382630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.