

Abstract
Lysosomal Ca2+ emerges as a critical component of receptor-evoked Ca2+ signaling and plays a crucial role in many lysosomal and physiological functions. Lysosomal Ca2+ release is mediated by the transient receptor potential (TRP) family member TRPML1, mutations that cause the lysosomal storage disease mucolipidosis type 4. Lysosomes play a key role in osteoclast function. However, nothing is known about the role of lysosomal Ca2+ signaling in osteoclastogenesis and bone metabolism. In this study, we addressed this knowledge gap by studying the role of lysosomal Ca2+ signaling in osteoclastogenesis, osteoclast and osteoblast functions, and bone homeostasis in vivo. We manipulated lysosomal Ca2+ signaling by acute knockdown of TRPML1, deletion of TRPML1 in mice, pharmacological inhibition of lysosomal Ca2+ influx, and depletion of lysosomal Ca2+ storage using the TRPML agonist ML-SA1. We found that knockdown and deletion of TRPML1, although it did not have an apparent effect on osteoblast differentiation and bone formation, markedly attenuated osteoclast function, RANKL-induced cytosolic Ca2+ oscillations, inhibited activation of NFATc1 and osteoclastogenesis-controlling genes, suppressed the formation of tartrate-resistant acid phosphatase (TRAP)-positive multinucleated cells (MNCs), and markedly reduced the differentiation of bone marrow–derived macrophages into osteoclasts. Moreover, deletion of TRPML1 resulted in enlarged lysosomes, inhibition of lysosomal secretion, and attenuated the resorptive activity of mature osteoclasts. Notably, depletion of lysosomal Ca2+ with ML-SA1 similarly abrogated RANKL-induced Ca2+ oscillations and MNC formation. Deletion of TRPML1 in mice reduced the TRAP-positive bone surfaces and impaired bone remodeling, resulting in prominent osteopetrosis. These findings demonstrate the essential role of lysosomal Ca2+ signaling in osteoclast differentiation and mature osteoclast function, which play key roles in bone homeostasis.
Keywords: TRPML1, LYSOSOME, BONE REMODELING, OSTEOCLASTOGENESIS, CA2+ SIGNALING
Introduction
Bone remodeling is a basic and essential process in maintaining bone homeostasis, which is delicately controlled by a balance between the activity of osteoclasts and osteoblasts.(1) The major function of osteoclasts in bone homeostasis is demineralization of the bone matrix. Abnormal differentiation of osteoclasts or aberrant activity of mature osteoclasts are the major causes of bone disorders, including osteoporosis and osteopetrosis.(2) Considering that fully matured osteoclasts no longer have the ability to replicate and mature osteoclasts have a relatively short lifespan, modulating the differentiation of precursor cells into osteoclasts to induce bone resorptive activity has been regarded as key to treating bone disorders caused by the disruption of bone homeostasis. Bone homeostasis can also be modulated by altering the function of the bone-forming osteoblasts.
Ca2+ signaling is the main pathway mediating osteoclastogenesis and bone remodeling.(3–5) A key regulator of osteoclast differentiation and function is the receptor activator of NF-κB ligand (RANKL) that acts on the RANK receptor.(6) Long-term RANKL treatment (24 to 72 hours) induces cytosolic Ca2+ ([Ca2+]i) oscillations that require several components of the Ca2+ signaling pathway.(7–10) The core Ca2+ signaling pathway activated by Gq, Gi, and tyrosine kinase-coupled receptors entails the activation of phospholipase C to hydrolyze phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] and generation of inositol triphosphate (IP3) in the cytosol. IP3 activates the IP3 receptors Ca2+ channels to release Ca2+ from the endoplasmic reticulum (ER), which is followed by stromal interaction molecule (STIM) 1-dependent activation of the plasma membrane store-operated Orai1 and TRPC Ca2+ influx channels.(11) The increase in [Ca2+]i levels inhibits the Ca2+ influx channels and activates both the ER-localized sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) and the plasma membrane-localized plasma membrane Ca2+-ATPase (PMCA) Ca2+ pumps to restore basal [Ca2+]i level.(12) This cycle is repeated to evoke Ca2+ oscillations.
Another, much less defined and understood component of the Ca2+ signal is Ca2+ release from intracellular organelles, primarily the lysosomes.(13) With identification of the lysosomal Ca2+ release channels, recent studies began to reveal the features and roles of lysosomal Ca2+ homeostasis. Two Ca2+ permeable channels have been identified in the lysosomes: the two pore channels (TPCs) TPC1 and TPC2; and transient receptor potential mucolipin subfamily 1 (TRPML1), also known as MCOLN1, a member of the TRP channel superfamily.(14) The TPCs are required for nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated Ca2+ release from endosomes and lysosomes,(13) whereas TRPML1 is the primary channel mediating lysosomal Ca2+ release.(14)
TRPML1 plays crucial roles in lysosomal trafficking and various lysosomal functions.(15,16) Loss-of-function mutations in MCOLN1 cause mucolipidosis type IV (MLIV), a lysosomal storage disorder.(17) MLIV cells are characterized by dysfunction of the lysosomes due to lysosomal accumulation of unprocessed lipids and polysaccharides.(18,19) Consequently, MLIV cells display defects in the fusion of late endosomes to lysosomes, lysosomal degradation, lysosomal exocytosis,(20–22) phagocytosis,(23) membrane repair,(24) and metabolic sensing,(25) all of which are essential cellular functions for bone remodeling. However, the role of lysosomal Ca2+ signaling and TRPML1 in bone remodeling remains unclear.
The binding of RANKL to the RANK receptor initiates a signaling cascade that mediates the differentiation of hematopoietic stem cells into osteoclasts.(6) A key step in the cascade is the RANKL-evoked Ca2+ oscillations through co-stimulatory signals of the Fc receptor common γ subunit (FcRγ) and the DNAX-activating protein 12 (DAP12).(4) [Ca2+]i oscillations subsequently activate nuclear factor of activated T cells, cytoplasmic 1 (NFATc1), which orchestrates the late stage of osteoclastogenesis.(3) Numerous studies revealed that RANKL-induced [Ca2+]i oscillations involve mobilization of internal Ca2+ stores,(7,8,26) with the main Ca2+ store assumed to be exclusively that in the ER.(27) However, recent work revealed that the ER Ca2+ store communicates with the lysosomal Ca2+ store to trigger the Ca2+ signal and modulate its functions.(28,29) Osteoclast precursor cells and mature osteoclasts are rich in lysosomes and the role and function of the osteoclastic lysosomal Ca2+ store in cell differentiation and function is unknown. Elucidating the role of the lysosomal Ca2+ stores in RANKL-mediated Ca2+ oscillations, NFATc1 activation and osteoclastogenesis is essential for understanding bone homeostasis. In the present study, we determined the role of lysosomal Ca2+ signaling in osteoclastogenesis and bone remodeling in vivo, using a mouse model of MLIV, generated by deletion of TRPML1. Multiple assays of osteoclast differentiation and function and analysis of bone structure reveal the crucial role of lysosomal Ca2+ signaling in osteoclastogenesis and bone-resorptive activity, which are essential processes in bone remodeling.
Materials and Methods
Animals and reagents
All animal experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee of Wonkwang and Yonsei Universities. Mice (C57BL/6×129SV mixed background) were housed at 3 to 4 animals per cage under specific pathogen-free conditions (12/12 hours light/dark cycles, 22±2°C temperature, and 50% to 60% humidity). Mice were euthanized by brief exposure to CO2 and cervical dislocation. TRPML1−/− mice were generated as described.(30) TRPML1−/− mice and matching littermates (4 to 8 weeks old) were used in all experiments. Proximal femoral bones were removed and used to prepare bone marrow–derived macrophages (BMMs) and bone sections. BMMs were cultured in α-modified minimum essential medium (α-MEM; Gibco BRL, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (FBS) and incubated in 5% CO2 atmosphere. Soluble RANKL and macrophage colony stimulating factor (M-CSF) were purchased from KOMA Biotech (Seoul, Republic of Korea). Glycyl-phenylalanine-2-naphthylamide (GPN), bafilomycin A1, Fura-2/AM, ML-SA1, cyclopiazonic acid (CPA), and anti-LAMP1 were purchased from Sigma Aldrich (St. Louis, MO, USA). Anti-TRPML1 was obtained from Abcam (Cambridge, MA, USA). Anti-NFATc1 and -β-actin antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Acute knockdown of TRPML1, using retroviral system
RNAi oligonucleotides (TRPML1, 5′-CCTCACACTGAAATTCCAC-3′ and GFP as negative control, 5′-CATGGAT GAACTATACAAA-3′; Integrated DNA Technologies, Coralville, IA, USA) were inserted into the retroviral short hairpin RNA vector (pSUPER-retro-puro; OligoEngine, Seattle, WA, USA). These retroviral vectors were transiently transfected into the packaging cell line, Plate-E (Cell Biolabs, San Diego, CA, USA), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and incubated for 24 hours. Supernatants containing virus particles were harvested and used for acute knockdown of TRPML1. Cultured BMMs were infected with the virus and incubated for 2 days before treatment with RANKL for the indicated times to induce osteoclastogenesis.
Preparation of BMMs and evaluation of osteoclast formation
Murine BMMs were prepared from the proximal femoral bones of 4-week-old to 8-week-old mice, as described.(26) Briefly, the bone marrow was flushed out with α-MEM and collected. After removal of red blood cells, whole marrow cells were plated on non-coated Petri dishes with low concentration of M-CSF (10 ng/mL). BMMs were collected on the next day, seeded on culture dishes, and supplemented with 30 ng/mL M-CSF. BMMs were then stimulated with RANKL (50 ng/mL) for the indicated times to induce the differentiation of BMMs to osteoclasts.
Osteoclast formation was evaluated by seeding BMMs on 24-well culture dishes at a density of 1.2×105 cells per well and culturing under the conditions detailed in “Animals and reagents” and subsequent cytochemical staining for tartrate resistant acid phosphatase (TRAP), as described.(31) Briefly, cells were fixed with 10% formalin and permeabilized with 1:1 mixture of methanol and acetone. The cells were then treated with TRAP staining buffer (Sigma Aldrich, St. Louis, MO, USA) for 30 min at 37°C.
Measurement of secreted TRAP activity
BMMs were seeded on 24-well culture dishes at a density of 1.2×105 cells per well. The following day, cells were treated with RANKL (50 ng/mL) for a total of 9 days. Every 3 days, the culture medium was collected and exchanged with fresh medium. The collected medium, which contained the secreted TRAP, was used to measure TRAP activity. To calculate the percentage of secreted TRAP, at the end of each incubation interval of 3, 6, and 9 days, cells in parallel wells, which were seeded and incubated under the same conditions, were lysed and used for measuring total TRAP, in addition to measuring secreted TRAP in the medium. Secreted TRAP is calculated as a percentage of the respective total and is presented as fold change relative to 3-day wild-type (WT) without RANKL. TRAP activity was determined by incubating the samples with p-nitrophenyl phosphate (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at 37°C and measuring optical density (OD) at 405 nm.
[Ca2+]i measurement
[Ca2+]i was determined using Ca2+-sensitive fluorescence dye Fura-2, as described.(26) Briefly, BMMs were plated on cover glass at 80% confluence and incubated for 48 hours with or without RANKL (50 ng/mL). The cells were then loaded with Fura-2 by incubation with 5 μM Fura-2 AM for 50 min at 37°C. The cover glasses were assembled into a perfusion chamber, and cells were continuously perfused with HEPES-buffered medium containing 10 mmol/L HEPES, 140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1 mmol/L CaCl2, and 10 mmol/L glucose, adjusted to pH 7.4 and 310 mOsm (designated as HEPES buffer). Test compounds were diluted with the HEPES buffer. Fura-2 fluorescence was measured using excitation wavelengths of 340 and 380 nm and the emission wavelength of 510 nm using a charge coupled device (CCD) camera. Captured images were digitized and analyzed using MetaFluor software (Molecular Devices Corporation, Downingtown, PA, USA). The fluorescence is presented as F340/F380 ratio.
RT-PCR and real-time qPCR
BMMs were incubated with RANKL for the indicated time and total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA). One microgram of total RNA was transcribed to first-strand cDNA with random hexamers. Real-time qPCR was performed using the VeriQuest SYBR Green qPCR master mix (Affymetrix, Santa Clara, CA, USA) and StepOnePlus Real-Time PCR system (Applied Biosystems, Inc., Foster City, CA, USA). Results were normalized to those of the housekeeping gene GAPDH. Primers used in this study are the following; (TRPML1: forward 5′-ATC TAC CTG GGC TAT TGC TTC TGT G-3′, reverse 5′-TGT CGT TC CGT TGA TGA GTG A-3′; TRAP forward 5′-CTG GAG TGC ACG ATG CCAGCG ACA-3′, reverse 5′-TCC GTG CTC GGC GAT GGA CCA GA-3′; c-Fos forward 5′-CTG GTG CAG CCC ACT CTG GTC-3′, reverse 5′-CTT TCA GCA GAT TGG CAA TCT C-3′; Oscar forward 5′-CTG CTG GTA ACG GAT CAG CTC CCC AGA-3′, reverse 5′-CCA AGG AGC CAG AAC CTT CGA AAC T-3′; GAPDH forward 5′-TGC CAG CCT CGT CCC GTA GAC-3′, reverse 5′-CCT CAC CCC ATT TGA TGT TAG-3′).
Pit formation assay
BMMs were seeded on Osteo assay plates (Corning, Corning, NY, USA) coated with a proprietary hydroxyapatite mineral surface. Following RANKL stimulation of the BMMs for the indicated time, the plates were washed with sodium hypochlorite solution. Pits formed on the surface of the hydroxyapatite mineral were measured and analyzed by ImageJ software (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/).
Western blot
BMMs were seeded onto 60-mm dishes at a density of 1×106 cells/dish. Following incubation under the indicated conditions, cells were lysed in RIPA buffer (25 mM Tris-HCL pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitors. The cell lysates were centrifuged at 14,000g for 10 min at 4°C. Proteins in the supernatants were separated by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were incubated over-night with antibodies against NFATc1 (1:1000), TRPML1 (1:1000), and β-actin (1:2000) and immunoreactive proteins were detected using an enhanced chemiluminescence (ECL) detection system.
Visualizing lysosomes in live cells
BMMs were seeded on 35-mm cover-glass-bottom dishes and incubatedwithor without RANKL (50 ng/mL)for 48 hours. The cells were then loaded with 1 μM Lysotracker (Invitrogen, Carlsbad, CA, USA) for 30 min and used to visualize the lysosomes. Images were captured by confocal microscopy and the lysosomal perimeters in randomly captured fluorescence images were measured.
Histological analysis
Tibias were isolated and immediately fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS), and were then decalcified in EDTA. Processed tissues were embedded in paraffin and sectioned to 12 μm thickness. Tissue sections were stained for TRAP. Briefly, tissue sections were sequentially deparaffinized, rehydrated, permeabilized, and treated with TRAP staining buffer for 30 min at 37°C. Tissue sections were counterstained using Fast Green (Sigma Aldrich, St. Louis, MO, USA) and mounted after brief dehydration and clearing.
Bone analysis by μCT
Proximal femoral bones and tibias were isolated from 8-week-old TRPML1−/− and WT littermates. To avoid differences resulting from setting up the threshold or resolution, samples from WT and TRPML1−/− bones were set in one polystyrene holder. Bone density was then measured by three-dimensional μCT (SkyScan-1076 high resolution in vivo μCT system; SkyScan, Aartselaar, Belgium). Each parameter, including bone volume (BV), bone volume per tissue volume (BV/TV), bone mineral density (BMD), trabecular number (Tb.N), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), and structure model index (SMI) were calculated by CT analyzer (CTAn) analysis, using cone beam reconstruction software.
Dynamic histomorphometry of bone formation
Dynamic histomorphometry was performed as described.(26) Briefly, 4-week-old mice were injected twice with calcein (15 mg/kg intraperitoneally), 2 days apart. Calcein-injected mice were euthanized on day 4, and un-decalcified bones were embedded in methyl methacrylate. Longitudinal sections (10 μm thick) of the tibias and femurs were prepared and new bone formation was assessed by recording calcein fluorescence by confocal microscopy (model LSM 700; Zeiss, Inc., Thornwood, NY, USA). The bone formation rate (BFR/BS, μm3/μm2/day), mineralizing surface (MS/BS, %), and mineral apposition rate (MAR, μm/day) were then evaluated.
In vitro osteoblastogenesis assay
Murine bone marrow–derived mesenchymal stromal (BMSC) cells were isolated from the femurs of 4-week-old to 6-week-old mice, as described.(32) Briefly, bone marrow was flushed out with culture medium (α-MEM) and plated in 48-well plates at a density of 1.5×105 cells. After 6 days of incubation, the cells were treated with 10 mM β-glycerol phosphate and 50 μg/mL L-ascorbic acid (both from Sigma Aldrich, St. Louis, MO, USA) to induce osteoblastogenesis. For alkaline phosphate (ALP) staining, cells were fixed with 70% ethanol for 15 min at room temperature, and then treated for 15 min with a staining solution containing 1% N,N-dimethyl formamide, 0.01% naphthol AS-MX (Sigma Aldrich, St. Louis, MO, USA) phosphate, and 0.06% fast blue BB. To measure ALP activity, culture media was removed, cells were washed with PBS and incubated in lysis buffer (Tris-HCl, 150 mM NaCl, 1% Triton X-100) for 1 hour at room temperature. The lysates were incubated with 100 μL of ALP substrate (Sigma Aldrich, St. Louis, MO, USA), and absorbance resulting from ALP activity was measured at the wavelength of 405 nm.
Statistical analysis
Results were analyzed using SPSS version 14.0 (SPSS Inc., Chicago, IL, USA) and data presented as mean±SD of the stated number of observations obtained from the indicated number of independent experiments. Statistical differences were analyzed by one-way ANOVA followed by Tukey’s post hoc test. Values of p < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01).
Results
Acute deletion of TRPML1 suppresses TRAP-positive MNC formation
TRPML1 is one of the lysosomal Ca2+ release channels that initiates the receptor-evoked Ca2+ signal, which is required for many lysosomal functions(15) that are crucial for the differentiation and physiological functions of osteoclasts.(3,33) Among them are the RANKL-elicited [Ca2+]i oscillations that activate the Ca2+/calmodulin–dependent protein kinase, the phosphatase, calcineurin, and NFATc1.(3) Moreover, lysosomal exocytosis by membrane fusion of lysosomes with the ruffled border membrane mediates acidification of the bone surface and releases lysosomal hydrolases that are critical for the demineralization of the bone matrix.(33–35) Yet the importance and role of lysosomal Ca2+ signaling in osteoclastogenesis and bone remodeling have not been examined. To address this biological question, we manipulated lysosomal Ca2+ signaling by acute knockdown and chronic deletion of TRPML1 in mice. We first examined whether TRPML1 is expressed in BMMs and the expression level of TRPML1 in response to RANKL-induced osteoclastogenesis. Figure 1A, B show that TRPML1 is expressed in BMMs and the expression does not change in response to RANKL stimulation. We then acutely knocked down TRPML1 (Fig. 1C) and evaluated osteoclast formation induced by RANKL-mediated differentiation of BMMs in culture into osteoclasts. Figure 1D shows that knockdown of TRPML1 reduced TRAP-positive MNC formation by about 50% (n=3) compared to control (shGFP). These findings indicated acute requirement for lysosomal Ca2+ release for osteoclastogenesis and prompted us to determine the in vivo role of TRPML1 in RANKL-mediated osteoclastogenesis, using TRPML1−/− mice.
Fig. 1.
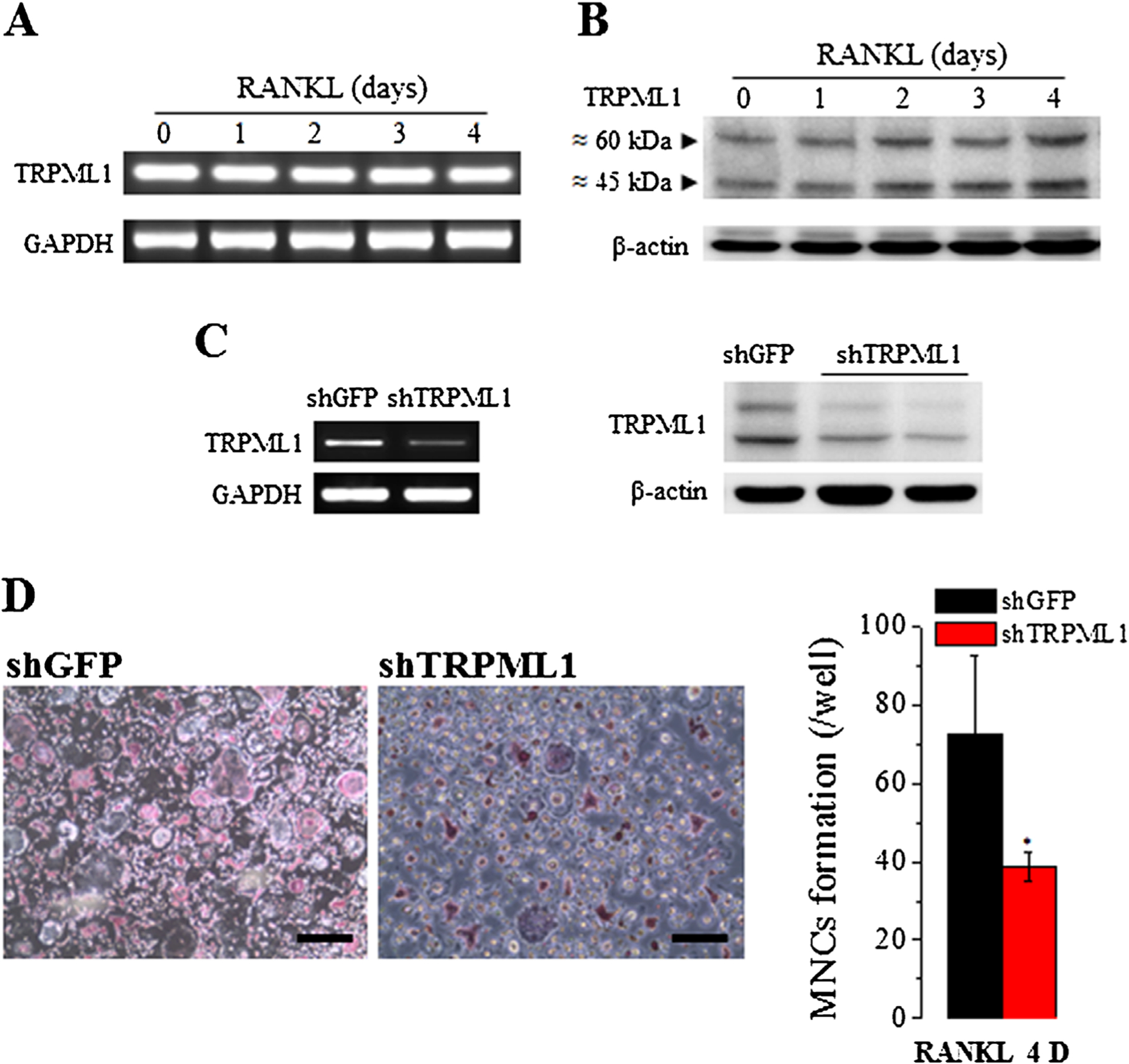
Acute deletion of TRPML1 inhibits osteoclastogenesis. (A, B) BMMs in primary culture were incubated with 50 ng/mL RANKL for the indicated times and the levels of TRPML1 mRNA (A) and protein (B) were analyzed. (C) BMMs were treated with shTRPML1, and shGFP as a negative control, for 72 hours, and the level of TRPML1 mRNA (left blots) and protein (right blots) were determined. (D) Osteoclastogenesis was induced by treating BMMs that were treated with shGFP or shTRPML1 with 50 ng/mL RANKL for 3 days. Shown are representative images (Scale bar = 100 μm) from 3 experiments. Depletion of TRPML1 suppressed osteoclastogenesis as revealed by reduced TRAP staining and multinucleated cells. *p < 0.05.
Lysosomal Ca2+ is required for RANKL-induced [Ca2+]i oscillations
We used pharmacological agents to examine the role of lysosomal Ca2+ in RANKL-induced [Ca2+]i oscillations. Figure 2A, B show that rupturing the lysosomes by exposing to 200 μM GPN and depleting lysosomal Ca2+ by inhibiting the lysosomal V-type H+ pump with 100 nM bafilomycin A1, efficiently inhibited the [Ca2+]i oscillations. To directly examine the involvement of TRPML1 in RANKL-induced [Ca2+]i oscillations, BMMs from WT and TRPML1−/− mice were cultured for 48 hour in the presence of RANKL. Figure 2C shows that deletion of TRPML1 resulted in 47% and 26% reduction in the frequency and amplitude of the Ca2+ spikes, respectively. Reduction in the frequency and amplitude of Ca2+ oscillations can be secondary to reduction in ER Ca2+ content or inhibition of store-operated Ca2+ influx. The results in Fig. 2D, E exclude these possibilities. Thus, releasing the ER Ca2+ pool by inhibiting the SERCA pump with CPA (25 μM) in the absence of extracellular Ca2+ showed no difference between WT and TRPML1−/− BMMs, both untreated (Fig. 2D) and treated with RANKL (Fig. 2E). Similarly, deletion of TRPML1 had no effect on the store-mediated Ca2+ influx, assayed by Ca2+ re-addition (Fig. 2D, E). Together, the results in Fig. 2 indicate that lysosomal Ca2+ mobilization is essential for RANKL-induced [Ca2+]i oscillations.
Fig. 2.
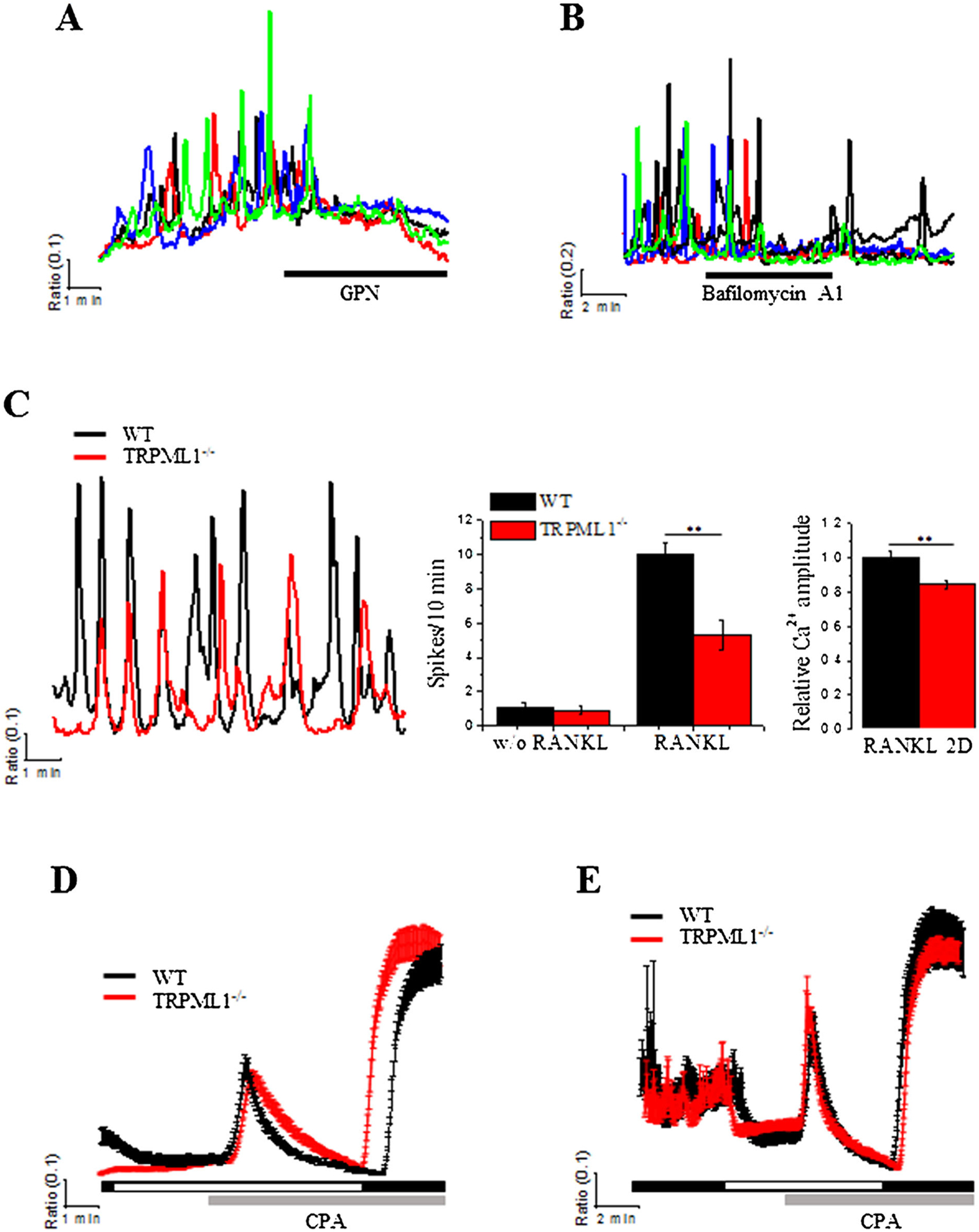
RANKL-induced [Ca2+]i oscillations are attenuated by deletion of TRPML1 and inhibition of lysosomal Ca2+ storage without affecting ER Ca2+ content. (A, B) Bursting lysosomes with GPN (200 μM) and inhibition of lysosomal Ca2+ influx by treatment with bafilomycin A1 (100 nM) eliminate RANKL-induced Ca2+ oscillations in BMMs cultured with RANKL for 48 hours. (C) RANKL-induced [Ca2+]i oscillations were measured in BMMs isolated from WT and TRPML1−/− mice and cultured for 48 hours in the presence of RANKL. Cells were continuously perfused with HEPES buffer throughout the experiments. The columns show the average frequency and amplitude of the Ca2+ oscillations. **p < 0.01. (D, E) ER Ca2+ content was evaluated in WT (black traces) and TRPML1−/− BMMs (red traces) following 48 hours of incubation with (E) or without (D) RANKL (50 ng/mL). ER Ca2+ was measured by releasing it to the cytoplasm by exposing the BMMs to 10 μM of the SERCA pump inhibitor, CPA. Ca2+ influx mediated by store-operated Ca2+ channels was then evaluated by addition of 1 mM Ca2+ to the perfusate. The traces are the mean±SD of at least 3 experiments.
Lysosomal Ca2+ is required for RANKL-mediated osteoclastogenesis
Reduction in RANKL-induced [Ca2+]i oscillations in TRPML1−/− BMMs may affect RANKL-mediated activation of NFATc1 expression and osteoclastogenesis. Indeed, as shown in Fig. 3A, B, deletion of TRPML1 suppressed RANKL-mediated NFATc1 expression and reduced TRAP-positive MNC formation by 46% relative to WT BMMs. Accordingly, deletion of TRPML1 reduced the expression of RANKL-mediated induction of the osteoclastic gene differentiation, TRAP, c-Fos, and Oscar (Fig. 3C).
Fig. 3.
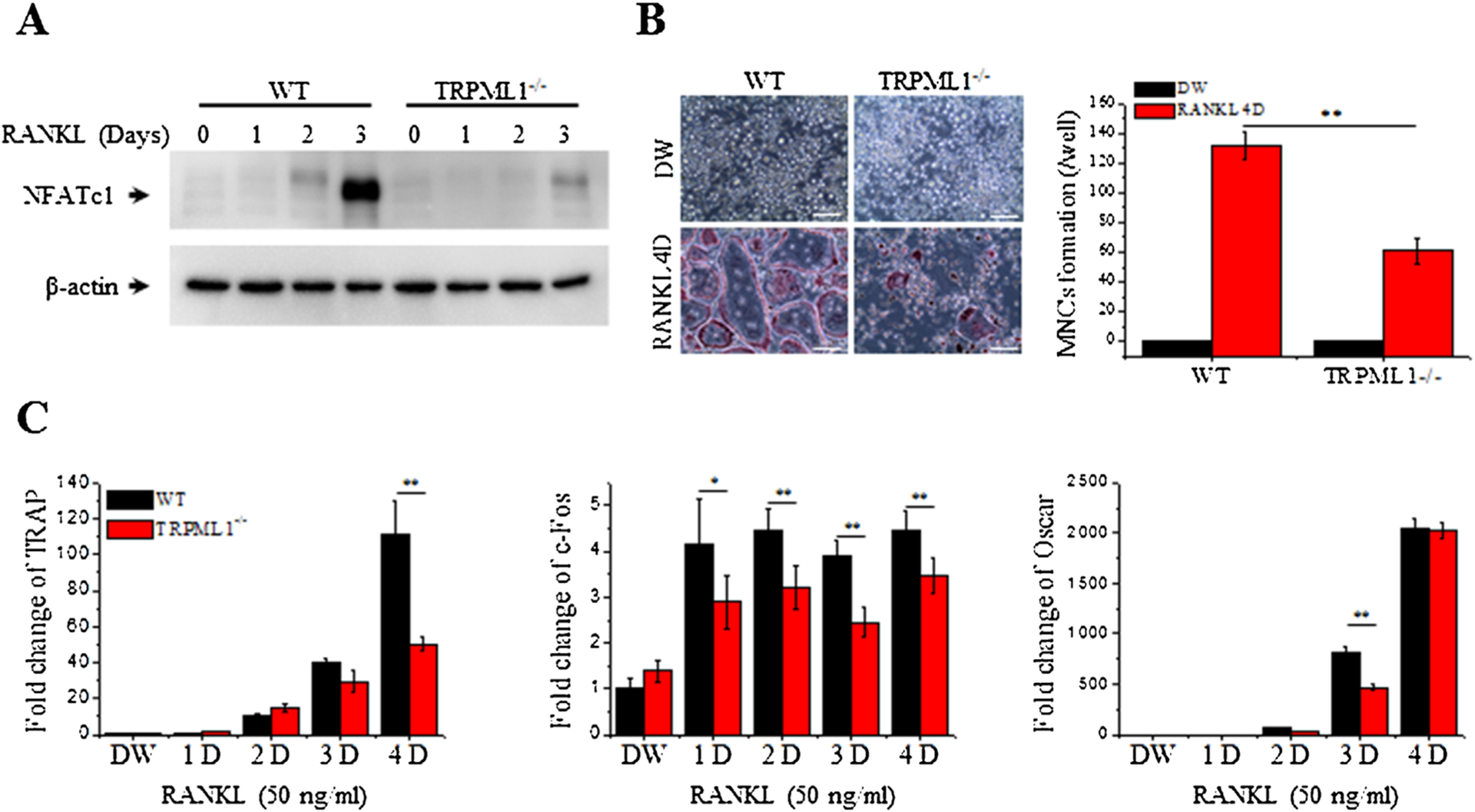
Aberrant NFATc1 activation and RANKL-mediated osteoclastogenesis in TRPML1−/− mice. BMMs isolated from WT and TRPML1−/− mice were cultured and used for the experiments below. (A) RANKL-mediated NFATc1 expression is suppressed in TRPML1−/− BMMs. β-actin was used as a loading control. (B) Deletion of TRPML1 reduces RANKL-mediated MNC formation. The number of TRAP-positive MNCs, which are identified by the presence of more than 3 nuclei and cell size larger than 100 μm in diameter, present in each well were counted. The columns are the mean±SD of 5 experiments. (Scale bar = 100 μm). (C) Deletion of TRPML1 alters expression of differentiation marker genes for RANKL-mediated osteoclastogenesis. Results are presented as fold increase compared to WT cells treated with DW. *p < 0.05, **p < 0.01.
Depletion of lysosomal Ca2+ impairs RANKL-mediated osteoclastogenesis similar to deletion of TRPML1
To further probe the role of lysosomal Ca2+ homeostasis in osteoclastogenesis, we depleted lysosomal Ca2+ with the TRPML activator ML-SA1.(36) The specificity of ML-SA1 in the BMMs is shown in Fig. 4A. Treating the cells with 20 μM ML-SA1 resulted in oscillatory Ca2+ signals only in WT BMMs and had no effect in TRPML1−/− cells. Importantly, Fig. 4B shows that exposing RANKL-treated WT BMMs to ML-SA1 reduced the frequency of Ca2+ oscillations similar to that observed in TRPML1−/− BMMs, while having no effect on Ca2+ oscillations in TRPML1−/− BMMs. This further establishes the specificity of ML-SA1 and allowed us to test the effect of depleting lysosomal Ca2+ on osteoclastogenesis. Notably, Fig. 4C shows that depleting lysosomal Ca2+ with ML-SA1 suppressed RANKL-induced TRAP-positive (TRAP+) MNC formation by about 42% in WT BMMs, while having no effect in TRPML1 deficient BMMs. These findings provide additional evidence for the importance of lysosomal Ca2+ in RANKL-induced [Ca2+]i oscillations and differentiation of osteoclasts.
Fig. 4.
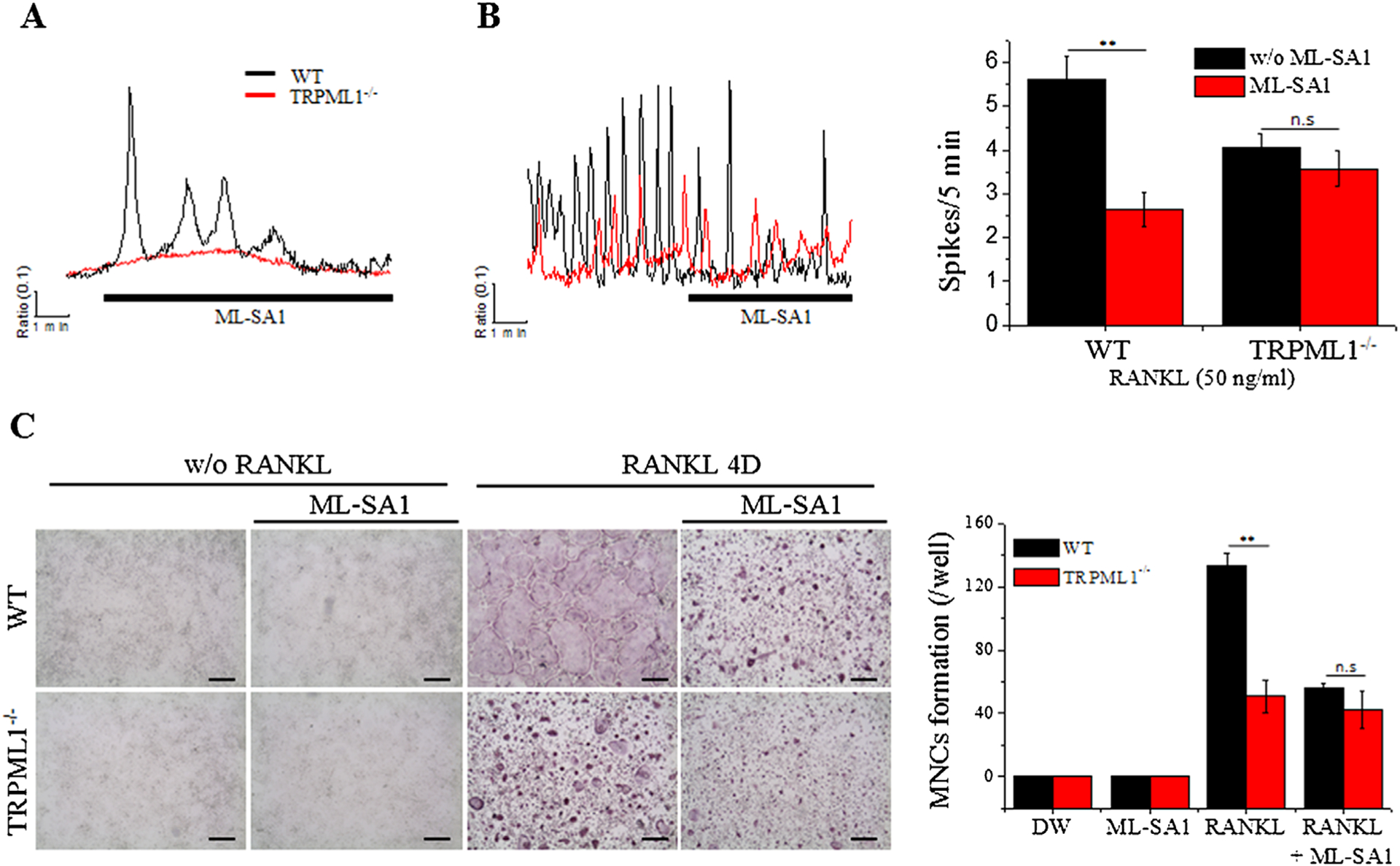
Depletion of lysosomal Ca2+ by activation of TRPML1 abrogates RANKL-induced [Ca2+]i oscillations and osteoclastogenesis. (A, B) BMMs from WT and TRPML1−/− mice were cultured for 48 hours with (B) or without (A) RANKL stimulation (50 ng/mL). The cells were then exposed to the specific TRPML activator, ML-SA1 (20 μM), to selectively deplete lysosomal Ca2+. The columns are the mean±SD of Ca2+ oscillation frequency recorded in 7 experiments. (C) BMMs from WT and TRPML1−/− mice were cultured with and without RANKL and in the presence or absence of 20 μM MA-SA1 for 3 days. The cultures were then stained for TRAP and TRAP+ MNCs were counted. The columns show the mean±SD from 3 experiments. (Scale bar = 200 μm). **p < 0.01.
Impaired lysosomal size and bone-resorptive activity in TRPML1−/− BMMs
The human mucolipidosis type 4 (MLIV) disease and the MLIV mouse model are typified by impaired lysosomal function and trafficking.(21) Recently, we reported that the lysosomes in secretory cells fuse with secretory organelles resulting in enlarged lysosomal hybrids.(37) To determine if the same occurs in the secretory BMMs, we measured lysosomal size in undifferentiated and RANKL-differentiated WT and TRPML1−/− BMMs. The lysosomes were visualized in live cells treated with or without RANKL for 2 days, loaded with Lysotracker, and the perimeter of the lysosomes was measured. Figure 5A shows that the lysosomes are significantly larger in TRPML1−/− compared to WT BMMs, whereas RANKL treatment slightly increased the size of the lysosomes in both WT and TRPML1−/− BMMs.
Fig. 5.
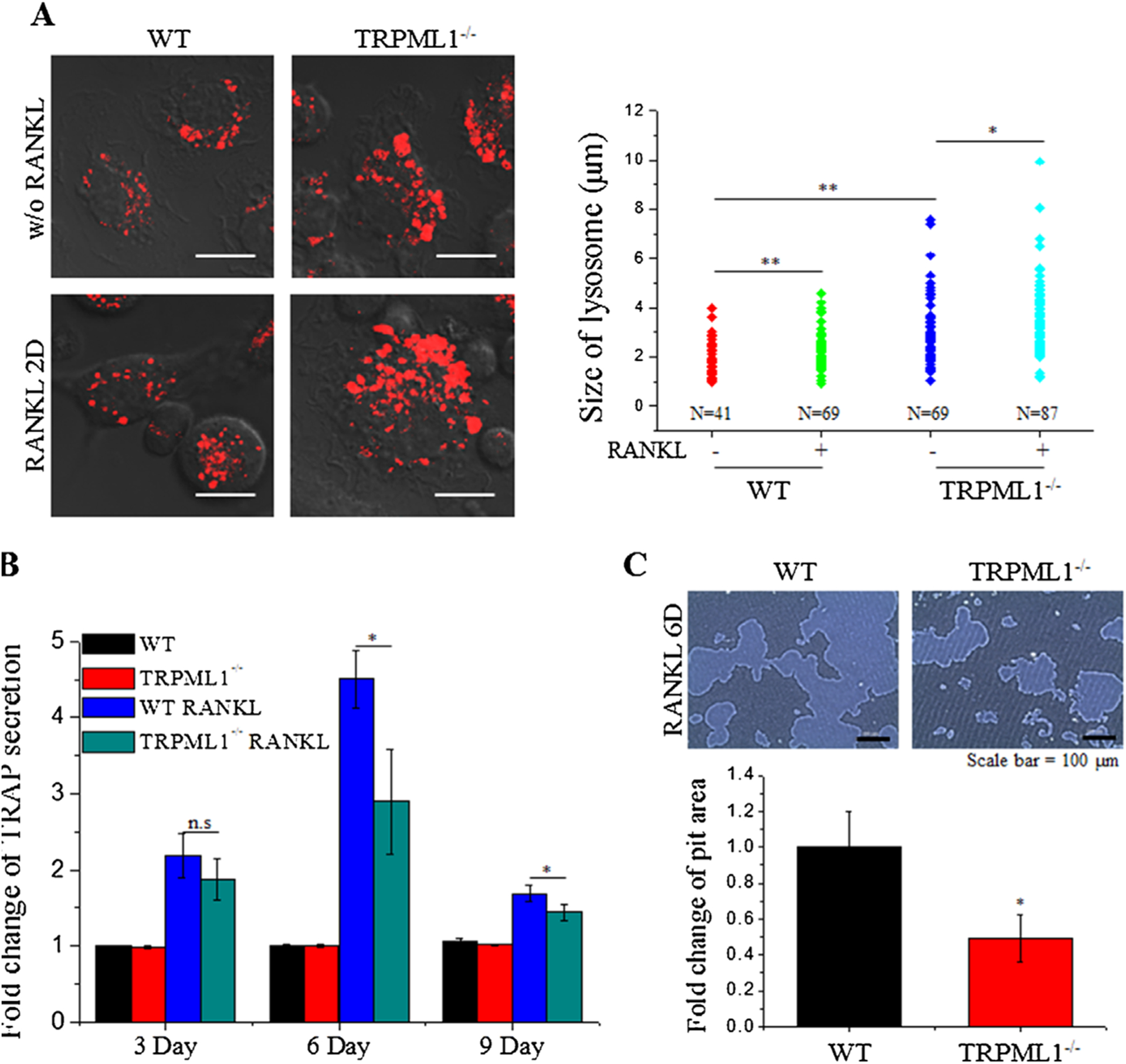
Deletion of TRPML1 enlarges osteoclastic lysosome size and impairs bone resorption. (A) BMMs isolated from WT and TRPML1−/− mice were cultured with and without RANKL for 2 days, as indicated. The lysosomes were then loaded with Lysotracker and imaged by confocal microscopy by random selection of images. Lysosome sizes were then analyzed based on lysosome circumference. The figure shows representative images (Scale bar = 10 μm) and lysosome size under each condition. The results are from 3 experiments and 4 images were randomly chosen from each experiment. (B) Effects of TRPML1 deficiency on the TRAP secretion was determined by its release into culture medium. The media were collected at the indicated times and used to measure TRAP activity. TRAP secretion was normalized to the activity of total TRAP collected by lysing the cells. Fold change in TRAP activities compared to WT without RANKL, is shown as the mean±SD of 3 experiments. *p < 0.05, **p < 0.01 respectively. (C) Bone-resorptive activity of WT and TRPML1−/− osteoclasts was evaluated by seeding BMMs on hydroxyapatite-coated dishes and culturing for 6 days with or without RANKL. After removal of the cells, pits formed by mature osteoclasts were measured using ImageJ software. Images of RANKL-treated WT and TRPML1−/− cells are shown. (Scale bar = 100 μm). Pit area relative to RANKL-treated WT cells is shown as the mean±SD of 3 experiments.
The enlarged size of the lysosomes in TRPML1−/− BMMs may impair lysosomal secretion, which is essential for osteoclast function and bone remodeling.(38) To test this, we evaluated lysosomal secretion during bone resorption by measuring TRAP secretion in RANKL-stimulated WT and TRPML1−/− BMMs grown on hydroxyapatite-coated plates for 9 days. TRAP activity in the culture media was measured every 3 days. Figure 5B shows that after 6 days in culture, RANKL-stimulated TRAP secretion is significantly reduced in TRPML1−/− BMMs (WT 4.505±0.376 fold, TRPML1−/− 2.894±0.688 fold, p < 0.05, n=3). TRAP secretion was similar in WT and TRPML1−/− BMMs at 3 and 9 days, when the bone resorption was low.
The effect of impaired lysosomal secretion on bone resorption, due to impaired lysosomal Ca2+ signaling, is shown in Fig. 5C. WT and TRPML1−/− BMMs were seeded on hydroxyapatite-coated plates and the area of the pits formed by mature osteoclasts was evaluated. Deletion of TRPML1 resulted in 0.51±0.133 fold (p < 0.05, n=6) reduction in pit formation. Hence, impaired lysosomal Ca2+ signaling diminishes both differentiation of osteoclasts and the resorbing function of mature osteoclasts.
Reduced osteoclast number and defective bone remodeling in TRPML1−/− mice
Analysis of RANKL-mediated osteoclastogenesis showed that inhibition of lysosomal Ca2+ signaling resulted in impaired osteoclastic differentiation and function. To determine the in vivo consequences of impaired lysosomal Ca2+ signaling, we evaluated the proportion of TRAP+ surface in bone surface as an indicator of in vivo osteoclastogenesis. Figure 6A shows that deletion of TRPML1 significantly reduced the osteoclast positive surface (Oc.S/BS) by 0.64±0.107 fold (p < 0.01, n=4), indicating reduced osteoclast population. The effect of reduced number of osteoclasts on bone remodeling and quality was determined by imaging the bone microstructure by high-resolution μCT. Figure 6B shows that bone volume, trabecular bone volume per tissue volume (BV/TV), trabecular number (Tb.N), and trabecular thickness (Tb.Th) are increased, whereas trabecular spacing (Tb.Sp) and structure model index are reduced without a change in bone mineral density (BMD) in TRPML1−/− mice, indicating abnormal bone remodeling in vivo. Thus, all parameters, except BMD, indicate osteopetrosis in TRPML1−/− mice.
Fig. 6.
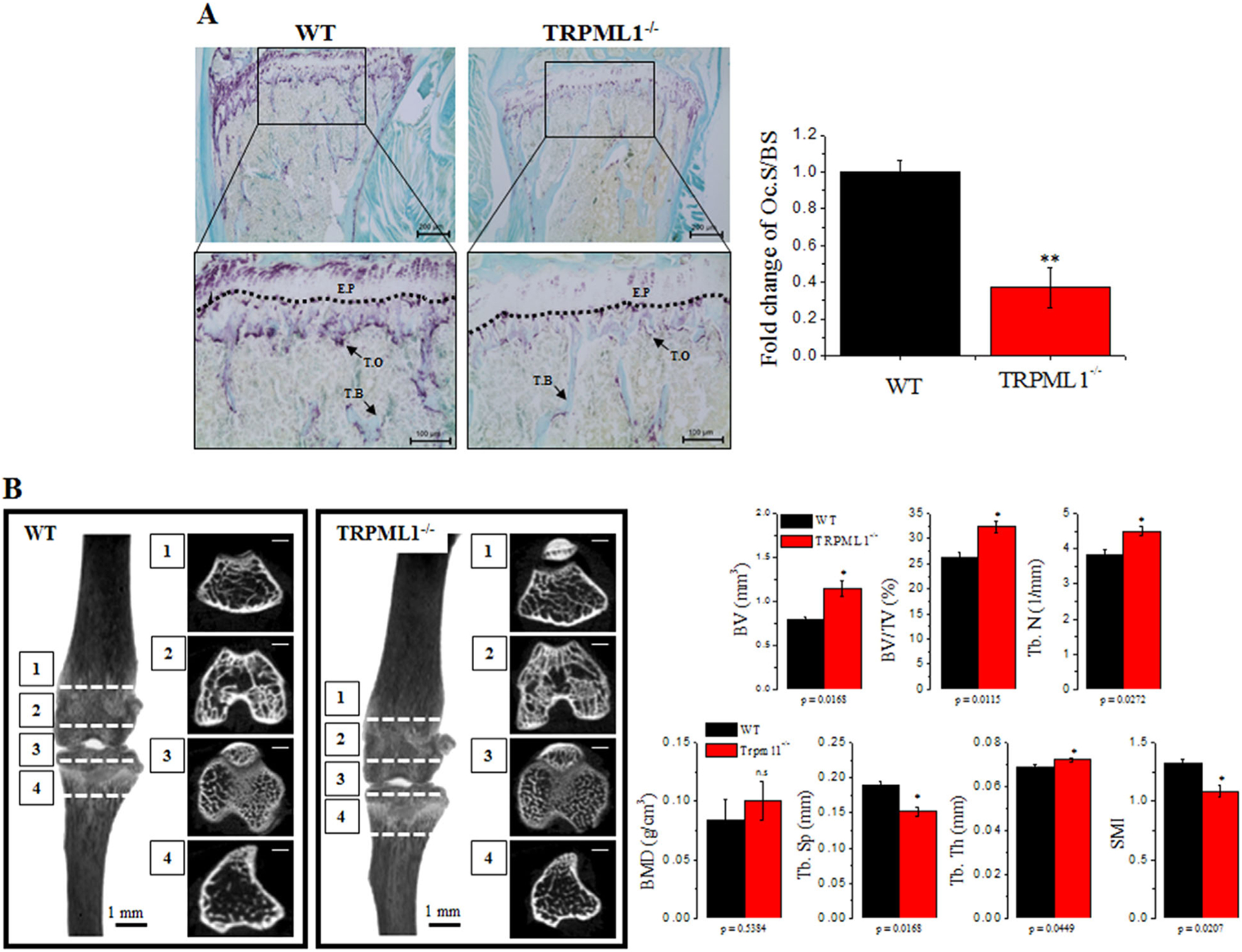
Pathological osteoclast function and bone remodeling in TRPML1−/− mice. For the in vivo experiments, proximal femoral bones and tibias were isolated from WT and TRPML1−/− mice and used for histological analysis and μCT. (A) Bone sections were stained for TRAP. The Oc.S/BS (%) was analyzed, and results are presented as fold change relative to WT. (Scale bar = 100 μm). Dashed line demarcates the E.P. (B) μCT images of proximal femur obtained from WT and TRPML1−/− mice. Each parameter, including BV, BV/TV, BMD, Tb.N, Tb.Th, Tb.Sp, and SMI were calculated and are presented. *p < 0.05, **p < 0.01. (Scale bar = 500 μm). Oc.S/BS = osteoclast surface; E.P = epiphyseal plate; T.O = TRAP+ osteoclasts; T.B = trabecular bone; BV = bone volume; BV/TV = bone volume per tissue volume; BMD = bone mineral density; Tb.N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; SMI = structure model index.
Abnormally increased osteoblast function in TRPML1−/− mice may contribute to osteopetrosis. To test this hypothesis, we measured bone formation in vivo according to the calcein method, and osteoblast differentiation in vitro. The results in Fig. 7 show that bone formation rate (BFR/BS), mineral apposition rate (MAR), mineralizing surface (MS/BS) (Fig. 7A) and osteoblastic differentiation (Fig. 7B, C) are not altered by deletion of TRPML1.
Fig. 7.
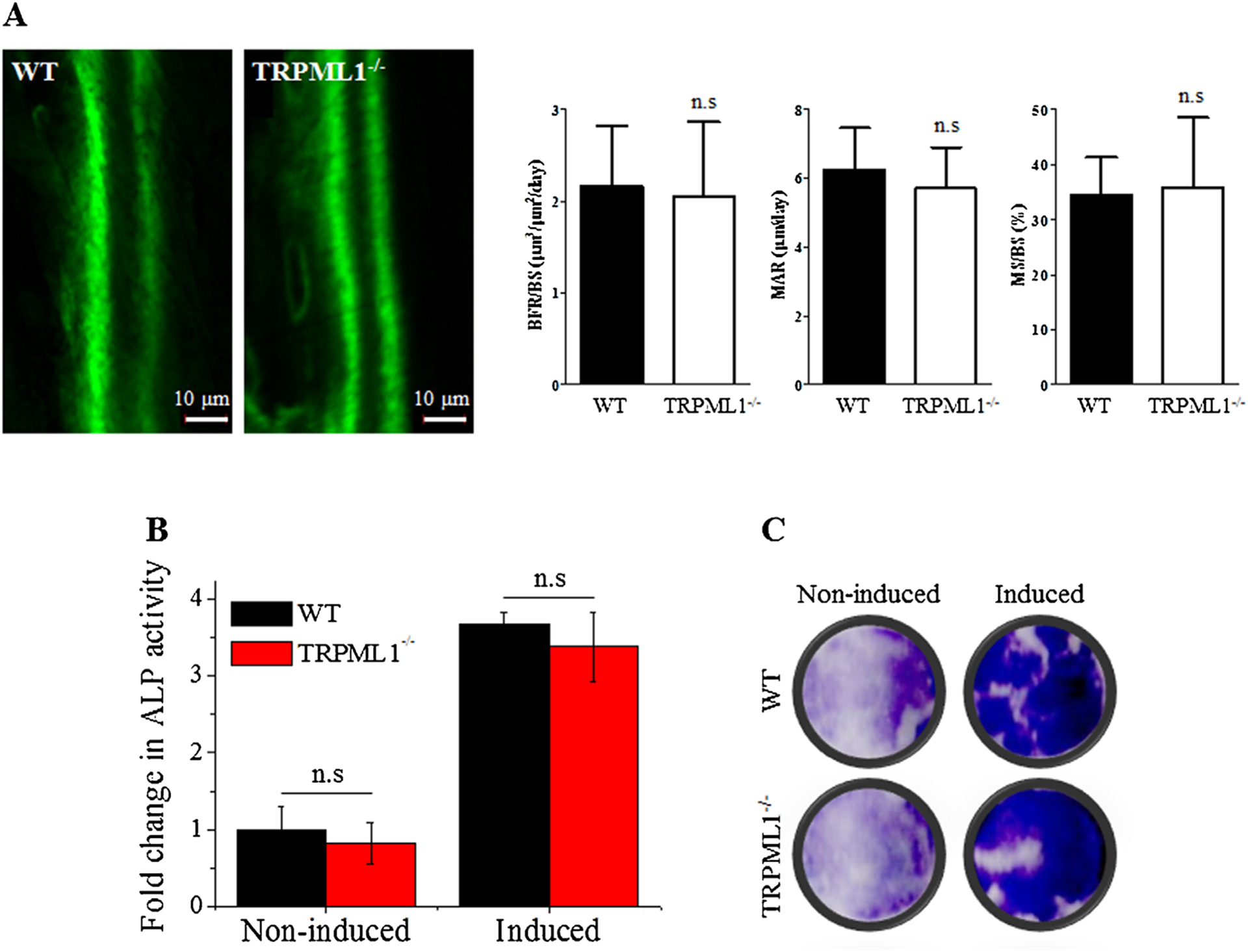
Deletion of TRPML1 has no effect on bone formation in vivo and osteoblastogenesis in vitro. (A) Active bone formation assayed by the calcein method showing similar formation in WT and TRPML1−/− mice. The columns on the right present the BFR/BS, MAR, and MS/BS (n = 3 mice). (B, C) Osteoblastogenesis in vitro was evaluated by measuring total ALP activity (B), and intracellular ALP in fixed and stained cells of differentiated osteoblasts (C) (n = 83). BFR/BS = bone formation rate; MAR = mineral apposition rate; MS/BS = mineralizing surface.
Discussion
A growing body of evidence indicates that lysosomal Ca2+ homeostasis and signaling is essential for many lysosomal functions,(39) including membrane trafficking, lysosomal exocytosis, and receptor evoked Ca2+ signaling.(40) Physiological receptor stimulation induces Ca2+ oscillations rather than peak/plateau type response, which is a pathological Ca2+ signal.(41) Lysosomal Ca2+ release is the trigger for Ca2+ release from ER during Ca2+ oscillations,(42) and thus, lysosomal Ca2+ release is involved in the receptor-dependent Ca2+ signal and Ca2+-regulated cell functions. Lysosomal Ca2+ release is mediated by the TRP family channel, TRPML1.(20–22,43,44) Hence, TRPML1 function is expected to regulate lysosomal function. Another function of the lysosomes that require lysosomal Ca2+ release, which is of particular significance to the present work, is lysosomal exocytosis, which is required for surface membrane remodeling, as observed in osteoclast activation,(45) phagocytosis,(23) and membrane repair.(46) Osteoclastogenesis and bone remodeling is regulated by RANKL interaction with RANK and generation of [Ca2+]i oscillations in the early stage of osteoclastogenesis. Subsequent fusion of lysosomes with the ruffled membrane mediates bone resorptive activity of mature osteoclasts.(45) It was thus of interest to determine the role of lysosomal Ca2+ homeostasis and function in osteoclast differentiation and function and the consequent bone metabolism, in an effort to further understand the molecular mechanism of these processes.
Ca2+ signaling by RANKL acting on osteoclasts has been studied extensively,(3,4,7,8) including by us,(26) which showed that the key events in RANKL-induced Ca2+ oscillations involve the interaction of the regulator of G-protein signaling 10 (RGS10) with phosphatidylinositol 3,4,5-triphosphate (PIP3) and PLCγ2 with immune receptor tyrosine activation motif (ITAM) proteins, leading to Ca2+ release from the ER and subsequent Ca2+ influx by the store-operated Ca2+ channels.(47,48) In this study, we extend this conclusion by providing multiple lines of evidence, which show that lysosomal Ca2+ release is essential for the RANKL-induced [Ca2+]i oscillations, probably by sensitizing and triggering the ER-mediated Ca2+ release. Thus, depletion of lysosomal Ca2+ content by bursting the lysosomes with GPN, inhibiting the lysosomal H+ pump to collapse the lysosomal pH, sustainably activating TRPML1 with ML-SA1, and deleting TRPML1 inhibits the RANKL-mediated Ca2+ oscillations.
Disruption of RANKL-mediated Ca2+ oscillations by deletion of TRPML1 inhibited all downstream functions involved in osteoclastogenesis and bone remodeling. This included induction and activation of NFATc1, activation of genes involved in osteoclast differentiation, and formation of MNCs. In MLIV and in TRPML1 knockout mice, the disease includes achlorhydria, aberrant neuromuscular junctions, neurodegeneration,(49–52) and chronic pancreatitis.(37) The neurodegeneration can have multiple effects on various tissues. Therefore, it is important to show that the results obtained upon acute knockdown of TRPML1 in vitro replicated those obtained by inhibition of osteoclastogenesis in vivo by deletion of TRPML1. Indeed, acute deletion of TRPML1 in BMMs suppressed TRAP+MNC formation (Fig. 1).
In addition to its role in osteoclastogenesis, lysosomal Ca2+ signaling regulates the function of mature osteoclasts. Lysosomal exocytosis plays an important role in several membrane trafficking events, such as plasma membrane repair(46) and phagocytosis,(23) which require lysosomal Ca2+ release. Lysosomal exocytosis is also an important step in osteoclastic bone resorption, requiring fusion of lysosomes with the ruffled membrane and secretion of lysosomal hydrolases to the bone surface.(45) Our findings show that lysosomal exocytosis is impaired in mature TRPML1−/− osteoclasts, which probably contributed to the impairment of bone resorption. When bone resorption and exocytosis are relatively slow (at 3 and 9 days in culture), differences between WT and TRPML1−/− cells were not observed, but at high bone resorption period (day 6 in culture), lysosomal exocytosis in TRPML1−/− osteoclasts was reduced. Several altered lysosomal functions likely account for the reduced exocytosis. The lysosomes in TRPML1−/− osteoclasts are markedly enlarged (Fig. 5), as occurs in lysosomal storage diseases,(53) which impairs lysosomal trafficking. Once at the plasma membrane, lysosomal fusion requires lysosomal Ca2+ release. The lack of lysosomal Ca2+ release is expected to prevent lysosomal fusion with the ruffled membrane.
The impaired osteoclastogenesis and mature osteoclast function due to aberrant lysosomal Ca2+ signaling resulted in osteopetrosis without a change in bone mineral density (Fig. 6). Osteopetrosis is a heterogeneous disease with multiple phenotypes.(54) Bone density is affected by the function of both osteoclasts and osteoblasts and is regulated by many factors. The processes that determine bone density appear to be independent of lysosomal Ca2+ signaling and function. However, this requires further in-depth probing.
Notably, the phenotype reported here is specific to MLIV. In MLIV, the function of osteoclasts was impaired whereas the function of osteoblasts and bone formation remained unaltered (Fig. 7), resulting in osteopetrosis. In mucolipidosis type II the function of osteoblasts and osteoblastogenesis is markedly impaired, whereas osteoclastogenesis is dramatically increased and their bone resorbing function is unaltered, resulting in osteoporosis.(55) The etiology of the two mucolipidoses is different; mucolipidosis type II is a metabolic disease with altered lysosomal hydrolytic functions, whereas MLIV is associated with impaired lysosomal trafficking. This may account for the opposite phenotypes of these two lysosomal storage diseases.
The findings presented in this study reveal the critical roles of lysosomal Ca2+ signaling in all aspects of osteoclast functions and bone metabolism, and unravel a new aspect of the regulation of bone remodeling. Our findings should have implications for management of MLIV. Patients with MLIV present muscle weakness that is in part due to impaired neuromuscular junctions(52) and in part due to impaired muscle repair(24) and loss of mobility at an early age. Our findings indicate that defective skeletal structure could be an additional contributor to the loss of mobility and muscle function.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grants (NRF-2015R1D1A1A01058272 and NRF-2015R1A2A1A15054157); Ministry of Education, Science and Technology and Ministry of Science, ICT, and Future Planning in Republic of Korea; and by NIH, NIDCR intramural grant DE000735-06.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
References
- 1.Ikeda K, Takeshita S. Factors and mechanisms involved in the coupling from bone resorption to formation: how osteoclasts talk to osteoblasts. J Bone Metab. 2014;21(3):163–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng X, McDonald JM. Disorders of bone remodeling. Annu Rev Pathol. 2011;6:121–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. [DOI] [PubMed] [Google Scholar]
- 4.Koga T, Inui M, Inoue K, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428(6984):758–63. [DOI] [PubMed] [Google Scholar]
- 5.Hwang SY, Putney JW Jr. Calcium signaling in osteoclasts. Biochim Biophys Acta. 2011;1813(5):979–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu H, Lacey DL, Dunstan CR, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96(7):3540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ, Prasad V, Hyung SW, et al. Plasma membrane calcium ATPase regulates bone mass by fine-tuning osteoclast differentiation and survival. J Cell Biol. 2012;199(7):1145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang YM, Kim MS, Son A, et al. Alteration of RANKL-induced osteoclastogenesis in primary cultured osteoclasts from SERCA2+/− mice. J Bone Miner Res. 2009;24(10):1763–9. [DOI] [PubMed] [Google Scholar]
- 9.Kim H, Kim T, Jeong BC, et al. Tmem64 modulates calcium signaling during RANKL-mediated osteoclast differentiation. Cell Metab. 2013;17(2):249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Notomi T, Ezura Y, Noda M. Identification of two-pore channel 2 as a novel regulator of osteoclastogenesis. J Biol Chem. 2012;287(42): 35057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao X, Choi S, Maleth JJ, Park S, Ahuja M, Muallem S. The ER/PM microdomain, PI(4,5)P(2) and the regulation of STIM1-Orai1 channel function. Cell Calcium. 2015;58(4):342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krapivinsky G, Krapivinsky L, Stotz SC, Manasian Y, Clapham DE. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc Natl Acad Sci U S A. 2011;108(48): 19234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel S Function and dysfunction of two-pore channels. Sci Signal. 2015;8(384):re7. [DOI] [PubMed] [Google Scholar]
- 14.Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77: 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiselyov K, Chen J, Rbaibi Y, et al. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280(52):43218–23. [DOI] [PubMed] [Google Scholar]
- 16.Thompson EG, Schaheen L, Dang H, Fares H. Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol. 2007;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bargal R, Avidan N, Ben-Asher E, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26(1):118–23. [DOI] [PubMed] [Google Scholar]
- 18.Cheng X, Shen D, Samie M, Xu H. Mucolipins: intracellular TRPML1–3 channels. FEBS Lett. 2010;584(10):2013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altarescu G, Sun M, Moore DF, et al. The neurogenetics of mucolipidosis type IV. Neurology. 2002;59(3):306–13. [DOI] [PubMed] [Google Scholar]
- 20.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, et al. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281(11):7294–301. [DOI] [PubMed] [Google Scholar]
- 21.Miedel MT, Rbaibi Y, Guerriero CJ, et al. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008;205(6):1477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaPlante JM, Sun M, Falardeau J, et al. Lysosomal exocytosis is impaired in mucolipidosis type IV. Mol Genet Metab. 2006;89(4):339–48. [DOI] [PubMed] [Google Scholar]
- 23.Samie M, Wang X, Zhang X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell. 2013;26(5):511–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng X, Zhang X, Gao Q, et al. The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent+muscular dystrophy. Nat Med. 2014;20(10):1187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Gao Q, Yang M, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A. 2015;112(11):E1373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MS, Yang YM, Son A, et al. RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J Biol Chem. 2010;285(10):6913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1(1):11–21. [DOI] [PubMed] [Google Scholar]
- 28.Morgan AJ, Davis LC, Wagner SK, et al. Bidirectional Ca(2)(+) signaling occurs between the endoplasmic reticulum and acidic organelles. J Cell Biol. 2013;200(6):789–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez-Sanjurjo CI, Tovey SC, Prole DL, Taylor CW. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J Cell Sci. 2013; 126(Pt 1):289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandra M, Zhou H, Li Q, Muallem S, Hofmann SL, Soyombo AA. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology. 2011;140(3): 857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu DR, Hwang JK, Erkhembaatar M, et al. Inhibitory effect of Chrysanthemum zawadskii Herbich var. latilobum Kitamura extract on RANKL-induced osteoclast differentiation. Evid Based Complement Alternat Med. 2013;2013:509482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoemann CD, El-Gabalawy H, McKee MD. In vitro osteogenesis assays: influence of the primary cell source on alkaline phosphatase activity and mineralization. Pathol Biol (Paris). 2009;57(4):318–23. [DOI] [PubMed] [Google Scholar]
- 33.Mulari MT, Zhao H, Lakkakorpi PT, Vaananen HK. Osteoclast ruffled border has distinct subdomains for secretion and degraded matrix uptake. Traffic. 2003;4(2):113–25. [DOI] [PubMed] [Google Scholar]
- 34.Palokangas H, Mulari M, Vaananen HK. Endocytic pathway from the basal plasma membrane to the ruffled border membrane in bone-resorbing osteoclasts. J Cell Sci. 1997;110(Pt 15):1767–80. [DOI] [PubMed] [Google Scholar]
- 35.Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nat Rev Mol Cell Biol. 2008;9(11): 833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen D, Wang X, Li X, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park S, Ahuja M, Kim MS, et al. Fusion of lysosomes with secretory organelles leads to uncontrolled exocytosis in the lysosomal storage disease mucolipidosis type IV. EMBO Rep. 2016;17(2):266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Touaitahuata H, Blangy A, Vives V. Modulation of osteoclast differentiation and bone resorption by Rho GTPases. Small GTPases. 2014;5:e28119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lloyd-Evans E, Platt FM. Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium. 2011;50(2): 200–5. [DOI] [PubMed] [Google Scholar]
- 40.LaPlante JM, Ye CP, Quinn SJ, et al. Functional links between mucolipin-1 and Ca2+-dependent membrane trafficking in mucolipidosis IV. Biochem Biophys Res Commun. 2004;322(4):1384–91. [DOI] [PubMed] [Google Scholar]
- 41.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–29. [DOI] [PubMed] [Google Scholar]
- 42.Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci. 2013;126(Pt 1):60–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CS, Bach G, Pagano RE. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc Natl Acad Sci U S A. 1998;95(11):6373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vergarajauregui S, Connelly PS, Daniels MP, Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum Mol Genet. 2008;17(17):2723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H Membrane trafficking in osteoblasts and osteoclasts: new avenues for understanding and treating skeletal diseases. Traffic. 2012;13(10):1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106(2):157–69. [DOI] [PubMed] [Google Scholar]
- 47.Yang S, Li YP. RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation. Genes Dev. 2007;21(14):1803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest. 2006;116(11):2869–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiffmann R, Dwyer NK, Lubensky IA, et al. Constitutive achlorhydria in mucolipidosis type IV. Proc Natl Acad Sci U S A. 1998;95(3):1207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gordon RM, Marchese T. Mucolipidosis type IV a rare genetic disorder: new addition to the Ashkenazi Jewish panel. J Midwifery Womens Health. 2004;49(4):359–60. [DOI] [PubMed] [Google Scholar]
- 51.Geer JS, Skinner SA, Goldin E, Holden KR. Mucolipidosis type IV: a subtle pediatric neurodegenerative disorder. Pediatr Neurol. 2010;42(3):223–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venkatachalam K, Long AA, Elsaesser R, Nikolaeva D, Broadie K, Montell C. Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell. 2008;135(5):838–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bargal R, Bach G. Mucolipidosis type IV: abnormal transport of lipids to lysosomes. J Inherit Metab Dis. 1997;20(5):625–32. [DOI] [PubMed] [Google Scholar]
- 54.Aggarwal S Skeletal dysplasias with increased bone density: evolution of molecular pathogenesis in the last century. Gene. 2013;528(1):41–5. [DOI] [PubMed] [Google Scholar]
- 55.Kollmann K, Pestka JM, Kuhn SC, et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol Med. 2013;5(12):1871–86. [DOI] [PMC free article] [PubMed] [Google Scholar]