

Abstract
Neutral sphingomyelinase 2 (nSMase2), a key enzyme in ceramide biosynthesis, is a new therapeutic target for the treatment of neurological disorders and cancer. Using 2,6-dimethoxy-4-[4-phenyl-5-(2-thienyl)-1H-imidazol-2-yl]phenol (DPTIP), our initial hit compound (IC50 = 30 nM) from nSMase2 screening efforts, as a molecular template, a series of 4-(1H-imidazol-2-yl)-2,6-dialkoxyphenol derivatives were designed, synthesized, and evaluated. Systematic examination of various regions of DPTIP identified the key pharmacophore required for potent nSMase2 inhibition as well as a number of compounds with the 4-(1H-imidazol-2-yl)-2,6-dialkoxyphenol scaffold with similar or higher inhibitory potency against nSMase2 as compared to DPTIP. Among them, 4-(4,5-diisopropyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol (25b) was found to be metabolically stable against P450 metabolism in liver microsomes and displayed higher plasma exposure following oral administration as compared to DPTIP. Analysis of plasma samples identified an O-glucuronide as the major metabolite. Blockade of the phase II metabolism should further facilitate our efforts to identify potent nSMase2 inhibitors with desirable ADME properties.
Keywords: phosphodiesterase, neutral sphingomyelinase 2, sphingomyelin, ceramide, glucuronidation
Graphical Abstract
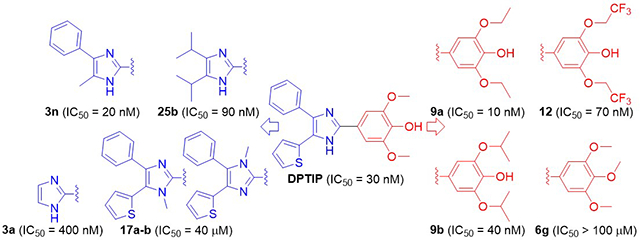
1. Introduction
Neutral sphingomyelinase 2 (nSMase2) is a membrane associated enzyme that catalyzes the hydrolysis of sphingomyelin (SM) into phosphorylcholine and ceramide (Figure 1) in response to extracellular stimuli [1, 2]. Formation of ceramide enriched areas due to the action of nSMase2 is associated with changes in mechanical properties of the membrane [3] as well as organization and function of various membrane bound receptors [4, 5]. Moreover, ceramide is involved in formation of exosomes, a specific type of extracellular vesicle, which participate in intercellular communication in both physiological and pathological processes through encapsulation and transfer of diverse types of substances (proteins, lipids, and RNAs) [6, 7]. For instance, exosomes have been shown to carry pathogenic molecules such as amyloid-β [8] and tau protein [9] and upregulated ceramide production has been implicated in various neurological disorders including Alzheimer’s disease, HIV-associated neurocognitive disorders, Parkinson’s disease, and ALS [1, 10–12]. Furthermore, nSMase2-regulated release of miRNAs was reported to play an important role in cancer cell metastasis [13, 14] while production of extracellular vesicles containing hepatitis B virus (HBV) DNA was implicated as a separate pathway for transmission of HBV.[15] These findings collectively suggest that pathogenic extracellular vesicles generated through the action of nSMase2 are involved in a broad range of diseases. Thus, inhibition of nSMase2 may offer unique therapeutic opportunity in these diseases by reducing ceramide levels and subsequent exosome formation.
Fig. 1.
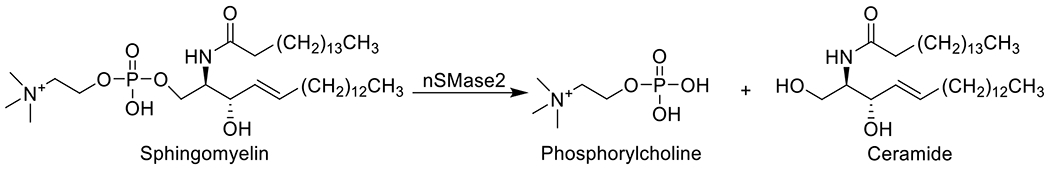
Reaction catalyzed by nSMase2.
To date, a number of nSMase2 inhibitors have been reported. Unfortunately, these compounds inhibit nSMase2 only at micromolar concentrations and display unfavorable physicochemical and molecular properties, limiting their ability to serve as molecular templates for lead optimization efforts [16–18]. For instance, the most widely used tool inhibitor GW4869 (Figure 2) [19], exhibits low nSMase2 inhibitory activity (IC50 = 1 μM) in biochemical assays. In addition, it has negligible aqueous solubility and very poor solubility in organic solvents (e.g. 0.2 mg/ml in DMSO) which has limited its potential as a lead nSMase2 inhibitor. Cambinol, an inhibitor identified by our group from a pilot screening of commercially available small chemical libraries (Figure 2) [18], showed better solubility compared to GW4869, but it was metabolically unstable and exhibited low potency (IC50 = 5 μM).
Fig. 2.
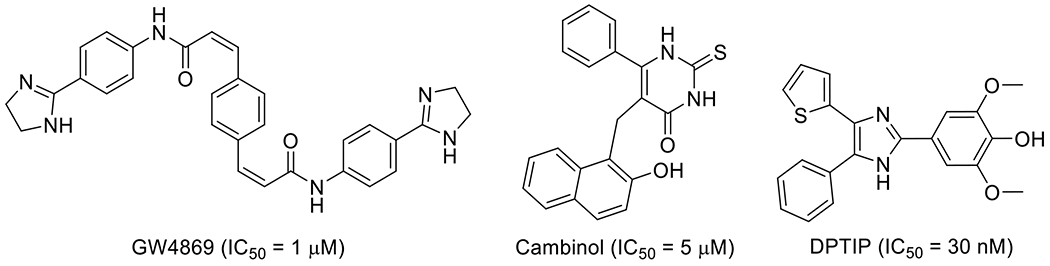
Examples of nSMase2 inhibitors.
Our large-scale screening efforts have recently identified a highly potent nSMase2 inhibitor (IC50 = 30 nM), 2,6-dimethoxy-4-[4-phenyl-5-(2-thienyl)-1H-imidazol-2-yl]phenol (DPTIP, Figure 2), which is structurally distinct from the existing nSMase2 inhibitors [20]. Although the phenol moiety in DPTIP raises concerns about the promiscuous activity towards multiple targets, DPTIP showed weak activity (2 - 50 μM) in only 19 (2.5%) of 759 different bioassays conducted at NCATS [20]. In addition, DPTIP was found to dose-dependently inhibit extracellular vesicle release in primary astrocyte cultures, an orthogonal assay of high physiological relevance [20]. Furthermore, DPTIP was reported to attenuate IL-1β-induced astrocyte-derived extracellular vesicle release in an animal model of brain injury conducted in GFAP-GFP mice [20]. In order to further establish structure-activity relationship (SAR) of this new promising series of nSMase2 inhibitors, we conducted a systematic examination of various regions of DPTIP with the primary focus on determining the key pharmacophore required for potent nSMase2 inhibition. Since DPTIP is already highly potent in inhibiting nSMase2, the objective of the lead optimization efforts was to improve ADME properties rather than to further improve inhibitory potency. We divided the molecule into three distinct components, namely, the para-substituted phenol, the central imidazole ring, and two substituents at the 4- and 5-positions of the imidazole ring. Performing chemical modifications in each of the three areas independently allowed us to evaluate its relative importance for nSMase2 inhibition. Herein, we report the design, synthesis, SAR, and ADME profile of DPTIP derivatives as potent nSMase2 inhibitors.
2. Results and discussion
2.1. Chemistry
Compounds 3a-n containing various substituents at the 4 and 5-positions of the imidazole ring were prepared by condensation of syringaldehyde (1) and α-dicarbonyl compounds 2a-n in the presence of ammonium acetate (Scheme 1) as previously reported [21] with some minor modifications.
Scheme 1.
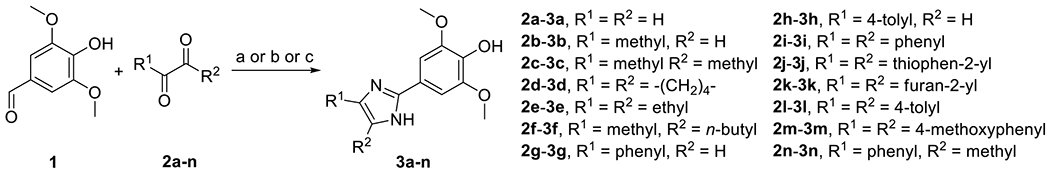
Synthesis of compounds 3a-3n. Reagents and conditions: (a) NH4OAc, AcOH, reflux, 3-11%; (b) NH4OAc, EtOH, 60-85 °C, 10-72%; (c) NH4OAc, EtOH, rt, 11-26%.
DPTIP derivatives 6a-e containing an altered phenolic moiety were prepared from aldehydes 5a-e and 1-phenyl-2-(thiophen-2-yl)ethane-1,2-dione 4 (Scheme 2) using condensation reactions as described in Scheme 1. Additional DPTIP derivatives 9a-d retaining the para-phenol group were prepared from 3,4,5-trihydroxybenzaldehyde 5d in three steps. After conversion of 5d to its 4-(p-methoxybenzyl) (PMB) derivative 7 using modified literature procedure [22], Mitsunobu reaction was performed to afford 3,5-dialkoxy derivatives 8a-d. The advantage of using PMB protecting group was its spontaneous cleavage during the subsequent condensation with 4, providing the desired final products 9a-d (Scheme 3).
Scheme 2.
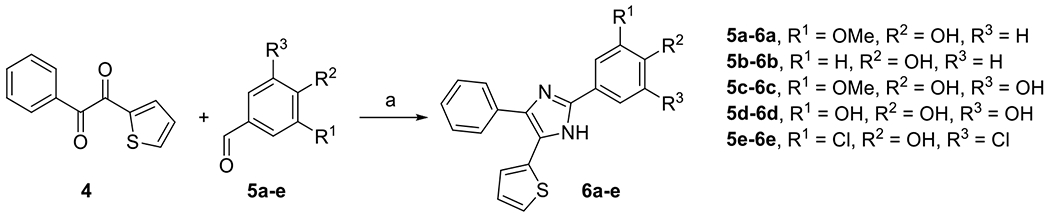
Synthesis of compounds 6a-e. Reagents and conditions: (a) NH4OAc, AcOH, 120 °C, 19-35%.
Scheme 3.
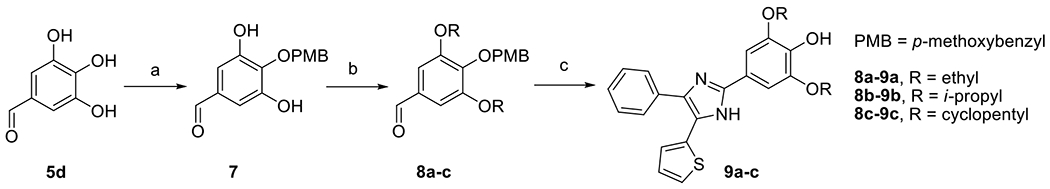
Synthesis of compounds 9a-c. Reagents and conditions: (a) NaH, PMB-Cl, DMF, 0 °C to rt, 44 %; (b) DIAD, PPh3, ROH, THF, 0 °C to rt, 20-90 %; (c) 4, NH4OAc, AcOH, 120 °C, 13-44 %.
Di-2,2,2-trifluoroethoxy derivative 12 could not be obtained by the Mitsunobu method described above. Instead, we converted dibromide 10 into 4-hydroxy-3,5-bis(2,2,2-trifluoroethoxy)benzaldehyde 11 using freshly prepared sodium 2,2,2-trifluoroethoxide in the presence of CuCl2 (Scheme 4) [23]. Compound 11 was subsequently condensed with 4 in the presence of ammonium acetate to give compound 12 (Scheme 4).
Scheme 4.
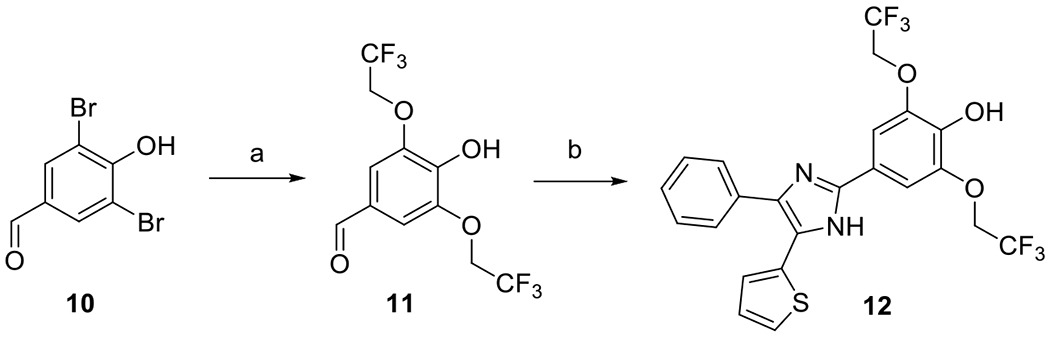
Synthesis of compound 12. Reagents and conditions: (a) CF3CH2OH, Na, CuCl2, DMF, 115 °C, 53 %; (b) 4, NH4OAc, AcOH, 120 °C, 36 %.
Di-tert-butoxy derivative 16 was synthesized using 3,5-dihydroxy-4-methoxybenzaldehyde 13 [24] as a starting material (Scheme 5). Reaction of 13 with N,N-dimethylformamide di-tert-butyl acetal [25] yielded 3,5-di-tert-butoxy-4-methoxybenzaldehyde 14, which was subsequently condensed with 4 in the presence of ammonium acetate to form compound 15. Demethylation of 15 was achieved by heating in a mixture of water and piperidine [26], giving the desired product 16.
Scheme 5.
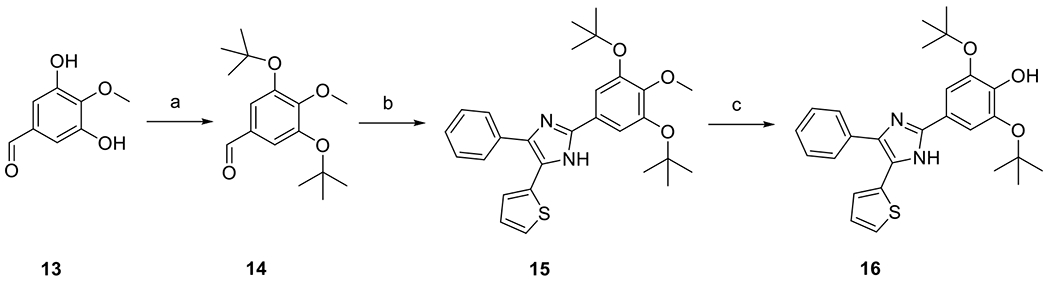
Synthesis of compound 16. Reagents and conditions: (a) N,N-Dimethylformamide di-t-butyl acetal, PhMe, 85 °C, 30 %; (b) 4, NH4OAc, AcOH, 120 °C, 19 %; (c) piperidine-H2O (v/v 50%), sealed tube, 150 °C, 67 %.
N-Methyl analogues of DPTIP (a mixture of two regioisomers 17a and 17b) were prepared by condensation of aldehyde 1 and diketone 4 in the presence of ammonium acetate and methylamine as previously described (Scheme 6) [27]. Compounds 17a and 17b were not separated. They were characterized and tested as mixture.
Scheme 6.
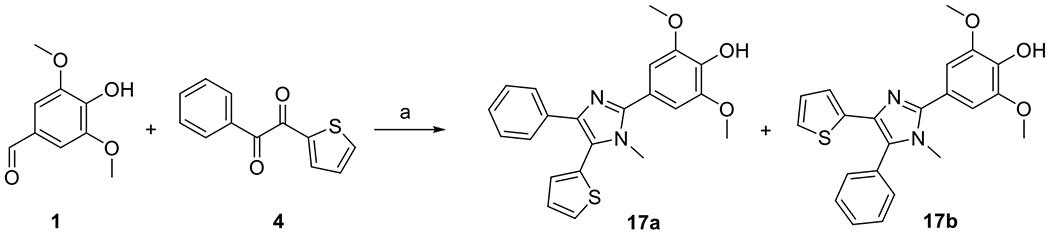
Synthesis of compounds 17a and 17b. Reagents and conditions: (a) NH4OAc, NH2Me, NaH2PO4, THF, sealed tube, 150 °C, 17 %.
Thiazole analog 19 lacking substituents at the 4- and 5-positions was synthesized by demethylation of the central methoxy group of compound 18 [28] using boron trichloride (Scheme 7) [29].
Scheme 7.
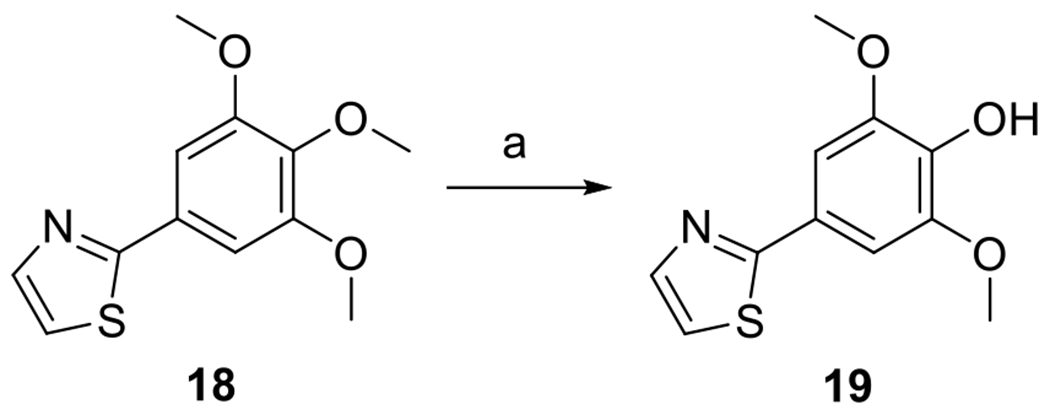
Synthesis of compound 19. Reagents and conditions: (a) BCl3, DCM, rt, 15 %.
Imidazoline analog 22 lacking 4- and 5-substituents was synthesized from aldehyde 1. Aldehyde 1 was first converted into the corresponding methanesulfonate 20, which was subsequently condensed with ethylenediamine under oxidative conditions [30] to give compound 21. Hydrolysis of the methanesulfonate ester 21 under basic conditions [31] gave compound 22 (Scheme 8).
Scheme 8.
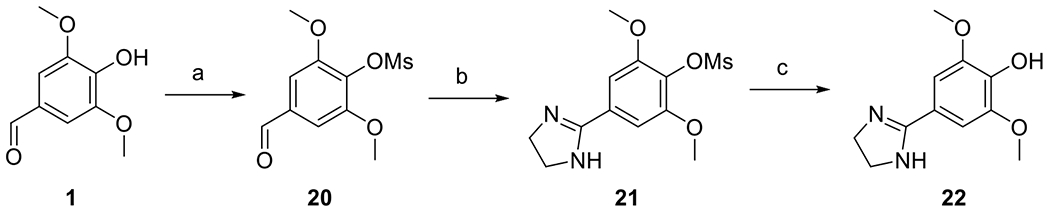
Synthesis of compound 22. Reagents and conditions: (a) MsCl, Et3N, DCM, 0 °C to rt, 71 %; (b) ethane-1,2-diamine, NBS, DCM, 0 °C to rt, 89 %; (c) KOH, MeOH, rt, 15 %.
Compounds 25a and 25b were prepared from α-hydroxy ketones 24a and 24b, respectively (Scheme 9) [32]. The oxidative formation of the imidazole ring appears to be mediated by dissolved oxygen in the solvent as the products did not form in the absence of oxygen.
Scheme 9.
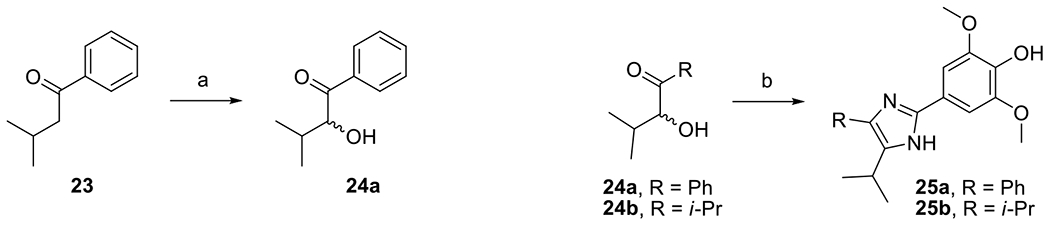
Synthesis of compounds 25a and 25b. Reagents and conditions: (a) (diacetoxyiodo)benzene, KOH, MeOH, rt; (b) 1, NH4OAc, O2, EtOH, 65 °C, 16 % for 25a, 43 % for 25b.
Synthesis of compound 29 is illustrated in Scheme 10. First, aldehyde 20 was converted to nitrile 26 with sulfamic acid [33]. Treatment of nitrile 26 with dry HCl followed by ammonolysis [34] afforded amidine 27, which was subsequently reacted with 2-bromo-1-(thiophen-2-yl)propan-1-one [35] to form 28. The methanesulfonyl protective group of 28 was removed by LDA [36] to yield compound 29.
Scheme 10.
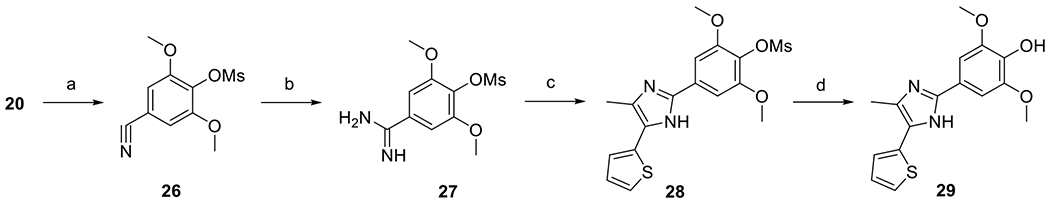
Synthesis of compound 29. Reagents and conditions: (a) H2NSO3H, AcOH, H2O, 100 °C, 98 %; (b) (i) HCl/dioxane, MeOH, rt, (ii) NH3/MeOH, rt, 20 %; (c) 2-bromo-1-(thiophen-2-yl)propan-1-one, K2CO3, EtOH, 78 °C, 48 %; (d) (i) LDA, THF, −78 °C to 0 °C, (ii) HCl, H2O, 0 °C, 14 %.
Glucuronide 30 was prepared from compound 25b by following a previously reported procedure (Scheme 11) [37]. Reaction afforded the β-anomer of O-glucuronide as the sole product. The low overall yield (two steps) was presumably caused by the poor reactivity of the sterically hindered phenolic OH of compound 25b in the first step. The formation of β-anomer was confirmed by 1H NMR coupling constant of anomeric hydrogen (J = 7.5 Hz). Equivalence of C4 and C5 carbons of imidazole ring (one signal in 13C NMR) suggests the symmetricity of the imidazole ring and rules out the formation of N-glucuronide. Another evidence supporting O-glucuronide formation was provided by the chemical shift of C-1’ anomeric carbon (101.4 ppm) of compound 30, which is in good agreement with those of other O-glucuronides (~100 ppm) derived from phenols[37] and a downfield shift of approximately 15 ppm with respect to the C-1’ (~85 ppm) of N-glucuronides derived imidazoles [38].
Scheme 11.
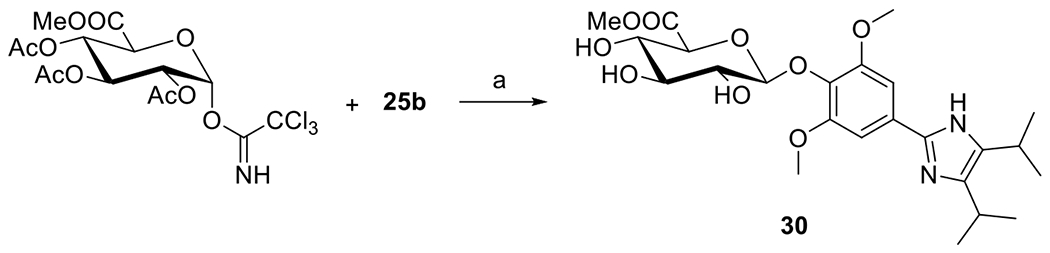
Synthesis of compound 30. Reagents and conditions: (a) (i) BF3∙OEt2, DCM, −10 °C; (ii) Na2CO3, MeOH, H2O, rt, 9 %.
2.2. Evaluation of nSMase2 inhibitory potency
The inhibitory potencies of the synthesized compounds were determined using a fluorescence-based coupled assay involving human nSMase2, alkaline phosphatase (AP), and choline oxidase as previously reported [18]. In order to assure that potent inhibitors identified in this coupled assay directly inhibit nSMase2, we performed a counter screen assay in which test compounds were incubated with AP, choline oxidase, and horseradish peroxidase in the presence of phosphorylcholine. Some caution needs to be taken in interpreting the inhibitory potency data when IC50 values from the primary assay are comparable to those from the counter screening as that may indicate interference of AP, choline oxidase, and/or horseradish peroxidase in addition to (or instead of) nSMase2 inhibition. We considered that compounds with greater than 5-fold separation in IC50 values in favor of nSMase2 assay over the counter assay predominantly inhibit nSMase2. The nSMase IC50 values for those with less than 5-fold separation from the counter assay are denoted with a double asterisk (**), indicating that those compounds may have a higher intrinsic IC50 value against nSMase2 than the listed value. The assay results are summarized in Tables 1-3.
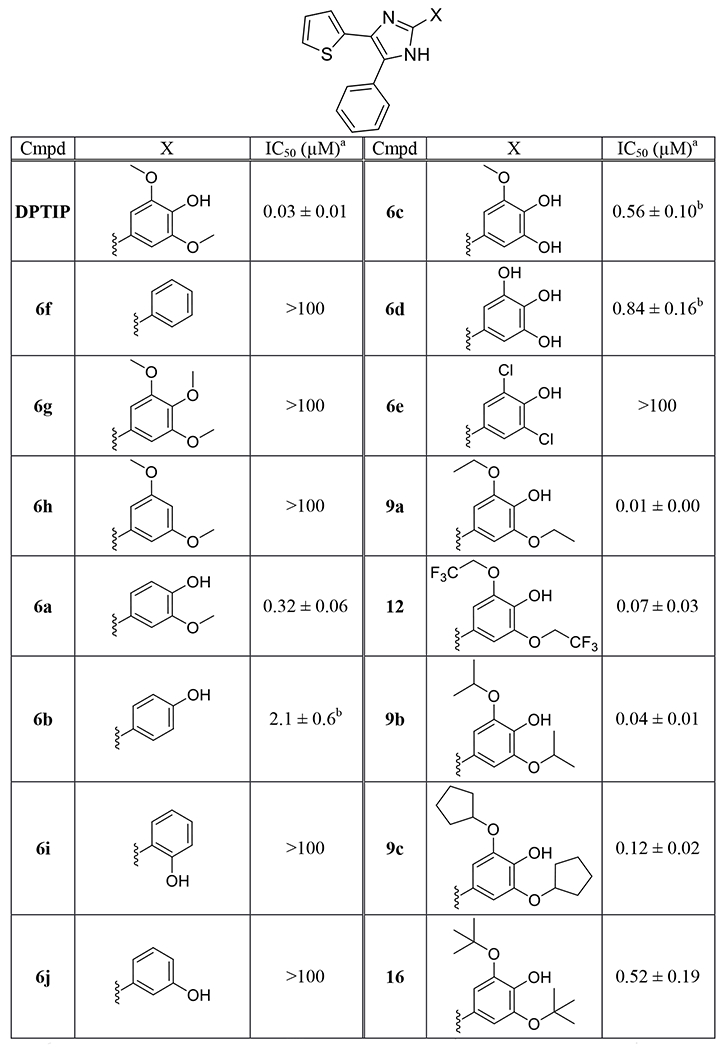
Table 1.
Inhibition of nSMase2 by compounds 6a-6j, 9a-9c, 12 and 16.
|
Values are mean ± SD of at least three experiments.
Separation of IC50 values in nSMase2 and counter assays is ≤ 5-fold.
Table 1 shows biological data obtained from DPTIP analogs in which its 4-hydroxy-3,5-dimethoxyphenyl group is modified. Compounds 6f and 6g bearing plain benzene ring and 3,4,5-trimethoxybenzene respectively, were shown to be completely inactive in the nSMase2 assay. Previously, we reported that 3,5-dimethoxyphenyl derivative 6h was also inactive as a nSMase2 inhibitor [20], clearly indicating the essential role played by the phenolic hydrogen of DPTIP in binding to nSMase2. The removal of one methoxy group from DPTIP resulted in 10-fold loss of inhibitory potency as seen in compound 6a. When both methoxy groups were removed (compound 6b), not only more profound loss of potency (100-fold) was observed but also IC50 values in nSMase2 and counter screen assay were no longer distinguishable. Moving the hydroxyl group of 6b to the position 2 or 3 (compounds 6i and 6j) led to complete loss of potency, underscoring the importance of para-hydroxyl relative to the imidazole ring. Replacing one or both methoxy substituents of DPTIP by hydroxyl group(s) (compounds 6c and 6d) led to moderate loss of potency compared to DPTIP. These compounds also displayed potent inhibition in the counter assay. Replacement of the two methoxy groups of DPTIP by chlorine atoms (compound 6e) caused complete loss of activity. In contrast, some of the compounds with methoxy moieties replaced by different alkoxy groups exhibited very potent nSMase2 inhibitory activity, including diethoxy derivative 9a with an IC50 value of 10 nM. Small alkyl chains were well tolerated though decrease in potency was seen with more sterically demanding branched alkyl groups such as cyclopentyl (compound 9c) and t-butyl (compound 16).
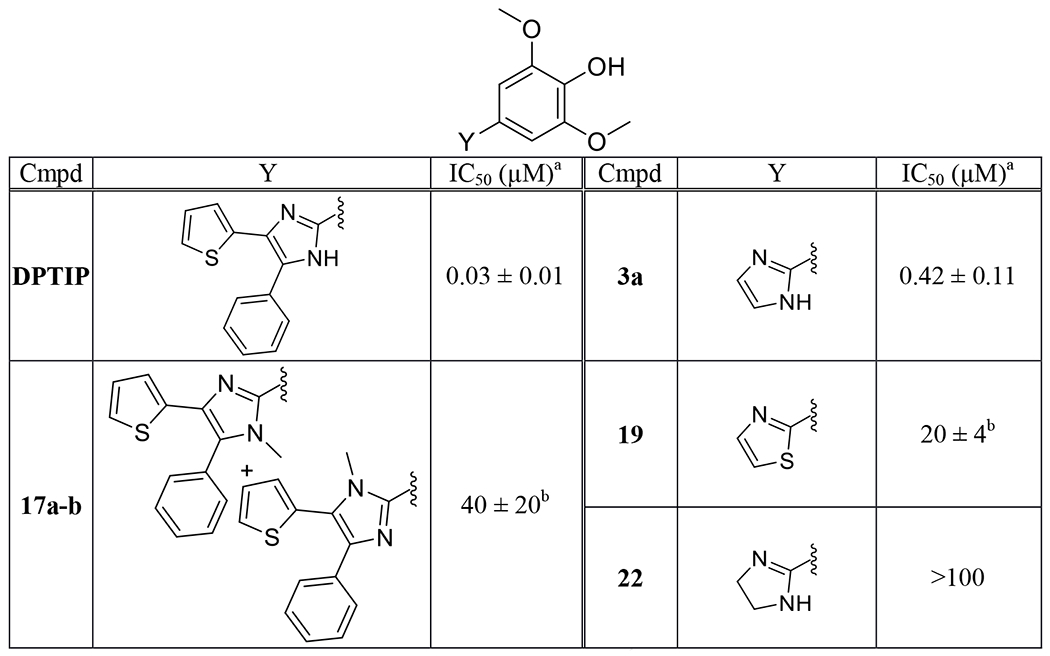
Table 2 summarizes inhibitory data of DPTIP analogs where its imidazole core is altered. Significant loss of potency was observed upon methylation of the imidazole NH (compounds 17a and 17b, tested as a mixture). On the other hand, the removal of the two aryl substituents from the imidazole core of DPTIP resulted in only 10-fold decrease in potency as seen in compound 3a. Replacement of the imidazole ring of 3a with a thiazole ring (compound 19) led to substantial decrease in potency presumably due to the loss of the hydrogen bond donor. Interestingly, conversion of the imidazole ring of 3a into an imidazoline 22 led to a complete loss of potency despite the presence of the critical hydrogen donor. These SAR findings indicate that 3a represents the key pharmacophore required for potent nSMase2 inhibition. Therefore, subsequent efforts were focused on analogs of DPTIP, where its two aryl substituents are altered while retaining the 3a pharmacophore.
Table 2.
Inhibition of nSMase2 by compounds 3a, 17a and 17b, 19, 22.
|
Values are mean ± SD of at least three experiments.
Separation of IC50 values in nSMase2 assay and counter assay is ≤ 5-fold.
Table 3 shows inhibitory data of DPTIP analogs in which its phenyl and thienyl substituents at positions 4 and 5 of imidazole ring are altered. All of the compounds within this structural series potently inhibited nSMase2 with IC50 values ranging from 0.02 to 0.5 μM although compounds 3b-3d displayed comparable inhibitory potency in the counter assay. In particular, di-substituted derivatives, including hybrid of alkyl and aryl groups, are consistently potent and it appears that a variety of substituents are tolerated at the 4- and 5-positions of the imidazole ring.
Table 3.
Inhibition of nSMase2 by compounds 3b-6n, 24a, 25b and 29.
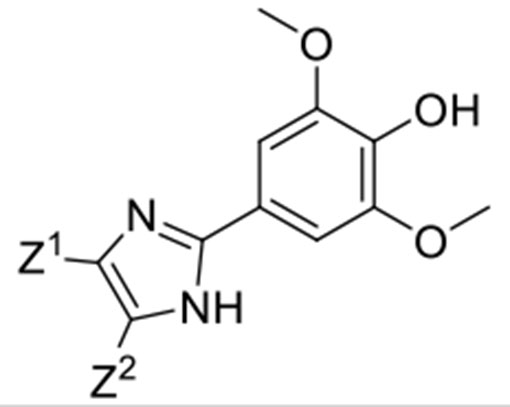
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | Z1 | Z2 | IC50 (μM)a | Cmpd | Z1 | Z2 | IC50 (μM)a |
| DPTIP | Ph | 2-Thienyl | 0.03 ± 0.01 | 3i | Ph | Ph | 0.02 ± 0.00 |
| 3b | H | CH3 | 0.22 ± 0.06b | 3j | 2-Thienyl | 2-Thienyl | 0.02 ± 0.00 |
| 3c | CH3 | CH3 | 0.52 ± 0.14b | 3k | 2-Furyl | 2-Furyl | 0.04 ± 0.02 |
| 3d | −(CH2)4- | 0.42 ± 0.08b | 3l | p-Tolyl | p-Tolyl | 0.07 ± 0.02 | |
| 3e | CH3CH2 | CH3CH2 | 0.12 ± 0.03 | 3m | p-CH3OPh | p-CH3OPh | 0.02 ± 0.00 |
| 3f | CH3 | n-Butyl | 0.06 ± 0.01 | 3n | CH3 | Ph | 0.02 ± 0.01 |
| 25b | (CH3)2CH | (CH3)2CH | 0.09 ± 0.03 | 25a | (CH3)2CH | Ph | 0.07 ± 0.01 |
| 3g | H | Ph | 0.07 ± 0.02 | 29 | CH3 | 2-Thienyl | 0.02 ± 0.01 |
| 3h | H | p-Tolyl | 0.13 ± 0.03 | ||||
Values are mean ± SD of at least three experiments.
Separation of IC50 values in nSMase2 and counter assays is ≤ 5-fold.
2.3. Evaluation of metabolic stability in liver microsomes
Selected compounds were tested in mouse and human liver microsomes for phase I metabolic stability (Table 4). As previously reported DPTIP was found to be stable against phase I metabolism in both human and mouse microsomes. The majority of DPTIP derivatives showed good metabolic stability in both mouse and human microsomes (>70% after 1 h incubation). The two exceptions are compounds 9a and 9b containing two ethoxy or isopropoxy groups instead of methoxy groups of DPTIP. Therefore, we suspect that the oxidation took place at the modified alkoxy groups of these compounds. Indeed, when trifluoroethoxy moieties were introduced in an attempt to enhance resistance to oxidation, metabolic stability was completely regained as seen in compound 12.
Table 4.
Metabolic stability of selected nSMase2 inhibitors in mouse and human liver microsomes.
| Compd | % Remaining at 1 h | Compd | % Remaining at 1 h | ||
|---|---|---|---|---|---|
| mouse | human | mouse | human | ||
| DPTIP | 95 ± 4 | 95 ± 7 | 9a | 35 ± 0 | NDa |
| 3e | 75 ± 0 | >95 | 9b | 4 ± 0 | NDa |
| 3i | >95 | >95 | 12 | >95 | 94 ± 3 |
| 3l | >95 | >95 | 25a | 94 ± 1 | >95 |
| 3m | 73 ± 1 | 75 ± 1 | 25b | 82 ± 2 | >95 |
| 3n | 82 ± 1 | 95 ± 1 | 29 | 95 ± 1 | 73 ± 1 |
Not Determined.
2.4. Evaluation of oral pharmacokinetics in mice
Subsequent to the in vitro metabolic stability studies, we selected compound 25b for preliminary in vivo pharmacokinetics studies in mice. Although compound 25b is not the most potent nSMase2 inhibitor, it exhibits a substantial structural difference from DPTIP with the two aliphatic substituents on the imidazole ring (as opposed to two aryl groups in DPTIP). Comparison of these two compounds allowed us to assess the impact of the two substituents on the pharmacokinetic properties. Figure 3 shows plasma levels of DPTIP and 25b at 0.5 and 2 hour following oral administration (10 mg/kg). Interestingly, plasma levels of compound 25b were higher than those of DPTIP at both of the time points (3-fold at 0.5 h and 6-fold at 2 h). Compound 25b also exhibited 3-fold higher brain levels at 2 h following oral administration compared to DPTIP (Figure 3). The brain/plasma ratio of 25b at 2 h, however, was only 9 % (17 % for oral DPTIP).
Fig. 3.
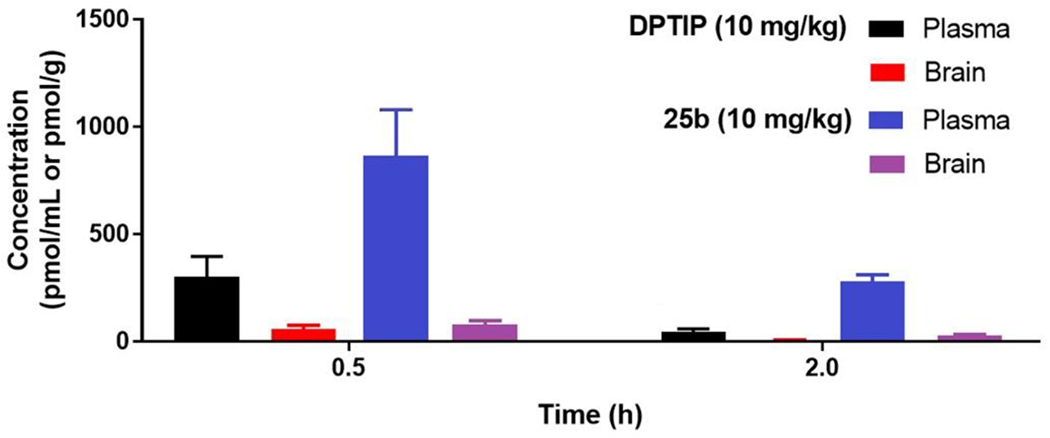
Plasma and brain levels of DPTIP and 25b in mice following oral administration of DPTIP (10 mg/kg) and 25b (10 mg/kg), respectively.
2.5. Identification of O-glucuronide metabolite from plasma samples
Subsequent metabolite identification analysis of the plasma samples identified a glucuronide as the major metabolite. Glucuronidation of 25b can possibly take place at either its phenolic OH or imidazole NH group [39]. We found that the retention time and mass spectrum of the glucuronidated metabolite of 25b from mouse plasma samples (2 h after oral administration of 25b) matched with those of O-glucuronide 30 chemically synthesized from 25b (Figure S1 in Supporting Information). Subsequently, we assessed the metabolic stability of 25b in mouse and human liver microsomes in the presence of UDPGA. Consistent with the mouse plasma pharmacokinetic data, compound 25b was found to be substantially glucuronidated with less than 20% of the parent compound remaining intact after 1 h incubation in mouse liver microsomes. Compound 25b also underwent substantial glucuronidation in human microsomes (32% of the parent compound remaining intact after 1 h incubation). In order to investigate whether the O-glucuronidation can be minimized with bulky alkoxyl groups introduced in the proximity of the phenol group, we assessed the stability of DPTIP and its analog 9b containing two isopropoxy groups in liver microsomes in the presence of UDPGA. As shown in Table 5, DPTIP was found to undergo a moderate degree of glucuronidation, resulting in the loss of the parent compound by 33% (mouse liver microsomes) and 50% (human liver microsomes), respectively. In contrast, compound 9b was completely resistant to glucuronidation in mouse liver microsomes, demonstrating that two sterically hindered alkoxy groups adjacent to the phenolic OH group can completely block the glucuronidation.
Table 5.
Metabolic stability of DPTIP, 9b and 25b in mouse and human liver microsomes in the presence of UDPGA.
| Compd | % Remaining at 1 h | |
|---|---|---|
| mouse | human | |
| 25b | 14 ± 0 | 32 ± 0 |
| DPTIP | 67 ± 0 | 50 ± 7 |
| 9b | >95 | NDa |
Not Determined.
3. Conclusion
Cumulative evidence indicates that ceramide plays a key role in the pathophysiology of various neurological disorders and cancer. Development of small molecule inhibitors of nSMase2 will enable us to further investigate the pathogenic role played by ceramide as well as the therapeutic utility of blocking nSMase2-mediated production of ceramide pharmacologically. DPTIP represents one of the most potent nSMase2 inhibitors and our systematic SAR studies on this scaffold presented herein revealed the key pharmacophore essential for the potent nSMase2 inhibition as well as the structural modifications that can be tolerated by the enzyme. In particular, the 4- and 5-positions of the imidazole ring and the two alkoxy groups offer ample opportunities to introduce structural diversity in an effort to improve ADME properties while retaining potent nSMase2 inhibitory activity. The binding mode of DPTIP and its analogs presented herein has not yet been determined. While the crystal structure of the human nSMase2 catalytic domain recently determined at 1.85-Å resolution is highly informative [2], further structural investigation is required to identify the binding site given the noncompetitive mechanism of inhibition shown by DPTIP with respect to sphingomyelin [20]. In this regard, the allosterically gated “DK switch” of nSMase2 is of particular interest as a potential binding site and has been recently implicated as the target of cambinol by docking studies [40]. Despite the substantial degree of glucuronidation, compound 25b displayed superior plasma pharmacokinetics as compared to DPTIP following oral administration. Given that compound 9b showed complete resistance to glucuronidation, this metabolic liability can be circumvented while retaining the key pharmacophore. Future efforts will be centered on identifying alkoxy groups that would be metabolically stable against oxidation yet prevent glucuronidation of the phenolic OH group. Although compound 25b exhibited negligible CNS penetration, its therapeutic potential can be realized in non-CNS disease in which a minimum brain exposure is preferred.
4. Experimental
4.1. Chemical Synthesis
4.1.1. General
All solvents were reagent grade or HPLC grade. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Compounds 6f and 6h were obtained from ChemDiv Inc. Compounds 6i and 6j were obtained from InterBioScreen Ltd. Compound 6g was obtained from ChemBridge Corp. Melting points were obtained on a Mel-Temp apparatus and are uncorrected. 1H NMR spectra were recorded at 400 or 500 MHz. 13C NMR spectra were recorded at 100 or 125 MHz. In many cases signals of quaternary carbons in 13C NMR were not observable. With unsymmetrically substituted imidazoles, tautomers could be observed in NMR leading to difficulties during spectra interpretation. This could be overcome by converting given sample to protonated form (HCl or TFA-d salt) where only one protonated species exists. In such case “NMR measured as salt” is noted prior experimental data. The HPLC solvent system consisted of deionized water and acetonitrile, both containing 0.1% formic acid. Preparative HPLC purification was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector using a Phenomenex Luna 5 μm C18 column (21.2 mm × 250 mm, 5 μm). Analytical HPLC was performed on an Agilent 1200 series HPLC system equipped with an Agilent G1315D DAD detector (detection at 220 nm) and an Agilent 6120 quadrupole MS detector. Unless otherwise specified, the analytical HPLC conditions involve: (A) for nonpolar compounds 20% acetonitrile/80% water for 0.25 min followed by gradient to 85% acetonitrile/15% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min with a Luna C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 1.25 mL/min; (B) for polar compounds 5% acetonitrile/95% water for 0.25 min followed by gradient to 40% acetonitrile/60% water over 1.5 min and continuation of 85% acetonitrile/15% water for 2.25 min with a Luna C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 1.25 mL/min. Unless otherwise noted, all final compounds biologically tested were confirmed to be of ≥ 95% purity by the HPLC methods described above.
4.1.2. 4-(1H-Imidazol-2-yl)-2,6-dimethoxyphenol (3a).
A mixture of 4-hydroxy-3,5-dimethoxybenzaldehyde (0.50 g, 2.74 mmol), ammonium acetate (2.12 g, 27.45 mmol) and glyoxal (40% solution in water, 5.49 mmol, 0.63 mL) in glacial acetic acid (15 mL) was stirred at 120 °C for 16 hours. The solvent was removed in vacuo and the residue was partitioned between 10% aq. NaHCO3 (15 mL) and EtOAc (15mL). The water layer was washed with EtOAc (2 x 10 mL) and combined organic layers were washed with brine (30 mL), dried over MgSO4, filtered and solvents evaporated. The residue was purified using a Biotage Isolera One flash purification system with a silica gel cartridge (5-10 % MeOH/DCM). Dark red product was further purified using preparative HPLC (0% MeCN/100 % water for 5 min followed by an increase to 20 % MeCN over 35 min and an increase to 40% MeCN over 10 min; flow rate 10 mL/min) to give 18 mg (3 %) of title compound as brown solid; mp 99 °C. 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 6H), 7.07 (s, 2H), 7.23 (s, 2H), 8.15 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 56.0, 102.5, 121.2, 135.7, 146.1, 148.1. LCMS: (B) retention time 0.29 min, m/z = 221 [M + H]+.
4.1.3. 2,6-Dimethoxy-4-(5-methyl-1H-imidazol-2-yl)phenol (3b).
A mixture of 4-hydroxy-3,5-dimethoxybenzaldehyde (0.182 g, 1.0 mmol), ammonium acetate (0.769 g, 10.0 mmol) and methylglyoxal (35-45% solution in water, 0.226 g, ca. 1.1 mmol) in EtOH (10 mL) was stirred at RT for 16 hours. Solvents were evaporated and the residue was partitioned between water (10 mL) and EtOAc (30 mL). Organic phase was washed with saturated NaHCO3, brine, dried over MgSO4, filtered and solvent was evaporated. Residue was purified using Biotage Isolera One flash purification system with a silica gel cartridge (5-10 % MeOH/DCM) to give 25 mg (11 %) of title compound as brown solid; mp > 200 °C (dec). NMR measured as HCl salt: 1H NMR (500 MHz, DMSO-d6) δ 2.34 (s, 3H), 3.86 (s, 6H), 7.40 (s, 1H), 7.61 (s, 2H), 9.36 (bs, 1H), 14.59 (bs, 1H), 14.79 (bs, 1H); 13C NMR (126 MHz, DMSO-d6) δ 9.7, 56.5, 104.4, 112.8, 115.7, 129.2, 138.9, 143.4, 148.3. LCMS: (B) retention time 0.74 min, m/z = 235 [M + H]+.
4.1.4. 4-(4,5-Dimethyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol hydrochloride (3c)
A solution of 4-hydroxy-3,5-dimethoxybenzaldehyde (0.182 g, 1.0 mmol), ammonium acetate (0.769 g, 10.0 mmol) and dimethylglyoxal (0.437 mL, 5.0 mmol) in glacial acetic acid (15 mL) was stirred at 80 °C. Workup as described for 3a. The product was further purified by forced precipitation of HCl salt from MeOH solution with Et2O to give 32 mg (11 %) of title compound as beige powder; mp > 265 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 2.26 (s, 6H), 3.86 (s, 6H), 7.54 (s, 2H), 9.31 (s, 1H), 14.42 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 8.6, 56.5, 104.0, 112.8, 124.5, 138.7, 141.7, 148.3. LCMS: (B) retention time 1.61 min, m/z = 249 [M + H]+.
4.1.5. 2,6-Dimethoxy-4-(4,5,6,7-tetrahydro-1H-benzo[d]imidazol-2-yl)phenol (3d)
The compound was prepared as described for 3b except cyclohexane-1,2-dion (0.123 g, 1.10 mmol) was used instead of methylglyoxal. Obtained 71 mg (26 %) of title compound as yellow solid; mp > 120 °C (dec). 1H NMR (500 MHz, methanol-d4) δ 1.86 (bs, 4H), 2.61 (bs, 4H), 3.90 (s, 6H), 7.15 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 23.2, 26.6, 101.9, 121.9, 135.4, 148.1. LCMS: (B) retention time 1.76 min, m/z = 275 [M + H]+.
4.1.6. 4-(4,5-Diethyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol (3e)
Compound 3e was prepared as described for the preparation of 3b except hexane-3,4-dione (94 %, 0.142 mL, 1.1 mmol) was used in place of methylglyoxal and the reaction was stirred 16 hours at 65 °C. Obtained 76 mg (28 %) of title compound as brown solid. M.P. > 225 °C (dec); 1H NMR (500 MHz, Acetone-d6) δ 1.18 (t, J = 7.5 Hz, 6H), 2.57 (q, J = 7.5 Hz, 4H), 3.77 (s, 6H), 7.26 (s, 2H); 13C NMR (126 MHz, Acetone-d6) δ 15.5, 19.5, 56.5, 103.3, 123.2, 136.8, 144.9, 148.9. LCMS: (B) retention time 1.84 min, m/z = 277 [M + H]+.
4.1.7. 4-(5-Butyl-4-methyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol (3f)
Compound 3f was prepared as described for the preparation of 3b except heptane-2,3-dione (97 %, 0.158 mL, 1.1 mmol) was used in place of methylglyoxal and the reaction was stirred 16 hours at 65 °C. Obtained 52 mg (18 %) of title compound as brown solid; mp > 105 °C (dec). 1H NMR (500 MHz, Acetone-d6) δ 0.89 (t, J = 7.4 Hz, 3H), 1.29-1.35 (m, 2H), 1.551-1.61 (m, 2H), 2.17 (s, 3H), 2.54 (t, J = 7.6 Hz, 2H), 3.77 (s, 6H), 7.28 (s, 2H); 13C NMR (126 MHz, Acetone-d6) δ 11.0, 14.2, 23.0, 25.8, 33.1, 56.5, 103.3, 122.7, 137.0, 144.9, 148.9. LCMS: (B) retention time 2.02 min, m/z = 291 [M + H]+.
4.1.8. 2,6-Dimethoxy-4-(5-phenyl-1H-imidazol-2-yl)phenol hydrochloride (3g)
Compound 3g was prepared as described for the preparation of 3b except phenylglyoxal monohydrate (0.167 g, 1.1 mmol) was used in place of methylglyoxal. After workup the free base was converted to HCl salt using excess of 4 N HCl in dioxane and purified using preparative HPLC (10% MeCN/90 % water for 5 min followed by an increase to 50 % MeCN over 35 min and an increase to 70% MeCN over 10 min; flow rate 15 mL/min) to give 60 mg (18 %) of title compound as brown solid; mp > 110 °C (dec). 1H NMR (500 MHz, DMSO-d6) δ 3.85 (s, 6H), 7.20 (t, J = 7.5 Hz, 1H), 7.30 (s, 2H), 7.37 (t, J = 7.6 Hz, 2H), 7.65 (bs, 1H), 7.83 (d, J = 7.6 Hz, 2H), 8.61 (bs, 1H), 12.46 (bs, 1H); 13C NMR (126 MHz, DMSO-d6) δ 56.1, 102.7, 121.01, 128.5, 136.0, 148.1. LCMS: (B) retention time 1.89 min, m/z = 297 [M + H]+.
4.1.9. 2,6-Dimethoxy-4-(4-p-tolyl-1H-imidazol-2-yl)phenol (3h).
Compound 3h was prepared as described for the preparation of 3b except 4-tolylglyoxal hydrate (95 %, 0.183 g, 1.1 mmol) was used in place of methylglyoxal. Obtained 35 mg (11 %) of title compound as yellow-green solid; mp > 130 °C (dec). 1H NMR (500 MHz, Methanol-d4) δ 2.35 (s, 3H), 3.94 (s, 6H), 7.21 (d, J = 7.7 Hz, 2H), 7.29 (s, 2H), 7.35 (s, 1H), 7.63 (d, J = 7.7 Hz, 2H); 13C NMR (126 MHz, Methanol-d4) δ 21.2, 56.8, 104.2, 122.3, 126.0, 130.3, 137.8, 149.6. LCMS: (B) retention time 2.05 min, m/z = 311 [M + H]+.
4.1.10. 4-(4,5-Diphenyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol (3i)
Compound 3i was prepared as described for the preparation of 3b except benzil (0.236 g, 1.10 mmol) was used in place of methylglyoxal and reaction mixture was stirred at 80 °C for 6 hours. Obtained 40 mg (15 %) of title compound as pale brown solid; mp 293 °C. 1H NMR (500 MHz, DMSO-d6) δ 3.85 (s, 6H), 7.21 (t, J = 7.3 Hz, 1H), 7.28-7.31 (m, 2H), 7.37-7.39 (m, 3H), 7.43-7.50 (m, 4H), 7.54-7.55 (m, 2H), 8.62 (s, 1H), 12.44 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 56.1, 102.8, 120.7, 126.4, 127.0, 127.5, 127.7, 128.1, 128.4, 128.7, 131.3, 135.3, 136.0, 136.6, 146.0, 148.1. LCMS: (B) retention time 2.38 min, m/z = 273 [M + H]+.
4.1.11. 4-(4,5-Di(thiophen-2-yl)-1H-imidazol-2-yl)-2,6-dimethoxyphenol (3j)
Compound 3j was prepared as described for the preparation of 3b except 2,2`-thenil (0.250 g, 1.1 mmol) was used in place of methylglyoxal and the reaction was stirred at 65 °C. Product was further purified by trituration (2 × 2 mL MeOH) to give 40 mg (10 %) of title compound as white solid; mp 218 °C. 1H NMR (500 MHz, DMSO-d6) δ 3.84 (s, 6H), 7.00 (t, J = 4.1 Hz, 1H), 7.14 (d, J = 3.5 Hz, 1H), 7.19-7.25 (m, 1H), 7.32 (s, 2H), 7.40 (dd, J = 8.1, 4.3 Hz, 2H), 7.71 (d, J = 5.0 Hz, 1H), 8.69 (s, 1H), 12.63 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 56.1, 102.9, 119.9, 120.1, 123.3, 124.6, 127.3, 127.5, 127.6, 128.5, 131.0, 133.3, 136.4, 136.5, 146.3, 148.11. LCMS: (B) retention time 2.47 min, m/z = 385 [M + H]+.
4.1.12. 4-(4,5-Di(furan-2-yl)-1H-imidazol-2-yl)-2,6-dimethoxyphenol hydrochloride (3k)
Compound 3k was prepared as described for the preparation of 3b except 2,2`-furil (97 %, 0.215 g, 1.1 mmol) was used in place of methylglyoxal and the reaction was stirred 6 hours at 85 °C. Product was further purified by forced precipitation with Et2O of HCl salt from its MeOH solution. After filtration and drying 67 mg (17 %) of title compound was obtained as white solid; mp > 220 °C (dec). 1H NMR (500 MHz, Methanol-d4) δ 3.97 (s, 6H), 6.69 (dd, J = 3.6, 1.9 Hz, 2H), 7.15 (d, J = 3.5 Hz, 2H), 7.44 (s, 2H), 7.80 (s, 2H); 13C NMR (126 MHz, Methanol-d4) δ 57.1, 106.4, 112.7, 113.3, 121.3, 142.5, 145.7, 147.4, 150.0. LCMS: (B) retention time 2.35 min, m/z = 353 [M + H]+.
4.1.13. 4-(4,5-Di-p-tolyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol (3l)
Compound 3l was prepared as described for the preparation of 3b except 4,4`-dimethylbenzil (95 %, 0.276 g, 1.10 mmol) was used in place of methylglyoxal and the reaction was stirred at 60 °C. Crude product was purified by trituration with Et2O/CHCl3 4/1 to give 0.287 g (72 %) of title compound as pink solid; mp > 260 °C (dec). 1H NMR (500 MHz, DMSO-d6) δ 2.29 (s, 3H), 2.35 (s, 3H), 3.84 (s, 6H), 7.10 (d, J = 7.7 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.32-7.40 (m, 4H), 7.43 (d, J = 7.8 Hz, 2H), 8.59 (s, 1H), 12.33 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 20.8, 20.9, 56.1, 102.8, 120.9, 126.9, 127.1, 128.2, 128.5, 128.7, 129.2, 132.5, 135.3, 136.0, 136.4, 136.8, 145.7, 148.1. LCMS: (B) retention time 2.70 min, m/z = 401 [M + H]+.
4.1.14. 4-(4,5-Bis(4-methoxyphenyl)-1H-imidazol-2-yl)-2,6-dimethoxyphenol (3m)
Compound 3m was prepared as described for the preparation of 2b except 4,4`-dimethoxybenzil (0.303 g, 1.1 mmol) was used in place of methylglyoxal and the reaction was stirred 6 hours at 65 °C. Obtained 90 mg (21 %) of title compound as pink solid; mp > 300 °C. 1H NMR (500 MHz, DMSO-d6) δ 3.74 (s, 3H), 3.80 (s, 3H), 3.84 (s, 6H), 6.87 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 7.34 (s, 2H), 7.39 (d, J = 8.8 Hz, 2H), 7.45 (d, J = 8.8 Hz, 2H), 8.58 (s, 1H), 12.27 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 55.0, 55.1, 56.1, 102.7, 113.6, 114.1, 121.0, 123.8, 126.4, 128.0, 128.1, 129.7, 135.8, 135.9, 145.3, 148.1, 157.8, 158.7. LCMS: (B) retention time 2.46 min, m/z = 433 [M + H]+.
4.1.15. 2,6-Dimethoxy-4-(4-methyl-5-phenyl-1H-imidazol-2-yl)phenol (3n)
Compound 3n was prepared as described for the preparation of 3b except 1-phenylpropane-1,2-dione (98 %, 0.166 g, 1.1 mmol) was used in place of methylglyoxal and the reaction was stirred at 65 °C. Obtained 123 mg (35 %) of title compound as brown solid; mp > 135 °C (dec). NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 2.46 (s, 3H), 3.87 (s, 6H), 7.32-7.41 (m, 1H), 7.47 (s, 2H), 7.48-7.53 (m, 2H), 7.67-7.73 (m, 2H), 9.03 (s, 1H), 13.68 (bs, 2H); 13C NMR (101 MHz, DMSO-d6) δ 11.0, 56.3, 102.7, 127.1, 127.4, 128.7, 137.6, 143.7, 148.2. LCMS: (B) retention time 2.02 min, m/z = 353 [M + H]+.
4.1.16. 2-Methoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (6a)
A mixture of aldehyde 5a (0.045 g, 0.30 mmol), ammonium acetate (0.178 g, 2.31 mmol) and diketone 4 (0.050 g, 0.23 mmol) in glacial acetic acid (5 mL) was stirred at 120 °C for 16 hours. The solvent was removed in vacuo and the residue was purified using a Biotage Isolera One flash purification system with a silica gel cartridge (EtOAc/hexane) to give title compound 6a (0.015 g, 19 %) as a beige solid; mp 224 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 3.91 (s, 3H), 7.04 (d, J = 8.4 Hz, 1H), 7.19 (dd, J = 5.1, 3.6 Hz, 1H), 7.57-7.51 (m, 3H), 7.66 (ddd, J = 7.8, 4.7, 1.9 Hz, 3H), 7.87-7.77 (m, 2H), 8.04 (d, J = 2.2 Hz, 1H), 10.20 (s, 1H), 14.92 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 56.2, 111.5, 113.7, 115.9, 121.3, 123.2, 126.9, 127.7, 128.5, 128.8, 128.9, 129.56, 129.64, 129.9, 144.4, 148.0, 150.5. LCMS: (A) retention time 1.38 min, m/z 349 [M + H]+.
4.1.17. 4-(4-Phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (6b)
Compound 6b was prepared as described for the preparation of 6a except benzaldehyde 5b was used in place of benzaldehyde 5a. Yellow solid foam (23 % yield); mp 122 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 7.02 (d, J = 8.8 Hz, 2H), 7.19 (dd, J = 3.8, 5.1 Hz, 1H), 7.51-7.55 (m, 3H), 7.62-7.65 (m, 2H), 7.67 (d, J = 4.6 Hz, 2H), 8.09-8.13 (m, 2H), 10.58 (bs, 1H), 14.69 (bs, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.05, 144.51, 129.84, 129.42, 129.11, 129.08, 128.89, 128.37, 127.70, 127.10, 116.08. LCMS: (A) retention time 1.35 min, m/z 319 [M + H]+.
4.1.18. 3-Methoxy-5-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)benzene-1,2-diol (6c)
Compound 6c was prepared as described for the preparation of 6a except benzaldehyde 5c was used in place of benzaldehyde 5a. Dark brown solid (35 % yield); mp 133 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 3.89 (s, 3H), 7.19 (dd, J = 3.6, 5.1 Hz, 1H), 7.36 (d, 1H), 7.54 (dt, J = 2.8, 4.2 Hz, 4H), 7.61-7.65 (m, 2H), 7.68 (dd, J = 1.2, 5.2 Hz, 1H), 7.71 (d, J = 3.6 Hz, 1H), 9.51 (bs, 2H), 14.73 (bs, 2H); 13C NMR (101 MHz, DMSO-d6) δ 56.4, 103.6, 108.8, 127.7, 128.9, 129.6, 129.9, 138.4, 144.6, 146.2, 148.6. LCMS: (A) retention time 1.30 min, m/z 365 [M + H]+.
4.1.19. 5-(4-Phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)benzene-1,2,3-triol (6d)
Compound 6d was prepared as described for the preparation of 6a except benzaldehyde 5d was used in place of benzaldehyde 5a. Grey solid (20 % yield); mp > 250 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 7.14 (s, 2H), 7.19 (m, 1H), 7.50-7.54 (m, 3H), 7.59-7.62 (m, 3H), 7.68 (dd, J = 1.2, 5.1 Hz, 1H), 9.54 (bs, 2H), 10.69 (bs, 1H), 14.61 (bs, 2H); 13C NMR (101 MHz, DMSO-d6) δ 107.3, 126.8, 127.6, 127.7, 128.6, 128.8, 128.2, 129.9, 137.7, 144.8, 146.5. LCMS: (B) retention time 0.88 min, m/z 351 [M + H]+.
4.1.20. 2,6-Dichloro-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (6e)
Compound 6e was prepared as described for 6a except benzaldehyde 5e was used in place of benzaldehyde 5a, and the compound was purified by a reversed phase preparative HPLC (40% MeCN/100 % water followed by an increase to 100 % MeCN over 60 min and maintaining 100% MeCN over 10 min; flow rate 15 mL/min). Beige solid (31 % yield); mp >250 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 7.16 (dd, J = 3.6, 5.1 Hz, 1H), 7.49-7.59 (m, 4H), 7.61-7.68 (m, 3H), 8.34 (s, 2H), 11.16 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 122.8, 127.2, 127.7, 127.9, 128.9, 129.1, 129.5, 142.1, 151.4. LCMS: (A) retention time 1.99 min, m/z 388 [M + H]+.
4.1.21. 3,5-Dihydroxy-4-(4-methoxybenzyloxy)benzaldehyde (7)
To a cooled solution of 3,4,5-trihydroxybenzaldehyde (5d) (1.0 g, 6.49 mmol) in DMF (15 mL) was added sodium hydride (60% w/w, 0.26 g, 6.49 mmol). After 20 min stirring at 0 °C, 4-methoxybenzyl chloride (0.71 g, 4.54 mmol) was added. The mixture was stirred at 0 °C for an additional 20 min, then ice bath was removed and the reaction was allowed to stir at rt over weekend. Water was added followed by 3 mL of 10% aqueous KHSO4 solution. The product was extracted with EtOAc (x2). The organic layer was washed with brine, dried over sodium sulfate and concentrated. The resulting residue was purified by Biotage Isolera One flash system with a silica gel cartridge (30-50% EtOAc/hexanes with 2% AcOH) to give 0.78 g (44%) of title compound as a brown solid cake. 1H NMR (400 MHz, chloroform-d): δ 3.83 (s, 3H), 5.10 (s, 2H), 6.91 (d, J = 8.8 Hz, 2H), 7.03 (s, 2H), 7.31 (d, J = 8.6, 2H), 9.80 (s, 1H).
4.1.22. 3,5-Diethoxy-4-(4-methoxybenzyloxy)benzaldehyde (8a)
To a solution of triphenylphosphine (0.96 g, 3.65 mmol) in THF (10 mL) was slowly added DIAD (0.74 g, 3.65 mmol) via syringe at 0 °C. White precipitate was formed. The mixture was allowed to stir at 0 °C for 1 h, upon which a solution of compound 7 (0.25 g, 0.91 mmol) and ethanol (0.16 mL, 2.73 mmol) in THF (5 mL) was added via syringe. The reaction was stirred at 0 °C, brought up to rt and stirred overnight. After removal of solvent, the resulting residue was purified by Biotage Isolera One (20% EtOAc/hexanes) to give title compound in quantitative yield as a light yellow oil. 1H NMR (400 MHz, chloroform-d): δ 1.47 (t, J = 7.1 Hz, 6H), 3.81 (s, 3H), 4.11 (q, J = 7.1, 13.9 Hz, 4H), 5.08 (s, 2H), 6.85 (d, J = 8.6 Hz, 2H), 7.08 (s, 2H), 7.39 (d, J = 8.6 Hz, 2H), 9.83 (s, 1H).
4.1.23. 3,5-Diisopropoxy-4-(4-methoxybenzyloxy)benzaldehyde (8b)
Compound 8b was prepared as described for the preparation of 8a, with the exception that 2-propanol was used in place of ethanol. Bright yellow oil (85%). 1H NMR (400 MHz, chloroform-d): δ 1.36 (d, J = 6.1 Hz, 12H), 3.81 (s, 3H), 4.62 (m, 2H), 5.04 (s, 2H), 6.86 (d, J = 8.6 Hz, 2H), 7.08 (s, 2H), 7.40 (d, J = 8.6 Hz, 2H), 9.83 (s, 1H).
4.1.24. 3,5-Bis(cyclopentyloxy)-4-(4-methoxybenzyloxy)benzaldehyde (8c)
Compound 8c was prepared as described for the preparation of 8a, with the exception that cyclopentanol was used in place of ethanol. Light yellow oil (90%). The compound was used as is without further characterization.
4.1.25. 2,6-Diethoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (9a)
Diketone 4 (0.2 g, 0.92 mmol), aldehyde 8a (0.31 g, 0.92 mmol) and ammonium acetate (0.71 g, 9.25 mmol) were heated together in acetic acid (10 mL) at 120 °C overnight. The reaction was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The crude material was purified by Biotage Isolera One flash chromatography (EtOAc/hexane) to give a solid, which was further triturated in 15% EtOAc/hexanes to afford 50 mg (13%) of title compound as a purple solid; mp 245 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 1.38 (t, J = 7.0 Hz, 6H), 4.16 (q, J = 7.0 Hz, 4H), 7.19 (dd, J = 3.6, 5.1 Hz, 1H), 7.54 (dd, J = 1.8, 5.0 Hz, 3H), 7.61-7.70 (m, 5H), 7.75 (d, J = 3.6 Hz, 1H), 9.17 (bs, 1H), 17.75 (bs, 2H); 13C NMR (101 MHz, DMSO-d6) δ 14.8, 64.7, 106.1, 127.2, 127.7, 128.4, 128.9, 129.1, 129.3, 129.5, 129.9, 139.8, 144.5, 147.4. LCMS: (A) retention time 1.69 min, m/z 407 [M+H]+.
4.1.26. 2,6-Diisopropoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (9b)
Compound 9b was prepared as described for the preparation of 9a, with the exception that compound 8b was used in place of 8a. Purple solid (44%); mp 217 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 1.31 (d, J = 6.8 Hz, 12H), 4.72 (sept, J = 6.0 Hz, 2H), 7.19 (t, J = 4.3 Hz, 1H), 7.51-7.58 (m, 3H), 7.61-762 (m, 5H), 7.77 (s, 1H), 8.94 (bs, 1H), 14.76 (bs, 2H); 13C NMR (126 MHz, DMSO-d6) δ 22.0, 71.5, 108.4, 127.1, 127.6, 128.4, 128.9, 129.6, 129.9, 144.4, 146.4. LCMS: (A) retention time 1.83 min, m/z 435 [M+H]+.
4.1.27. 2,6-Bis(cyclopentyloxy)-4-(5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (9c)
Compound 9c was prepared as described for the preparation of 9a, with the exception that compound 8c was used in place of 8a. Grey solid (32%); mp 230 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 1.52-1.65 (m; 4H), 1.78 (tdd, J = 2.2, 6.3, 9.4 Hz, 8H), 1.987-1.91 (m, 4H), 4.93-4.97 (m, 2H), 7.19 (dd, J = 3.5, 5.1 Hz, 1H), 7.52-7.56 (m, 3H), 7.60 (s, 2H), 7.62-7.66 (m, 1H), 7.67 (dd, J = 1.2, 5.1 Hz, 1H), 7.73 (d, J = 3.6 Hz, 1H), 8.85 (bs, 1H), 14.71 (bs, 2H); 13C NMR (101 MHz, DMSO-d6) δ 23.6, 32.3, 80.4, 107.4, 127.7, 128.9, 129.6, 141.1, 144.4, 146.6. LCMS: (A) retention time 2.06 min, m/z 487 [M+H]+.
4.1.28. 4-Hydroxy-3,5-bis(2,2,2-trifluoroethoxy)benzaldehyde (11)
In a flask equipped with a Claisen distillation apparatus, freshly cut sodium (0.69 g, 30 mmol) was added to trifluoroethanol (10 mL) at rt and the mixture was stirred until a complete dissolution of sodium. The mixture was then heated at 80-90 °C to distill off portion of trifluoroethanol with the help of in house vacuum. Then, a solution of 3,5-dibromo-4-hydroxybenzaldehyde (10, 0.5 g, 1.79 mmol) and copper (II) chloride (0.20 g, 1.49 mmol) in DMF (4mL) was added in one portion. The blue mixture was heated and distilled at 110-115 °C overnight. The reaction was cooled to RT, then water was added and the undesired precipitate was filtered off. The filtrate was partitioned between EtOAc and water. The organic layer was washed with brine, dried over sodium sulfate and concentrated in vacuo to give 0.30 g (53%) of title compound as a brown solid, which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ 4.62 (q, J = 9.6, 19.0 Hz, 4H), 6.96 (s, 2), 9.20 (s, 1H).
4.1.29. 4-(4-Phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)-2,6-bis(2,2,2-trifluoroethoxy)phenol (12)
Diketone 4 (90 mg, 0.42 mmol), aldehyde 11 (155 mg, 0.49 mmol) and ammonium acetate (320 mg, 4.16 mmol) were heated together in acetic acid (6 mL) at 120 °C overnight. The reaction was concentrated in vacuo. The resulting residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over sodium sulfate and concentrated to give a brown oil. Purification by reverse phase preparative HPLC (40% ACN/60% H2O for 5 minutes followed by an increase to 100% ACN/0% H2O over 40 minutes and a continuation of 100% ACN/0% H2O until 50 minutes; flow rate 15 mL/min) afforded 75 mg (36%) of title compound as a light purple solid; mp 205 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 4.86 (q, J = 8.9 Hz, 4H), 7.19 (dd, J = 3.6, 5.1 Hz, 1H), 7.53-7.56 (m, 3H), 7.64-7.68 (m, 3H), 7.79 (bs, 1H), 7.97 (s, 2H), 10.00 (s, 1H), 14.84 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 66.3 (q, J = 34 Hz), 109.2, 123.9 (q, J = 277 Hz), 127.7, 128.9, 129.5, 129.9, 140.3, 143.6, 146.7. LCMS: (A) retention time 2.06 min, m/z 515 [M+H]+.
4.1.30. 3,5-Di-tert-butoxy-4-methoxybenzaldehyde (14)
Aldehyde 13 (0.2 g, 1.19 mmol) was heated at 85 °C in toluene (6 mL) for 30 min. N,N-Dimethylformamide di-tert-butyl acetal (3 mL) was slowly added and heating continued at 85 °C overnight. The next day, additional 1.5 mL of N,N-Dimethylformamide di-tert-butyl acetal was added and the reaction was completed after 3 h stirring at 85 °C. The reaction was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The crude material was purified by Biotage Isolera One flash chromatography (10% EtOAc/hexanes) to give 0.1 g (30%) of title compound as an oil. 1H NMR (400 MHz, chloroform-d) δ 1.39 (s, 18H), 3.95 (s, 3H), 7.31 (s, 2H), 9.83 (s, 1H).
4.1.31. 2-(3,5-Di-t-butoxy-4-methoxyphenyl)-4-phenyl-5-(thiophen-2-yl)-1H-imidazole (15)
Diketone 4 (80 mg, 0.37 mmol), aldehyde 14 (104 mg, 0.37 mmol) and ammonium acetate (285 mg, 3.70 mmol) were heated together in acetic acid (6 mL) at 120 °C for 2 h. The reaction was concentrated in vacuo. The product was partitioned between EtOAc and water. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The crude material was purified by Biotage Isolera One flash chromatography (20-50% EtOAc/hexanes with 2% AcOH) to give 35 mg (19%) of title compound as a yellow solid cake. 1H NMR (400 MHz, chloroform-d) δ 1.40 (s, 18H), 3.89 (s, 3H), 7.01 (m, 1H) 7.20 (m, 1H), 7.30 (s, 2H), 7.42 (m, 4H), 7.62 (m, 2H).
4.1.32. 2,6-Di-t-butoxy-4-(4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (16)
A solution of compound 15 (35 mg, 0.07 mmol) in a 1:1 mixture of piperidine-water (10 mL) was heated at 150 °C in a sealed tube for 10 days. Upon completion, the reaction was concentrated in vacuo. The product was partitioned between EtOAc and water. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The crude material was purified by Biotage Isolera One flash chromatography (10-30 % EtOAc/hexanes) to give 20 mg (67%) of title compound as a white solid; mp > 250 °C. NMR measured as TFA-d salt: 1H NMR (400 MHz, DMSO-d6) δ 1.36 (s, 18H), 7.22 (dd, J = 5.1, 3.6 Hz, 1H), 7.53-7.56 (m, 4H), 7.60 (m, 2H), 7.60-7.62 (m, 4H), 7.73 (dd, J = 5.1, 1.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 28.7, 81.3, 111.9, 119.8, 123.2, 127.0, 127.4, 128.2, 129.4, 129.48, 129.54, 129.9, 130.2, 130.5, 144.0, 144.5, 150.8. LCMS: (A) retention time 1.96 min, m/z 463 [M+H]+.
4.1.33. 2,6-Dimethoxy-4-(1-methyl-4-phenyl-5-(thiophen-2-yl)-1H-imidazol-2-yl)phenol and 2,6-Dimethoxy-4-(1-methyl-5-phenyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenol (17a, 17b).
Diketone 4 (100 mg, 0.46 mmol), syringaldehyde (1, 84 mg, 0.46 mmol), 2M methylamine solution in THF (0.23 mL, 0.46 mmol), ammonium acetate (36 mg, 0.46 mmol) and sodium phosphate monobasic (17 mg, 0.14 mmol) were heated together in a sealed tube at 150 °C for 5 h. The reaction was cooled to rt and stirred over weekend, then it was concentrated in vacuo. The residue was purified by Biotage Isolera One flash chromatography (40-50% EtOAc/hexanes) to afford 30 mg (17%) of mixture of title compounds 17a and 17b as a light brown solid. Regioisomers 17a and 17b were not separated and were characterized as mixture; mp > 250 °C. NMR of mixture of regioisomers in 1.5:1 ratio, *denotes minor isomer: 1H NMR (400 MHz, DMSO-d6) δ 3.44 (s, 1.8H), 3.51 (s, 1.2H)*, 3.84 (s, 6H), 6.77 (dd, J = 3.6, 1.2 Hz, 0.6H), 6.88 (dd, J = 5.1, 3.6 Hz, 0.6H), 6.94 (s, 1.2H), 6.98 (s, 0.8H)*, 7.14-7.20 (m, 0.4H)*, 7.27 (dd, J = 5.1, 1.1 Hz, 0.6H), 7.23-7.31 (m, 1.6H)*, 7.49-7.54 (m, 0.8H)*, 7.48-7.64 (m, 2H), 7.83 (dd, J = 4.7, 1.7 Hz, 0.4H)*, 8.78 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 32.7*, 33.0, 56.2, 106.6, 120.3, 120.5*, 121.6, 121.8*, 123.6, 126.3*, 126.5*, 127.2, 128.1*, 128.2*, 129.0, 129.1, 129.4*, 130.1, 130.6*, 130.8*, 130.9, 131.7, 134.4*, 136.45*, 136.48, 138.4*, 138.5, 147.3, 147.8, 148.2*. LCMS: (A) retention time 1.64 min, m/z 393 [M+H]+.
4.1.34. 2,6-Dimethoxy-4-(thiazol-2-yl)phenol (19)
Compound 18 (0.032 g, 0.18 mmol) was dissolved in DCM (1 mL), cooled to 0 °C and BCl3/DCM solution (1M, 0.42 mL, 0.42 mmol) was added dropwise. The reaction was allowed to warm to rt and stirred at rt for 2 hours. The reaction was quenched with MeOH/water (1:1, 1 mL), then solvents were evaporated. The resulting residue was purified using reversed phase preparative HPLC (20% MeCN/80 % water followed by an increase to 70 % MeCN over 40 min and an increase to 100 % MeCN over 10 min; flow rate 15 mL/min) to give title compound (6 mg, 15 %) as a brown solid; mp 136 °C. 1H NMR (400 MHz, DMSO-d6) δ 3.84 (s, 6H), 7.17 (s, 2H), 7.68 (d, J = 3.2 Hz, 1H), 7.83 (d, J = 3.3 Hz, 1H), 8.97 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 56.1, 103.7, 119.3, 123.7, 137.8, 143.3, 148.2, 167.6. LCMS: (A) retention time 0.95 min, m/z 238 [M + H]+.
4.1.35. 4-Formyl-2,6-dimethoxyphenyl methanesulfonate (20)
A solution of 4-hydroxy-3,5-dimethoxybenzaldehyde (10.0 g, 54.90 mmol) in DCM (200 mL) and Et3N (23.0 mL, 163.70 mmol) was cooled to 0 °C, then methanesulfonyl chloride (6.670 mL, 94.60 mmol) was added dropwise and reaction was stirred at RT 16 hours. Reaction mixture was poured onto ice; after melting phases were separated and water phase was extracted with DCM (3 × 50 mL). Combined organic phases were sequentially washed with saturated NaHCO3 solution and brine, dried over MgSO4, filtered and solvent was evaporated. Crude product was crystalized from MeOH/CHCl3 to give 10.10 g (71 %) of title compound as beige solid. 1H NMR (400 MHz, DMSO-d6) δ 3.46 (s, 3H), 3.92 (s, 6H), 7.35 (s, 2H), 9.96 (s, 1H); m/z = 261 [M + H]+.
4.1.36. 4-(4,5-Dihydro-1H-imidazol-2-yl)-2,6-dimethoxyphenyl methanesulfonate (21).
To a solution of methansulfonate 20 (0.260 mg, 1.0 mmol) in DCM (10 mL) cooled to 0 °C was added 1,2-ethylenediamine (70 μL, 1.05 mmol) and reaction was stirred at 0 for 30 min, then N-bromosuccinimide (0.187 g, 1.05 mmol) was added in one portion. The reaction was allowed to reach RT and was stirred overnight. Reaction was diluted with EtOAc, washed with sat. NaHCO3, dried over MgSO4 and solvents were evaporated to give crude title compound (0.267 g, 89 %) which was used in next step without further purification. 1H NMR (400 MHz, chloroform-d) δ 3.30 (s, 3H), 3.79 (s, 4H), 3.90 (s, 6H), 7.05 (s, 2H).
4.1.37. 4-(4,5-Dihydro-1H-imidazol-2-yl)-2,6-dimethoxyphenol hydrochloride (22)
Imidazoline 21 (0.150 g, 0.50 mmol) was dissolved in MeOH (10 mL) and saturated solution of KOH (3.0 mL) was added in one portion. After stirring at RT for 1 hour the reaction was carefully neutralized with 1.5 N HCl to pH = 7-8 then it was diluted with MeOH (250 mL) and formed precipitates were filtered off. Solvents were evaporated and the residue was filtered through short column of SiO2 with 30 % MeOH in CHCl3. Solvents were evaporated and crude product was purified using reversed phase preparative HPLC (0% MeCN/100 % water for 5 min followed by an increase to 20 % MeCN over 35 min and an increase to 40 % MeCN over 10 min; flow rate 10 mL/min). The product was further purified by forced precipitation with Et2O of its HCl salt from MeOH solution to give title compound (20 mg, 15 %) as white powder; mp > 250 °C (dec). 1H NMR (400 MHz, DMSO-d6) δ 3.84 (s, 4H), 3.95 (s, 6H), 7.53 (s, 2H), 9.78 (bs, 1H), 10.65 (bs, 2H); 13C NMR (101 MHz, DMSO-d6) δ 44.0, 56.6, 106.9, 111.1, 141.5, 147.9, 164.5. LCMS: (B) retention time 0.42 min, m/z = 223 [M + H]+.
4.1.38. 2-Hydroxy-3-methyl-1-phenylbutan-1-one (24a)
To a solution KOH (0.308 g, 5.5 mmol) in MeOH (2 mL) was added ketone 23 (0.171 g, 1.0 mmol) and the mixture was cooled to 0 °C. Then (diacetoxyiodo)benzene (0.354 g, 1.1 mmol) was added and reaction was stirred 48 hours at RT. Reaction was diluted with water (10 mL) and extracted with Et2O (3 × 5 mL). Combined organic phases were sequentially washed with saturated NaHCO3 solution and brine, dried over MgSO4 and solvent was evaporated to give crude title compound (95 mg) which was used in next step without further purification. m/z = 179 [M + H]+, 161 [M − H2O + H]+.
4.1.39. 4-(5-Isopropyl-4-phenyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol (25a)
Crude compound 24a (0.095 g) was dissolved in EtOH (5 mL) and aldehyde 1 (0.111 g, 0.6 mmol) and NH4OAc (0.457 g, 6.0 mmol) were added. The reaction was stirred 16 hours at 65 °C. Workup as described for 3b. Crude product was purified using column chromatography on silica (DCM + 1-2 % MeOH) to give 55 mg (16 %) of title compound as black solid. Purity (HPLC) = 90 %; mp 239 °C. NMR measured as HCl salt: 1H NMR (400 MHz, DMSO-d6) δ 1.44 (d, J = 7.2 Hz, 6H), 3.15-3.22 (m, 1H), 3.91 (s, 6H), 7.44-7.71 (m, 5H), 7.82 (s, 2H), 9.38 (bs, 1H), 14.44 (bs, 1H), 14.96 (bs, 1H); 13C NMR (101 MHz, DMSO-d6) δ 21.8, 24.4, 56.7, 105.4, 112.6, 126.9, 127.4, 129.0, 129.1, 129.2, 135.3, 139.1, 143.9, 148.2. LCMS: (B) retention time 2.21 min, m/z = 339 [M + H]+.
4.1.40. 4-(4,5-Diisopropyl-1H-imidazol-2-yl)-2,6-dimethoxyphenol hydrochloride (25b)
4-Hydroxy-2,5-dimethylhexan-3-one (0.152 g, 1.0 mmol) was dissolved in EtOH (6 mL) and aldehyde 1 (0.185 g, 1.0 mmol) and NH4OAc (0.770 g, 10.0 mmol) were added. The reaction was stirred 16 hours at RT. Workup as described for 3b. Purification using Biotage Isolera One flash chromatography (5-20 % MeOH/DCM) followed by forced precipitation with Et2O of HCl salt from MeOH solution afforded title compound (0.147g, 43 %) as deep purple solid; mp > 230 °C (dec); NMR (400 MHz, DMSO-d6) δ 1.34 (d, J = 6.9 Hz, 12H), 3.19 (hept, J = 6.9 Hz, 2H), 3.90 (s, 6H), 7.63 (s, 2H), 9.31 (s, 1H), 14.05 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 21.9, 24.0, 56.7, 105.4, 112.8, 132.8, 139.0, 143.2, 148.2. LCMS: (B) retention time 2.43 min, m/z = 305 [M + H]+.
4.1.41. 4-Cyano-2,6-dimethoxyphenyl methanesulfonate (26)
To a suspension of compound 20 (5.20 g, 20.0 mmol) in H2O (100 mL) was added AcOH (1.21 mL, 21.10 mmol) and hydroxylamine sulfonic acid (2.38 g, 21.1 mmol) and reaction was stirred 16 hours at 100 °C. The precipitate was filtered and thoroughly washed with water to give 4.88 g (98 %) of title compound as white solid. 1H NMR (400 MHz, chloroform-d) δ 3.33 (s, 3H), 3.93 (s, 6H), 6.91 (s, 2H); m/z = 258 [M + H]+.
4.1.42. 4-Carbamimidoyl-2,6-dimethoxyphenyl methanesulfonate (27)
To a solution of compound 26 (4.88 g, 18.99 mmol) in dry MeOH (30 mL) was added solution of HCl in dioxane (4 N, 60 mL) and reaction was stirred at RT under N2 atmosphere 4 days. Solvents were evaporated, residue was dried on high vacuum 4 hours and then it was dissolved in solution of NH3 in MeOH (7N, 100 mL) and reaction was stirred at RT under N2 atmosphere 7 days. LCMS showed most of the product was converted to amide probably due to wet solution of NH3/MeOH. Solvents were evaporated and residue was chromatographed on silica (CHCl3/MeOH) to give 1.02 g (20 %) of title compound as white foam. 1H NMR (500 MHz, DMSO-d6) δ 3.46 (s, 3H), 3.93 (s, 6H), 7.30 (s, 2H), 9.40 (s, 3H); m/z = 275 [M + H]+.
4.1.43. 2,6-Dimethoxy-4-(5-methyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenyl methanesulfonate (28)
To a solution of amidine 27 (0.274 g, 1.0 mmol) and NaHCO3 (0.252 g, 3.0 mmol) in EtOH (10 mL) was added 2-bromo-1-(thiophen-2-yl)propan-1-one (0.438 g, 2.0 mmol) and the reaction was stirred 16 hours at 60 °C. Reaction was cooled to RT, solids were filtered off and solvent was evaporated. The residue was purified using Biotage Isolera One flash purification system with a silica gel cartridge (CHCl3) to give 0.190 g, (48 %) of title compound as pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 2.47 (s, 3H), 3.41 (s, 3H), 3.91 (s, 6H), 7.09 (dd, J = 5.1, 3.6 Hz, 1H), 7.19-7.22 (m, 1H), 7.31 (s, 2H), 7.38-7.41 (m, 1H), 12.57 (s, 1H); m/z = 395 [M + H]+.
4.1.44. 2,6-Dimethoxy-4-(5-methyl-4-(thiophen-2-yl)-1H-imidazol-2-yl)phenol hydrochloride (29)
To a solution of diisopropyl amine (0.092 mL, 0.64 mmol) in dry THF (3 mL) coled to −78 °C was under N2 atmosphere added solution of n-butyllithium in hexanes (2.5M, 0.244 mL, 0.61 mmol) and mixture was let to reach 0 °C. Then solution of methansulfonate 28 (0.096 g, 0.24 mmol) in dry THF (2 mL) was added in one portion and reaction was stirred under N2 atmosphere 1 minute at 0 °C. Reaction was quenched by addition of HCl solution (5%, 10 mL). Reaction was diluted with water (20 mL) and extracted with EtOAc (3 × 15 mL). Combined organic layers were washed with brine, dried over MgSO4, filtered and solvents were evaporated. The residue was purified using preparative HPLC (10% MeCN/90 % water for 5 min followed by an increase to 50 % MeCN over 35 min and an increase to 70 % MeCN over 10 min; flow rate 15 mL/min) and further by forced precipitation with Et2O of HCl salt from its MeOH solution. After filtration and drying 12 mg (14 %) of title compound as pale brown-gray solid was obtained. Purity (HPLC) = 90 %; mp > 125 °C (dec). 1H NMR (500 MHz, Methanol-d4) δ 2.47 (s, 3H), 3.93 (s, 6H), 7.13-7.15 (m, 1H), 7.27 (s, 2H), 7.34 (d, J = 3.6 Hz, 1H), 7.44 (d, J = 5.0 Hz, 1H), 8.27 (s, 1H); 13C NMR (126 MHz, Methanol-d4) δ 11.2, 56.9, 104.5, 119.4, 125.8, 125.9, 127.7, 128.5, 128.6, 134.7, 146.6, 149.7. LCMS: (B) retention time 2.04 min, m/z = 316 [M + H]+.
4.1.45. (2S,3S,4S,5R,6S)-6-(4-(4,5-Diisopropyl-1H-imidazol-2-yl)-2,6-dimethoxyphenoxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid (30)
To a solution of 25b (130 mg, 0.43 mmol) and 2,3,4-Tri-O-acetyl-α-D-glucuronide methyl ester trichloroacetimidate (362 mg, 0.76 mmol) in dry DCM (6.0 mL) cooled to −10 °C was under N2 atmosphere added BF3∙OEt2 complex (29 μL, 0.22 mmol) and the reaction was stirred 4 hours at −10 °C until all starting material was consumed as indicated by LCMS analysis. Then the reaction was quenched by addition of Et3N (200 μL), solvents were evaporated and the residue was purified using flash chromatography (DCM + MeOH, 0-15 %). Obtained brow glassy solid (52 mg) was dissolved in MeOH and solution of Na2CO3 (25 mg, 0.27 mmol) in H2O (0.5 mL) was added. The reaction was stirred at RT 3 hours until complete consumption of intermediate was confirmed by LCMS analysis. Solvents were evaporated and the residue was purified using preparative HPLC (10% MeCN/90 % water for 5 min followed by an increase to 50 % MeCN over 35 min and an increase to 70 % MeCN over 10 min; flow rate 15 mL/min) to give title compound (18 mg, 9 %) as white solid; mp > 180 °C (dec). 1H NMR (500 MHz, Methanol-d4) δ 1.37 (dd, J = 16.8, 7.0 Hz, 12H), 3.21 (hept, J = 7.1 Hz, 2H), 3.42-3.58 (m, 3H), 3.53 (s, 3H), 3.64 (d, J = 9.7 Hz, 1H), 5.76 (d, J = 7.4 Hz, 1H), 7.04 (s, 2H). 13C NMR (125 MHz, Methanol-d4) δ 22.5, 22.6, 26.1, 57.1, 73.4, 75.4, 75.7, 77.7, 101.4, 105.4, 118.7, 134.8, 136.7, 143.2, 153.2, 176.4. LCMS: (B) retention time 2.08 min, m/z = 418 [M + H]+.
4.2. In Vitro nSMase2 assay
The inhibitory potencies of the synthesized compounds were determined using a fluorescence-based coupled assay involving human recombinant nSMase2, alkaline phosphatase (AP), and choline oxidase as previously reported [18]. Briefly, human nSMase2 recombinant enzyme was used to catalyze the hydrolysis of sphingomyelin into ceramide and phosphorylcholine. Phosphorylcholine undergoes dephosphorylation by AP to produce choline which is subsequently oxidized by choline oxidase. H2O2 generated in this oxidative step is detected by horseradish peroxidase-catalyzed conversion of Amplex Red® into resorufin (ex/em 530/590 nm). In order to assure that potent inhibitors identified in this coupled assay directly inhibit nSMase2, they were subsequently assessed in a counter assay using AP, choline oxidase, and horseradish peroxidase (but devoid of nSMase2 and sphingomyelin) in the presence of phosphorylcholine as previously described [18].
4.3. In Vitro Metabolic Stability Studies.
The metabolic stability was evaluated using mouse liver microsomes. For the cytochrome P450 (CYP)-mediated metabolism, the reaction was carried out with 100 mM potassium phosphate buffer, pH 7.4, in the presence of NADPH regenerating system (1.3 mM NADPH, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2, 0.4 U/mL glucose-6-phosphate dehydrogenase, 50 μM sodium citrate). Reactions, in triplicate, were initiated by addition of the liver microsomes to the incubation mixture (compound final concentration was 5 μM; 0.5 mg/mL microsomes). For the glucuronidation reaction, a test compounds were added to TRIS-HCl buffer (50 mM, pH 7.5) with microsomes (0.5 mg/mL), along with MgCl2 (8 mM) and alamethicin (25 μg/mL), and preincubated at 37 °C. The reaction was initiated (in triplicate) with UDPGA (2 mM; final concentration). Controls in the absence of cofactors were carried out to determine the specific cofactor-free degradation. After 60 min of incubation, aliquots of the mixture were removed and the reaction quenched by addition of three times the volume of ice-cold acetonitrile spiked with the internal standard. Compound disappearance was monitored over time using a liquid chromatography and tandem mass spectrometry (LC/MS/MS) method.
4.4. In Vivo Pharmacokinetics
All procedures involving mice were approved by and conformed to the guidelines of the Institutional Animal Care Committee of the Johns Hopkins University. Male CD-1 mice between 25 and 30 g were obtained from Harlan, and maintained on a 12-h light-dark cycle with ad libitum access to food and water. Three animals were used per time-point for each treatment group. DPTIP and 25b were prepared immediately prior to dosing in 5% DMSO, 5% polysorbate 80, 90% saline and administered to male mice as a single oral dose by oral gavage of 10 mg/kg. The mice were sacrificed at specified time points (0.5 and 2 h) post-drug administration. For collection of plasma and brain tissue, animals were euthanized with CO2, and blood samples were collected in heparinized microtubes by cardiac puncture. Brains were dissected and immediately flash frozen (−80 °C). Blood samples were spun at 2,000 × g for 15 min, plasma was removed and stored at −80 °C until LC-MS analysis. Prior to extraction, frozen samples were thawed on ice. The calibration curves were developed using plasma and brain from naïve animals as a matrix. Plasma samples (50 μL), were processed using a single liquid extraction method by addition of 300 μL of acetonitrile as with internal standard (losartan: 0.5 μM), followed by vortex mixing for 30 sec and then centrifugation at 10,000 × g for 10 min at 4 °C. Fifty microliter of the supernatant is diluted with 50 μL of water and transferred to 250 μL polypropylene autosampler vials sealed with a Teflon cap. For brain tissue analysis, brain samples were weighed in the range of (80-100 mg) to which 2-volumes of acetonitrile as with internal standard (losartan: 0.5 μM) was added and homogenized for extraction of the analyte. Samples were vortex mixed for 1 min and centrifuged as above. A 20 μL aliquot of supernatant was diluted with 20 μL of water and transferred to 250 μL polypropylene autosampler vials sealed with a Teflon cap. A volume of 3 μL was injected onto the ultra-performance liquid chromatography (UPLC) instrument for quantitative analysis by LC-MS/MS. Plasma and brain samples were analyzed on a Thermo Scientific Accela UPLC system coupled to Accela open autosampler at ambient temperature with an Agilent Eclipse Plus column (100 × 2.1mm i.d.) packed with a 1.8 μm C18 stationary phase. The autosampler was temperature controlled and operated at 10 °C. The mobile phase used for the chromatographic separation was composed of acetonitrile/water containing 0.1% formic acid and with a flow rate of 0.4 mL/min for 4.5 min using gradient elution. The column effluent was monitored using TSQ Vantage triple-quadrupole mass-spectrometric detector, equipped with an electrospray probe set in the positive ionization mode. Samples were introduced into the ionization source through a heated nebulized probe (350 °C).
Supplementary Material
Acknowledgment
This research was supported by NIH grants R01MH107659 (CR), R01AG059799 (CR), and P30MH075673 (BSS). OS is supported by a joint postdoctoral fellowship from Johns Hopkins Drug Discovery and the Institute of Organic Chemistry and Biochemistry (IOCB) of the Czech Academy of Sciences.
Abbreviations Used:
- ADME
absorption, distribution, metabolism, and excretion
- ALS
amyotrophic lateral sclerosis
- AP
alkaline phosphatase
- nSMase2
neutral sphingomyelinase 2
- DPTIP
2,6-dimethoxy-4-[4-phenyl-5-(2-thienyl)-1H-imidazol-2-yl]phenol
- SAR
structure-activity relationship
- UDPGA
uridine diphosphate glucuronic acid
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2019.03.015.
References
- [1].Shamseddine AA, Airola MV, Hannun YA, Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes, Adv Biol Regul, 57 (2015) 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Airola MV, Shanbhogue P, Shamseddine AA, Guja KE, Senkal CE, Maini R, Bartke N, Wu BX, Obeid LM, Garcia-Diaz M, Hannun YA, Structure of human nSMase2 reveals an interdomain allosteric activation mechanism for ceramide generation, Proc Natl Acad Sci U S A, 114 (2017) E5549–E5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Catapano ER, Natale P, Monroy F, Lopez-Montero I, The enzymatic sphingomyelin to ceramide conversion increases the shear membrane viscosity at the air-water interface, Adv Colloid Interface Sci, 247 (2017) 555–560. [DOI] [PubMed] [Google Scholar]
- [4].Eich C, Manzo C, de Keijzer S, Bakker GJ, Reinieren-Beeren I, Garcia-Parajo MF, Cambi A, Changes in membrane sphingolipid composition modulate dynamics and adhesion of integrin nanoclusters, Sci Rep, 6 (2016) 20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bollinger CR, Teichgraber V, Gulbins E, Ceramide-enriched membrane domains, Biochim Biophys Acta, 1746 (2005) 284–294. [DOI] [PubMed] [Google Scholar]
- [6].Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M, Ceramide triggers budding of exosome vesicles into multivesicular endosomes, Science, 319 (2008) 1244–1247. [DOI] [PubMed] [Google Scholar]
- [7].Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T, Secretory mechanisms and intercellular transfer of microRNAs in living cells, J Biol Chem, 285 (2010) 17442–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E, Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease, Neurobiol Aging, 35 (2014) 1792–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T, Depletion of microglia and inhibition of exosome synthesis halt tau propagation, Nat Neurosci, 18 (2015) 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].van Echten-Deckert G, Walter J, Sphingolipids: critical players in Alzheimer’s disease, Prog Lipid Res, 51 (2012) 378–393. [DOI] [PubMed] [Google Scholar]
- [11].Mielke MM, Bandaru VV, McArthur JC, Chu M, Haughey NJ, Disturbance in cerebral spinal fluid sphingolipid content is associated with memory impairment in subjects infected with the human immunodeficiency virus, J Neurovirol, 16 (2010) 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP, Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis, Ann Neurol, 52 (2002) 448–457. [DOI] [PubMed] [Google Scholar]
- [13].Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, Lotvall J, Nakagama H, Ochiya T, Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier, Nat Commun, 6 (2015) 6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T, Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis, J Biol Chem, 288 (2013) 10849–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sanada T, Hirata Y, Naito Y, Yamamoto N, Kikkawa Y, Ishida Y, Yamasaki C, Tateno C, Ochiya T, Kohara M, Transmission of HBV DNA Mediated by Ceramide-Triggered Extracellular Vesicles, Cell Mol Gastroenterol Hepatol, 3 (2017) 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G, Inhibitors of sphingolipid metabolism enzymes, Biochim Biophys Acta, 1758 (2006) 1957–1977. [DOI] [PubMed] [Google Scholar]
- [17].Canals D, Perry DM, Jenkins RW, Hannun YA, Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases, Br J Pharmacol, 163 (2011) 694–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Figuera-Losada M, Stathis M, Dorskind JM, Thomas AG, Bandaru VV, Yoo SW, Westwood NJ, Rogers GW, McArthur JC, Haughey NJ, Slusher BS, Rojas C, Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties, PLoS One, 10 (2015) e0124481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luberto C, Hassler DF, Signorelli P, Okamoto Y, Sawai H, Boros E, Hazen-Martin DJ, Obeid LM, Hannun YA, Smith GK, Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase, J Biol Chem, 277 (2002) 41128–41139. [DOI] [PubMed] [Google Scholar]
- [20].Rojas C, Barnaeva E, Thomas AG, Hu X, Southall N, Marugan J, Chaudhuri AD, Yoo SW, Hin N, Stepanek O, Wu Y, Zimmermann SC, Gadiano AG, Tsukamoto T, Rais R, Haughey N, Ferrer M, Slusher BS, DPTIP, a newly identified potent brain penetrant neutral sphingomyelinase 2 inhibitor, regulates astrocyte-peripheral immune communication following brain inflammation, Sci Rep, 8 (2018) 17715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lombardino JG, Wiseman EH, Preparation and antiinflammatory activity of some nonacidic trisubstituted imidazoles, J Med Chem, 17 (1974) 1182–1188. [DOI] [PubMed] [Google Scholar]
- [22].Yu MJ, McCowan JR, Phebus LA, Towner RD, Ho PP, Keith PT, Luttman CA, Saunders RD, Ruterbories KJ, Lindstrom TD, et al. , Benzylamine antioxidants: relationship between structure, peroxyl radical scavenging, lipid peroxidation inhibition, and cytoprotection, J Med Chem, 36 (1993) 1262–1271. [DOI] [PubMed] [Google Scholar]
- [23].Rao DV, Stuber FA, An Efficient Synthesis of 3,4,5-Trimethoxybenzaldehyde from Vanillin, Synthesis, 1983 (1983) 308–308. [Google Scholar]
- [24].Node M, Kodama S, Hamashima Y, Katoh T, Nishide K, Kajimoto T, Biomimetic synthesis of (+/−)-galanthamine and asymmetric synthesis of (−)-galanthamine using remote asymmetric induction, Chem Pharm Bull, 54 (2006) 1662–1679. [DOI] [PubMed] [Google Scholar]
- [25].Corcoran JPT, Kalindjian SB, Borthwick AD, Adams DR, Brown JT, Taddei DMA, Shiers JJ, Therapeutic aryl-amido-aryl compounds and their use, PCT Int. Pat. Appl. WO 2011/027106, 2011. [Google Scholar]
- [26].Ren X, Chen X, Peng K, Xie X, Xia Y, Pan X, First enantioselective synthesis of daphneticin and its regioisomer, Tetrahedron: Asymmetry, 13 (2002) 1799–1804. [Google Scholar]
- [27].Zhang NT, Zeng CC, Lam CM, Gbur RK, Little RD, Triarylimidazole redox catalysts: electrochemical analysis and empirical correlations, J Org Chem, 78 (2013) 2104–2110. [DOI] [PubMed] [Google Scholar]
- [28].Tani S, Uehara TN, Yamaguchi J, Itami K, Programmed synthesis of arylthiazoles through sequential C–H couplings, Chem Sci, 5 (2014) 123–135. [Google Scholar]
- [29].Carvalho C, Russo A, Sargent M, Boron, Trichloride as a Selective Demethylating Agent for Hindered Ethers: a Synthesis of the Phytoalexins α- and β-Pyrufuran, a Synthesis of Tri-O-methylleprolomin and its Demethylation, Aust J Chem, 38 (1985) 777–792. [Google Scholar]
- [30].Fujioka H, Murai K, Ohba Y, Hiramatsu A, Kita Y, A mild and efficient one-pot synthesis of 2-dihydroimidazoles from aldehydes, Tetrahedron Lett, 46 (2005) 2197–2199. [Google Scholar]
- [31].Lantos I, Loev B, The total synthesis of (±)-decinine, Tetrahedron Letters, 16 (1975) 2011–2014. [Google Scholar]
- [32].Xu Y, Wan L-F, Salehi H, Deng W, Guo Q-X, Microwave-assisted One-Pot Synthesis of Tri-substituted Imidazoles on Solid Support, Heterocycles, 63 (2004) 1613–1618. [Google Scholar]
- [33].Quinn DJ, Haun GJ, Moura-Letts G, Direct synthesis of nitriles from aldehydes with hydroxylamine-O-sulfonic acid in acidic water, Tetrahedron Lett, 57 (2016) 3844–3847. [Google Scholar]
- [34].Balo C, Lopez C, Brea JM, Fernandez F, Caamano O, Synthesis and evaluation of adenosine antagonist activity of a series of [1,2,4]triazolo[1,5-c]quinazolines, Chem Pharm Bull (Tokyo), 55 (2007) 372–375. [DOI] [PubMed] [Google Scholar]
- [35].Li B, Chiu CKF, Hank RF, Murry J, Roth J, Tobiassen H, An Optimized Process for Formation of 2,4-Disubstituted Imidazoles from Condensation of Amidines and α-Haloketones, Org Proc Res Dev, 6 (2002) 682–683. [Google Scholar]
- [36].Ritter T, Stanek K, Larrosa I, Carreira EM, Mild cleavage of aryl mesylates: methanesulfonate as potent protecting group for phenols, Org Lett, 6 (2004) 1513–1514. [DOI] [PubMed] [Google Scholar]
- [37].Lucas R, Alcantara D, Morales JC, A concise synthesis of glucuronide metabolites of urolithin-B, resveratrol, and hydroxytyrosol, Carbohydr Res, 344 (2009) 1340–1346. [DOI] [PubMed] [Google Scholar]
- [38].Kumar P, Wiebe LI, Mannan RH, Zhang Z, Xia H, McEwan AJ, [99mTc]Technetium labelled PnAo-azomycin glucuronides: a novel class of imaging markers of tissue hypoxia, Appl Radiat Isot, 57 (2002) 719–728. [DOI] [PubMed] [Google Scholar]
- [39].Kaivosaari S, Finel M, Koskinen M, N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases, Xenobiotica, 41 (2011) 652–669. [DOI] [PubMed] [Google Scholar]
- [40].Bilousova T, Elias C, Miyoshi E, Alam MP, Zhu C, Campagna J, Vadivel K, Jagodzinska B, Gylys KH, John V, Suppression of tau propagation using an inhibitor that targets the DK-switch of nSMase2, Biochem Biophys Res Commun, 499 (2018) 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.