

ABSTRACT
Mitophagy is an essential mitochondrial quality control mechanism that eliminates damaged mitochondria and the production of reactive oxygen species (ROS). The relationship between mitochondria oxidative stress, ROS production and mitophagy are intimately interwoven, and these processes are all involved in various pathological conditions of acute kidney injury (AKI). The elimination of damaged mitochondria through mitophagy in mammals is a complicated process which involves several pathways. Furthermore, the interplay between mitophagy and different types of cell death, such as apoptosis, pyroptosis and ferroptosis in kidney injury is unclear. Here we will review recent advances in our understanding of the relationship between ROS and mitophagy, the different mitophagy pathways, the relationship between mitophagy and cell death, and the relevance of these processes in the pathogenesis of AKI.
Abbreviations: AKI: acute kidney injury; AMBRA1: autophagy and beclin 1 regulator 1; ATP: adenosine triphosphate; BAK1: BCL2 antagonist/killer 1; BAX: BCL2 associated X, apoptosis regulator; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; BH3: BCL2 homology domain 3; BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; CASP1: caspase 1; CAT: catalase; CCCP: carbonyl cyanide m-chlorophenylhydrazone; CI-AKI: contrast-induced acute kidney injury; CISD1: CDGSH iron sulfur domain 1; CL: cardiolipin; CNP: 2’,3’-cyclic nucleotide 3’-phosphodiesterase; DNM1L/DRP1: dynamin 1 like; E3: enzyme 3; ETC: electron transport chain; FA: folic acid; FUNDC1: FUN14 domain containing 1; G3P: glycerol-3-phosphate; G6PD: glucose-6-phosphate dehydrogenase; GPX: glutathione peroxidase; GSH: glutathione; GSK3B: glycogen synthase kinase 3 beta; GSR: glutathione-disulfide reductase; HIF1A: hypoxia inducible factor 1 subunit alpha; HUWE1: HECT, UBA and WWE domain containing 1; IL1B: interleukin 1 beta; IMM: inner mitochondrial membrane; IPC: ischemic preconditioning; IRI: ischemia-reperfusion injury; LIR: LC3-interacting region; LPS: lipopolysaccharide; MA: malate-aspartate; MPT: mitochondrial permeability transition; MUL1: mitochondrial E3 ubiquitin protein ligase 1; mtROS: mitochondrial ROS; NLR: NOD-like receptor; NLRP3: NLR family pyrin domain containing 3; NOX: NADPH oxidase; OGD-R: oxygen-glucose deprivation-reperfusion; OMM: outer mitochondrial membrane; OPA1: OPA1 mitochondrial dynamin like GTPase; OXPHOS: oxidative phosphorylation; PARL: presenilin associated rhomboid like; PINK1: PTEN induced kinase 1; PLSCR3: phospholipid scramblase 3; PMP: peptidase, mitochondrial processing; PRDX: peroxiredoxin; PRKN: parkin RBR E3 ubiquitin protein ligase; RPTC: rat proximal tubular cells; ROS: reactive oxygen species; SLC7A11/xCT: solute carrier family 7 member 11; SOD: superoxide dismutase; SOR: superoxide reductase; SQSTM1/p62: sequestosome 1; TCA: tricarboxylic acid; TIMM: translocase of inner mitochondrial membrane; TOMM: translocase of outer mitochondrial membrane; TXN: thioredoxin; VDAC: voltage dependent anion channel; VCP: valosin containing protein.
KEYWORDS: Acute kidney injury, cell death, mitochondria, mitophagy, reactive oxygen species
Introduction
Maintaining mitochondrial homeostasis through balanced mitochondrial biogenesis and clearance of damaged mitochondria is a crucial determinant of cellular function. Defective mitochondria can be selectively removed through a specific form of autophagy, known as mitophagy which is responsible for the basal mitochondrial turnover to eliminate dysfunctional mitochondria [1]. Mitophagy can be induced in various pathological process, such as oxidative stress and inflammation, as a stress-response mechanism to inhibit mitochondria-dependent cell death [2,3]. The pathogenesis of acute kidney injury (AKI) also involves multiple stressors including hypoxia, inflammation, oxidant injury, and other damaging insults, all of which are known to drive mitophagy induction [4]. In most cases, mitophagy promotes cellular adaptation protecting the cell through a variety of mechanisms including the elimination of damaged mitochondria which are a major source of reactive oxygen species (ROS) generation [5]. Therefore, mitophagy is important in kidney injury and is a potential therapeutic target in the pathogenesis of AKI. A growing number of studies have revealed the role of mitophagy in kidney diseases [6]. Dysregulation of mitophagy results in diverse pathophysiology including cardiac injury, kidney fibrosis and neurodegenerative disease [6–8]. However, the role mitophagy induction in AKI is not completely understood. Here, we summarized the role of activation of mitophagy during different causes of kidney injury and the mechanism of mitophagy activation in the kidney, discuss the pharmacologic induction of mitophagy as a potential therapeutic strategy, and provide suggestions for future perspectives in this field.
The generation and elimination of mitochondria ROS
The main sources of cellular ROS are mitochondria and NOX (NADPH oxidase) [9]. Mitochondria utilize oxygen to generate adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) which is one of sources of mitochondrial ROS (mtROS) [10]. The electron transport chain (ETC) is a series of electron transporters embedded in the inner mitochondrial membrane (IMM) that shuttle hydrogen ions (H+) across the mitochondrial membrane from the mitochondrial matrix into the intermembrane space to generate a potential ionic gradient that contains the energy necessary to synthesize ATP. Complex V allows H+ to enter the mitochondrial matrix in favor of a concentration gradient, releasing the necessary energy to couple phosphate into adenosine and synthesize ATP [11]. In this process, electrons are transported through the ETC and are finally shuttled to molecular oxygen by complex IV [12]. Leak of electrons at complexes I and III interact with oxygen to produce the superoxide anion, the most important and dangerous mtROS [13,14]. It is now known that cells produce mtROS as important signaling molecules that participate in physiologic functions. However, abnormal increments in the production of mtROS are known to induce cell injury through oxidative stress. Besides ATP production, mitochondria are also involved in heme and iron sulfur center biosynthesis which induce ROS production and play important roles in oxidative damage [15,16].
NOXs are central components for regulating the cellular redox balance [17]. There are seven isoforms of the NADPH oxidases have been identified: NOX1, CYBB/NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2 [18]. NADPH is derived from the metabolism of glucose through glycolysis wherein G6PD (glucose-6-phosphate dehydrogenase) is the rate-limiting enzyme [19]. Pyruvate, the end-product of glycolysis, is ultimately transported into mitochondria to undergo further metabolism through the tricarboxylic acid (TCA) cycle generating reduced equivalents in the form of NADH and FADH2 which will then enter the electron transport chain in the last step of oxidative phosphorylation [20]. Complex I catalyzes the oxidation of NADPH/NADH generated in the TCA cycle [21,22]. Cytosolic and mitochondrial NADH are exchanged through the malate-aspartate (MA) shuttle and the G3P (glycerol-3-phosphate) shuttle which has been reviewed in previous studies [23–25]. All NOX family members share six highly conserved transmembrane domains. The cytoplasmic COOH terminus contains conserved NADPH binding domains [26]. The active enzyme complex transports electrons to oxygen from NADPH to produce superoxide free radical [26]. ROS produced by NOXs can also cause mitochondrial ROS production which is an important mechanistic pathways of ROS amplification or propagation (summarized in Figure 1) [27].
Figure 1.
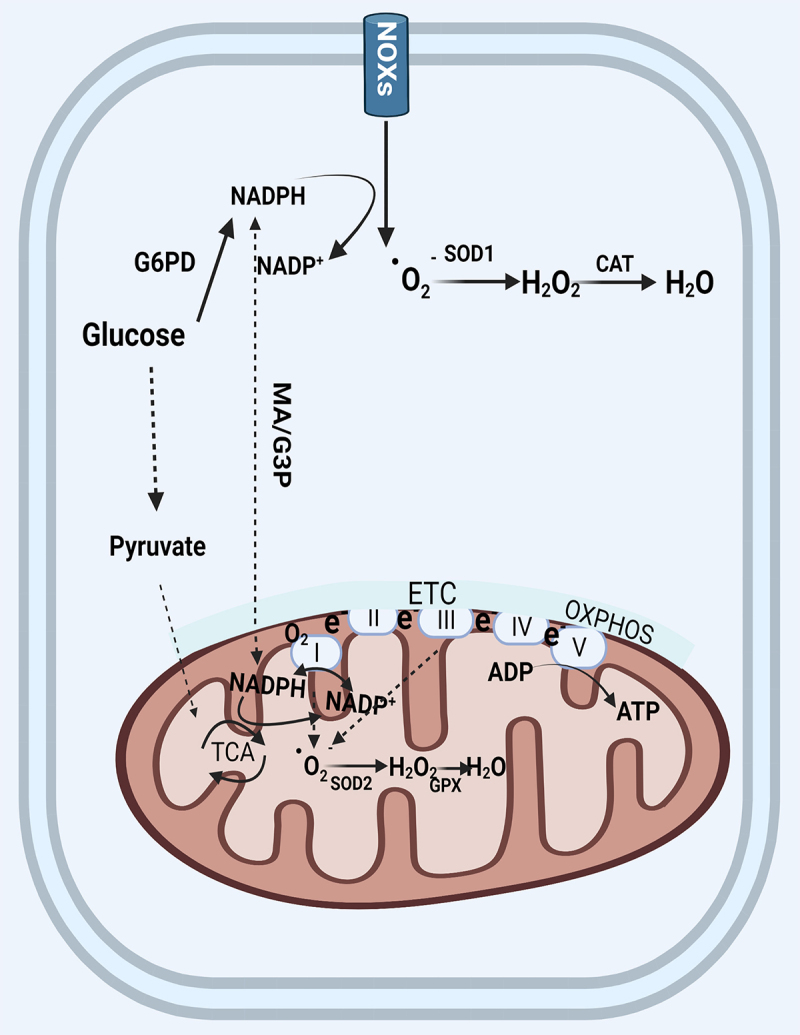
The generation and elimination of mitochondria ROS. In the cytosol, NADPH is primarily produced by G6PD in the glycolysis pathway. The cytosolic and mitochondrial NADPH is exchanged through two shuttles. NOXs transports electrons to oxygen from NADPH to produce superoxide free radical which was converted by SOD (SOD2 in mitochondria) to hydrogen peroxide, and finally converted by catalase to harmless H2O. Mitochondrial ROS are produced from the leakage of electrons to form superoxide at complex I and complex III in the electron transport chain. Mitochondria utilize oxygen to generate ATP through OXPHOS. Abbreviations: ATP: adenosine triphosphate; CAT: catalase; ETC: electron transport chain; G6PD: glucose-6-phosphate dehydrogenase; MA/G3P: the malate-aspartate (MA) and the glycerol-3-phosphate(G3P) shuttle; NOX: NADPH oxidase; OXPHOS: oxidative phosphorylation; SOD: superoxide dismutase; TCA: tricarboxylic acid.
To protect cells from the oxidative stress, enzymatic and non-enzymatic defense systems in mitochondria eliminate excess ROS [28]. The non-enzymatic defense system includes flavonoids, vitamins, and glutathione [29]. SOD (superoxide dismutase), SOR (superoxide reductase), CAT (catalase), GPX (glutathione peroxidase), GSR (glutathione-disulfide reductase), PRDX (peroxiredoxin), and TXN (thioredoxin) constitute scavenging enzymes systems that are involved in the regulation of mitochondrial ROS [29]. SOD is in charge of converting the superoxide anion, the most dangerous ROS produced [30], into hydrogen peroxide, which can be then converted to H2O by CAT [30].
The relationship between ROS and mitophagy
Mitophagy is considered a bona fide strategy to limit mtROS production by removing the aged and damaged mitochondria via the specific sequestration and engulfment of mitochondria in lysosome [31]. Mitophagy may function more broadly to limit the deleterious effects of ROS on cellular function [32]. ROS induces mtDNA damage, decreases the mitochondrial membrane potential, and induces oxidation of proteins and lipids [30]. Mitophagy after DNA damage is a vital cellular response to maintain mitochondrial functions and DNA repair. A previous study reported that suppression of mitophagy disturbs mitochondrial Ca2+ homeostasis, affects ATP production, and attenuates DNA repair [33]. Mitochondrial proteins and lipids also play important roles in maintaining mitochondrial function. Specific mitochondrial lipids are critical for proper assembly of the electron transport chain complexes and for effective responses to mitophagy [34]. Under stress conditions, cardiolipin (CL), which constitutes almost 20% of the lipid content in the IMM, translocates to the outer mitochondrial membrane (OMM) where, together with ceramide, it binds LC3 and LC3B-II to recruit phagophores to damaged organelles (discussed further in the next section) [35]. Damage-induced ROS disrupt mitochondrial proteins and damage existing macromolecules. Furthermore, these ROS oxidize ergosterol to ergosterol peroxide in the OMM [36], which acts as a signal to recruit VCP (valosin containing protein) associated mitochondrial stress responsive 1, a component of a mitochondrial surveillance system [37], to damaged mitochondria for the subsequent signal transduction to protein degradation by the proteasome which is involved in maintaining mitochondrial protein homeostasis [35]. These data suggest that, oxidation of proteins and lipids should have the presence of adaptive mitophagy for cell to survivor from oxidative damage.
Enhanced mitophagy is usually an early response to promote survival while overwhelming or prolonged mitochondrial damage can induce excessive, pathological activation of mitophagy, thereby inducing cell death and tissue injury [38]. While complex I inhibition stimulates the activation of mitophagy through mtROS generation, subsequent cell death is ultimately a consequence of mtROS that are mitophagy- dependent [39]. These data suggest that the process of mitophagy may, in some cases, increase mtROS levels which could trigger a cell to further induce mitophagy and therefore propagate the elevation in mtROS levels through a positive feedback loop [40].
Molecular regulation of mitophagy
The best described pathway inducing mitophagy is driven by the enzyme 3 (E3) ubiquitin ligase PRKN (parkin RBR E3 ubiquitin protein ligase) and the kinase PINK1 (PTEN induced kinase 1). The vast majority of the studies investigating the role of mitophagy in physiological or pathological conditions focus on the PINK1-PRKN pathway [41]. However, there are PRKN-independent mechanisms that can trigger the activation of mitophagy including those driving ubiquitin ligases, receptor and CL.
PRKN-dependent mitophagy
PRKN, a member of the RING-between-RING family of E3 ligases, composed of 14 complex multidomain enzymes which are mutated in recessive familial forms of Parkinson disease, is a key mediator of mitochondrial quality control processes [42]. PINK1, a serine/threonine kinase containing a mitochondrial targeting signal at the N terminus and also plays an important role in this process [42]. Normally, PINK1 is constitutively imported to the inner membrane via the TIMM (translocase of inner mitochondrial membrane)-TOMM (translocase of outer mitochondrial membrane) complex. PINK1 is then cleaved by several proteases including PMP (peptidase, mitochondrial processing) which removes PINK1’s N-terminal mitochondrial targeting signal and the inner membrane PARL (presenilin associated rhomboid like), cleaves PINK1 between amino acids A103 and F104 in its hydrophobic domain spanning the IMM, which ultimately results in proteasomal degradation [43–46]. Mitochondrial membrane potential (ΔΨm) is a key indicator of mitochondrial health and injury. PINK1 is imported as normal to the OMM of depolarized mitochondria, however the loss of ΔΨm prevents its import and subsequent cleavage in the IMM [47]. When mitochondrial import is disrupted by depolarization, unprocessed PINK1 accumulates specifically at the OMM of dysfunctional mitochondria. In response to various stressors, PINK1 accumulates at OMM bound to the TOMM complex where it is activated through dimerization and autophosphorylation [48]. PINK1 can stabilize on the outer membrane to recruit PRKN from the cytosol to damaged mitochondria in response to decreased ΔΨm. PINK1 will phosphorylate PRKN on the Ub-like domain on the Ser resulting in an increase of its E3 activity (Ub ligase activity) and the formation of polyubiquitin chains on the surface of depolarized mitochondrial membranes [49,50]. Activated PRKN polyubiquitinates numerous substrates of OMM proteins, leading to the recruitment of the autophagy machinery including the Ub- and LC3-binding receptor SQSTM1/p62 (sequestosome 1) (summarized in Figure 2) [51].
Figure 2.
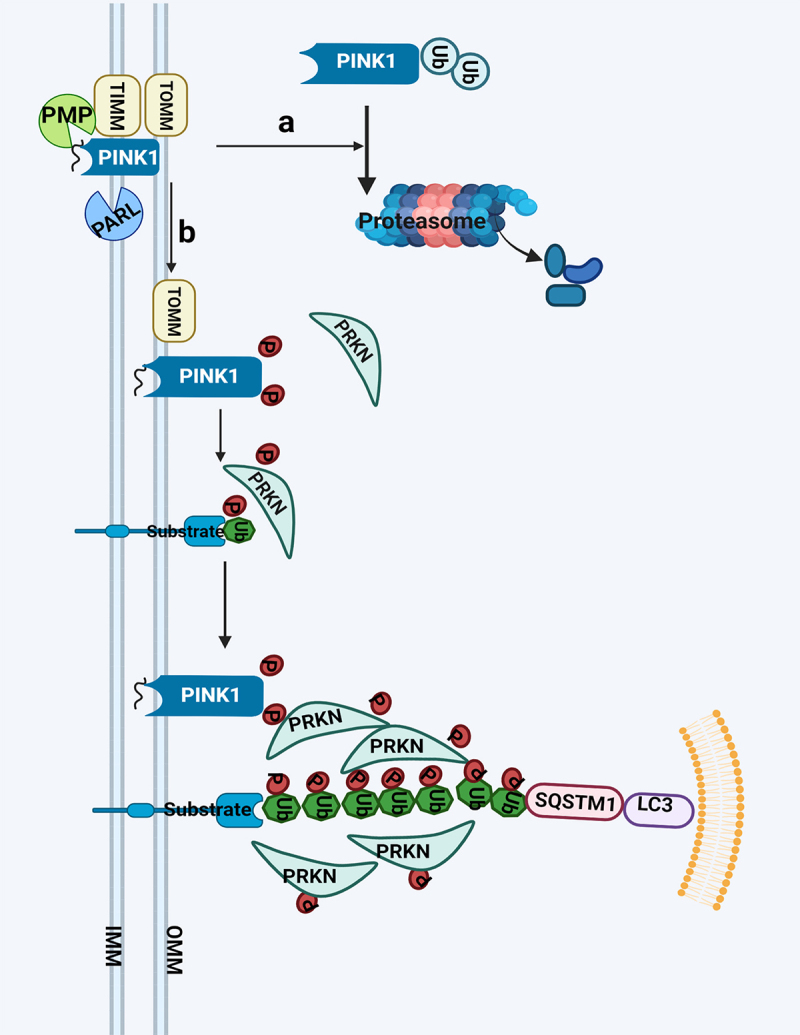
PRKN-dependent mitophagy. PINK1 is constitutively processed by mitochondrial proteases, PMP and PARL, resulting in its proteasomal degradation in normal condition (a); In damaged mitochondria, PINK1 accumulates at OMM bound to the TOMM complex where it is activated through auto-phosphorylation. Activated PINK1 subsequently phosphorylates ubiquitin, which triggers recruitment of PRKN recruitment to mitochondria and activation of its E3 ligase activity. It further ubiquitinates mitochondrial substrates and initiate autophagosome formation. PRKN acts as an enhancer of this signaling through further ubiquitination of mitochondrial proteins (b). Abbreviations: IMM: inner mitochondrial membrane; OMM: outer mitochondrial membrane; PARL: presenilin associated rhomboid like; PINK1: PTEN induced kinase 1; PMP: peptidase, mitochondrial processing; PRKN: parkin RBR E3 ubiquitin protein ligase; SQSTM1/p62: sequestosome 1; TIMM: translocase of inner mitochondrial membrane; TOMM: translocase of outer mitochondrial membrane.
PRKN-independent mitophagy
Receptor-mediated mitophagy
The OMM exists of several LC3-interacting regions (LIR) containing autophagic receptors anchored in the membrane of the phagophore [52]. Unlike PINK1-PRKN mediated mitophagy requiring the translocation of PRKN to the damaged mitochondria, some receptors, like BNIP3 (BCL2 interacting protein 3) and FUNDC1 (FUN14 domain containing 1), can bind to LC3 proteins, thereby linking the phagophore to the targeted mitochondria, directly inducing mitophagy [52]. This process is known as receptor-mediated mitophagy. A growing number of OMM proteins containing LIR domains have also been identified including BNIP3, its homolog BNIP3L/NIX (BCL2 interacting protein 3 like), and FUNDC1, which have been reviewed previously [41,53]. Proteins in the IMM can also act as crucial mitophagy receptors involved in targeting mitochondria for autophagic degradation. PHB2 (prohibitin 2), an IMM protein, binds the phagophore membrane-associated protein LC3 through a LIR domain which is required for PRKN-induced mitophagy in mammalian cells [54]. Some receptors, like activating molecule in AMBRA1 (autophagy and beclin 1 regulator 1) which is a BCL2 homology domain 3 (BH3)-like protein, can play a role in the selective degradation of ubiquitylated mitochondria, transducing both canonical PINK1-PRKN-dependent and -independent mitophagy [55].
Cardiolipin-mediated mitophagy
CL is a hallmark mitochondrial lipid which is almost exclusively found at the IMM [56]. CL can have different roles in mitochondrial quality control and morphology depending on its location. When located in the IMM, CL cooperates with OPA1 (OPA1 mitochondrial dynamin like GTPase) to induce IMM fusion. In response to stress, the mitochondrial PLSCR3 (phospholipid scramblase 3) allows the translocation of CL from the inner to the outer leaflet of the membrane to bind directly to LC3 to induce mitophagy [57]. During mitophagy, CL can also interact with BECN1 (beclin 1), a central regulator of autophagy, and recruit the autophagic machinery by its interaction with LC3 [58].
Ubiquitin-mediated mitophagy
Mitochondria ubiquitination plays a central role in mitophagy due to the E3 ligase activity of protein PRKN. Several ubiquitin ligases other than PRKN have been proved to have common effect with PRKN. MUL1 (mitochondrial E3 ubiquitin protein ligase 1) is a resident mitochondrial ubiquitin E3 ligase inserted in the OMM and has a multifunction including mitochondrial fusion, interplay between the endoplasmic reticulum and mitochondria and mitophagy [59,60]. MUL1 acts in parallel with the PINK1-PRKN pathway in compensate for PINK1-PRKN loss in their mutant phenotypes [60]. MUL1 can act not only as a ubiquitin ligase but also as a mitophagy receptor. It can directly recruit the autophagy machinery by interaction of GABARAP (GABA type A receptor-associated protein (Atg8-family protein) [61]. HUWE1 (HECT, UBA and WWE domain containing E3 ubiquitin protein ligase 1) has been also identified play a role in AMBRA1-mediated mitophagy in a PRKN-independent pathway (summarized in Figure 3) [62].
Figure 3.
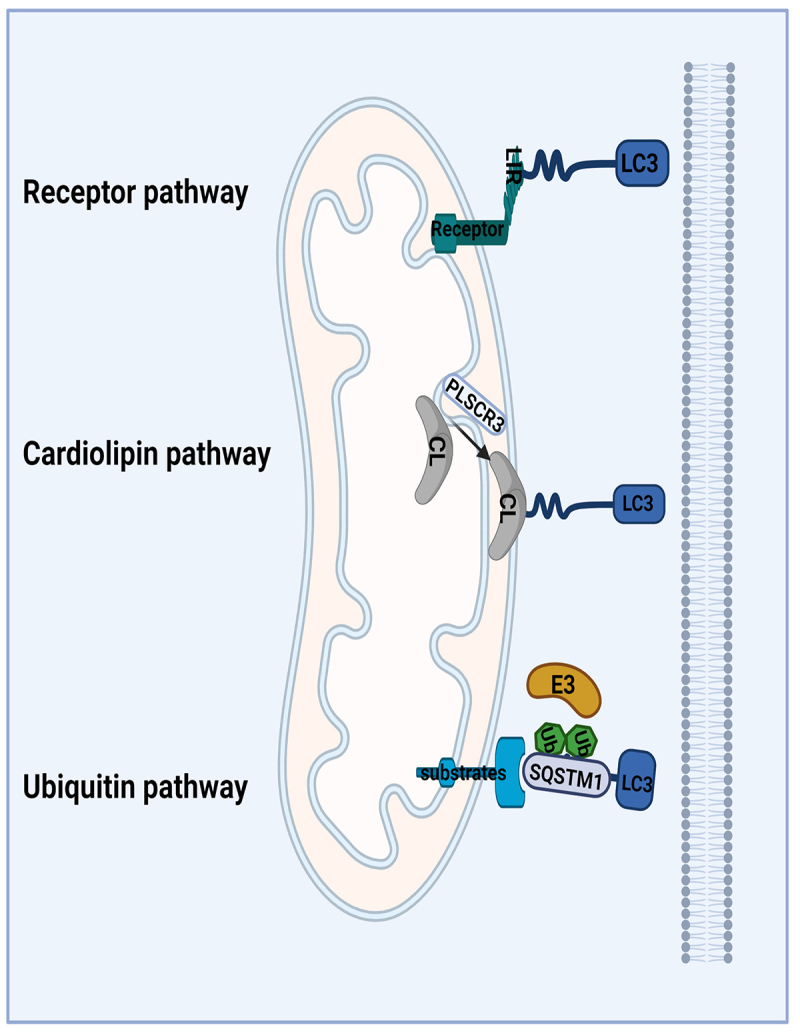
PRKN-independent mitophagy. Receptor mediated mitophagy: BNIP3, BNIP3L and FUNDC1, are OMM receptors containing an LIR-domain that directly binds to LC3 proteins to recruit the phagophore to the damaged mitochondria and leading to its degradation; cardiolipin-mediated mitophagy: IMM CL can be translocated to the OMM through the action of PLSCR3. Once at the OMM, CL binds to LC3A to recruit the phagophore and to remove the damaged mitochondria; Ubiquitin mediated mitophagy: Some proteins have E3 ubiquitin ligases activities which can be located at damaged mitochondria to ubiquitinate OMM proteins, and subsequently recruit the phagophore to damaged mitochondria. Abbreviations: BNIP3: BCL2 interacting protein 3; BNIP3L/NIX: BCL2 interacting protein 3 like; CL: cardiolipin; FUNDC1: FUN14 domain containing 1; E3: enzyme 3; IMM: inner mitochondrial membranes; OMM: outer mitochondrial membranes; LIR: LC3-interacting region; PLSCR3: phospholipid scramblase 3.
Mitophagy in acute kidney injury
The kidney is only second to the heart in mitochondrial density and oxygen consumption. This is due to the high-energy demand necessary to reabsorb ~70% of the solute load filtered through the glomerulus and for excretion [63]. Increased oxidative stress, inflammation, and uncoupling of oxygen consumption from ATP, all of which are associated with AKI, promote mitochondrial damage which can trigger mitophagy [63]. While the role of mitophagy in AKI has been controversial, many studies have now demonstrated the protective effect of mitophagy in AKI, including PRKN-independent pathways, such as BNIP3-mediated mitophagy [64] and PRKN-dependent pathways [65]. A few studies have also reported a damage function of mitophagy in kidney injury [66]. Some recent reviews have summarized mitochondrial biogenesis in kidney injury and repair including mitophagy [67,68]. The role of mitophagy in various pathological conditions of acute kidney injury is summarized in Table 1.
Table 1.
The role of mitophagy in various pathological conditions of acute kidney injury.
| Pathways | Factors | Model | Cells and Animals | Mechanisms | Effect | Reference |
|---|---|---|---|---|---|---|
| PRKN-dependent | IRI | CCCP for HK-2 cells and IRI for mice | PINK1 siRNA, PRKN siRNA, or double siRNA in cells; pink1-KO, prkn-KO or double-KO in mice. | 1. Reduce mitochondrial damage 2. Modulate the removal of ROS 3. Relieve inflammatory response 4. Modulate cell death |
Activation of mitophagy protects against AKI | [65] |
| IRI | H/R for HK-2 cells and IRI for mice | PmirGLO-Dual-luciferase reporter vector of MEG3 and RTKN in cells; AAV-sh-MEG3 vector in mice via tail vein. |
Promoting apoptosis | Activation of mitophagy aggravates AKI | [66] | |
| IRI | CCCP for RPTC cells and IRI for mice |
Pink1 shRNA in cells PT-atg7-KO in mice |
1.Suppressed mitochondrial depolarization 2.Improved ATP production 3.Inhibited the generation of ROS |
Activation of mitophagy protects against AKI | [72] | |
| Sepsis | LPS for RPTC cells and LPS or CLP for mice | Pink1 siRNA, Prkn siRNA, or Optn siRNA in cells; pink1-KO or prkn KO in mice | 1. Mitochondrial quality control 2. Reduce cells apoptosis |
Activation of mitophagy protects against AKI | [81] | |
| Sepsis | LPS for RPTC cells | Pink1 siRNA and Prkn siRNA in cells | 1. Inhibited the apoptosis 2. Remove damaged mitochondria |
Activation of mitophagy protects against AKI | [82] | |
| Sepsis | LPS for HK-2 cells and CLP for rats | PRKN siRNA or SIRT1 inhibitor in HK-2 cells; Prkn silencing lentivirus and SIRT1 inhibitor EX527 in rats via tail vein | 1. Inhibited the apoptosis 2. Inhibited the pyroptosis 3. Remove damaged mitochondria |
Activation of mitophagy protects against AKI | [83] | |
| Sepsis | CLP for mice | prkn-KO in mice | 1. Inhibition of mitochondrial dysfunction 2. Inhibition of NLRP3 inflammasome activation |
Activation of mitophagy protects against AKI | [84] | |
| Cisplatin | Cisplatin injected intraperitoneally for mice | pink1-KO or prkn-KO in mice | Reduce cells apoptosis | Activation of mitophagy protects against AKI | [87] | |
| Cisplatin | Cisplatin for RTECs and cisplatin injected intraperitoneally for mice | - | 1. Reversed cellular ROS induced by cisplatin 2. Reversed mitochondrial membrane potential level induced by cisplatin |
Activation of mitophagy protects against AKI | [88] | |
| Cisplatin | Cisplatin for HK-2 | PINK1 siRNA, PRKN siRNA and PINK1 or PRKN overexpression plasmids in HK-2 | 1. Protected against mitochondrial dysfunction 2. Protected against cell injury |
Activation of mitophagy protects against AKI | [89] | |
| Cisplatin | Intraperitoneal injection of cisplatin for rats | pink1-KO in rats | 1. PINK1 deficiency inhibited DNM1L-mediated mitochondrial fission 2. PINK1 deficiency inhibited excessive mitophagy |
Excessive mitophagy aggravates AKI | [90] | |
| Contrast | Iohexol for HK-2 and iohexol administration for mice | PINK1 siRNA or PRKN siRNA in cells; pink1-KO or prkn-KO in mice | 1. Reduce mitochondrial ROS 2. Reduce NLRP3 inflammasome activation |
Activation of mitophagy protects against AKI | [91] | |
| Contrast | Ioversol for HK-2 and ioversol for rats | - | 1. Reduce oxidative stress 2. Reduce mitochondrial damage |
Activation of mitophagy protects against AKI | [94] | |
| PRKN-independent | IRI | OGD-R for BUMPT cells and IRI for mice | Bnip3 silence in cells; bnip3-KO in mice | 1. Eliminate damaged mitochondria 2. Modulate the removal of ROS 3. Modulate cell death 4. Relieve inflammatory response |
Activation of mitophagy protects against AKI | [64] |
| IRI | IRI for rats | Mdivi-1, an inhibitor of DNM1L, in rats | 1. Mitophagic clearance of damaged mitochondria 2. Protects cells apoptosis. |
Activation of mitophagy protects against AKI | [73] | |
| IRI | IRI for mice | cnp-KO in mice. | Aggressive removal of injured mitochondria | Activation of mitophagy protects against AKI | [74] | |
| IRI | H/R for HK-2 cells and IRI for mice | HIF1A siRNA, BNIP3 siRNA, and BNIP3-overexpression plasmid for cell; hif1a-CKO and bnip3-overexpression adenovirus in mice | 1. Inhibited apoptosis 2. Inhibited ROS production |
Activation of mitophagy protects against AKI | [75] | |
| IRI | IRI for mice and rotenone for primary tubule cells | fundc1-PTKO, dnm1l-PTKO, ulk1-PTKO and fundc1-dnm1l-PTKO in mice | 1. Mitochondrial quality control 2. Reduce ROS oxidative stress 3. Reduce mitochondrial apoptosis |
Activation of mitophagy protects against AKI | [76] | |
| Contrast | Iohexol for HK-2 and iohexol administration for mice | BNIP3 siRNA in cells; bnip3-KO in mice | Reduce cells apoptosis | Activation of mitophagy protects against AKI | [93] |
Abbreviations: AKI: acute kidney injury; BNIP3: BCL2 interacting protein 3; BUMPT cells: Boston University mouse proximal tubule cells; CCCP: carbonyl cyanide m-chlorophenylhydrazone; CKO: conditional knockout; CLP: cecal ligation and puncture; CNP: 2’,3’-cyclic nucleotide 3’phosphodiesterase; DNM1L: dynamin 1 like; FUNDC1: FUN14 domain containing 1; H/R: hypoxia/reoxygenation; HIF1A: hypoxia inducible factor 1 subunit alpha; HK-2: human proximal tubular cells; IRI: ischemia-reperfusion injury; KO: knockout; LPS: lipopolysaccharide; MEG3: maternally expressed 3; NLRP3: NLR family pyrin domain containing 3; OGD-R: oxygen-glucose deprivation-reperfusion; OPTN: optineurin; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; PTKO: proximal tubule-specific knockout; ROS: reactive oxygen species; RPTC: rat proximal tubular cells; RTEC: renal tubular epithelial cell; RTKN: rhotekin; ULK1: unc-51 like autophagy activating kinase 1
Mitophagy in IR injury
The pathophysiology of renal ischemia-reperfusion injury (IRI) appears to be characterized by a complex cascade of oxidative damage contributing to tubular injury with ultrastructural changes in mitochondria [69,70]. Many studies have reported the role of mitophagy during IRI-induced AKI. The beneficial effect of mitophagy during renal IR was revealed by bnip3 and pink1 knockout mice. BNIP3 has dual functions of regulating cell death and mitophagy [71]. Tang et al. demonstrated that bnip3 deletion have reduced mitophagy resulting in the accumulation of damaged mitochondria, increasing production of reactive oxygen species, cell death and inflammation in oxygen-glucose deprivation-reperfusion (OGD-R) and IRI model [64]. Tang and his co-authors proved that PINK1-PRKN-mediated mitophagy plays an important role in mitochondrial quality control, tubular cell survival, and renal function in both in vitro and in vivo models of ischemic AKI [65]. Knockdown of pink1 suppressed mitophagy and reduced the cytoprotective effect of ischemic preconditioning (IPC) when treated carbonyl cyanide m-chlorophenylhydrazone (CCCP) in the rat proximal tubular cells (RPTC) cell and mice induced IRI model, which suggesting that mitophagy plays an important role in the protective effect of IPC [72]. DNM1L/DRP1 (dynamin 1 like) translocated to mitochondria and was phosphorylated at S616 in response to IRI [73]. Inhibiting DNM1L phosphorylation significantly suppressed without affecting general autophagy suggesting that DNM1L was involved the process of mitochondrial fragmentation and downregulation of mitophagy significantly aggravated kidney dysfunction indicating that mitophagy was activated via DNM1L-dependent pathway to protect cells from IRI-induced apoptosis [73]. Similar results were obtained with cnp 2’,3’-cyclic nucleotide 3’-phosphodiesterase) deletion mice which attenuates IRI-induced AKI, in part by accelerating mitophagy with targeted removal of damaged mitochondria [74]. Fu et al reported that BNIP3 overexpression reversed the inhibitory effect of HIF1A (hypoxia inducible factor 1 subunit alpha) knockout on mitophagy and prevented enhanced kidney damage in vivo and vitro [75]. Wang and his co-authors demonstrated that FUNDC1-dependent mitophagy, primarily driven by IPC, confers resistance to AKI through reconciliation of mitochondrial fission in a rotenone treated model in vitro and IRI model in vivo [76]. However, Zhou et al reported that activation of mitophagy also aggravates kidney ischemia-reperfusion injury by trigger the WNT-CTNNB1/beta-catenin pathway in vitro and vivo [66].
Mitophagy in response to sepsis
Sepsis is defined as organ dysfunction resulting from the host’s deleterious response to infection and one of the most common organs affected is the kidney [77]. Sepsis and inflammation at the tissue and cellular level are associated with decreased levels of intracellular ATP and with mitochondrial injury in various organs, including the kidney [78]. Ultrastructural changes are observed in mitochondria during sepsis-induced AKI including decreased mitochondrial mass, disruption of cristae, and extensive mitochondrial swelling [79]. Sepsis also induces profound alterations in kidney tubular epithelial cell metabolism and in peritubular capillary flow distribution, all of which can result in significant energy imbalance and further mitochondrial injury [80]. Importantly, mitophagy is activated early in the course of sepsis, providing the cell with a mechanism to remove damaged mitochondria which is important to minimize cell injury and accelerate recovery [78]. Several studies demonstrated that PRKN-dependent pathway of mitophagy was induced in in mouse models of septic AKI induced by lipopolysaccharide (LPS) treatment or by cecal ligation and puncture or in cultured proximal tubular epithelial cells exposed to LPS, which plays an important role in mitochondrial quality control, tubular cell survival, and renal function in septic AKI [81–84]. These protective effect of mitophagy in sepsis induced AKI could be attributed to a decreasing inflammation [85].
Mitophagy in response to cisplatin
Cisplatin is a widely used chemotherapeutic drug with notorious toxicity to the kidneys, which involves mitochondrial dysfunction and damage in renal tubular cells. Mitophagy induction and its cytoprotective role have been demonstrated both in vitro and in vivo in models of cisplatin-induced AKI [86–88]. Both pink1 and prkn knockout mice showed more severe renal functional loss, tissue damage, and apoptosis during cisplatin treatment [87]. In vitro, knockdown of PINK1-PRKN induced the aggravation of mitochondrial function, leading to the increase of cell injury, while the overexpression of PINK1-PRKN shown the contrary phenomenon suggesting mitophagy plays a cytoprotective role against cisplatin injury [89]. However, Liu et al reported that pink1 deficiency ameliorated cisplatin-induced AKI in rats, possibly via inhibiting DNM1L-mediated mitochondrial fission and excessive mitophagy [90].
Mitophagy in response to contrast
Iodinated intravascular contrast is widely used for vessel and chamber imaging in coronary angiography and percutaneous intervention. Despite advancements in imaging and interventional techniques, contrast-induced acute kidney injury (CI-AKI) occurs in more than 30% of patients after intravenous iodinated [91,92]. Mitophagy has been shown to play an important role in CI-AKI. Lin et al reported that PINK1-PRKN-mediated mitophagy prevented apoptosis and tissue damage in CI-AKI in vitro and vivo through reducing mitochondrial ROS and subsequent NLRP3 (NLR family, pyrin domain containing 3) inflammasome activation [91]. BNIP3-mediated mitophagy also has the protective function in CI-AKI [93]. Some drugs have the protective effect in CI-AKI by regulating mitophagy. Bae et al reported that paricalcitol, an active vitamin D analogue, could play a renoprotective role against CI-AKI through the PRKN-dependent pathway [94]. Tetramethylpyrazine can be protective against CI-AKI because it can ameliorate renal oxidative stress and aberrant mitochondrial dynamics, and modulate mitophagy in tubular cells in vitro and vivo [95]. Ward et al showed that the radiocontrast agent diatrizoic acid could induce mitophagy and oxidative stress via calcium dysregulation, however, the role of the mitophagy in this process has not been further investigated [96].
Mitophagy in the other drug-induced AKI
Many drugs can induce AKI, including some that are classically used as models of AKI such as folic acid (FA) and oxalate. The role of mitophagy in the pathobiology of AKI from these drugs is poorly understood. However, previous studies indicated that mitochondria play a critical role in their cytotoxicity to kidney. Zhang et al reported that inhibition of mitochondrial complex I activity aggravated renal tubular injury, mitochondrial damage and oxidative stress in FA-induced AKI [97]. This cytotoxicity effect of FA may due to trigger a mitophagy dependent ROS increase leading to cell death [39]. Mitophagy interfaces with mitochondrial biogenesis to regulate mitochondrial content and longevity [98]. During the FA-induced kidney injury, the mitochondrial biogenesis was suppressed and mitochondrial dysfunction is persistent to promote the progression of kidney injury [99]. Acute oxalosis displayed calcium oxalate crystals inside distal tubular epithelial cells caused AKI associated with mitochondrial changes characteristic of mitochondrial permeability transition [100], which can finally lead to mitophagy [101].
Interplay of mitophagy, apoptosis, pyroptosis and ferroptosis in AKI
Mitophagy is mainly thought of as a survival mechanism by removing damaged mitochondria and related ROS from these damaged mitochondria. In addition to elimination of damaged mitochondria, mitophagy prevents cell death by regulating genes associated with programmed cell death [32], which is a general mechanism of interaction between mitophagy and various cell death forms.
Both mitophagy and apoptosis are generally induced in response to a common stimulus. The crosstalk between mitophagy and apoptosis is mediated by several key molecules including members of the BCL2 (BCL2 apoptosis regulator) family and its interacting protein like BNIP3 and FUNDC1, and other mitophagy-related proteins. BCL2 family proteins regulate apoptosis by controlling the assembly of multimeric BAX (BCL2 associated X, apoptosis regulator)-BAK1 (BCL2 antagonist/killer 1) channels and thereby, modulating the permeability of the OMM [102]. BCL2 regulates macroautophagy through binding to the autophagy regulator BECN1 by interacting with its BH3-only domain and blocking its participation in the triggering of autophagosome formation. BCL2 family proteins have also been proved to inhibit PINK1-PRKN-dependent mitophagy [103]. Some BCL2-interacting proteins like BNIP3 and FUNDC1 that disrupt interaction between the BECN1 and antiapoptotic BCL2 family to promote positive regulation of mitophagy [104,105]. In a renal IRI model, BNIP3 in kidney cell induced light chain 3 expression and formation of autophagosomes which were mainly localized to the mitochondria, suggesting that mitophagy is induced in renal tubules by BNIP3, which may be activated to protect the renal tubules during IRI-AKI [106]. BCL2 binding to BECN1 is known to inhibit autophagy [107]. Disruption of BECN1-BCL2 interactions can enhance of autophagy activity to protect kidney from ischemia-reperfusion injury [108]. BCL2 family proteins inhibits apoptosis and autophagy in kidney injury has been largely investigated [109–111]. BNIP3, inhibit mitophagy, and enhancing apoptosis and ROS production during IRI [75].
Pyroptosis is a pro-inflammatory form of regulated cell death and is dependent on the enzymatic activity of inflammatory proteases that belong to the family of cysteine-dependent aspartate-specific proteases (caspases) [112]. Mitochondrial components are recognized as danger-associated molecular patterns by cytosolic pattern recognition receptors such as NOD-like receptors family member of NLRP3 inflammasomes [113]. They process pro-CASP1 (caspase 1) to active CASP1, which cleaves pro-inflammatory IL1B (interleukin 1 beta) to mature IL1B causing inflammation and cell death by pyroptosis. NOD-like receptor (NLR) family member with a mitochondrial targeting sequence, contains a LIR and binding of its LIR motif to LC3 induce mitophagy [114,115]. Another study demonstrated that NLRP3 activators induced mitochondrial damage, leading to their PRKN-dependent mitophagy [116]. Inhibition of NLRP3 inflammasome activation was dependent on PRKN-mediated mitophagy in sepsis-induced acute injury [84]. CASP1 inhibits mitophagy to amplify mitochondrial damage, mediated in part by cleavage of the key mitophagy regulator PRKN [117]. Mitochondria dysfunction induced NLRP3 activation. Both ROS generation and inflammasome activation are suppressed when mitochondrial activity is dysregulated by inhibition of the VDAC (voltage dependent anion channel) [118].These studies indicated that mitophagy removed damage mitochondria caused by pyroptosis is a compensatory mechanism to the pyroptotic cell death through the interplay protein of NLRP3 and CASP1.
Ferroptosis is a form of cell death that results from the catastrophic accumulation of lipid ROS caused by inactive GPX4 and the accumulation of iron [119]. Erastin, a special ferroptosis inducer, targets cystine/glutamate antiporter SLC7A11/xCT (solute carrier family 7 member 11), to inhibit extracellular cystine and intracellular glutamate exchange [120]. Cystine is a source of the synthesis of the glutathione (GSH) which is a cofactor of the GPX4 [119]. Thus, on the one hand, erastin targets the SLC7A11 receptor on membrane to influence the activity of GPX4 to induce ferroptosis. On the other hand, it can also activate VDAC receptor located at mitochondrial outer membrane to increase Ca2+ transport which controls energy production and metabolism by modulating critical enzymes in the TCA cycle [121]. Besides, glutamate can transfer to α-Ketoglutarate through alanine aminotransferase which is involved in TCA cycle [122]. Thus, mitochondria may play an important role in the crosstalk between mitophagy and ferroptosis.
Increasing evidence shows that mitochondrial dysfunction has an important role in ferroptosis [123–125]. GSK3B (glycogen synthase kinase 3 beta) resides at the nexus of multiple signaling pathways implicated in the regulation of mitochondrial permeability transition (MPT) which finally caused mitochondrial clearance though mitophagy [39,126]. GSK3B increased VDAC phosphorylation, key MPT regulators located at OMM, and sensitized cells to MPT during drug-induced oxidant stress kidney injury [127]. VDAC is an important protein in the crosstalk between mitophagy and ferroptosis, and serves as a mitochondrial docking site to recruit PRKN from the cytosol to defective mitochondria to induce mitophagy [128]. The translocator protein interacts with VDAC1 which plays an important role in PINK1-PRKN-dependent mitophagy [129]. This procedure may be enrolled during the kidney injury. VDAC was reported as the only channel‐forming protein and allowed the metabolites and irons across the outer membrane, erastin-induced VDAC opening mediated mitochondrial iron uptake may accelerate ferroptosis [130]. MitoNEET, also referred to as CISD1 (CDGSH iron sulfur domain 1), a redox-sensitive (2Fe-2S) cluster protein, is an OMM protein essential for sensing and regulation of iron and ROS homeostasis [131]. CISD gates VDAC when oxidized in a redox-dependent manner in cells, closing the pore and likely disrupting flow of iron [132]. Mitochondrial TCA plays an important role in mitophagy and ferroptosis. The flux of respiratory substrates, ADP, and Pi into mitochondria and the release of mitochondrial ATP to the cytosol occur through voltage-dependent anion channels [39]. Thus, inhibition VDAC caused mitochondrial TCA cycle inhibition which mitigate mitochondrial membrane potential hyperpolarization, lipid peroxide accumulation and triggers a mitophagy-dependent ROS increase leading to ferroptosis (summarized in Figure 4) [39,133,134].
Figure 4.
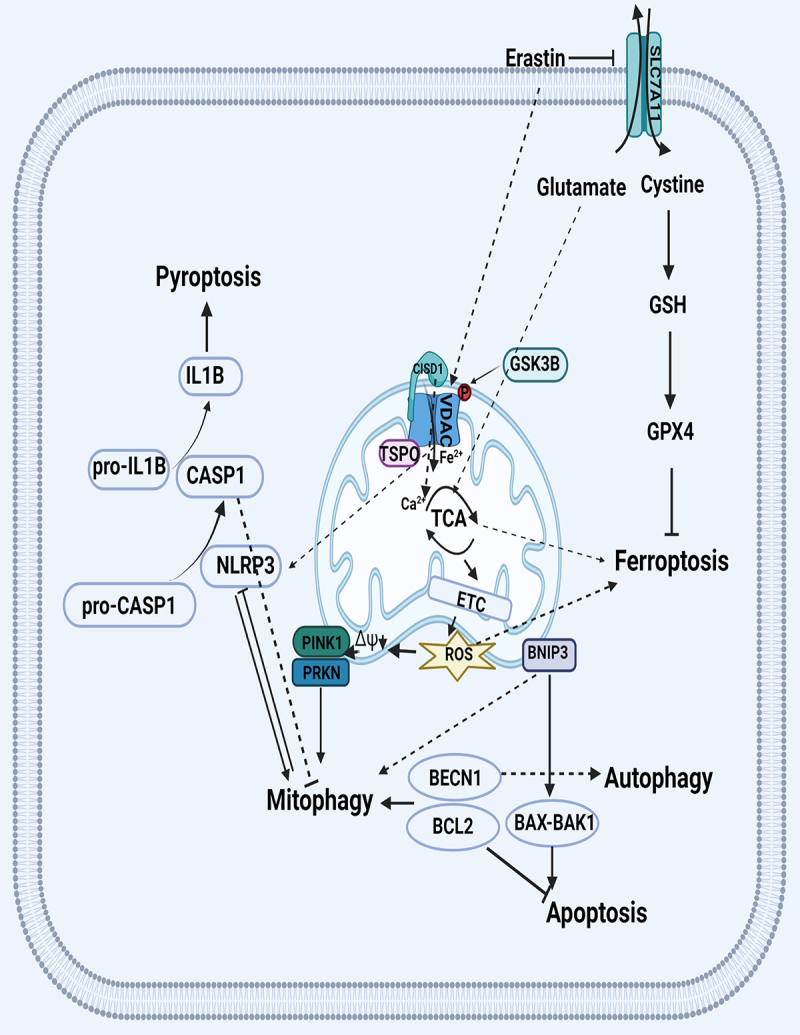
Crosstalk between mitophagy and other type of cell death. VDAC regulates mitochondria iron homeostasis through CISD1 which may affect the iron-dependent cell death of ferroptosis. Translocator protein interacts with VDAC in PINK-PRKN-dependent mitophagy. VDAC gate the Ca2+ transport to control energy production and metabolism by modulating enzyme activity of TCA which impacts the production of mitochondria ROS in the electron transport chain. GSK3B increased VDAC phosphorylation to control MPT. SLC7A11/xCT regulates extracellular cystine and intracellular glutamate exchanging which influence the TCA and GSH metabolism through glutamate and cystine, respectively. BCL2 family proteins regulate apoptosis through controlling the assembly of multimeric BAX-BAK1 channels. The interaction of BECN1-BCL2 inhibits autophagy and mitophagy but increase apoptosis. BCL2 family proteins, like BNIP3, can disrupt interaction between the BECN1 and antiapoptotic BCL2 family. NLRP3 process pro-CASP1 to active CASP1, which cleaves pro-inflammatory IL1B to mature IL1B causing pyroptosis. NLRP3 binding of its LIR motif to LC3 induce mitophagy while CASP1 inhibits mitophagy to amplify mitochondrial damage. VDAC activates NLRP3 to induce pyroptosis. Abbreviations: BAK1: BCL2 antagonist/killer 1; BAX: BCL2 associated X, apoptosis regulator; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; CASP1: caspase 1; CISD1: CDGSH iron sulfur domain 1; GPX: glutathione peroxidase; GSH: glutathione; GSK3B: glycogen synthase kinase 3 beta; IL1B: interleukin 1 beta; LIR: LC3-interacting region; MPT: mitochondrial permeability transition; NLRP3: NLR family pyrin domain containing 3; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; SLC7A11/xCT: solute carrier family 7 member 11; TCA: tricarboxylic acid; VDAC: voltage dependent anion channel.
Conclusion
Mitophagy is a key cellular homeostatic mechanism that is activated early during AKI. The effect of mitophagy and the relationship between mtROS levels and mitophagy are perhaps more complicated. During AKI, mitophagy is activated early through PRKN-dependent and independent signaling pathways. The activation of mitophagy is protective in this context, removing dysfunctional mitochondria from TEC and decreasing thereby local inflammation and oxidative damage. The crosstalk between mitophagy and forms of cell death such as apoptosis and pyroptosis is an important contributor to some forms of AKI, and emerging evidence suggests an important link between iron metabolism, mitophagy and a novel form of cell death known as ferroptosis. Further study could explore and validate the interplay of mitophagy and ferroptosis.
Acknowledgments
Not applicable.
Funding Statement
This work was supported by the National Natural Science Foundation of China [82102298]; National Natural Science Foundation of China [81772046]; National Natural Science Foundation of China [81971816]; Innovation Cultivation Foundation of Wuhan University/Zhongnan Hospital [413000345/CXPY2020017].
Disclosure statement
None of the authors declared any conflict of interest in this work.
Data availability statement
The data supporting the findings of this study are available within the article [and/or] its supplementary materials.
References
- [1].Yang X, Zhang R, Nakahira K, et al. Mitochondrial DNA mutation, diseases, and nutrient-regulated mitophagy. Annu Rev Nutr. 2019;39(1):201–226. 10.1146/annurev-nutr-082018-124643 [DOI] [PubMed] [Google Scholar]
- [2].VanderVeen BN, Fix DK, Carson JA.. Disrupted skeletal muscle Mitochondrial dynamics, Mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid Med Cell Longev. 2017;2017:3292087. 2017. 10.1155/2017/3292087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Xiong H, Chen S, Lai L, et al. Modulation of miR-34a/SIRT1 signaling protects cochlear hair cells against oxidative stress and delays age-related hearing loss through coordinated regulation of mitophagy and mitochondrial biogenesis. Neurobiol Aging. 2019;79:30–42. [DOI] [PubMed] [Google Scholar]
- [4].Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int. 2016;89(4):779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Song SB, Jang SY, Kang HT, et al. Modulation of Mitochondrial membrane potential and ROS generation by nicotinamide in a manner independent of SIRT1 and Mitophagy. Mol Cells. 2017;40(7):503–514. 10.14348/molcells.2017.0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Choi ME. Autophagy in kidney disease. Annu Rev Physiol. 2019;82:297–322. [DOI] [PubMed] [Google Scholar]
- [7].Fivenson EM, Lautrup S, Sun N, et al. Mitophagy in neurodegeneration and aging. Neurochem Int. 2017;109:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gong G, Song M, Csordas G, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350(6265):aad2459. 10.1126/science.aad2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cristofano MD, F A, Giacomo MD, et al. Mechanisms underlying the hormetic effect of conjugated linoleic acid: focus on Nrf2, mitochondria and NADPH oxidases. Free Radic Biol Med. 2021;167:276–286. [DOI] [PubMed] [Google Scholar]
- [10].Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cadonic C, Sabbir MG, Albensi BC. Mechanisms of Mitochondrial dysfunction in Alzheimer’s disease. Mol Neurobiol. 2016;53(9):6078–6090. [DOI] [PubMed] [Google Scholar]
- [12].Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020;37:101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sumegi K, Fekete K, Antus C, et al. BGP-15 Protects against oxidative stress- or lipopolysaccharide-induced Mitochondrial destabilization and reduces mitochondrial production of reactive oxygen species. PLoS One. 2017;12(1):e0169372. : 10.1371/journal.pone.0169372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Iwasaki Y, Takeshima Y, Fujio K. Basic mechanism of immune system activation by mitochondria. Immunol Med. 2020;43(4):142–147. [DOI] [PubMed] [Google Scholar]
- [15].Nogueira NP, Saraiva FMS, Oliveira MP, et al. Heme modulates trypanosoma cruzi bioenergetics inducing mitochondrial ROS production. Free Radic Biol Med. 2017;108:183–191. [DOI] [PubMed] [Google Scholar]
- [16].Dong D, Hao Q, Zhang P, et al. Endoplasmic reticulum Ca2+ release causes rieske iron-sulfur protein-mediated mitochondrial ROS generation in pulmonary artery smooth muscle cells. Biosci Rep. 2019;39(12). 10.1042/BSR20192414 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [17].Rudolf J, Raad H, Taieb A, et al. NADPH oxidases and their roles in skin homeostasis and carcinogenesis. Antioxid Redox Signal. 2018;28(13):1238–1261. 10.1089/ars.2017.7282 [DOI] [PubMed] [Google Scholar]
- [18].Hahner F, Moll F, Schroder K. NADPH oxidases in the differentiation of endothelial cells. Cardiovasc Res. 2020;116(2):262–268. [DOI] [PubMed] [Google Scholar]
- [19].Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64(5):362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71(14):2577–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].SP A, AJ M, J R. Mammalian NADH:ubiquinone oxidoreductase (complex I) and nicotinamide nucleotide transhydrogenase (Nnt) together regulate the mitochondrial production of H(2)O(2)–implications for their role in disease,especially cancer. J Bioenerg Biomembr. 2011;43(5):541–564. [DOI] [PubMed] [Google Scholar]
- [22].Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15(1):2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Glancy B, Kane DA, Kavazis AN, et al. Mitochondrial lactate metabolism: history and implications for exercise and disease. J Physiol. 2021;599(3):863–888. 10.1113/JP278930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McKenna MC, Waagepetersen HS, Schousboe A, et al. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol. 2006;71(4):399–407. 10.1016/j.bcp.2005.10.011 [DOI] [PubMed] [Google Scholar]
- [25].Bakker BM, Overkamp KM, van Maris AJ, et al. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2001;25(1):15–37. 10.1111/j.1574-6976.2001.tb00570.x [DOI] [PubMed] [Google Scholar]
- [26].Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. [DOI] [PubMed] [Google Scholar]
- [27].Zhang Y, Murugesan P, Huang K, et al. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. 2020;17(3):170–194. 10.1038/s41569-019-0260-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Czarna M, Jarmuszkiewicz W. Role of mitochondria in reactive oxygen species generation and removal; relevance to signaling and programmed cell death. Postepy Biochem. 2006;52(2):145–156. [PubMed] [Google Scholar]
- [29].Ismail T, Kim Y, Lee H, et al. Interplay between Mitochondrial peroxiredoxins and ROS in cancer development and progression. Int J Mol Sci. 2019;20(18):4407. 10.3390/ijms20184407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Suski JM, Lebiedzinska M, Bonora M, et al. Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 2012;810:183–205. [DOI] [PubMed] [Google Scholar]
- [31].Evans CS, Holzbaur EL. Degradation of engulfed mitochondria is rate-limiting in optineurin-mediated mitophagy in neurons. Elife. 2020;9. 10.7554/eLife.50260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ma K, Chen G, Li W, et al. Mitophagy, Mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. 2020;8:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dan X, Babbar M, Moore A, et al. DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res. 2020;48(12):6611–6623. 10.1093/nar/gkaa393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nielson JR, Rutter JP. Lipid-mediated signals that regulate mitochondrial biology. J Biol Chem. 2018;293(20):7517–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bocking T, Barrow KD, Netting AG, et al. Effects of singlet oxygen on membrane sterols in the yeast Saccharomyces cerevisiae. Eur J Biochem. 2000;267(6):1607–1618. 10.1046/j.1432-1327.2000.01179.x [DOI] [PubMed] [Google Scholar]
- [37].Heo JM, Nielson JR, Dephoure N, et al. Intramolecular interactions control Vms1 translocation to damaged mitochondria. Mol Biol Cell. 2013;24(9):1263–1273. 10.1091/mbc.e13-02-0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2016;311(2):L433–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Basit F, van Oppen LM, Schockel L, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8(3):e2716. 10.1038/cddis.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schofield JH, Schafer ZT. Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid Redox Signal. 2021;34(7):517–530. [DOI] [PubMed] [Google Scholar]
- [41].Villa E, Marchetti S, Ricci JE. No parkin zone: mitophagy without Parkin. Trends Cell Biol. 2018;28(11):882–895. [DOI] [PubMed] [Google Scholar]
- [42].Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- [43].Takatori S, Ito G, Iwatsubo T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett. 2008;430(1):13–17. [DOI] [PubMed] [Google Scholar]
- [44].Zhang C, Wang R, Liu Z, et al. The plant triterpenoid celastrol blocks PINK1-dependent mitophagy by disrupting PINK1’s association with the mitochondrial protein TOM20. J Biol Chem. 2019;294(18):7472–7487. 10.1074/jbc.RA118.006506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jin SM, Lazarou M, Wang C, et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191(5):933–942. 10.1083/jcb.201008084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Deas E, Plun-Favreau H, Gandhi S, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet. 2011;20(5):867–879. 10.1093/hmg/ddq526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lazarou M, Jin SM, Kane LA, et al. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22(2):320–333. 10.1016/j.devcel.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Okatsu K, Uno M, Koyano F, et al. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem. 2013;288(51):36372–36384. 10.1074/jbc.M113.509653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–383. 10.1073/pnas.0911187107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shiba-Fukushima K, Inoshita T, Hattori N, et al. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014;10(6):e1004391. 10.1371/journal.pgen.1004391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu L, Sakakibara K, Chen Q, et al. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7):787–795. 10.1038/cr.2014.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chu CT. Multiple pathways for mitophagy: a neurodegenerative conundrum for Parkinson’s disease. Neurosci Lett. 2019;697:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wei Y, Chiang WC, Sumpter R Jr., et al. Prohibitin 2 is an inner Mitochondrial membrane mitophagy receptor. Cell. 2017;168(1–2):224–238 e210. 10.1016/j.cell.2016.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Strappazzon F, Nazio F, Corrado M, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22(3):419–432. 10.1038/cdd.2014.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Pangborn MC. A simplified preparation of cardiolipin, with note on purification of lecithin for serologic use. J Biol Chem. 1945;161(1):71–82. [PubMed] [Google Scholar]
- [57].Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol. 2017;5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Huang W, Choi W, Hu W, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22(3):473–489. 10.1038/cr.2012.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Puri R, Cheng XT, Lin MY, et al. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat Commun. 2019;10(1):3645. 10.1038/s41467-019-11636-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yun J, Puri R, Yang H, et al. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014;3:e01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ambivero CT, Cilenti L, Main S, et al. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal. 2014;26(12):2921–2929. 10.1016/j.cellsig.2014.09.004 [DOI] [PubMed] [Google Scholar]
- [62].Di Rita A, Peschiaroli A, DA P, et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat Commun. 2018;9(1):3755. 10.1038/s41467-018-05722-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Duann P, Lin PH. Mitochondria damage and kidney disease. Adv Exp Med Biol. 2017;982:529–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tang C, Han H, Liu Z, et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019;10(9):677. 10.1038/s41419-019-1899-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tang C, Han H, Yan M, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14(5):880–897. 10.1080/15548627.2017.1405880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu D, Liu Y, Zheng X, et al. c-MYC-induced long noncoding RNA MEG3 aggravates kidney ischemia-reperfusion injury through activating mitophagy by upregulation of RTKN to trigger the Wnt/beta-catenin pathway. Cell Death Dis. 2021;12(2):191. 10.1038/s41419-021-03466-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tang C, Cai J, Yin XM, et al. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol. 2021;17(5):299–318. 10.1038/s41581-020-00369-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tang C, Livingston MJ, Liu Z, et al. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol. 2020;16(9):489–508. 10.1038/s41581-020-0309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].de Braganca Ac, Volpini RA, Mehrotra P, et al. Vitamin D deficiency contributes to vascular damage in sustained ischemic acute kidney injury. Physiol Rep. 2016;4(13). 10.14814/phy2.12829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hall AM, Rhodes GJ, Sandoval RM, et al. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013;83(1):72–83. 10.1038/ki.2012.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lampert MA, Orogo AM, Najor RH, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15(7):1182–1198. 10.1080/15548627.2019.1580095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Livingston MJ, Wang J, Zhou J, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019;15(12):2142–2162. 10.1080/15548627.2019.1615822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Li N, Wang H, Jiang C, et al. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp Cell Res. 2018;369(1):27–33. 10.1016/j.yexcr.2018.04.025 [DOI] [PubMed] [Google Scholar]
- [74].Jackson EK, Menshikova EV, Mi Z, et al. Renal 2’,3’-Cyclic Nucleotide 3’-phosphodiesterase is an important determinant of aki severity after ischemia-reperfusion. J Am Soc Nephrol. 2016;27(7):2069–2081. 10.1681/ASN.2015040397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Fu ZJ, Wang ZY, Xu L, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020;36:101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang J, Zhu P, Li R, et al. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hsiao HW, Tsai KL, Wang LF, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37(3):289–296. 10.1097/SHK.0b013e318240b52a [DOI] [PubMed] [Google Scholar]
- [79].Tran M, Tam D, Bardia A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121(10):4003–4014. 10.1172/JCI58662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gomez H, Kellum JA, Ronco C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat Rev Nephrol. 2017;13(3):143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang Y, Zhu J, Liu Z, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chen ZD, Hu BC, Shao XP, et al. Ascorbate uptake enables tubular mitophagy to prevent septic AKI by PINK1-PARK2 axis. Biochem Biophys Res Commun. 2021;554:158–165. [DOI] [PubMed] [Google Scholar]
- [83].Guo J, Wang R, Liu D. Bone marrow-derived mesenchymal stem cells ameliorate sepsis-induced acute kidney injury by promoting mitophagy of renal tubular epithelial cells via the SIRT1/Parkin axis. Front Endocrinol (Lausanne). 2021;12:639165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gao Y, Dai X, Li Y, et al. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J Transl Med. 2020;18(1):114. 10.1186/s12967-020-02283-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Harris J, Deen N, Zamani S, et al. Mitophagy and the release of inflammatory cytokines. Mitochondrion. 2018;41:2–8. [DOI] [PubMed] [Google Scholar]
- [86].Ichinomiya M, Shimada A, Ohta N, et al. Demonstration of Mitochondrial damage and mitophagy in cisplatin-mediated nephrotoxicity. Tohoku J Exp Med. 2018;246(1):1–8. 10.1620/tjem.246.1 [DOI] [PubMed] [Google Scholar]
- [87].Wang Y, Tang C, Cai J, et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018;9(11):1113. 10.1038/s41419-018-1152-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Qi J, Xue Q, Kuang L, et al. Berberine alleviates cisplatin-induced acute kidney injury by regulating mitophagy via PINK 1/Parkin pathway. Transl Androl Urol. 2020;9(4):1712–1724. 10.21037/tau-20-1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhao C, Chen Z, Xu X, et al. Pink1/Parkin-mediated mitophagy play a protective role in cisplatin induced renal tubular epithelial cells injury. Exp Cell Res. 2017;350(2):390–397. 10.1016/j.yexcr.2016.12.015 [DOI] [PubMed] [Google Scholar]
- [90].Zhou L, Zhang L, Zhang Y, et al. PINK1 deficiency ameliorates cisplatin-induced acute kidney injury in rats. Front Physiol. 2019;10:1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lin Q, Li S, Jiang N, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].McCullough PA, Choi JP, Feghali GA, et al. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2016;68(13):1465–1473. 10.1016/j.jacc.2016.05.099 [DOI] [PubMed] [Google Scholar]
- [93].Lin Q, Li S, Jiang N, et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. 2020;16(1):1–16. 10.1080/15548627.2019.1665293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bae E, Kim JH, Jung MH, et al. Paricalcitol attenuates contrast-induced acute kidney injury by regulating mitophagy and senescence. Oxid Med Cell Longev. 2020;2020:7627934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gong X, Duan Y, Zheng J, et al. Tetramethylpyrazine prevents contrast-induced nephropathy via modulating tubular cell mitophagy and suppressing Mitochondrial fragmentation, CCL2/CCR2-mediated inflammation, and intestinal injury. Oxid Med Cell Longev. 2019;2019:7096912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ward DB, Brown KC, Valentovic MA. Radiocontrast agent diatrizoic acid induces mitophagy and oxidative stress via calcium dysregulation. Int J Mol Sci. 2019;20(17). 10.3390/ijms20174074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zhang W, Yang Y, Gao H, et al. Inhibition of Mitochondrial complex I aggravates folic acid-induced acute kidney injury. Kidney Blood Press Res. 2019;44(5):1002–1013. 10.1159/000501934 [DOI] [PubMed] [Google Scholar]
- [98].Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521(7553):525–528. [DOI] [PubMed] [Google Scholar]
- [99].Stallons LJ, Whitaker RM, Schnellmann RG. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol Lett. 2014;224(3):326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mulay SR, Honarpisheh MM, Foresto-neto O, et al. mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J Am Soc Nephrol. 2019;30(10):1857–1869. 10.1681/ASN.2018121218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rodriguez-Enriquez S, Kai Y, Maldonado E, et al. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy. 2009;5(8):1099–1106. 10.4161/auto.5.8.9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399(6735):483–487. [DOI] [PubMed] [Google Scholar]
- [103].Hollville E, Carroll RG, Cullen SP, et al. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell. 2014;55(3):451–466. 10.1016/j.molcel.2014.06.001 [DOI] [PubMed] [Google Scholar]
- [104].Mazure NM, Pouyssegur J. Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy. 2009;5(6):868–869. [DOI] [PubMed] [Google Scholar]
- [105].Springer MZ, Macleod KF. In brief: mitophagy: mechanisms and role in human disease. J Pathol. 2016;240(3):253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ishihara M, Urushido M, Hamada K, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013;305(4):F495–509. 10.1152/ajprenal.00642.2012 [DOI] [PubMed] [Google Scholar]
- [107].Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. 10.1038/cdd.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Li P, Shi M, Maique J, et al. Beclin 1/Bcl-2 complex-dependent autophagy activity modulates renal susceptibility to ischemia-reperfusion injury and mediates renoprotection by Klotho. Am J Physiol Renal Physiol. 2020;318(3):F772–F792. 10.1152/ajprenal.00504.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Borkan SC. The role of bcl-2 family members in acute kidney injury. Semin Nephrol. 2016;36(3):237–250. [DOI] [PubMed] [Google Scholar]
- [110].Chien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation. 2007;84(9):1183–1190. [DOI] [PubMed] [Google Scholar]
- [111].Isaka Y, Suzuki C, Abe T, et al. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant Proc. 2009;41(1):52–54. 10.1016/j.transproceed.2008.10.026 [DOI] [PubMed] [Google Scholar]
- [112].Xu YJ, Zheng L, Hu YW, et al. Pyroptosis and its relationship to atherosclerosis. Clin Chim Acta. 2018;476:28–37. [DOI] [PubMed] [Google Scholar]
- [113].Singh LP, Devi TS, Yumnamcha T. The role of txnip in mitophagy dysregulation and inflammasome activation in diabetic retinopathy: a new perspective. JOJ Ophthalmol. 2017;4(4). 10.19080/JOJO.2017.04.555643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Zhang Y, Yao Y, Qiu X, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019;20(4):433–446. 10.1038/s41590-019-0324-2 [DOI] [PubMed] [Google Scholar]
- [115].Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol. 2019;10:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. 10.1016/j.cell.2015.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yu J, Nagasu H, Murakami T, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111(43):15514–15519. 10.1073/pnas.1414859111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- [119].Su LJ, Zhang JH, Gomez H, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. 10.1038/cdd.2015.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].V S-B, De S MVDAC. the Na(+)/Ca(2+) exchanger, and the Ca(2+) uniporter in ca(2+) dynamics and signaling. Adv Exp Med Biol. 2017;981:323–347. [DOI] [PubMed] [Google Scholar]
- [122].Quan J, Fitch MD, Fleming SE. Rate at which glutamine enters TCA cycle influences carbon atom fate in intestinal epithelial cells. Am J Physiol. 1998;275(6):G1299–1308. [DOI] [PubMed] [Google Scholar]
- [123].Neitemeier S, Jelinek A, Laino V, et al. Bid links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017;12:558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Abdalkader M, Lampinen R, Kanninen KM, et al. Targeting Nrf2 to suppress ferroptosis and Mitochondrial dysfunction in neurodegeneration. Front Neurosci. 2018;12:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Battaglia AM, Chirillo R, Aversa I, et al. Ferroptosis and cancer: Mitochondria meet the “Iron Maiden” cell death. Cells. 2020;9(6):1505. 10.3390/cells9061505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Tanaka T, Saotome M, Katoh H, et al. Glycogen synthase kinase-3beta opens mitochondrial permeability transition pore through mitochondrial hexokinase II dissociation. J Physiol Sci. 2018;68(6):865–871. 10.1007/s12576-018-0611-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Wang Z, Ge Y, Bao H, et al. Redox-sensitive glycogen synthase kinase 3beta-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic Biol Med. 2013;65:849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Sun Y, Vashisht AA, Tchieu J, et al. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012;287(48):40652–40660. 10.1074/jbc.M112.419721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Moras M, Hattab C, Gonzalez-Menendez P, et al. Downregulation of Mitochondrial tspo inhibits mitophagy and reduces enucleation during human terminal erythropoiesis. Int J Mol Sci. 2020;21(23):9066. 10.3390/ijms21239066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Wang H, Liu C, Zhao Y, et al. Mitochondria regulation in ferroptosis. Eur J Cell Biol. 2020;99(1):151058. 10.1016/j.ejcb.2019.151058 [DOI] [PubMed] [Google Scholar]
- [131].Geldenhuys WJ, TC L, Carroll RT. Carroll RT. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov Today. 2014;19(10):1601–1606. [DOI] [PubMed] [Google Scholar]
- [132].Lipper CH, Stofleth JT, Bai F, et al. Redox-dependent gating of VDAC by mitoNEET. Proc Natl Acad Sci U S A. 2019;116(40):19924–19929. 10.1073/pnas.1908271116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].McElroy GS, Reczek CR, Reyfman PA, et al. NAD+ regeneration rescues lifespan, but not ataxia, in a mouse model of brain Mitochondrial complex i dysfunction. Cell Metab. 2020;32(2):301–308 e306. 10.1016/j.cmet.2020.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Gao M, Yi J, Zhu J, et al. Role of Mitochondria in ferroptosis. Mol Cell. 2019;73(2):354–363 e353. 10.1016/j.molcel.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of this study are available within the article [and/or] its supplementary materials.