

ABSTRACT
SQSTM1/p62 (sequestosome 1) is a well-established indicator of macroautophagic/autophagic flux. It was initially characterized as the ubiquitin-binding autophagic receptor in aggrephagy, the selective autophagy of ubiquitinated protein aggregates. Recently, several studies correlated its levels with the abundance of intracellular lipid droplets (LDs). In the absence of a bona fide receptor for the selective autophagy of LDs (lipophagy), a few studies demonstrated the role of SQSTM1 in lipophagy. Our analysis of these studies shows that SQSTM1 colocalizes with LDs, bridges them with phagophores, is co-degraded with them in the lysosomes, and affects LD abundance in a variety of cells and under diverse experimental conditions. Although only one study reported all these functions together, the overwhelming and complementary evidence from other studies suggests that the role of SQSTM1 in lipophagy via tagging, movement, aggregation/clustering and sequestration of LDs is rather a common phenomenon in mammalian cells. As ubiquitination of the LD-associated proteins under stress conditions is increasingly recognized as another common phenomenon, some other ubiquitin-binding autophagic receptors, such as NBR1 and OPTN, might soon join SQSTM1 on a list of the non-exclusive lipophagy receptors.
Abbreviations: LD: lipid droplet; LIR: LC3-interacting region; PAT: Perilipin, ADRP and TIP47 domain; SAR: selective autophagy receptor.
KEYWORDS: Lipid droplet, lipophagy, lipophagy receptor, p62, phagophore, SAR, selective autophagy, selective autophagy receptor, SQSTM1, ubiquitin
SQSTM1/p62 (sequestosome 1) is a macroautophagic/autophagic receptor that bridges ubiquitinated proteins and the growing autophagic membrane, the phagophore, via its ubiquitin-associated (UBA) domain and LC3-interacting region (LIR). SQSTM1 was originally found to mediate aggrephagy, the selective autophagic degradation of ubiquitinated protein aggregates [1,2]. It was the first example of receptor-mediated selective autophagy in mammals making SQSTM1 the first mammalian selective autophagy receptor (SAR). Later, SQSTM1 was found to oligomerize via its Phox and Bem1 (PB1) domain and result in aggregation of its substrates. The roles of the SQSTM1 receptor in autophagy and the ubiqutin-proteasome system have been reviewed [3,4]. Importantly, SQSTM1 itself is an autophagic substrate and accumulates when autophagy is blocked. Therefore, its levels, in combination with the levels of lipidated MAP1LC3B (microtubule associated protein 1 light chain 3 beta), which reports about the abundance of autophagosomes, can be used to measure autophagic flux [5]. Since 2009, SQSTM1 was used as an autophagic marker in numerous studies with some of them also showing the accumulation of both SQSTM1 and intracellular lipid droplets (LDs). Like SQSTM1, these lipid storage organelles can be degraded by selective autophagy (lipophagy [6]; reviewed in [7–12]) and not only by cytosolic lipolysis. Interestingly, some cytosolic lipases that act on LDs, such as PNPLA2/ATGL (patatin like phospholipase domain containing 2) and LIPE/HSL (lipase E, hormone sensitive type), have multiple putative LIRs [13] and are proposed to act as lipophagy receptors [14]. However, they bind mostly to the cytosolic (not lumenal) face of autophagosomes [13]. Moreover, PNPLA2 localizes to LDs in a LIR-dependent manner [13] suggesting that autophagic membranes mediate the recruitment of these cytosolic lipases to LDs and not vice versa. Therefore, a question of the LD-specific SAR remains open and of great interest to the field. Because a few recent studies suggested the involvement of SQSTM1 in lipophagy, we reviewed this one aspect of SQSTM1 function and evaluated a possibility of SQSTM1 acting as a non-exclusive lipophagy receptor.
To be considered a SAR, a protein should meet several requirements: 1) it must colocalize and physically interact with its substrate, 2) it has to bridge its substrate with the phagophore, 3) it must be degraded together with its substrate in the lysosome, and 4) lack or overexpression of this protein should result in accumulation or disappearance of its substrate in the cytosol, respectively. We applied these criteria to SQSTM1 in relation to LDs and summarized our findings in Table 1. Several studies observed colocalization of SQSTM1 with LDs in different models and under a variety of LD induction conditions [15–19]. The recruitment of SQSTM1 to LDs is mediated by the LD-associated protein, PLIN1 (perilipin 1) [17]. Because LDs are ubiquitinated under those conditions, it is not clear if PLIN1 recruits SQSTM1 directly or via ubiquitin. Interestingly, SQSTM1 co-immunoprecipitates with PLIN1 [20] and two other LD proteins, PLIN2/ADRP (perilipin 2) [15] and PLA2G4A/cPLA2-alpha (phospholipase A2 group IVA) [18]. However, the nature of these interactions was not clarified. Because ubiquitination of LDs is rather a common phenomenon in mammalian cells [17,19], these interactions might have been mediated by ubiquitin. Indeed, both PLIN1 and PLIN2 are polyubiquitinated and degraded by the proteasome in the delipidated culture medium [21,22]. Also, PLIN1 is polyubiquitinated [20] and degraded by the autophagy-lysosomal pathway induced with TNF (tumor necrosis factor) [20] or nelfinavir [23]. In addition, lipophagy in mammalian cells depends on HTT (huntingtin), the selective autophagy scaffold that binds SQSTM1 and promotes its attachment to (1) polyubiquitinated proteins with the K63-linked ubiquitin chains and (2) MAP1LC3B [24]. Therefore, it seems very likely that the SQSTM1-LD interactions are at least partially mediated by ubiquitin (Figure 1).
Table 1.
The summary of SQSTM1 and LD relations regarding the role of SQSTM1 as a lipophagy SAR.
| Model | LD inducer | SQSTM1 |
Ref | |||
|---|---|---|---|---|---|---|
| Does it colocalize with LDs? | Does it bridge LDs and phagophores? | Is it co-degraded with LDs in lysosomes? | Does it affect LD abundance? | |||
| Rat L6 myocytes | Oleate and palmitate | Yes, it colocalizes with LDs; also, it co-IPs* with PLIN2 | n/a | Yes (modulated by Rapa and BafA1) | n/a | [15] |
| Male Wistar rat livers | Ethanol | Yes, it cofractionates with LDs, especially on ethanol diet | Maybe; it cofractionates with MAP1LC3B on LDs | n/a | n/a | [16] |
| Mouse AML12 hepatocytes | Ethanol | Yes, it colocalizes with LDs via PLIN1; also, it colocalizes with PLIN1 and Ub on LDs | Yes, it is required (together with PLIN1) for LDs-MAP1LC3B colocalization | Yes (modulated by Rapa, CBZ, 3-MA, Wort, CQ and ATG5) | Yes, its KD leads to lipid accumulation | [17] |
| Human C13, HeyA8MDR and OVCAR5 cells | Increased p-PFKFB3 | Yes, it colocalizes with LDs and PLA2G4A; also, it co-IPs with PLA2G4A | n/a | Yes (modulated by PFK158 and BafA1) | n/a | [18] |
| Human THP-1 and mouse peritoneal or bone marrow-derived macrophages | agLDL or oleate | Yes, it colocalizes with LDs; Ub also colocalizes with LDs | Maybe; it colocalizes with MAP1LC3B on LDs | n/a | Maybe; its KD reduces cholesterol efflux | [19] |
| Human HepG2 cells | Oleate | n/a | Yes, its LIR is required for LD degradation | n/a | Yes, its OE promotes LD degradation | [25] |
| Human H4 cells | None | No, but it forms contact sites with LDs via DYNLRB1 and moves together with LDs | n/a | n/a | Yes, its OE promotes LD degradation | [27] |
*3-MA: 3-methyladenine; agLDL: aggregated low-density lipoprotein; BafA1: bafilomycin A1; CBZ: carbamazepine; co-IP: co-immunoprecipitate; CQ: chloroquine; KD: knockdown; OE: overexpression; Rapa: rapamycin; Ub: ubiquitin; Wort: wortmannin.
Figure 1.
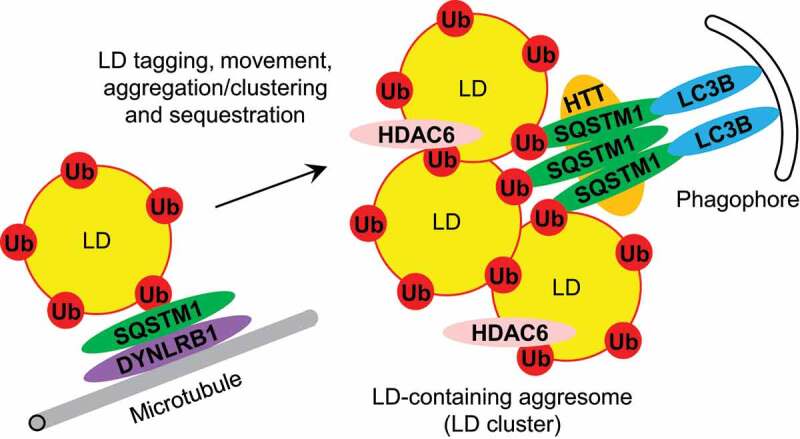
The role of SQSTM1 in lipophagy via tagging, movement, aggregation/clustering and sequestration of LDs. See text for details. LC3B: MAP1LC3B; Ub: ubiquitin.
SQSTM1 also satisfies the second requirement of a lipophagy SAR by bridging LDs with the phagophore. It has been shown that both PLIN1 and SQSTM1 are required for colocalization of LDs with the phagophore/autophagosome marker, MAP1LC3B [17]. In addition, SQSTM1 and MAP1LC3B were found to cofractionate [16] and colocalize [19] with each other on LDs. As mentioned above, the SQSTM1-MAP1LC3B interaction is facilitated by the HTT scaffold [24]. Also, it has been established that the LIR motif of SQSTM1, which is responsible for the SQSTM1-MAP1LC3B binding, is required for LD degradation upon SQSTM1 overexpression [25]. However, it might not be just a simple bridging of individual LDs and phagophores. Using its UBA and PB1 domains, SQSTM1 can form the LD-containing aggresomes rich in HDAC6 (histone deacetylase 6). HDAC6 is recruited to the aggresomes via its binder of ubiquitin zinc finger (BUZ) domain and this recruitment is required for LD degradation upon SQSTM1 overexpression [25]. In conclusion, SQSTM1 does bridge the LDs or LD-containing aggresomes with the phagophores.
But is SQSTM1 co-degraded with LDs in the autophagy-lysosomal pathway, as is the case with other SARs? To study this, autophagy was modulated either pharmacologically or genetically and the effects on SQSTM1 and LDs were monitored [15,17,18]. Activation of autophagy with rapamycin [15,17], carbamazepine [17] or PFK158 [18] causes the decline in both SQSTM1 and LDs. The opposite is true when autophagy is inhibited with bafilomycin A1 [15,18], 3-methyladenine, wortmannin, chloroquine or knockdown of ATG5 (autophagy related 5) [17]: SQSTM1 and LDs accumulate. The co-accumulation of SQSTM1 and LDs in the presence of autophagosome-lysosome fusion inhibitors, bafilomycin A1 and chloroquine, suggests that they are indeed co-degraded in the lysosomes. Thus, SQSTM1 meets the third requirement of a lipophagy SAR.
The forth criterium (role in lipophagy) is also important, because as we have recently shown, the ubiquitin-binding SARs, such as Cue5 in yeast, can (1) accumulate on LDs in a ubiquitin-dependent manner, and (2) follow LDs to the lytic compartment for degradation, but be dispensable for it [26]. Therefore, it is essential to test if the LD-associated SAR is indeed “driving” lipophagy, and is not just one of many LD “passengers”. Importantly, the knockdown of SQSTM1 does increase lipid abundance [17] and decreases the lipophagy-dependent cholesterol efflux [19]. In addition, overexpression of SQSTM1 promotes LD degradation [25,27]. Interestingly, the knockdown or overexpression of DYNLRB1 (dynein light chain roadblock-type 1) has the same effect on LD abundance as the knockdown or overexpression of SQSTM1 [27] suggesting that SQSTM1 might cooperate with its binding partner, the dynein motor complex, for LD movement and degradation.
In conclusion, various studies showed that SQSTM1 has all the characteristics of a lipophagy SAR (Table 1). Although the four requirements of a SAR together were addressed for SQSTM1 only in a single study [17], the cumulative evidence from other studies suggests that SQSTM1 might act as a lipophagy receptor in different mammalian cells and under a variety of experimental conditions. Moreover, when fused to the LD-binding PAT (Perilipin, ADRP and TIP47) domain of PLIN3/TIP47 (perilipin 3) the SQSTM1T352A variant with disabled KEAP1 (kelch like ECH associated protein 1) binding induces lipophagy in both mammalian cells and fertilized mouse embryos [28]. The PAT-SQSTM1T352A fusion also causes LD clustering and their movement to the cell periphery during early embryonic development. Surprisingly, the non-mutated SQSTM1 without PAT domain does not localize to LDs or cause these effects, but the PAT domain itself also fails to localize to LDs during embryonic development [28]. Therefore, it is not clear how the PAT-SQSTM1T352A fusion localizes to LDs and drives lipophagy. Nevertheless, the studies discussed in this article implicate SQSTM1 in LD tagging, movement, aggregation/clustering, and sequestration (Figure 1). However, more in vivo work is needed to strengthen this conclusion.
This paradigm is consistent with other findings in the field, e.g. (1) accumulation of PLIN2 and PLIN3 in the mouse embryonic fibroblasts lacking NFKB1/p50 (nuclear factor kappa B subunit 1) due to reduced expression of SQSTM1 [29] and (2) overaccumulation of LDs in the mouse primary adipocytes lacking EIF2A (eukaryotic translation initiation factor 2A) due to their inability to express SQSTM1 upon differentiation [30]. Excitingly, the knockdown of some other ubiquitin-binding SARs, such as NBR1 (NBR1 autophagy cargo receptor) and OPTN (optineurin), also reduces the lipophagy-driven cholesterol efflux [19]. Moreover, both NBR1 and OPTN colocalize with ubiquitin on LDs suggesting that SQSTM1 might not be the only autophagic receptor that has lipophagy affairs with LDs. It will be interesting to see if these other ubiquitin-binding receptors also satisfy all the criteria of a lipophagy SAR.
Acknowledgments
We are grateful to Dr. Anton A. Komar for helpful discussion of the manuscript.
Funding Statement
This work was supported by the NIH grant, GM119571, to Taras Y. Nazarko.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005. Nov 21;171(4):603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007. Aug 17;282(33):24131–24145. [DOI] [PubMed] [Google Scholar]
- [3].Liu WJ, Ye L, Huang WF, et al. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kumar AV, Mills J, and Lapierre LR, et al. Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging. Front Cell Dev Biol. 2022;10:793328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bjorkoy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. [DOI] [PubMed] [Google Scholar]
- [6].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009. Apr 30;458(7242):1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schulze RJ, Sathyanarayan A, and Mashek DG, et al. Breaking fat: the regulation and mechanisms of lipophagy. Biochim Biophys Acta Mol Cell Biol Lipids. 2017. Oct;1862(10 Pt B):1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zechner R, Madeo F, and Kratky D, et al. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. 2017. Nov;18(11):671–684. [DOI] [PubMed] [Google Scholar]
- [9].Zhang X, Evans TD, Jeong SJ, et al. Classical and alternative roles for autophagy in lipid metabolism. Curr Opin Lipidol. 2018. Jun;29(3):203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kounakis K, Chaniotakis M, Markaki M, et al. Emerging roles of lipophagy in health and disease. Front Cell Dev Biol. 2019;7:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shin DW. Lipophagy: molecular mechanisms and implications in metabolic disorders. Mol Cells.2020. Aug 31;43(8):686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rahman MA, Kumar R, Sanchez E, et al. Lipid droplets and their autophagic turnover via the raft-like vacuolar microdomains. Int J Mol Sci. 2021. Jul 29;22(15):8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Martinez-Lopez N, Garcia-Macia M, Sahu S, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016. Jan 12;23(1):113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019. Oct 17;76(2):268–285. [DOI] [PubMed] [Google Scholar]
- [15].Lam T, Harmancey R, Vasquez H, et al. Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Discov. 2016;2(1):16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rasineni K, Donohue TM Jr., Thomes PG, et al. Ethanol-induced steatosis involves impairment of lipophagy, associated with reduced Dynamin2 activity. Hepatol Commun. 2017. Aug;1(6):501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang L, Zhou J, Yan S, et al. Ethanol-triggered lipophagy requires SQSTM1 in AML12 hepatic cells. Sci Rep. 2017. Sep 26;7(1):12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mondal S, Roy D, Sarkar Bhattacharya S, et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int J Cancer. 2019. Jan 1;144(1):178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Robichaud S, Fairman G, Vijithakumar V, et al. Identification of novel lipid droplet factors that regulate lipophagy and cholesterol efflux in macrophage foam cells. Autophagy. 2021. Nov;17(11):3671–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ju L, Han J, Zhang X, et al. Obesity-associated inflammation triggers an autophagy-lysosomal response in adipocytes and causes degradation of perilipin 1. Cell Death Dis. 2019. Feb 11;10(2):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu G, Sztalryd C, and Londos C, et al. Degradation of perilipin is mediated through ubiquitination-proteasome pathway. Biochim Biophys Acta. 2006. Jan;1761(1):83–90. [DOI] [PubMed] [Google Scholar]
- [22].Xu G, Sztalryd C, Lu X, et al. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J Biol Chem. 2005. Dec 30;280(52):42841–42847. [DOI] [PubMed] [Google Scholar]
- [23].Kovsan J, Ben-Romano R, Souza SC, et al. Regulation of adipocyte lipolysis by degradation of the perilipin protein: nelfinavir enhances lysosome-mediated perilipin proteolysis. J Biol Chem. 2007. Jul 27;282(30):21704–21711. [DOI] [PubMed] [Google Scholar]
- [24].Rui YN, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015. Mar;17(3):262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yan Y, Wang H, Wei C, et al. HDAC6 regulates lipid droplet turnover in response to nutrient deprivation via p62-mediated selective autophagy. J Genet Genomics. 2019. Apr 20;46(4):221–229. [DOI] [PubMed] [Google Scholar]
- [26].Kumar R, Shroff A, and Nazarko TY, et al. Komagataella phaffii Cue5 piggybacks on lipid droplets for its vacuolar degradation during stationary phase lipophagy. Cells. 2022. Jan 10;11(2):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tapia D, Jimenez T, Zamora C, et al. KDEL receptor regulates secretion by lysosome relocation- and autophagy-dependent modulation of lipid-droplet turnover. Nat Commun. 2019. Feb 13;10(1):735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tatsumi T, Takayama K, Ishii S, et al. Forced lipophagy reveals that lipid droplets are required for early embryonic development in mouse. Development. 2018. Feb 23;145(4):dev161893. [DOI] [PubMed] [Google Scholar]
- [29].Garcia-Macia M, Santos-Ledo A, Caballero B, et al. Selective autophagy, lipophagy and mitophagy, in the Harderian gland along the oestrous cycle: a potential retrieval effect of melatonin. Sci Rep. 2019. Dec 9;9(1):18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Anderson R, Agarwal A, Ghosh A, et al. eIF2A-knockout mice reveal decreased life span and metabolic syndrome. FASEB J. 2021. Nov;35(11):e21990. [DOI] [PMC free article] [PubMed] [Google Scholar]