

ABSTRACT
Neurons and astrocytes face unique demands on their proteome to enable proper function and survival of the nervous system. Consequently, both cell types are critically dependent on robust quality control pathways such as macroautophagy (hereafter referred to as autophagy) and the ubiquitin-proteasome system (UPS). We previously reported that autophagy is differentially regulated in astrocytes and neurons in the context of metabolic stress, but less is understood in the context of proteotoxic stress induced by inhibition of the UPS. Dysfunction of the proteasome or autophagy has been linked to the progression of various neurodegenerative diseases. Therefore, in this study, we explored the connection between autophagy and the proteasome in primary astrocytes and neurons. Prior studies largely in non-neural models report a compensatory relationship whereby inhibition of the UPS stimulates autophagy. To our surprise, inhibition of the proteasome did not robustly upregulate autophagy in astrocytes or neurons. In fact, the effects on autophagy are modest particularly in comparison to paradigms of metabolic stress. Rather, we find that UPS inhibition in astrocytes induces formation of Ub-positive aggregates that harbor the selective autophagy receptor, SQSTM1/p62, but these structures were not productive substrates for autophagy. By contrast, we observed a significant increase in lysosomal degradation in astrocytes in response to UPS inhibition, but this stimulation was not sufficient to reduce total SQSTM1 levels. Last, UPS inhibition was more toxic in neurons compared to astrocytes, suggesting a cell type-specific vulnerability to proteotoxic stress.Abbreviations: Baf A1: bafilomycin A1; CQ: chloroquine; Epox: epoxomicin; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; p-ULK1: phospho-ULK1; SQSTM1/p62: sequestosome 1; Ub: ubiquitin; ULK1: unc-51 like kinase 1; UPS: ubiquitin-proteasome system
KEYWORDS: Astrocytes, autophagy, LC3, lysosomes, neurons, proteasome, SQSTM1, ubiquitin
Introduction
Astrocytes and neurons are two key distinctive cell types of the brain that each have unique demands on their proteomes [1]. Both cell types collaborate to establish and maintain a complex network of trillions of synaptic connections in the brain. This intricate circuitry can exhibit remarkably high levels of synaptic activity that can reach up to ~50-100 action potentials per second to enable complex thought and behavior [2]. Furthermore, this system must be robust for the lifetime of the organism. For example, in humans, neurons are born during embryogenesis and must survive for nearly a century of time [3]. Moreover, the majority of astrocytes in healthy CNS tissue are also post-mitotic and long-lived, although astrocyte proliferation and turnover can accelerate in stress and disease [4]. Maintaining functionality over an extended lifetime requires that both cell types must regulate the dynamic composition and quality of the proteome. In fact, dysregulation of proteostasis and the accumulation of misfolded protein is a unifying hallmark of various neurodegenerative diseases [5]. Consequently, astrocytes and neurons are dependent on robust quality control pathways such as autophagy and the ubiquitin-proteasome system (UPS) to maintain function and survival, but how these processes are regulated in each cell type remains largely unknown.
Autophagy is a degradative pathway whereby cellular components (e.g. proteins and organelles) are targeted to lysosomes for destruction [6–8]. In this process, cargo are packaged into autophagosomes which fuse with lysosomes to enable degradation by resident hydrolases. The amino acids generated are then exported from the lysosome to fuel new biosynthetic reactions [9]. Selective forms of autophagy degrade specific cargo that are recognized by receptors (e.g. SQSTM1/p62) that engage ubiquitin and the autophagy machinery [10,11]. Autophagy plays critical roles in neurodevelopment, function, and survival [12–15]. Moreover, deficits in autophagy can cause neurodevelopmental disorders [16,17] and neurodegenerative disease in humans [18–21]. We have previously shown that autophagy is differentially regulated in astrocytes as compared with neurons [22]. Our study showed that astrocytes upregulate autophagy more robustly than neurons in response to metabolic stress induced by nutrient deprivation and MTOR inhibition [22,23]. However, it remains unclear how autophagy is regulated in astrocytes and neurons in response to proteotoxic stress induced by UPS inhibition.
The UPS is a system whereby ubiquitinated proteins are targeted for degradation in the proteolytic chamber of the proteasome [24–26]. Substrates are de-ubiquitinated, unfolded, and threaded into a chamber that houses proteolytic activity. Short peptides are then released by the proteasome, which are subsequently digested into free amino acids by downstream cytosolic peptidases. Protein degradation via the proteasome is a key regulator of synaptic function and plasticity [27–30]. Consequently, dysfunction of the proteasome has been linked to the progression of various neurodegenerative diseases [30,31], potentially via a common mechanism by which disease-associated protein oligomers bind and inhibit the proteasome [32].
Several studies have reported a complex interplay between autophagy and the UPS [33,34]. Seminal studies defined a compensatory relationship whereby inhibition of the UPS upregulates autophagy [35,36]. However, an elevation in autophagy levels in response to UPS inhibition has not always been observed [37–39]. Further, Ebrahimi-Fakhari et al. suggest that an elevation in autophagy may only occur upon additional proteotoxic stress [40]. Thus, this interaction may be highly nuanced and requires further exploration, particularly in the context of astrocytes and neurons.
In this study, we set out to explore the connection between autophagy and the proteasome in astrocytes and neurons. Understanding brain cell-type specific responses to proteotoxic stress will aid in our understanding of how insults to the proteome are managed in aging and neurodegenerative disease. To our surprise, inhibition of the proteasome did not lead to a robust upregulation of autophagy in astrocytes or neurons. Effects on autophagy are modest particularly in comparison to paradigms of metabolic stress. UPS inhibition, however, did lead to the formation of SQSTM1- and Ub-positive aggresomes in astrocytes, but these were not productive substrates for autophagy. Rather, we find that UPS inhibition stimulates lysosomal degradation, but this increase was not sufficient to reduce SQSTM1 levels. Lastly, UPS inhibition was more toxic in neurons compared to astrocytes, suggesting a cell type-specific vulnerability to proteotoxic stress.
Results
Short-term UPS inhibition with epoxomicin does not significantly upregulate autophagic flux but leads to the formation of SQSTM1-positive fibril-like structures in astrocytes
The degree of coordination between the UPS and autophagy has been characterized in various cell types, predominantly in cancer cell lines. The interplay between these systems in neural cells, such as astrocytes and neurons, however, is less well understood. Here, we define how the autophagy-lysosomal pathway is regulated in response to UPS inhibition in primary astrocytes and perform comparative studies in primary neurons. For this study, we isolated and cultured mouse cortical glia that are enriched for astrocytes and largely depleted of microglia and oligodendrocytes, as we previously established [22]. To inhibit UPS activity, we first incubated primary astrocytes for 4 h with 5 µM epoxomicin, a selective inhibitor that covalently binds to catalytic subunits of the proteasome [41]. Treatment with epoxomicin increased levels of ubiquitinated protein relative to the DMSO solvent control, as measured by immunoblot analysis, consistent with effective inhibition of the UPS (Figure 1A). To further validate our treatment conditions, we also measured proteasomal degradation using a GFP-based reporter developed by Dantuma et al. (2000). We transfected astrocytes with UbG76V-GFP, a GFP that is constitutively targeted to the proteasome for degradation by fusion with a mutated, uncleavable ubiquitin [42]. Because the half-life of GFP is reduced, few astrocytes expressed detectable levels of cytoplasmic UbG76V-GFP (Fig. S1). Treatment with epoxomicin, however, increased the percentage of astrocytes with cytoplasmic UbG76V-GFP (Fig. S1). By contrast, blocking lysosomal degradation with bafilomycin A1 (Baf A1), an inhibitor of the vacuolar proton pump, did not increase the percentage of astrocytes with cytoplasmic UbG76V-GFP relative to the solvent control (Fig. S1). Thus, treatment with epoxomicin effectively reduces proteasomal degradation in primary astrocytes.
Figure 1.
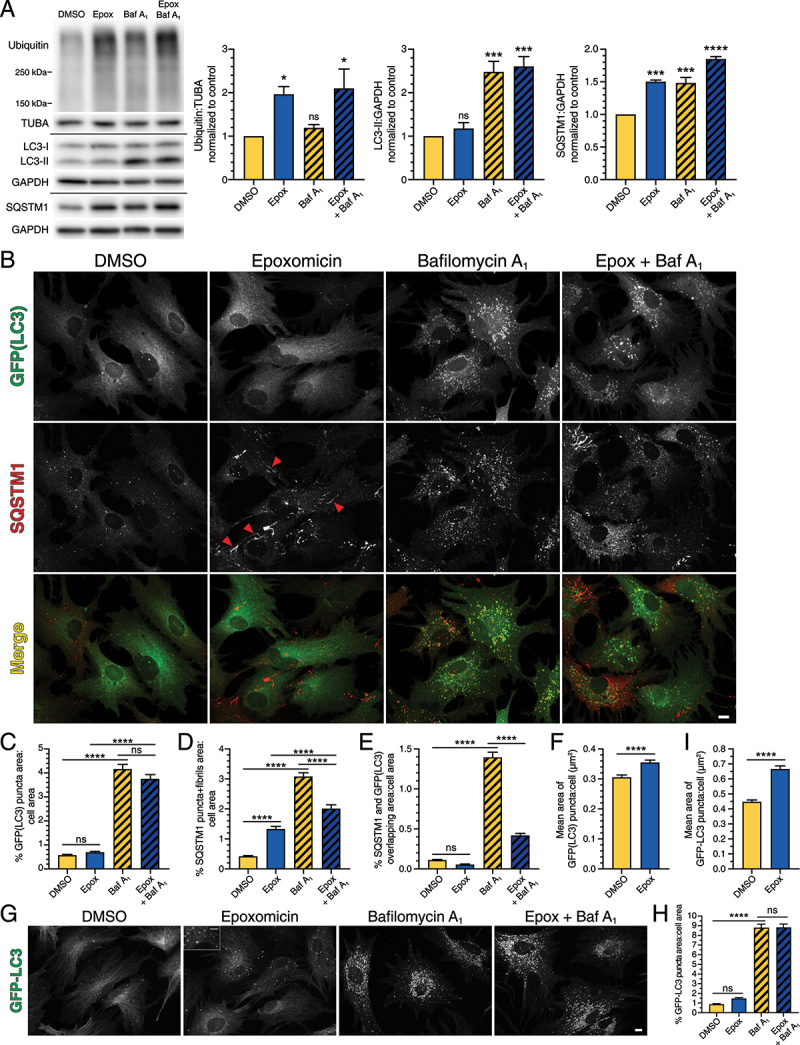
Short-term UPS inhibition with epoxomicin does not significantly upregulate autophagic flux, but leads to the formation of SQSTM1-positive fibril-like structures in primary astrocytes. Primary astrocytes were treated with 5 µM epoxomicin supplemented with 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) for 4 h and analyzed by (A) immunoblot, (B-F) immunostain, and (G-I) live cell imaging. (A) Immunoblot analysis and corresponding quantification of glial lysates. GAPDH and TUBA/α-tubulin serve as loading controls; horizontal lines designate individual blots. ubiquitin levels were normalized to TUBA/α-tubulin, and LC3-II and SQSTM1 levels were normalized to GAPDH (means ± SEM; one-way ANOVA with Dunnett’s post hoc test; n = 3–4 independent experiments; 4–9 DIV). (B) Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes immunostained for GFP and SQSTM1. SQSTM1 images are grayscale matched to facilitate direct comparisons. Red arrowheads denote SQSTM1-positive fibril-like structures. Bar: 10 µm. (C-F) Corresponding quantification of immunostain analysis shown in B. (C) Quantification of total GFP(LC3) puncta area normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 135–165 cells from 3 independent experiments; 3–5 DIV). (D) Quantification of total area occupied by SQSTM1 puncta and fibrils normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 131–160 cells from 3 independent experiments; 3–5 DIV). (E) Quantification of the percentage of overlapping area between GFP(LC3)-positive puncta and SQSTM1-positive structures normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 126–146 cells from 3 independent experiments; 3–5 DIV). (F) Mean area of GFP(LC3)-positive puncta per astrocyte (means ± SEM; unpaired t-test; DMSO, n = 148 astrocytes; Epoxomicin, n = 165 astrocytes; data from 3 independent experiments; 3–5 DIV). (G) Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes using live cell imaging. Bar: 10 µm. Inset bar: 5 µm. (H and I) Corresponding quantification of live cell imaging shown in G. (H) Quantification of total GFP-LC3 puncta area normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 101–116 cells from 3 independent experiments; 6–9 DIV). (I) Mean area of GFP-LC3-positive puncta per astrocyte (means ± SEM; unpaired t-test; DMSO, n = 110 astrocytes; Epoxomicin, n = 101 astrocytes; data from 3 independent experiments; 6–9 DIV).
To measure the effect of short-term UPS inhibition on autophagy, we treated astrocytes for 4 h with 5 µM epoxomicin and immunoblotted for endogenous LC3. LC3 has two key forms, cytosolic LC3-I and phagophore- and autophagosome-associated LC3-II [43]. To associate with phagophore membranes, cytosolic LC3-I becomes lipidated by the addition of phosphatidylethanolamine to form LC3-II [43], resulting in a shift in molecular weight that is detectable by immunoblot analysis. Treatment with epoxomicin resulted in a small but reproducible increase in LC3-II, ~1.2-fold higher than the DMSO control, which was not statistically significant (Figure 1A). Thus, we find that UPS inhibition induces only a very modest increase in steady state levels of LC3-II in primary astrocytes. Because steady state levels of autophagosomes are a balance between the antagonistic activities of autophagosome formation coupled with degradation, we blocked lysosomal degradation with Baf A1. In this way, we can measure the amount of material that is shuttling through the autophagy system, defined as autophagic flux, in response to UPS inhibition. Because Baf A1 blocks the terminal step of the autophagy pathway, alterations in autophagic flux revealed by Baf A1 treatment can be due to changes in autophagosome production and/or clearance in lysosomes. As expected, treatment with Baf A1 alone increased LC3-II levels (~2.5-fold increase relative to the DMSO solvent control), representing the amount of LC3-II that accumulates in response to lysosome inhibition under basal conditions. Co-treatment of Baf A1 with epoxomicin, however, did not significantly increase LC3-II levels relative to Baf A1 alone, suggesting that UPS inhibition does not robustly increase autophagic flux in primary astrocytes. We also noted that the effects of UPS inhibition on autophagy levels were muted as compared to the robust induction of autophagic flux we previously observed in astrocytes by immunoblot analysis in response to nutrient deprivation or pharmacological inhibition of MTOR [22]. Moreover, the latter models of autophagy induction resulted in a consistent reduction in cytosolic LC3-I levels as LC3-I is depleted during the conversion to LC3-II [22]. In the current study, however, UPS inhibition did not decrease LC3-I levels as compared to the DMSO control (Figure 1A). Combined, we find that short-term UPS inhibition does not significantly increase autophagic flux in primary astrocytes.
To further characterize autophagy levels, we also immunoblotted for SQSTM1, a receptor for selective forms of autophagy that targets ubiquitinated cargo to autophagosomes for degradation [10]. In response to UPS inhibition, we observed an increase in total SQSTM1 levels (Figure 1A). Because UPS inhibition increased SQSTM1 levels without a corresponding increase in autophagosome-associated LC3-II, we wanted to examine the cellular localization of SQSTM1 in astrocytes with respect to autophagosomes using confocal microscopy.
To that end, we isolated cortical glia from transgenic mice expressing GFP-LC3 [44], and immunostained for GFP(LC3) and SQSTM1. Autophagosomes are represented by GFP-LC3-positive puncta that are distributed throughout the cytoplasm (Figure 1B). To quantify levels of autophagy, we measured the area occupied by GFP-LC3-positive puncta using Ilastik, a machine learning-based program to identify and segment objects of interest [45]. For the immunofluorescence analysis, only cells that exhibited the characteristic morphology of monocultured GFAP-positive astrocytes, defined as polygonal cells often with finger-like projections, were analyzed. Thus, the immunofluorescence analysis represents a highly pure population of astrocytes. Consistent with the immunoblot, treatment with 5 µM epoxomicin for 4 h resulted in a small but reproducible increase in total area occupied by GFP(LC3)-positive puncta (~1.2-fold higher than the DMSO control), which was not statistically significant (Figure 1B, C). This effect was modest especially when compared to our previous reports of ~5-fold inductions in GFP(LC3)-positive puncta area in response to nutrient deprivation or MTOR inhibition for an equivalent time incubation [22]; representative image for starvation shown in Figure 8F. As expected, Baf A1 increased the total area of GFP(LC3)-positive puncta ~7.3-fold due to an accumulation of autophagosomes in the absence of lysosome-mediated degradation (Figure 1B, C). However, co-treatment of Baf A1 with epoxomicin did not have an additive effect on the accumulation of autophagosomes relative to the Baf A1-alone control (Figure 1B, C). Thus, UPS inhibition is not as potent a stimulator of autophagy in astrocytes as compared to an equivalent duration of nutrient deprivation or pharmacological inhibition of MTOR [22].
Figure 8.
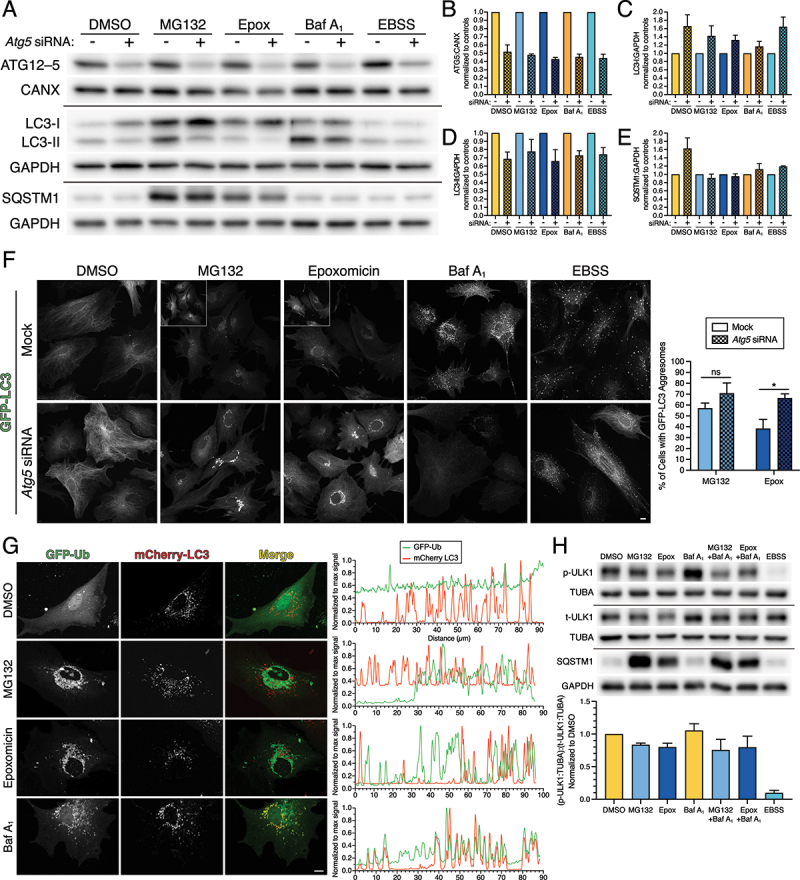
LC3 population on aggresomes is likely cytosolic LC3 rather than autophagosome-associated LC3. Primary astrocytes were treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h, 100 nM Baf A1 for 4 h, or EBSS for 4 h. (A-F) Astrocytes were also treated with siRNAs targeted against Atg5, or a mock treatment as a control. (A-E) Immunoblot analysis and corresponding quantification of glial lysates. CANX and GAPDH serve as loading controls; horizontal lines designate individual blots. (B) ATG5 levels (specifically in the context of the ATG12-ATG5 conjugate) were normalized to CANX; (C and D) LC3-I and LC3-II were normalized to GAPDH; and (E) SQSTM1 levels were normalized to GAPDH (means ± SEM; n = 3 independent experiments; 6 DIV). (F) Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes using live cell imaging. Images within the same column of treatment (e.g. DMSO mock and DMSO siRNA) are grayscale matched to facilitate direct comparisons; insets are grayscale adjusted individually. Bar: 10 µm. Corresponding quantification of the percentage of astrocytes with GFP-LC3-positive aggresomes (means ± SEM; unpaired t-test; n = 4 independent experiments; 6–7 DIV). (G) Primary astrocytes were treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h, or 100 nM Baf A1 for 4 h and analyzed using live cell imaging. Shown are maximum projections of z-stacks of astrocytes expressing mCherry-LC3 and GFP-Ub (shown are representative images from 3 independent experiments; 6 DIV). Bar: 10 µm. Corresponding line scans are drawn from the cell periphery toward the perinuclear region (left to right). Overlapping peaks for mCherry-LC3 and GFP-Ub are present in the basal conditions (DMSO and Baf A1). mCherry-LC3 and GFP-Ub are largely non-overlapping in treatments that block UPS function (MG132 and epoxomicin). (H) Primary astrocytes were treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; DMSO or 100 nM Baf A1 were included in the final 4 h. As a positive control, astrocytes were also starved in EBSS for 4 h. Immunoblot analysis and corresponding quantification of of glial lysates. TUBA/α-tubulin and GAPDH serve as loading controls; horizontal lines designate individual blots. p-ULK1 (S757) and total ULK1 (t-ULK1) levels were first normalized to their respective TUBA/α-tubulin controls, and then p-ULK1:TUBA/α-tub was then normalized to t-ULK1:TUBA/α-tub (means ± SEM; n = 3 independent experiments; 6–7 DIV).
By contrast, epoxomicin increased the area occupied by SQSTM1-positive structures ~3.2-fold relative to the DMSO control (Figure 1B, D), consistent with the increase in total SQSTM1 levels by immunoblot analysis (Figure 1A). These SQSTM1-positive structures included puncta, as well as fibril-like structures (Figure 1B, D). As expected, treatment with Baf A1 increased SQSTM1-positive puncta due to lysosomal inhibition, resulting in a 7.3-fold increase in the area occupied by SQSTM1-positive puncta relative to the DMSO control. (Figure 1B, D). Co-treatment of Baf A1 with epoxomicin, however, unexpectedly reduced SQSTM1 area ~1.5-fold relative to Baf A1 alone, suggesting a reduction in SQSTM1 targeted to lysosomes for degradation (i.e. SQSTM1 degradative flux). To further assess the effect of UPS inhibition on SQSTM1 degradative flux, we measured the percentage of overlapping area between SQSTM1 and GFP(LC3)-positive autophagosomes, and normalized this value to the respective total cell area. In the DMSO control, we observed colocalization between SQSTM1-positive puncta and GFP(LC3)-positive puncta representing ~0.1% of total cell area (Figure 1B, E). As expected, blocking autophagosome clearance with Baf A1 increased the area of overlap between SQSTM1- and GFP(LC3)-positive puncta ~12.6-fold relative to the DMSO control (Figure 1B, E). Blocking UPS activity with epoxomicin, however, decreased the area of overlap between GFP(LC3) and SQSTM1, i.e. a ~ 3.3-fold reduction with epoxomicin and Baf A1 relative to Baf A1 alone (Figure 1B, E). This finding suggests that UPS inhibition reduces SQSTM1 routed to autophagy and increases SQSTM1 that may be forming fibril-like structures (Figure 1B, E). Consistent with this possibility, we did not observe fibril-like structures positive for GFP(LC3) (Figure 1B). Combined, we find that UPS inhibition increases the formation of SQSTM1-positive fibrils without a corresponding induction in autophagic flux.
As a complementary approach, we also assessed autophagy levels in response to short-term UPS inhibition using live-cell imaging of GFP-LC3. Similar to the immunoblot and immunofluorescence results, live cell imaging detected a modest but consistent increase in total area occupied by GFP-LC3-positive puncta (~1.7-fold induction as compared to the DMSO control) in response to UPS inhibition, although this effect was not statistically significant (Figure 1G, H). We also did not observe fibril-like structures with GFP-LC3 as we did for SQSTM1 in response to UPS inhibition (Figure 1G). Furthermore, co-treatment of Baf A1 with epoxomicin did not stimulate autophagic flux beyond the Baf A1-alone control (Figure 1G, H). Although we did not observe a robust increase in total autophagosome area in astrocytes in response to UPS inhibition, we did observe changes in the size of individual autophagosomes. Treatment with epoxomicin increased the cross-sectional area of individual autophagosomes ~1.2- and ~1.4-fold relative to the DMSO solvent control, as measured by immunostain and live-cell imaging, respectively (Figure 1F, I). Because Baf A1 results in a perinuclear accumulation of autophagosomes, we were unable to accurately discern the size of individual autophagosomes in Baf A1-treated cells. Using live cell imaging, we also observed larger, ring-like autophagosomes with epoxomicin treatment (Figure 1G, inset). These results suggest that larger autophagosomes may not be fully preserved during fixation, consistent with the greater fold increase in cross-sectional area with UPS inhibition measured by live-cell imaging as compared to immunostain (Figure, 1F, I). In sum, short-term UPS inhibition has a modest activation of autophagy as evidenced by an increase in the size of individual autophagosomes, which supports our observations of a consistent trend in increased total autophagosome area.
We next investigated the nature of the SQSTM1-positive fibril-like structures that formed in response to short-term UPS inhibition. We immunostained astrocytes for ubiquitin using an antibody that detects both mono- and poly-ubiquitination. In the DMSO solvent control, ubiquitin was enriched in the nucleus as compared to the cytoplasm (Figure 2A), consistent with published results [46]. Treatment with epoxomicin redistributed ubiquitin from the nucleus to the cytoplasm, resulting in a striking increase in cytoplasmic ubiquitin levels relative to the DMSO control (Figure 2A-C; short-term and long-term UPS inhibition). These results are in line with observations from prior studies [46] and validate our antibody for specific detection of ubiquitin. Treatment with epoxomicin also increased ubiquitin localization to a population of SQSTM1-positive fibril-like structures as compared to SQSTM1-positive puncta (Figure 2A). Whereas nearly all ubiquitin-positive fibrils were co-positive for SQSTM1, not all SQSTM1-positive fibrils were co-positive for ubiquitin (Figure 2A). Blocking lysosomal degradation with Baf A1 increased SQSTM1-positive puncta and, to a lesser extent, ubiquitin-positive puncta in the cytoplasm, however, these markers largely did not overlap (Figure 2A). Co-treatment of epoxomicin with Baf A1 increased levels of ubiquitin in the cytoplasm and specific recruitment of ubiquitin to SQSTM1-positive fibrils relative to the DMSO control, similar to epoxomicin-alone (Figure 2A). Thus, short-term UPS inhibition with epoxomicin results in the formation of SQSTM1-positive fibrils that are partially co-positive for ubiquitin.
Figure 2.
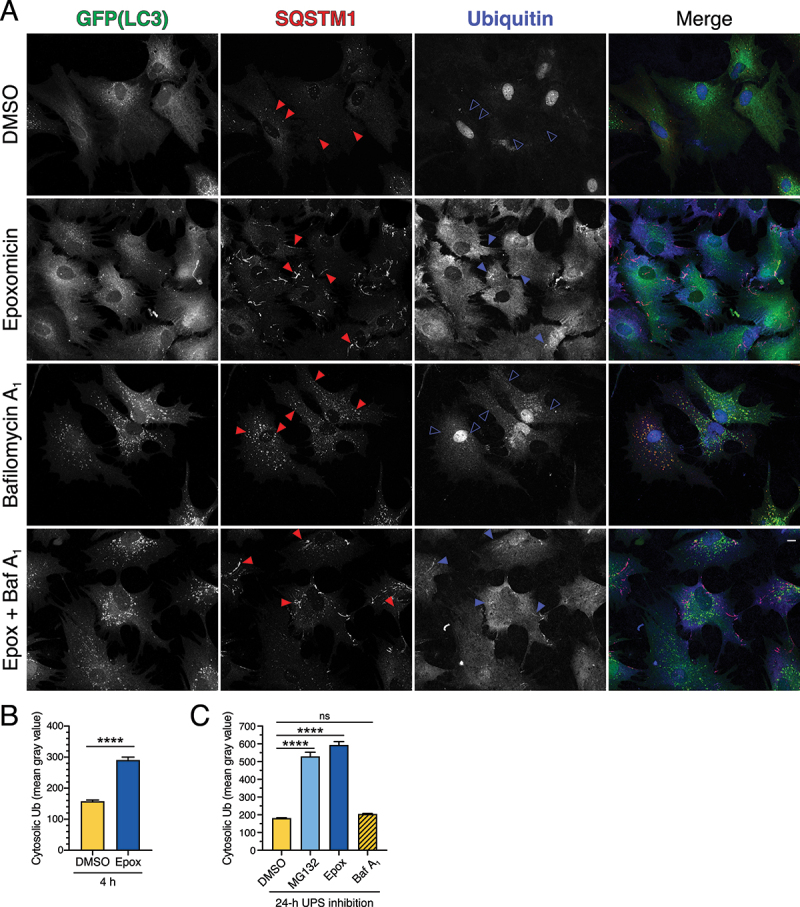
SQSTM1 fibril-like structures that form in response to short-term UPS inhibition are partially positive for ubiquitin. (A, B) Immunostain analysis and corresponding quantification of primary astrocytes treated with 5 µM epoxomicin supplemented with 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) for 4 h. (A) Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes immunostained for GFP, SQSTM1, and ubiquitin. Closed red arrowheads denote SQSTM1-positive puncta or fibril-like structures; closed blue arrowheads denote SQSTM1-positive fibril-like structures that are co-positive for ubiquitin; open blue arrowheads denote SQSTM1-positive puncta that are negative for ubiquitin. Bar: 10 µm. (B) Quantification of cytosolic ubiquitin signal intensity after 4 h of UPS inhibition (means ± SEM; unpaired t-test; n = 97–136 cells from 3 independent experiments; 6 DIV). (C) Quantification of cytosolic ubiquitin signal intensity after 24 h of UPS inhibition (0.5 µM MG132 or 10 nM epoxomicin) or after 4 h of 100 nM baf A1-treatment (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 82–116 cells from 3 independent experiments; 3–8 DIV).
Short-term UPS inhibition with MG132 does not significantly upregulate autophagic flux but leads to the formation of SQSTM1-positive fibril-like structures in astrocytes
Given that previous studies reported a stimulation of autophagy in response to UPS inhibition [35,36], we were surprised that similar treatments did not strongly upregulate autophagy in primary astrocytes. Thus, we used another inhibitor of the proteasome, MG132, a peptide aldehyde which effectively blocks the chymotrypsin-like activity of the 26S proteasome complex [47]. Treatment of astrocytes with 50 µM MG132 for 4 h increased levels of ubiquitinated protein, as measured by immunoblot, consistent with reduced UPS activity (Fig. S2A). Treatment with MG132 also increased the percentage of astrocytes with cytoplasmic UbG76V-GFP (Fig. S1), providing further validation that our treatment conditions effectively reduce UPS activity. Similar to our immunoblotting results with epoxomicin, MG132 resulted in a small but consistent increase in LC3-II levels (~1.4-fold increase relative to DMSO control) that was not statistically significant (Fig. S2A). Consistent across our experiments, Baf A1 increased LC3-II levels ~2.6-fold relative to the DMSO solvent control. However, co-treatment of MG132 with Baf A1 decreased LC3-II levels relative to Baf A1-alone, suggesting a reduction in autophagic flux (Fig. S2A). MG132 did not significantly affect total levels of SQSTM1, as measured by immunoblot analysis (Fig. S2A). Thus, we found that inhibiting the UPS with MG132 also did not strongly upregulate autophagy levels in astrocytes.
In parallel with the immunoblot analysis, we also analyzed the effects of MG132 treatment on autophagy in astrocytes by immunostain for GFP(LC3). Treatment with MG132 increased the total area of GFP(LC3)-positive puncta ~4.6-fold relative to the DMSO solvent control (Fig. S2B, C). However, co-treatment of MG132 with Baf A1 decreased the total area of GFP(LC3)-positive puncta ~1.7-fold relative to the Baf A1-alone control (Fig. S2B, C). These results reflect our observations with immunoblot analysis (Fig. S2A), and suggest that short-term treatment with MG132 may decrease autophagic flux in astrocytes. In fact, Baf A1 resulted in a larger accumulation of GFP(LC3) puncta in cells treated with DMSO as compared to MG132-treated cells (Fig. S2B, C). More specifically, Baf A1 increased GFP(LC3) puncta area by ~9.1-fold in control cells treated with DMSO alone (Fig. S2B, C). By contrast, Baf A1 added to MG132-treated cells increased GFP(LC3) puncta area by only ~1.2-fold relative to cells treated with MG132 alone (Fig. S2B, C). Thus, treatment with Baf A1 did not accumulate autophagosomes in MG132-treated cells to the large extent that it does in control cells treated with the DMSO solvent, suggesting a decrease in degradative autophagic flux with MG132 treatment. Furthermore, we noticed that the autophagosomes that accumulated in MG132-treated cells were tightly distributed in a perinuclear region. This perinuclear enrichment is striking when compared with our prior observations where autophagy is induced during nutrient deprivation [22]. In the latter case, starvation of astrocytes in EBSS results in the formation and distribution of autophagosomes throughout the cytoplasm (Fig. S2C’-C”’ and 8 F). By contrast, MG132 treatment results in a tight perinuclear enrichment relative to EBSS and even Baf A1-alone (Fig. S2C’-C”’). Combined with the decrease in autophagic flux, the distribution data suggest that MG132 may have off-target effects (i.e. effects on targets other than the proteasome), leading to possible alterations in intracellular trafficking (additional details provided below). Indeed, unlike epoxomicin, MG132 is also known to inhibit other proteases, such as calpains and lysosomal cathepsins [47].
Similar to epoxomicin, short-term treatment with MG132 increased SQSTM1-positive structures including puncta and fibril-like structures, although we observed fewer fibril-like structures with MG132 as compared with epoxomicin (Fig. S2B, D). Furthermore, MG132 increased cytoplasmic levels of SQSTM1 (Fig. S2B, E). Treatment with Baf A1 increased SQSTM1-positive puncta due to lysosomal inhibition (Fig. S2B, D), but co-treatment of MG132 with Baf A1 reduced SQSTM1 puncta area relative to Baf A1 alone, suggesting a reduction in SQSTM1 degradative flux. Accordingly, treatment with MG132 decreased the overlap between SQSTM1-positive structures and GFP(LC3)-positive autophagosomes (Fig. S2B, F), suggesting a reduction in SQSTM1 routed to autophagy for degradation.
To further define the effects of MG132 on autophagy, we also measured autophagic flux using chloroquine which captures autophagosomes by blocking their fusion with lysosomes [48]. Treatment with MG132 (50 µM MG132 for 4 h) effectively inhibited UPS activity as evidenced by an increase in ubiquitinated protein levels by immunoblot analysis (Fig. S3A). However, MG132 did not robustly stimulate autophagic flux when measured using chloroquine (Fig. S3A; immunoblot analysis). Using immunostain analysis, MG132-alone resulted in a perinuclear accumulation of GFP(LC3)-positive puncta, but did not significantly upregulate autophagic flux when measured using chloroquine (Fig. S3C). We also varied the time of MG132 treatment (from 30 min up to 6 h) and reduced MG132 concentration to minimize potential off-target effects (Fig. S3B, D, E). Across all permutations, ubiquitinated protein levels increased by immunoblot, confirming effective inhibition of the UPS (Fig. S3B, D, E). However, we did not observe a corresponding increase in autophagic flux (Fig. S3B, D, E). We note that only SQSTM1 levels increased with longer MG132 treatments (Fig. S3B). Thus, our results with MG132 largely corroborate our findings with epoxomicin in that short-term UPS inhibition does not strongly upregulate autophagy in astrocytes.
Long-term UPS inhibition does not significantly upregulate autophagic flux but leads to the formation of SQSTM1-positive aggresomes in astrocytes
Because short-term UPS inhibition did not significantly upregulate autophagy in astrocytes, we examined whether a long-term treatment with epoxomicin or MG132 might stimulate autophagy. For this experiment, we treated astrocytes with either 0.5 µM MG132, 10 nM epoxomicin, or an equivalent volume of DMSO solvent for 24 h. To measure autophagic flux, the final 4 h included 100 nM Baf A1 or DMSO as a solvent control. Immunoblot analysis revealed a significant accumulation of ubiquitinated protein with MG132 or epoxomicin treatment, indicating that our long-term treatment paradigm effectively inhibited UPS function (Figure 3A, B). Similar to our immunoblotting results with short-term UPS inhibition, long-term UPS inhibition resulted in a small but consistent increase in LC3-II levels, ~1.6-fold increase with either MG132 or epoxomicin as compared to the DMSO solvent control (Figure 3A, C). This effect, however, was not statistically significant and did not result in an increase in autophagic flux as measured with Baf A1 (Figure 3A, C). We also observed an increase in cytosolic LC3-I levels with UPS inhibition, albeit not statistically significant by immunoblot analysis (Figure 3A, D), but significant by immunostain as shown later in Figure 4 (Figure 4A, B). The most striking effect of UPS inhibition by immunoblot analysis was on SQSTM1 levels, which increased significantly with either MG132 or epoxomicin relative to the DMSO solvent control (Figure 3E). However, this increase in SQSTM1 with UPS inhibition did not appear to be targeted to lysosomes for degradation, as there is no further accumulation in SQSTM1 levels when lysosome function is blocked in UPS-inhibited cells with Baf A1 (Figure 3A, E). Combined, we find that long-term UPS inhibition does not elicit a strong stimulation of autophagic flux, but rather leads to an accumulation of SQSTM1 in astrocytes.
Figure 3.
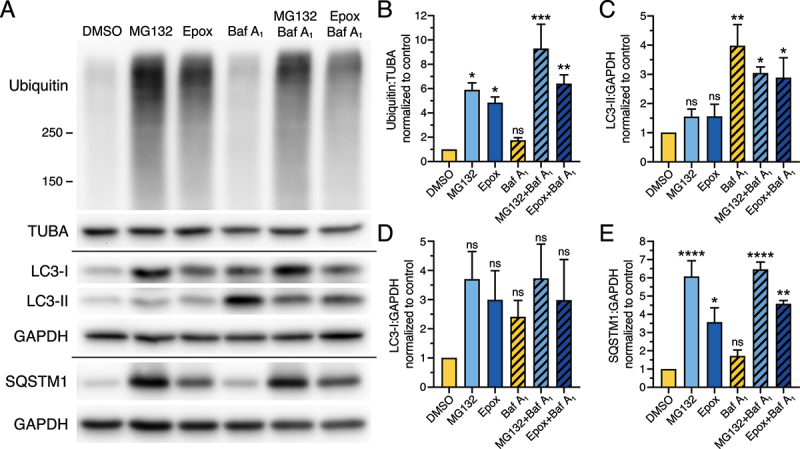
Long-term UPS inhibition does not significantly upregulate autophagic flux, but increases levels of SQSTM1 in primary astrocytes. (A-E) Immunoblot analysis and corresponding quantification of primary astrocytes treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. GAPDH and TUBA/α-tubulin serve as loading controls; horizontal lines designate individual blots. (B) Ubiquitin levels were normalized to TUBA/α-tubulin; (C, D) LC3-II and LC3-I were normalized to GAPDH; and (E) SQSTM1 levels were normalized to GAPDH (means ± SEM; one-way ANOVA with Dunnett’s post hoc test; n = 3 independent experiments; 4–6 DIV).
Figure 4.
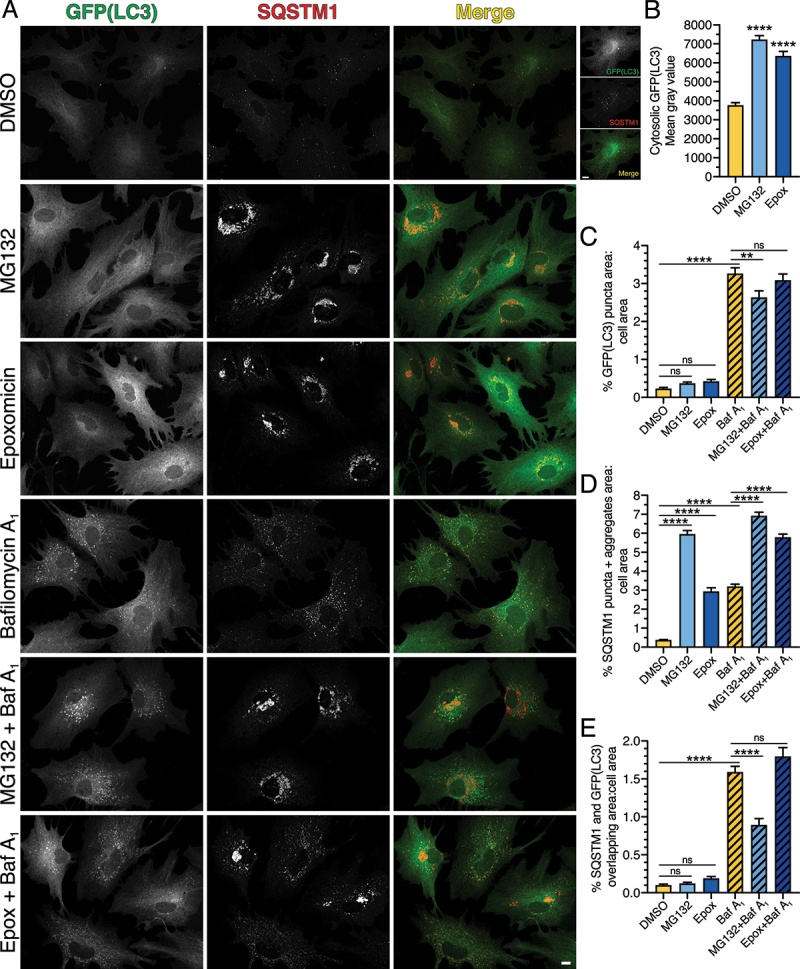
Long-term UPS inhibition does not significantly upregulate autophagic flux, but leads to the formation of SQSTM1-positive aggresomes in primary astrocytes. (A-E) Immunostain analysis and corresponding quantification of primary astrocytes treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. (A) Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes immunostained for GFP and SQSTM1. GFP(LC3) images for DMSO, MG132, and Epoxomicin are grayscale matched to show an increase in cytosolic LC3 upon UPS inhibition. Higher magnification images for DMSO are contrast enhanced for visualization. Bars: 10 µm. (B) Quantification of cytosolic GFP(LC3) signal intensity (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 143–182 cells from 3 independent experiments; 6–7 DIV). (C) Quantification of total GFP(LC3) puncta area normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 78–102 cells from 3 independent experiments; 6–7 DIV). (D) Quantification of total area occupied by SQSTM1-positive structures (puncta and aggresomes) normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 85–108 cells from 3 independent experiments; 6–7 DIV). (E) Quantification of the percentage of overlapping area between GFP(LC3)-positive and SQSTM1-positive structures normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 73–96 cells from 3 independent experiments; 6–7 DIV).
We also analyzed the effect of long-term UPS inhibition on autophagy by immunostain for GFP(LC3) and SQSTM1. Long-term treatment with MG132 or epoxomicin increased the total area of GFP(LC3)-positive puncta by ~1.6- and 1.8-fold, respectively, as compared to the DMSO control (Figure 4A, C). This effect was modest and not statistically significant, but it was consistently observed across our multiple paradigms of UPS inhibition (Figure 4A, C). Co-treatment of MG132 or epoxomicin with Baf A1, however, did not increase the total area of GFP(LC3)-positive autophagosomes above the level accumulated with Baf A1-alone, suggesting no robust induction of autophagic flux with UPS inhibition (Figure 4A, C). UPS inhibition did result in a significant increase in cytosolic GFP(LC3) levels (Figure 4A, B), corroborating our immunoblot results (Figure 3D). Thus, long-term UPS inhibition did not strongly upregulate autophagic flux, but increased levels of cytosolic LC3.
Treatment with MG132 and epoxomicin also resulted in a significant increase in SQSTM1-positive structures, including puncta and perinuclear aggregates (Figure 4A, D). This effect is similar to prior reports in non-neural cells where UPS inhibition leads to the collection of misfolded, ubiquitinated protein, into large perinuclear structures called aggresomes [35,49–52]. As expected, Baf A1 significantly increased SQSTM1-positive puncta in astrocytes relative to the DMSO control (Figure 4A, D). Co-treatment of Baf A1 with MG132 or epoxomicin increased SQSTM1 area as compared with Baf A1 alone. However, this increase in SQSTM1 area included SQSTM1-positive aggresomes that accounted for a large proportion of SQSTM1 area in the MG132 or epoxomicin-alone samples. Thus, to measure the population of SQSTM1 that is targeted to degradative compartments, we compared SQSTM1 area within each treatment plus or minus Baf A1. In DMSO-treated control cells, Baf A1 increased SQSTM1-positive structures ~8.5-fold, consistent with a robust targeting of SQSTM1 to degradative compartments under basal conditions (Figure 4A, D). However, in UPS-inhibited cells, Baf A1 had a more muted accumulation of SQSTM1-positive structures (i.e. ~1.2-fold increase in SQSTM1 with MG132 and Baf A1 versus MG132 alone, and ~2-fold increase in SQSTM1 with epoxomicin and Baf A1 versus epoxomicin alone) as compared to DMSO-treated controls. These results suggest that with long-term UPS inhibition, less SQSTM1 is routed to degradative compartments and this effect is concomitant with an increase in SQSTM1 routed to aggresomes.
Consistent with this result, whereas SQSTM1 puncta in Baf A1-treated cells are largely co-positive for GFP(LC3), SQSTM1-positive aggresomes in MG132 or epoxomicin-treated cells had reduced overlap with GFP(LC3)-positive puncta by immunostain (Figure 4A). In fact, co-treatment of Baf A1 with MG132 decreased colocalization between SQSTM1-positive structures and GFP(LC3)-positive puncta relative to Baf A1-treated cells (Figure 4E). Co-treatment of Baf A1 with epoxomicin, however, did not alter colocalization between SQSTM1-positive structures and GFP(LC3)-positive puncta relative to Baf A1-treated cells (Figure 4E). We later discover that this finding may be due to a heterogeneity in SQSTM1 phenotypes (described in the next section) with a population of astrocytes exhibiting increased colocalization and a population exhibiting decreased colocalization, leaving the average similar to Baf A1-alone. Combined, we find that long-term UPS inhibition does not significantly upregulate autophagic flux and diverts SQSTM1 away from degradative compartments and into aggresomes. Moreover, our results with long-term UPS inhibition are consistent with those from short-term UPS inhibition where we observed a decrease in SQSTM1 routed to autophagy for degradation and an increase in SQSTM1 that forms fibril-like structures.
Astrocytes exhibit diverse SQSTM1 structures with distinct differences in overlap with autophagosomes in response to UPS inhibition with epoxomicin
We noticed that area occupied by SQSTM1-positive structures (including puncta and aggresomes) increased more with MG132 than with epoxomicin treatment (Figure 4D). Upon closer examination we noticed that epoxomicin resulted in a heterogeneous population of SQSTM1 phenotypes. Some astrocytes exhibited only SQSTM1-positive puncta whereas other astrocytes displayed SQSTM1-positive aggresomes, or an intermediate phenotype of SQSTM1-positive small aggregates (Figure 5A). Therefore, we quantified the percentage of astrocytes with each SQSTM1 phenotype across each treatment of long-term UPS inhibition; representative images for each category are shown in Figure 5A. As expected, DMSO and Baf A1-treated astrocytes exhibited only SQSTM1-positive puncta (Figure 5B). By contrast, MG132 treatment resulted predominantly in astrocytes with almost only SQSTM1-positive aggresomes (Figure 5B). Epoxomicin, however, resulted in a mixed population of cells representing each SQSTM1 phenotype (Figure 5B). These differences in SQSTM1 phenotypes between MG132 and epoxomicin may be attributed to target specificity [47]. MG132 can inhibit other proteases like calpains and cathepsins, whereas epoxomicin is very specific for the proteasome [47].
Figure 5.
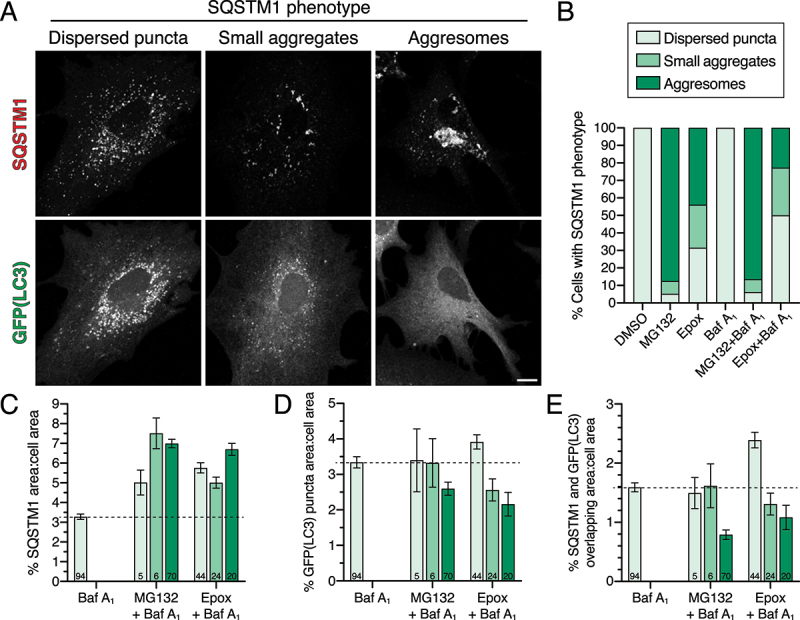
UPS inhibition with epoxomicin moderately elevates SQSTM1 incorporation into autophagosomes in astrocytes lacking aggresomes. (A-E) Immunostain analysis and corresponding quantification of primary astrocytes treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. (A) Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes immunostained for GFP and SQSTM1. Shown are images representing each cell category based on SQSTM1 phenotype; images shown are from the epoxomicin and Baf A1 co-treatment. Bar: 10 µm. (B) Quantification of the percentage of astrocytes displaying each category of SQSTM1 phenotype (same data used as in Figure 4E but Baf A1-treated samples only, n = 81–94 cells from 3 independent experiments; 6–7 DIV). (C) Quantification of total area occupied by SQSTM1-positive structures (includes puncta, smaller aggregates, and aggresomes) normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n-value [total number of cells from 3 independent experiments] is denoted at the base of each bar; 6–7 DIV). (D) Quantification of total GFP(LC3)-positive puncta area normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n-value [total number of cells from 3 independent experiments] is denoted at the base of each bar; 6–7 DIV). (E) Quantification of the percentage of overlapping area between GFP(LC3)-positive puncta and SQSTM1-positive structures normalized to cell area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n-value [total number of cells from 3 independent experiments] is denoted at the base of each bar; 6–7 DIV).
Next, based on the SQSTM1 phenotype, we parsed out the measurements for total area of SQSTM1-positive structures, GFP(LC3)-positive puncta, and SQSTM1-LC3 overlap. For this analysis, we focused on Baf A1-treated samples to more completely capture the different types of SQSTM1-positive structures present in astrocytes. Co-treatment of Baf A1 with epoxomicin or MG132 increased total SQSTM1 area relative to Baf A1 alone for all structures from puncta to aggresomes (Figure 5C). Interestingly, astrocytes treated with epoxomicin and Baf A1 that exhibited only SQSTM1 puncta had a slight increase in total GFP(LC3)-positive puncta area relative to Baf A1 alone, suggesting a modest induction of autophagic flux with UPS inhibition (Figure 5D). Furthermore, astrocytes treated with epoxomicin and Baf A1 that exhibited only SQSTM1 puncta had an increase in SQSTM1 colocalization with GFP(LC3)-positive puncta relative to Baf A1 alone (Figure 5E), suggesting that more SQSTM1 was routed to autophagy for degradation with UPS inhibition. Because MG132-treated astrocytes that exhibited only SQSTM1 puncta were few in number (5 out of 81 cells), the measurement error was too large to discern specific effects (Figure 5D). In contrast to astrocytes that displayed a punctate SQSTM1 phenotype, astrocytes with SQSTM1-positive aggresomes (with epoxomicin or MG132) exhibited a decrease in total GFP(LC3)-positive puncta area, suggesting a reduction in autophagic flux with UPS inhibition in cells with aggresomes (Figure 5D). Furthermore, astrocytes with SQSTM1-positive aggresomes (with epoxomicin or MG132) exhibited a decrease in overlap between SQSTM1 and LC3-positive puncta (Figure 5E), suggesting a routing of SQSTM1 to aggresomes rather than autophagosomes. Thus, we find that treatment with epoxomicin results in a heterogenous SQSTM1 phenotype that includes astrocytes that increase targeting of SQSTM1 to autophagosomes for degradation and astrocytes with reduced autophagic flux that divert SQSTM1 away from autophagosomes for accumulation in aggresomes.
Ubiquitin is enriched in SQSTM1-positive aggresomes as compared with SQSTM1-positive autophagosomes in astrocytes with UPS inhibition
We next wanted to further characterize astrocytes exhibiting different SQSTM1 phenotypes in response to UPS inhibition by immunostain for ubiquitin. We measured colocalization between GFP(LC3)-positive puncta, SQSTM1-positive structures, and ubiquitin using line scans of representative images (Figure 6). In the DMSO control, GFP(LC3)-positive puncta largely colocalized with SQSTM1-positive puncta (Figure 6A). Ubiquitin was enriched in the nucleus and not detected on autophagosomes in the cytoplasm that were co-positive for GFP(LC3) and SQSTM1 (Figure 6A). The same pattern of colocalization was observed with Baf A1-treatment, however more puncta co-positive for GFP(LC3) and SQSTM1 were detected as compared to the DMSO control (Figure 6E). Autophagosomes co-positive for SQSTM1 and ubiquitin were detected, albeit at a low frequency (Figure 6E). With UPS inhibition, ubiquitin redistributed from the nucleus to the cytoplasm (Figure 6B-D, F-H), as we observed with short-term UPS inhibition (Figure 2A). In astrocytes that formed SQSTM1 aggresomes in response to UPS inhibition (with MG132 or epoxomicin), ubiquitin colocalized with SQSTM1-positive aggresomes (Figure 6B, D, F, H). GFP(LC3)-positive puncta were found throughout the cytoplasm and in gaps within the perinuclear aggresome signal (Figure 6B, D, F, H). However, these GFP(LC3)-positive puncta exhibited a reduced overlap with SQSTM1-positive puncta and were, surprisingly, largely negative for ubiquitin. In fact, because cytosolic levels of ubiquitin increased dramatically with UPS inhibition, LC3 puncta were positioned in empty spaces, or in “holes”, in the cytoplasmic ubiquitin signal (Figure 6B, D, F-H). This effect was most apparent in astrocytes co-treated with UPS inhibitors and Baf A1 (Figure 6F, H). Thus, these results suggest that the SQSTM1-Ub-positive conjugates of the aggresome are not productive substrates for autophagy. These results corroborated our prior quantification showing reduced overlap between GFP(LC3) puncta and SQSTM1 in astrocytes with aggresomes (Figures 4 and 5E). In astrocytes that formed SQSTM1 puncta in response to UPS inhibition (with epoxomicin), we observed similar patterns of LC3-SQSTM1 puncta overlap as the DMSO solvent control, but at a higher frequency that is most evident in Baf A1-treated samples (Figure 6C, G). These GFP(LC3) and SQSTM1 co-positive puncta were largely negative for ubiquitin (Figure 6C, G). In fact, these autophagosomes were detected in the holes of the cytoplasmic ubiquitin signal (Figure 6C, G). Combined, these results reveal a differential regulation of SQSTM1-ubiquitin conjugates in response to UPS inhibition in astrocytes, and identify different populations of SQSTM1 that are either routed to autophagy for degradation or to aggresomes, the latter population being enriched for ubiquitin.
Figure 6.
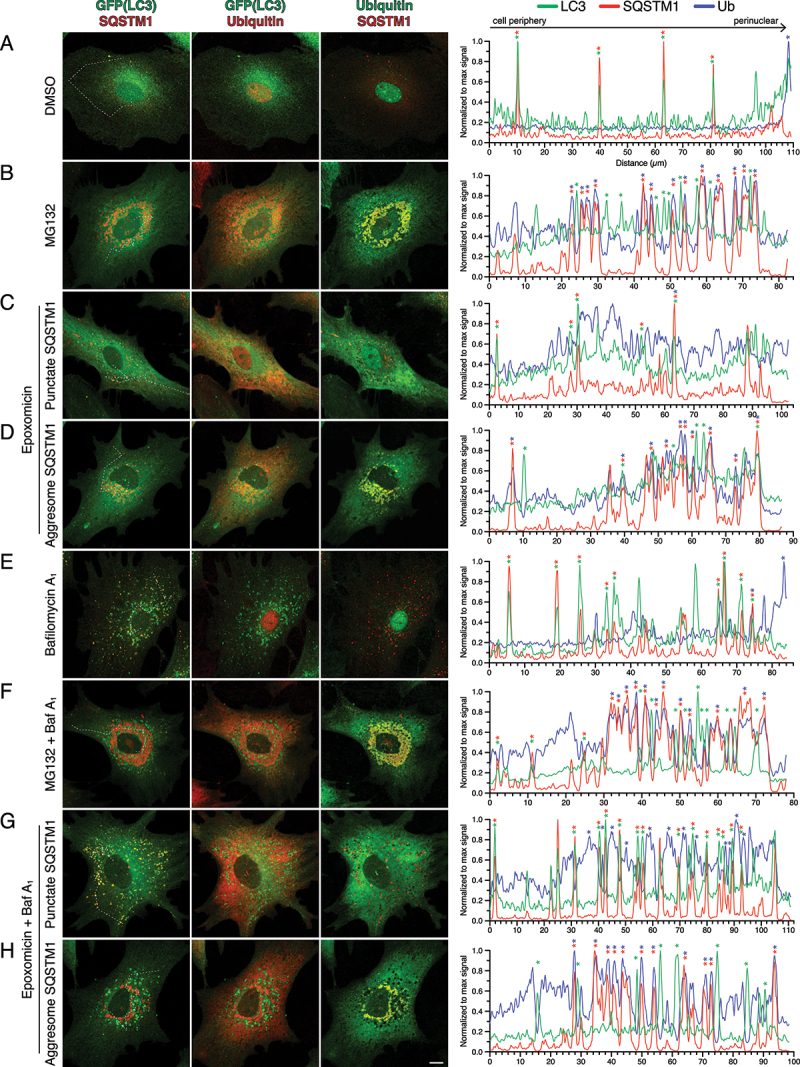
Ubiquitin is enriched on SQSTM1-positive aggresomes as compared with SQSTM1-positive puncta in astrocytes. (A-H) Immunostain analysis of primary astrocytes (3–8 DIV) treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes immunostained for GFP, SQSTM1, and ubiquitin. Corresponding line scans are drawn from the cell periphery toward the perinuclear region (left to right); asterisks indicate puncta or aggresomes with overlapping markers. Dashed line in maximum projection denotes location of line scan. Bar: 10 µm.
LAMP1 is enriched on SQSTM1-positive autophagosomes as compared with SQSTM1-positive aggresomes in astrocytes with UPS inhibition
We next set out to further assess whether SQSTM1 or the SQSTM1-Ub conjugates of the aggresome are reaching a degradative compartment in astrocytes during UPS inhibition by immunostaining for LAMP1, a marker for late endosomes and lysosomes (Figure 7). In the DMSO solvent, GFP(LC3)-positive puncta in the periphery were largely co-positive for SQSTM1 and negative for LAMP1, whereas autophagosomes co-positive for LAMP1 were enriched in perinuclear regions (Figure 7A). Compartments triple positive for GFP(LC3), LAMP1, and SQSTM1 were most effectively captured with Baf A1-treatment (Figure 7A, E). Combined, these results suggest that under basal conditions, a population of SQSTM1 is targeted to autophagosomes for degradation, and these compartments mature as autophagosomes move centripetally from the cell periphery to perinuclear regions. In astrocytes that developed aggresomes with UPS inhibition (with MG132 or epoxomicin), GFP(LC3)-positive puncta frequently overlapped with LAMP1-positive puncta, however, these compartments were largely negative for SQSTM1 (Figure 7B, D, F, H). These compartments did not overlap with SQSTM1-positive aggresomes and were distributed throughout cytoplasm and in the gaps of perinuclear SQSTM1-positive aggresomes (Figure 7B, D, F, H). This phenotype was most evident in astrocytes treated with MG132 or epoxomicin in combination with Baf A1 (Figure 7F, H). Thus, we find that SQSTM1-positive aggresomes were largely negative for LAMP1, indicating a compartment distinct from degradative organelles. In astrocytes that develop SQSTM1 puncta with UPS inhibition (with epoxomicin), we observed a significant population of puncta triple positive for GFP(LC3), SQSTM1, and LAMP1 in both epoxomicin alone, as well as in co-treatment with Baf A1 (Figure 7C, G). Combined, we find distinct trafficking itineraries for SQSTM1 in response to UPS inhibition; astrocytes that accumulate SQSTM1 puncta which are targeted to degradative compartments, and those that accumulate SQSTM1 in aggresomes which have a reduced targeting to degradative compartments.
Figure 7.
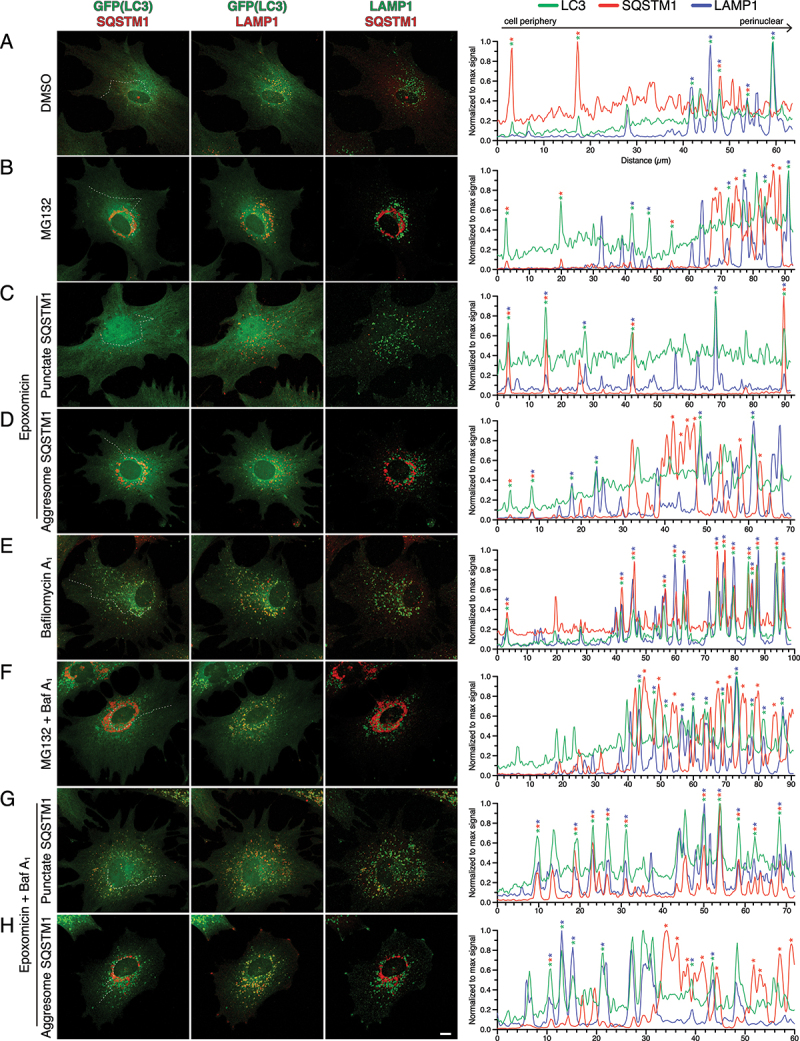
LAMP1 is enriched on SQSTM1-positive puncta as compared with SQSTM1-positive aggresomes in astrocytes. (A-H) Immunostain analysis of primary astrocytes (5–7 DIV) treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. Maximum projections of z-stacks of GFP-LC3 transgenic astrocytes immunostained for GFP, SQSTM1, and LAMP1. Corresponding line scans are drawn from the cell periphery toward the perinuclear region (left to right); asterisks indicate puncta or aggresomes with overlapping markers. Dashed line in maximum projection denotes location of line scan. Bar: 10 µm.
LC3 in aggresomes is likely cytosolic LC3 and not autophagosome-associated LC3
We did note that some images of GFP(LC3) showed an enrichment of LC3 in the perinuclear area in astrocytes with SQSTM1-positive aggresomes (Figure 4A; MG132-treated astrocytes). Thus, we were interested to further define these structures and to distinguish whether this LC3 population was cytosolic LC3-I or autophagosome-associated LC3-II. Thus, we knocked down expression of ATG5 using siRNAs to reduce cellular LC3-II levels. ATG5 is part of the conjugation machinery that leads to lipidation of cytosolic LC3-I to form autophagosome-associated LC3-II [53,54]. Immunoblot analysis of astrocytes transfected with siRNAs targeting Atg5 (a pool of two individual siRNAs) revealed a 50% reduction in ATG5 protein levels as compared to astrocytes treated with a mock transfection as a control (Figure 8A, B). This reduction in ATG5 expression was effectively maintained after 24 h incubation with MG132 or epoxomicin, and after 4 h incubation with Baf A1 or EBSS (a salt solution used to induce autophagy). Knockdown of ATG5 expression also elicited a reduction in LC3-II levels and a corresponding increase in LC3-I levels as compared to mock controls (Figure 8A, C, D). These effects were maintained across all treatment paradigms tested (Figure 8A, C, D). In fact, the reduction in LC3-II levels was maintained in astrocytes treated with MG132 or epoxomicin for 24 h (Figure 8A, C, D). Combined, these results indicate effective reduction in ATG5 protein expression and function in primary astrocytes. Interestingly, knockdown of ATG5 did not change SQSTM1 levels in astrocytes treated with MG132 or epoxomicin relative to their respective mock controls (Figure 8A, E). These results suggest that autophagy may not have a major role in SQSTM1 clearance in response to 24 h of UPS inhibition, or the remaining ATG5 protein is sufficient to shuttle SQSTM1 through the autophagy pathway.
To gain a higher resolution of the effects of ATG5 knockdown on autophagy levels in astrocytes, we performed live-cell imaging of GFP-LC3. First, we investigated several controls to further validate the effectiveness of the knockdown of ATG5. In the DMSO solvent control, knockdown of ATG5 increased cytosolic LC3 signal relative to the mock control, as evidenced by increased microtubule-localization, albeit a few GFP-LC3-positive puncta remained (Figure 8F). These results are consistent with the increase in LC3-I levels by immunoblot (Figure 8A, C). Next, we accumulated autophagosomes by blocking their clearance with Baf A1. We observed punctate structures in siRNA-treated cells with Baf A1, however, LC3 signal on the puncta was reduced dramatically as compared with the mock control (Figure 8F). These results are consistent with the reduction in LC3-II levels by immunoblot (Figure 8A, D). We also induced autophagy by starving cells in EBSS; a paradigm we previously showed to robustly activate autophagy in astrocytes [22]. ATG5 knockdown cells still generated punctate GFP-LC3-positive structures in response to EBSS (Figure 8F). However, these autophagosomes were often reduced in size and not as large as those generated in the mock control starved in EBSS (Figure 8F). Additionally, with EBSS treatment, we observed an increase in cytosolic LC3 (localized to microtubules) in siRNA-treated cells as compared with the mock control (Figure 8F). In total, our live-cell imaging data corroborates our immunoblot analysis in that Atg5 siRNA treatment reduces ATG5 function as evidenced by increased cytosolic LC3 and decreased punctate (autophagosome-associated) LC3.
Because ATG5 knockdown effectively reduced autophagosome-associated LC3 levels in astrocytes, we next wanted to determine whether the enrichment of LC3 in the perinuclear area in astrocytes with SQSTM1-positive aggresomes was cytosolic or autophagosome-associated LC3. With live-cell imaging of GFP-LC3, as compared to immunostain, we observed a clearer localization of GFP-LC3 to aggresomes in response to UPS inhibition, but aggresomes were present in only 40–50% of astrocytes (Figure 8F). ATG5 knockdown in UPS-inhibited astrocytes, however, significantly increased the intensity of GFP-LC3 signal on aggresomes, and increased the percentage of cells exhibiting GFP-LC3-positive aggresomes (Figure 8F). Because ATG5 knockdown in UPS-inhibited cells increased LC3-I and not LC3-II levels by immunoblot (Figure 8A, C, D), our results suggest that this increase in GFP-LC3 associated with aggresomes is likely cytosolic LC3.
To further investigate whether the LC3 localized to aggresomes is less mature, cytosolic forms of LC3 or more mature, autophagosome-associated forms of LC3, we performed live cell imaging of mCherry-LC3 (a more acid-stable version of LC3 that will label structures positive for GFP-LC3, but also mature, degradative autolysosomes where the GFP fluorescence of GFP-LC3 would be quenched). To label aggresomes, we performed dual-color imaging with GFP-Ubiquitin (GFP-Ub). Consistent with the immunostain for ubiquitin (Figures 2 and 6), GFP-Ub was enriched in the nucleus in the DMSO solvent control (Figure 8G). Moreover, UPS inhibition with MG132 or epoxomicin redistributed GFP-Ub from the nucleus to perinuclear aggresomes (Figure 8G). Thus, live-cell imaging with GFP-Ub corroborated results from the immunostain of ubiquitin. Next, we investigated the distribution of mCherry-LC3 relative to GFP-Ub in response to UPS inhibition. In the DMSO control, only a few mCherry-LC3 compartments colocalized with GFP-Ub; these compartments were more evident with Baf A1 (Figure 8G). With UPS inhibition, mCherry-LC3 did not localize to ubiquitin-positive aggresomes (Figure 8G). Rather, mCherry-LC3-positive compartments were distributed throughout the cytoplasm and in gaps of perinuclear aggresomes (Figure 8G). These results are distinct from our live-cell imaging of GFP-LC3 that exhibited a degree of localization to aggresomes (Figure 8F). Furthermore, treatment with MG132 or epoxomicin did not affect the levels of mCherry-LC3-positive puncta. Combined, these results suggest that the LC3 population that associates with aggresomes is likely immature cytosolic forms of LC3, and not processed, mature forms of LC3 that associate with autophagosomes and degradative autolysosomes. Incorporation of cytosolic LC3 into aggresomes may reduce the LC3-I pool available for lipidation and autophagosome formation, thereby reducing autophagic flux in astrocytes with aggresomes.
Lastly, to further investigate whether UPS inhibition stimulates autophagy, we measured the activity state of an upstream signaling kinase, ULK1, that initiates autophagosome production. Kim et al. demonstrated that ULK1 is phosphorylated at serine 757 (S757) by MTOR; phosphorylation at S757 inhibits ULK1 kinase and minimizes autophagy activation [55]. With nutrient deprivation, MTOR is inhibited which relieves phosphorylation at S757 and leads to activation of ULK1 and induction of autophagy [55]. Therefore, we assessed whether UPS inhibition affected the presence of the inhibitory phosphorylation at S757 on ULK1 by immunoblot analysis. Consistent with Kim et al. (2011), starvation of astrocytes in EBSS dramatically reduced the inhibitory phosphorylation on ULK1 at S757 (Figure 8H). Interestingly, UPS inhibition had only a modest reduction in phospho-ULK1 (S757), indicating that the inhibitory phosphorylation at S757 is largely present on ULK1 (Figure 8H). By contrast, UPS inhibition led to a dramatic increase in total SQSTM1 levels (Figure 8H), thus validating the effectiveness of the drug treatments. Combined, our results suggest that ULK1 kinase activity may not be robustly stimulated in UPS inhibition, particularly when compared to starvation, which may minimize autophagy induction.
Long-term UPS inhibition does not significantly upregulate autophagic flux in primary neurons
Next, we wanted to determine the effects of UPS inhibition on autophagy in primary neurons. For this experiment, we first performed a titration curve of epoxomicin concentrations from 1 to 10 nM for 24 h and analyzed primary cortical neurons by immunoblot. For comparison, we also loaded lysates generated from primary cortical astrocytes treated with 10 nM epoxomicin for 24 h or 100 nM Baf A1 for 4 h. With 1 nM epoxomicin, we observed no change in the levels of ubiquitinated protein of higher molecular weight as compared to the DMSO control in neurons (Figure 9A, B). With 2.5 nM epoxomicin, however, we observed a significant increase in ubiquitinated protein in neurons as compared to the DMSO control (Figure 9A, B). This effect was reduced with higher concentrations of 5 nM epoxomicin and further reduced with 10 nM epoxomicin (Figure 9A, B). In sum, these results indicate a very narrow range for the optimum concentration of epoxomicin for effective inhibition of the UPS in neurons. Strikingly, epoxomicin had significantly lower accumulation of ubiquitinated protein in neurons as compared to astrocytes (Figure 9A). These results suggest that neurons may have a reduced flux of material to the UPS for degradation as compared to astrocytes, or that the effects of UPS inhibition in neurons may be restricted to the soma and consequently masked when normalized to total protein distributed throughout the neuron. We also examined levels of autophagy in neurons with epoxomicin treatment and observed no change in autophagosome-associated LC3-II or cytosolic LC3-I levels, indicating no robust activation of autophagy in response to UPS inhibition (Figure 9A, C, D). We did, however, observe an increase in SQSTM1 with epoxomicin treatment as compared to the DMSO control in neurons, albeit a more modest effect as compared to that in astrocytes (Figure 9A, E). Combined, we find that epoxomicin reduces UPS activity in primary neurons, but does not elicit a robust upregulation of autophagy as measured by immunoblot analysis.
Figure 9.
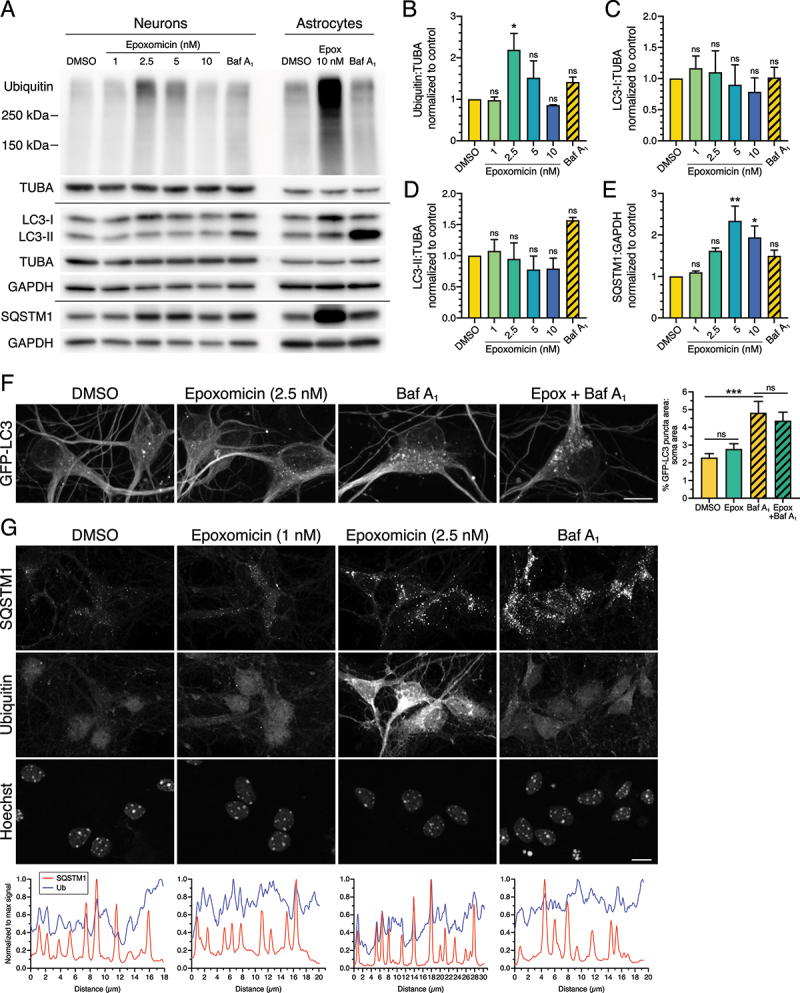
UPS inhibition has a reduced effect on SQSTM1 aggresome formation in primary cortical neurons. (A-E) Immunoblot analysis and corresponding quantification of increasing concentrations of epoxomicin (24-h treatment) as compared with 100 nM Baf A1 for 4 h in primary mouse cortical neurons. Astrocytes were treated with 10 nM epoxomicin for 24 h or 100 nM Baf A1 for 4 h; 3 DIV. GAPDH and TUBA/α-tubulin serve as loading controls; horizontal lines designate individual blots. ubiquitin, LC3-I, and LC3-II levels were normalized to TUBA/α-tubulin, and SQSTM1 levels were normalized to GAPDH (means ± SEM; one-way ANOVA with Dunnett’s post hoc test; n = 3 independent experiments; 8 DIV). Differences in Ub and SQSTM1 levels in the dosage response at 5 nM epoxomicin may reflect issues with toxicity and normalization to GAPDH. (F) Maximum projections of z-stacks of GFP-LC3 transgenic mouse cortical neurons using live cell imaging. Neurons were treated with 2.5 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. Bar: 10 µm. Corresponding quantification of total GFP-LC3 puncta area normalized to soma area (means ± SEM; one-way ANOVA with Tukey’s post hoc test; n = 53–64 neurons from 3 independent experiments; 7 DIV). (G) Immunostain and corresponding line scan analysis of primary mouse cortical neurons (7 DIV) treated with DMSO, 1 nM or 2.5 nM epoxomicin for 24 h, or 100 nM Baf A1 for 4 h. Images with the same marker (SQSTM1, Ub, or Hoechst) are grayscale matched to facilitate direct comparisons across treatments. Bar: 10 µm.
To more thoroughly confirm this lack of autophagy induction in neurons with UPS inhibition, we tested higher concentrations of epoxomicin ranging from 10 nM to 100 nM (Fig. S4). With higher concentrations, we did not observe any statistically significant changes in ubiquitin, LC3-II, LC3-I, or SQSTM1 in response to UPS inhibition as compared to the DMSO control (Fig. S4A-E). Only treatment with Baf A1 accumulated ubiquitinated protein and autophagosome-associated LC3-II as compared to the DMSO control, providing a positive control for the assay (Fig. S4A, B, D). Moreover, the effects of epoxomicin on ubiquitin and SQSTM1 levels in neurons were dramatically lower than those observed in astrocytes with UPS inhibition (Fig. S4A). Combined, we did not observe a significant activation of autophagy at higher concentrations of epoxomicin as measured by immunoblot analysis. We did note significant toxicity issues at concentrations of ≥10 nM epoxomicin, suggesting a higher sensitivity to epoxomicin in neurons as compared with astrocytes. Thus, because ubiquitin levels peaked with 2.5 nM epoxomicin, we utilized this concentration (and 1 nM epoxomicin as a negative control) for subsequent analysis by live-cell imaging and immunostain.
To achieve higher resolution analysis of autophagy levels, we performed live-cell imaging of GFP-LC3 in primary neurons in response to 24-h treatment with 2.5 nM epoxomicin plus or minus Baf A1 to measure autophagic flux. For this analysis, we focused on the somal compartment of primary neurons. We found that treatment with epoxomicin-alone only slightly increased the area of GFP-LC3-positive autophagosomes as compared to the DMSO solvent in neurons (Figure 9F). Across our study, we have consistently observed that UPS inhibition elicits only a modest increase in steady state levels, but this effect was not significant nor did it result in increased autophagic flux (Figure 9F).
Because we observed more dramatic effects on SQSTM1 and ubiquitin as compared to LC3 in astrocytes in response to UPS inhibition, we next analyzed neurons treated with epoxomicin by immunostain for SQSTM1 and ubiquitin. In the DMSO control, we observed SQSTM1 puncta in the cytoplasm, and ubiquitin was distributed largely to the nucleus (Figure 9G). We did note that nuclear enrichment of ubiquitin in the DMSO control was more evident in astrocytes as compared with neurons (Figure 9G). As expected based on the immunoblot results, 1 nM epoxomicin did not have a large effect on SQSTM1 or ubiquitin in neurons (Figure 9G). Treatment with 2.5 nM epoxomicin, however, increased SQSTM1 puncta and dramatically increased cytosolic ubiquitin signal relative to the DMSO control (Figure 9G), consistent with the immunoblot. Epoxomicin treatment, however, did not lead to the formation of SQSTM1-positive aggresomes in neurons (Figure 9G). Baf A1 also increased SQSTM1 puncta in neurons due to a block in degradative flux, and increased ubiquitin puncta in the cytoplasm (Figure 9G). Across treatments, SQSTM1 puncta were co-positive for ubiquitin, but the strongest overlapping peaks were observed with 2.5 nM epoxomicin (Figure 9G). Thus, UPS inhibition in neurons increases puncta co-positive for SQSTM1 and ubiquitin, but does not elicit aggresome formation.
In finding that 2.5 nM epoxomicin was the ideal concentration to inhibit the UPS in neurons, we wanted to perform a parallel study in astrocytes and determine the effects of 2.5 nM epoxomicin on autophagy in astrocytes. Treatment of astrocytes with 2.5 nM epoxomicin increased cytoplasmic ubiquitin, albeit not as dramatically as with 10 nM epoxomicin (Fig. S5A). Epoxomicin (2.5 nM) also increased cytosolic SQSTM1, the total area occupied by SQSTM1-positive puncta, and the intensity of SQSTM1 signal on puncta relative to the DMSO control (Fig. S5A, C). However, we did not observe SQSTM1-positive aggresomes in astrocytes treated with 2.5 nM epoxomicin (Fig. S5A). We also did not observe a corresponding increase in GFP(LC3)-positive puncta with 2.5 nM epoxomicin (Fig. S5A, B). Epoxomicin (2.5 nM) increased the degradative flux of SQSTM1, as evidenced by an increase in SQSTM1 puncta area with epoxomicin with Baf A1 relative to Baf A1-alone, but we did not observe a corresponding increase in autophagic flux (Fig. S5A, B, C). Consistent with a stimulation in SQSTM1 degradative flux, we observed an increase in area of puncta co-positive for SQSTM1 and GFP(LC3) when normalized to cell area, as well as to GFP(LC3) puncta area (Fig. S5A, D, E). Combined, these data suggest that treatment with 2.5 nM epoxomicin increases the routing of SQSTM1 to autophagosomes for degradation relative to the DMSO control. These results are reminiscent of astrocytes with a punctate SQSTM1 phenotype in response to 10 nM epoxomicin (Figures 5 and 6). Our results in astrocytes are similar to neurons in that 2.5 nM epoxomicin elicits the formation of SQSTM1-positive puncta and not aggresomes. However, these SQSTM1 puncta in neurons are more strongly positive for ubiquitin as compared with astrocytes (Figure 9 and S5A), suggesting a cell type-specific regulation of ubiquitination in response to UPS inhibition.
Long-term UPS inhibition stimulates lysosomal degradation in astrocytes
Next, we wanted to determine the effect of long-term UPS inhibition on lysosomal function in astrocytes. For this experiment, we inhibited UPS activity with 0.5 µM MG132 or 10 nM epoxomicin for 24 h and measured lysosomal function using a DQ-BSA substrate. DQ-BSA is an endocytosed BSA cargo that is fluorescently labeled at such a high density, it self-quenches. Proteolytic cleavage of the BSA in lysosomes relieves the self-quenching and activates fluorescence. Thus, fluorescence from DQ-BSA serves as a readout for proteolytically-active lysosomes.
In the DMSO control, we observed DQ-BSA-positive puncta distributed within the cytoplasm, concentrated in a perinuclear location (Figure 10A). Surprisingly, treatment with MG132 reduced fluorescence signal from DQ-BSA ~2.5-fold relative to the DMSO control (Figure 10A). Conversely, treatment with epoxomicin increased fluorescence signal from DQ-BSA ~1.4-fold relative to the DMSO control (Figure 10A). As a control, we also incubated astrocytes for 4 h in 100 nM Baf A1 to neutralize lysosomes and block proteolytic degradation of the DQ-BSA. Consistent with this effect, Baf A1 decreased fluorescence signal from DQ-BSA ~3.3-fold relative to the DMSO control (Figure 10A). In total, we observed opposing effects of MG132 versus epoxomicin on DQ-BSA fluorescence.
Figure 10.
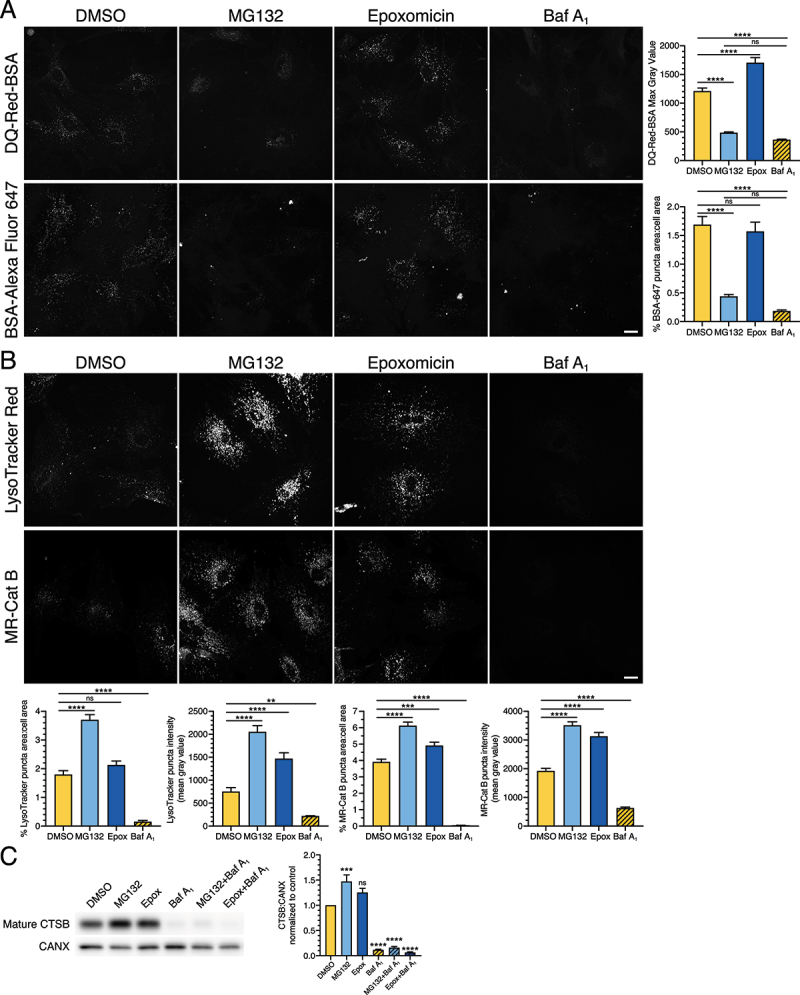
Long-term UPS inhibition stimulates lysosome function in astrocytes. (A and B) Live-cell imaging analysis of primary astrocytes treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h, or 100 nM Baf A1 for 4 h. All images within the same marker are grayscale matched to facilitate direct comparisons. (A) Maximum projections of z-stacks and corresponding quantification of astrocytes labeled with DQ-Red-BSA or BSA-Alexa Fluor 647 (BSA-647) for the final 1 h of the indicated treatment (means ± SEM; one-way ANOVA with Tukey’s post hoc test; DQ-Red-BSA, n = 95–121 cells from 3 independent experiments; BSA-647, n = 85–96 cells from 3 independent experiments; 5–8 DIV). Bar: 20 µm. (B) Maximum projections of z-stacks and corresponding quantitation of astrocytes labeled with LysoTracker Red or Magic-Red Cathepsin B substrate for the final 30 min of the indicated treatment (means ± SEM; one-way ANOVA with Tukey’s post hoc test; LysoTracker, n = 69–76 cells from 3 independent experiments; MR-Cat B, n = 72–97 cells from 3 independent experiments; 7–10 DIV). Bar: 20 µm. (C) Immunoblot analysis and corresponding quantification of primary astrocytes treated with DMSO, 0.5 µM MG132 or 10 nM epoxomicin for 24 h; 100 nM Baf A1 (or equivalent volume of DMSO as a solvent control) was included in the last 4 h. Mature CTSB levels were normalized to CANX (means ± SEM; one-way ANOVA with Dunnett’s post hoc test; n = 3–4 independent experiments; 6–9 DIV).
An increase in DQ-BSA fluorescence can be attributed to either a stimulation in lysosomal activity, or an increase in endocytic delivery of the BSA substrate to lysosomes. Conversely, a decrease in DQ-BSA fluorescence can be attributed to either a decrease in lysosomal activity, or a decrease in endocytic delivery of the BSA substrate to lysosomes. To distinguish among these possibilities, we labeled endolysosomal compartments by incubating astrocytes with BSA-Alexa Fluor 647, an endocytic cargo that fluoresces all along the endolysosomal pathway, labeling early endosomes to lysosomes. In the DMSO control, we observed BSA-Alexa Fluor 647-positive puncta distributed throughout the cytoplasm, concentrated in a perinuclear location (Figure 10A). Surprisingly, MG132 reduced the total area occupied by BSA-Alexa Fluor 647-positive puncta relative to the DMSO control, suggesting a decrease in endocytic uptake and/or maturation (Figure 10A). Thus, the loss in DQ-BSA signal with MG132 treatment was due to a decrease in substrate delivery to lysosomes. Combined with our earlier observations of decreased autophagic flux (Fig. S2), our data indicate that MG132 may have off-target effects causing defects in trafficking along the endocytic pathway. Consistent with this possibility, LAMP1 signal increased and appeared more aggregated with only MG132 (short-term and long-term treatment), but not with epoxomicin (Fig. S6). Surprisingly, Baf A1 also reduced BSA-Alexa Fluor 647 puncta in astrocytes (Figure 10A), suggesting that Baf A1 also decreases endocytic maturation and the loss in DQ-BSA signal was not simply attributed to a loss of lysosomal acidity. Epoxomicin, however, had no effect on the total area of BSA-Alexa Fluor 647-positive puncta relative to the DMSO control. Thus, the stimulation in DQ-BSA fluorescence with epoxomicin is likely due to an increase in lysosomal function, rather than an increase in substrate delivery to lysosomes.
To further define the effects of UPS inhibition on lysosome function, we measured the density of acidic compartments using the cell-permeant dyes, LysoTracker Red and Magic-Red Cathepsin B (MR-Cat B). Because these dyes are cell-permeant and not dependent on endocytosis for delivery to target compartments, they will more completely label acidic organelles in the endolysosomal pathway. For this analysis, we measured total area occupied by LysoTracker Red-positive or MR-Cat B-positive puncta, as well as the mean fluorescence intensity for LysoTracker Red-positive or MR-Cat B-positive puncta. With UPS inhibition, we observed an increase in total puncta area and/or the mean intensity of acidic compartments labeled with LysoTracker Red or MR-Cat B. MG132 increased the total area and mean intensity of LysoTracker Red puncta ~2.1-fold and 2.7-fold, respectively, relative to the DMSO control (Figure 10B). Epoxomicin increased the total area and mean intensity of LysoTracker Red puncta ~1.2-fold and ~1.9-fold, respectively, relative to the DMSO control; only the effect on puncta mean intensity was statistically significant (Figure 10B). MG132 also increased the total area and mean intensity of MR-Cat B puncta ~1.6-fold and 1.8-fold, respectively, relative to the DMSO control (Figure 10B). Epoxomicin increased the total area and mean intensity of MR-Cat B puncta ~1.3-fold and ~1.6-fold, respectively, relative to the DMSO control (Figure 10B). As expected, across all the measurements, neutralization of lysosomes with Baf A1 reduced the total area occupied by, and mean intensity of, acidic compartments (Figure 10B). Combined, we find that long-term inhibition of UPS activity increases acidic compartments in astrocytes. These alterations in lysosomes are also observed after short-term inhibition of UPS activity, however, the effects are not as large as those observed after long-term treatments (Fig. S7). Consistent with an increase in acidic compartments, long-term UPS inhibition also increased levels of mature CTSB (cathepsin B) by immunoblot analysis (Figure 10C). Because maturation of CTSB depends on proteolytic cleavage, eliminating acidic compartments with Baf A1 dramatically reduced levels of mature CTSB (Figure 10C). In sum, the effects of UPS inhibition on lysosomal function are modest, but they are consistently observed across multiple indicators of lysosomal activity. Moreover, we find that UPS inhibition preferentially stimulates lysosome function as compared with autophagic flux in primary astrocytes.
Discussion
The nature of the coordination between two key quality control pathways, the UPS and autophagy, in the main cell types of the brain, neurons and astrocytes, is not well understood. Most studies to date have focused on cellular models of cancerous origin. Here, we address this issue and investigate the balance between the UPS and autophagy in primary cortical astrocytes and perform comparative studies in primary cortical neurons. We find that short-term inhibition of the proteasome in astrocytes using epoxomicin elicits only a modest increase in autophagy as determined by an increase in the size of individual autophagosomes (Figure 1). However, when we measure global autophagosome levels, we observed only a small, but consistent increase in steady state levels of autophagosomes [~1.2-fold increase in GFP(LC3)-positive puncta area with epoxomicin as compared to the DMSO control]. Notably, this effect was not statistically significant, particularly in comparison to the induction of autophagy by starvation in EBSS or MTOR-inhibition for an equivalent duration of time [our prior reports of ~5-fold increases in GFP(LC3)-positive puncta area relative to controls at steady state [22]]. Importantly, epoxomicin did not increase autophagic flux, as measured with Baf A1 by immunoblot or fluorescence-based assays, suggesting that short-term UPS inhibition is not a potent stimulator of autophagy in astrocytes. The more striking effect of short-term UPS inhibition was on the selective autophagy receptor SQSTM1. Short-term UPS inhibition led to the accumulation of SQSTM1 in fibril-like structures that were co-positive for ubiquitin (Figure 2), possibly representing precursors for aggresomes that form in response to long-term UPS inhibition (Figure 4).
Short-term treatment with MG132 did elicit a significant increase in steady state levels of autophagosomes (Fig. S2). However, treatment with MG132 did not stimulate autophagic flux as measured with Baf A1 or chloroquine (Fig. S2 and S3). We noted that the autophagosomes in MG132-treated cells were concentrated in a perinuclear region, more tightly than those in Baf A1-treated cells. This distribution is also distinct from models of robust autophagy activation (e.g. starvation in EBSS) where autophagosome formation is induced throughout the cytoplasm (Fig. S2). Given that MG132 can have off-target effects and inhibit lysosomal proteases [47], we reasoned that the tight perinuclear accumulation of autophagosomes with MG132 might be due largely to a block in autophagosome clearance rather than a stimulation in autophagosome production. Furthermore, even at lower concentrations, MG132 had off-target effects on intracellular trafficking. We found that MG132 reduced compartments positive for the endocytic cargo, BSA-Alexa Fluor 647, consistent with a defect in endocytic uptake and/or maturation (Figure 10). Thus, the perinuclear accumulation of autophagosomes with MG132 may in part also be due to alterations in intracellular trafficking. Combined, our data suggest caution is needed when interpreting autophagy levels with MG132 treatment.
Long-term UPS inhibition resulted in multiple populations of cells defined by distinct structures and trafficking itineraries for SQSTM1 (summarized in Figure 11). On one end of the spectrum, UPS inhibition stimulated the targeting of SQSTM1 to autophagosomes, and ultimately to compartments co-positive for LAMP1 for proteolytic degradation (Figures 5, 6, 7). On the other end of the spectrum, UPS inhibition diverted SQSTM1 away from autophagosomes to accumulate in perinuclear aggresomes (Figures 4 and 5). Aggresomes were enriched for ubiquitin, but negative for LAMP1 (Figures 6 and 7). Surprisingly, astrocytes with SQSTM1-positive aggresomes exhibited a decreased autophagic flux as measured by reduced GFP-LC3-positive puncta by immunostain and LC3-II levels by immunoblot (Figures 3, 4, 5). Moreover, the inhibitory phosphorylation on S757 of ULK1 is largely present in UPS inhibition (Figure 8), which may also minimize autophagy activation. Interestingly, live-cell imaging revealed an enhanced accumulation of GFP-LC3 in aggresomes when autophagy was reduced via siRNA-mediated knockdown of ATG5 (Figure 8). However, despite this increase in GFP-LC3 signal in aggresomes, we did not observe a corresponding increase in LC3-II by immunoblot analysis (Figure 8). As expected, knockdown of ATG5 decreased lipidated autophagosome-associated LC3 and increased cytosolic LC3 (Figure 8), suggesting that the stimulation in LC3 association with aggresomes in siRNA-treated cells may be cytosolic LC3 rather than lipidated autophagosome-associated LC3. These results also raise the possibility that sequestration of cytosolic LC3 in aggresomes may reduce pools available for autophagosome formation, thereby reducing autophagic flux in astrocytes with aggresomes. In summary, UPS inhibition results in distinct trafficking itineraries for SQSTM1. SQSTM1 is either routed to autophagy for proteolytic degradation which results in only a modest upregulation of autophagy, or is diverted away from degradative compartments to accumulate in aggresomes, the latter population being enriched for ubiquitin (Figure 11).
Figure 11.
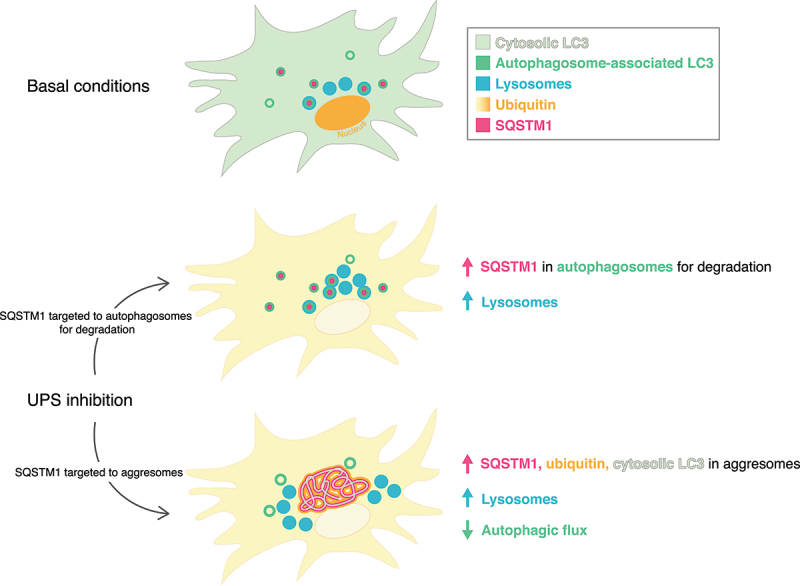
Model for long-term UPS inhibition on autophagy in primary astrocytes. Long-term UPS inhibition elicits multiple phenotypes defined by distinct structures and trafficking itineraries for SQSTM1 that may represent different stages in the cellular response. In one population of astrocytes, SQSTM1 is routed to autophagy for proteolytic degradation, resulting in only a modest activation of autophagy. In another population of astrocytes, SQSTM1 is diverted away from degradative compartments to accumulate in aggresomes that are enriched for ubiquitin and cytosolic LC3. Sequestration of cytosolic LC3 may contribute to the reduction in autophagic flux observed in astrocytes with aggresomes.
The basis for these distinct cellular phenotypes remains unclear, but these populations may represent different stages in the cellular response to UPS inhibition. While UPS inhibitors were applied as a bath treatment, a particular population of astrocytes may be more susceptible to the inhibitors than another, thus staggering the cellular response. Consequently, the initial response to UPS inhibition may be to shuttle SQSTM1 to phagophores for degradation. However, once the system is further challenged, SQSTM1 accumulates in aggresomes. Consistent with this model, MG132 which appears to be a more potent blocker of the proteasome with potential off-target effects that could contribute to cellular stress (as determined by a greater accumulation of ubiquitinated protein with MG132 as compared with epoxomicin; Figure 3A, B), resulted predominantly in astrocytes with aggresomes; intermediate phenotypes were revealed with only epoxomicin.
Interestingly, long-term UPS inhibition increased the presence of acidic, proteolytically-active lysosomes (Figure 10). Thus, astrocytes compensate for the decrease in degradation via proteasomes by upregulating degradation via lysosomes. This stimulation in degradative lysosomes, however, is not sufficient to clear SQSTM1-positive aggresomes within the time frame examined. In fact, blocking lysosome function with Baf A1 did not change total SQSTM1 levels, based on immunoblot analysis of cells treated with UPS-inhibitors (Figure 3A, E). Consistent with these results, reduction of autophagy with siRNAs targeting Atg5 did not increase total SQSTM1 levels by immunoblot analysis (Figure 8). Only when we categorized cells based on SQSTM1 phenotype, did we observe an increase in SQSTM1 degradative flux in cells that exhibited SQSTM1 puncta with epoxomicin treatment (Figure 5C). However, this phenotype represents only ~5% of MG132-treated cells. Although this phenotype represents a larger population of epoxomicin-treated cells, it is countered when pooled with astrocytes that exhibit SQSTM1-positive aggresomes and decreased targeting of SQSTM1 to degradative compartments. The dominant SQSTM1 phenotype in astrocytes in response to UPS inhibition is the formation of aggresomes. In astrocytes with aggresomes, we did observe an increase in SQSTM1 area in UPS-inhibited cells with Baf A1 as compared with Baf A1 alone (Figure 5C). However, this effect is dominated by the large area occupied by aggresomes that are present in steady state conditions, rather than an increase in degradative flux. Consistent with this interpretation, we observed a decrease in overlap between SQSTM1 and LC3 in astrocytes with aggresomes (Figure 5E). Lastly, one of our most striking observations is that while SQSTM1-positive aggresomes are strongly co-positive for ubiquitin, autophagosomes in UPS-inhibited cells are largely negative for SQSTM1-Ub conjugates (Figure 6). In fact, autophagosomes reside within the negative spaces of ubiquitin-enriched cytoplasm (Figure 6). Thus, SQSTM1-ubiquitin conjugates of the aggresome do not appear to be digested by autophagy within the time frame examined. Thus, the increase in lysosome function is not sufficient to eliminate aggresomes, suggesting that autophagy may be a rate limiting step.
Consistent with prior studies in other cell types [35,49–52], UPS inhibition leads to the formation of aggresomes positive for SQSTM1 and ubiquitin in astrocytes (Fig. 4–6). SQSTM1 levels have been shown to increase to concentrate ubiquitinated cargo in a perinuclear aggresome which may serve as a protective response to proteotoxic stress associated with UPS inhibition [49,51,52,56]. Several groups report degradation of SQSTM1-Ub conjugates via autophagy [49,51,57]. However, in primary astrocytes, we do not detect SQSTM1-Ub conjugates in LC3-positive autophagosomes (Figure 6), and the LC3 that localizes to SQSTM1-Ub-positive aggresomes is likely cytosolic LC3 rather than lipidated LC3 (Figure 8). Furthermore, total SQSTM1 levels do not change in the context of UPS inhibition with or without lysosomal degradation (Figure 3A, E) or autophagy (Figure 8A, E). Consistent with results in other studies, we observed a small increase in autophagosomes with UPS inhibition [51,52], but this effect is not accompanied by an increase in autophagic flux (Figure 3). Indeed, with long-term UPS inhibition, autophagic flux decreased (Figure 3). In many studies, flux is not considered. However, in cases where it is, some reports also observe a decrease in flux, although this finding is not emphasized [51,52]. Other studies in neuronal cells also observe an increase in SQSTM1-Ub aggregates in response to UPS inhibition, without a corresponding induction of autophagy [37,38]. It seems likely under our conditions, that at this stage of proteotoxic stress, the aggresome is largely refractory to autophagy. Indeed, some aggresomes cannot be readily degraded by autophagy [58]. It may be that the specific aggresomes that form in astrocytes are also difficult to resolve by autophagic mechanisms.
Neurons respond slightly differently as compared to astrocytes in response to UPS inhibition. UPS inhibition appears to be more toxic in neurons where only a lower threshold can be tolerated (Figure 9 and S4). Moreover, SQSTM1 does not appear to accumulate to the same extent in neurons as compared to astrocytes (Figure 9). At the concentrations tested, we did not observe the formation of SQSTM1-positive aggresomes in neurons. The molecular basis for this cell type-specific aggregation of substrates is unknown. It would be of great interest to define how other autophagy receptors that recognize ubiquitinated protein aggregates, including Alfy, optineurin, NBR1 and TAX1BP1, respond to UPS inhibition in astrocytes [11,56,59,60]. Moreover, proteomic studies of aggresomes isolated from astrocytes will enable a comprehensive atlas of protein composition and potential contributions from other autophagy receptors.
Similar to astrocytes, there is a small but not significant increase in GFP(LC3)-positive puncta in neurons that does not result in an increase in autophagic flux (Figure 9). In non-neural cells, transcription of autophagy and proteasomal genes can be coordinated [61], and induced by UPS inhibition [52,62]. The limited increase in steady state levels of autophagosomes in astrocytes and neurons in response to UPS inhibition may reflect a muted or delayed transcriptional response. Further studies will be needed to explore this possibility.
Combined, our results might indicate a selective vulnerability of neurons as compared to astrocytes with respect to proteotoxic stress. In fact, Tydlacka et al. revealed that UPS activity is lower in neurons versus astrocytes, and declines more rapidly as a function of time in neurons as compared with astrocytes [63]. A limitation of our study is that the observed phenotypes may be influenced by the in vitro nature of the studies which do not fully reflect the complexity of intercellular interactions between neurons and astrocytes in the brain. Nonetheless, in vitro systems have been valuable in gaining mechanistic insights into fundamental differences between neurons and astrocytes. Future studies can build the complexity of our in vitro studies by co-culturing neurons and astrocytes to define non-cell autonomous responses to proteotoxic stress. Elucidating how neurons and astrocytes collaborate to mitigate proteotoxic stress in the brain will enable strategies to combat neurodegenerative diseases associated with aberrant protein misfolding and aggregation [21,64].
Materials and methods
Reagents
Transgenic mice expressing GFP-LC3 (EGFP fused to the C terminus of rat LC3B) were obtained from the RIKEN BioResource Research Center (RBRC00806; strain B6.Cg-Tg(CAG-EGFP/LC3)53 Nmi/NmiRbrc; GFP-LC3#53) and maintained as heterozygotes. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Primary antibodies for immunofluorescence include chicken anti-GFP (Aves Labs, Inc., GFP-1020), mouse anti-SQSTM1/p62 (Abcam, Ab56416; Fig. 1, 4, 5, and S2), rabbit anti-SQSTM1/p62 (Abcam, Ab109012; Fig. 2, 6, 7, 9, and S5), rat anti-LAMP1 (Abcam, Ab25245), and mouse anti-ubiquitin antibodies that recognize mono- and poly-ubiquitination (Enzo Life Sciences, BML-PW8810-0100 for (Figures 2, 6, 9), ENZ-ABS840-0100 for Fig S5). Due to commercial availability, we had to use a different antibody against ubiquitin for Fig. S5. To validate the new antibody, we repeated our prior treatment condition of 10 nM epoxomicin (Enzo Life Sciences, PI127; Sigma, 324,800) as a positive control in Fig. S5. In the DMSO control, ubiquitin was enriched in the nucleus and treatment with 10 nM epoxomicin (plus or minus Baf A1 [Sigma, B1793]) increased cytoplasmic ubiquitin relative to nuclear ubiquitin, and led to the formation of ubiquitin-positive aggresomes (Fig. S5A). Thus, our results with ENZ-ABS840-0100 phenocopied those with BML-PW8810-0100. Secondary antibodies used for immunofluorescence assays include goat anti-chicken Cy2 (Jackson ImmunoResearch Laboratories, 103–225-155), goat anti-mouse Alexa Fluor 594 (Invitrogen, A11032), goat anti-rabbit Alexa Fluor 594 (Invitrogen, A11037), goat anti-rat Alexa Fluor 594 (Invitrogen, A11007), goat anti-mouse Alexa Fluor 647 (Invitrogen, A32728), and goat anti-rabbit Alexa Fluor 647 (Invitrogen, A21245). Primary antibodies for immunoblot analysis include rabbit anti-LC3 (Abcam, Ab48394 and Ab192890), mouse anti-GAPDH (Advanced Immunochemicals, 2-RGM2 clone 6C5), mouse anti-TUBA/α-tubulin (Sigma, T9026; clone DM1A), mouse anti-ubiquitin (Cell Signaling Technology, 3936), mouse anti-SQSTM1/p62 (Abcam, Ab56416), rabbit anti-CTSB/cathepsin B (Cell Signaling Technology, 31,718), rabbit anti-ATG5 (Cell Signaling Technology, 12,994), rabbit anti-CANX/calnexin (Enzo Life Sciences, ADI-SPA-860-D), rabbit anti-phospho-ULK1 at Serine 757 (Cell Signaling Technology, 6888), and rabbit anti-ULK1 (Cell Signaling Technology, 8054). Secondary antibodies for immunoblot analysis include peroxidase-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories, 715–035-151) and donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, 711–035-152). Other small molecules include MG132 (carbobenzoxy-Leu-Leu-leucinal) (Sigma, C2211) and chloroquine (Sigma, C6628). Bafilomycin A1, MG132, and epoxomicin were dissolved in DMSO. Chloroquine was dissolved in sterile milliQ deionized water. Cell permeant dyes include LysoTracker Red (ThermoFisher, L-7528) and Magic Red Cathepsin-B Substrate (Immunochemistry LLC, 937). BSA conjugates include DQ-Red BSA (ThermoFisher, D12051) and BSA-Alexa Fluor 647 (ThermoFisher, A34785). Constructs include mCherry-LC3B [23], GFP-Ubiquitin (Addgene 11,928; deposited by Nico Dantuma), and UbiquitinG76V-GFP (Addgene 11,941; deposited by Nico Dantuma). Lipofectamine 2000 (Thermo Fisher Scientific/Invitrogen, 11,668–027) was used to transfect glia on DIV2 following manufacturer’s protocol.
Primary cortical astrocyte culture
Cerebral cortices were dissected from brains of GFP-LC3 transgenic or non-transgenic neonatal mice of either sex at P0-P1 [65]. Cortical tissue was digested using 0.25% trypsin for 10 min at 37°C, then triturated with a 5-mL pipet to break up large pieces of tissue, and then triturated with a P1000 pipet until a homogenous suspension is achieved. Cells were passed through a 40-µm pore cell strainer (Falcon Brand Products, 352,340) and plated at a density of 2,000,000–3,000,000 cells per 10-cm dish. Glia were plated and grown in glial media (DMEM [Thermo Fisher Scientific/Gibco, 11,965–084] supplemented with 10% heat inactivated fetal bovine serum [Hyclone, SH30071.03], 2 mM Glutamax [Thermo Fisher Scientific/Gibco, 35,050–061], 100 U/ml penicillin and 100 µg/ml streptomycin [Thermo Fisher Scientific/Gibco, 15,140–122]) at 37°C in a 5% CO2 incubator. The day after plating, and every 4–5 days following, glia were fed by replacing the glial media. After reaching 75–90% confluence (~8-10 days after plating), astrocytes were trypsinized and plated for experiments. Prior to media changes, or splitting, the culture dishes were tapped to dislodge any microglia which were then discarded with the media change. Glia were plated at a density of 150,000–500,000 cells per well of a 6-well plate for immunoblotting, or per 35-mm fluorodish (World Precision Instruments, FD35-100) coated with 500 µg/ml poly-L-lysine (Sigma, P2636) for immunofluorescence and live-cell imaging. Astrocytes were grown in glial media for 3–10 DIV at 37°C in a 5% CO2 incubator.
Primary cortical neuron culture
Cortices were dissected from brains of GFP-LC3 transgenic (or non-transgenic littermate) mouse embryos of either sex at day 15.5. Cortical tissue was digested with 0.25% trypsin for 10 min at 37°C, and then triturated to achieve a homogeneous cell suspension. For immunofluorescence and live-cell imaging, neurons were plated at a density of 3,000,000 cells (750,000 GFP-LC3 transgenic cortical neurons diluted with 2,250,000 non-transgenic cortical neurons) per 10-cm dish filled with eight 25-mm acid-washed glass coverslips coated with 1 mg/mL poly-L-lysine (Sigma, P2636). For immunoblotting, 480,000 non-transgenic cortical neurons were plated per well of a 6-well plate. Cortical neurons were maintained for 7–8 DIV in neuron maintenance media (Neurobasal medium [Thermo Fisher Scientific/Gibco, 21–103-049] supplemented with 2% B-27 [Thermo Fisher Scientific/Gibco, 17,504–044], 37.5 mM NaCl, 33 mM glucose [Sigma, G8769], 2 mM glutaMAX, and 100 U/ml penicillin and 100 µg/ml streptomycin) at 37°C in a 5% CO2 incubator. Every 3–4 days, 25% media was replaced; 1 µM AraC (antimitotic drug; Sigma, C6645) was added to the first feed on DIV 3–4.
Proteasomal inhibition in astrocytes
For short-term treatments, astrocytes were incubated for 4 h at 37°C in a 5% CO2 incubator in glial media supplemented with 50 µM MG132, 5 µM epoxomicin, 10 nM epoxomicin, 100 nM bafilomycin A1, or co-treatment of 50 µM MG132 or 5 µM epoxomicin with 100 nM bafilomycin A1. Variations in these treatment conditions (and experiments that used 50 µM chloroquine instead of Baf A1) for Fig. S3 are specifically denoted directly in the figure and legend. Whenever indicated, an equivalent volume of DMSO was included as a solvent control. For long-term treatments, astrocytes were incubated for 20 h at 37°C in a 5% CO2 incubator in glial media supplemented with 2.5 nM or 10 nM epoxomicin, 500 nM MG132, or an equivalent volume of DMSO as a solvent control. After 20 h, the media was removed and replaced with fresh glial media supplemented with 2.5 nM or 10 nM epoxomicin, 500 nM MG132, 100 nM bafilomycin A1, 2.5 nM or 10 nM epoxomicin with 100 nM bafilomycin A1, or 500 nM MG132 with 100 nM bafilomycin A1. Whenever indicated, an equivalent volume of DMSO was included as a solvent control. Cells were then incubated for 4 h at 37°C in a 5% CO2 incubator.
Proteasomal inhibition in neurons
Neurons were treated with 1, 2.5, 5, 10, 25, 50, or 100 nM epoxomicin, or an equivalent volume of DMSO as a solvent control, in pre-equilibrated neuron maintenance media diluted 1:1 with conditioned neuron maintenance media for 20 h in a 37°C, 5% CO2 incubator. Following 20 h, the treatment media was removed and replaced with pre-equilibrated neuron maintenance media supplemented with the indicated concentration of epoxomicin, 100 nM bafilomycin A1, co-treatment of epoxomicin with 100 nM bafilomycin A1, or DMSO as a solvent control. Neurons were then incubated for 4 h at 37°C in a 5% CO2 incubator.
Treatment with siRNAs targeting Atg5
Two SilencerTM siRNAs targeting Atg5 were obtained from Ambion/Thermo Fisher Scientific (Assay IDs: 85,553 and 256,090) and pooled together for knockdown of ATG5 expression. siRNA Assay ID 85553: Sense sequence: 5’-GGUUAUGAGACAAGAAGAUTT-3’ and antisense sequence: 5’-AUCUUCUUGUCUCAUAACCTT-3’. siRNA Assay ID 256090: Sense sequence: 5’-GUCUAUUGGUAUGCAAACATT-3’ and antisense Sequence: 5’-UGUUUGCAUACCAAUAGACTG-3’.
Primary astrocytes were plated at a density of 250,000 cells per well of a 6-well plate for immunoblot, or per 35-mm fluorodish for live-cell imaging. At 2–3 DIV, each well of a 6-well plate or fluorodish of astrocytes was transfected with 45 pmol of siRNA diluted in Opti-MEM media (ThermoFisher: Gibco, 31,985–070) for 45 min using Lipofectamine RNAiMax (Invitrogen, 13,778,100). The mock treatment utilized nuclease-free water (ThermoFisher, AM9914G) instead of the siRNA. Astrocytes were then washed with glial media and fed fresh glial media for the next 96 h. At 6–7 DIV, astrocytes were harvested for immunoblot or live-cell imaging analysis. For starvation treatments, glia were washed once in EBSS (Sigma E3024), and incubated in EBSS for 4 h at 37°C in a 5% CO2 incubator prior to immunoblot or live-cell imaging analysis (imaged in EBSS).
Immunostaining
Astrocytes or neurons were fixed for 10 min in 4% PFA-4% sucrose (PFA, Sigma, P6148; D-sucrose, Fisher Scientific, BP220-1) in PBS (150 mM NaCl, 50 mM NaPO4, pH 7.4) previously warmed to 37°C. Cells were washed twice in PBS, and stored in PBS at 4°C until subsequent processing. On the day of imaging, cells were permeabilized for 5 min in 0.1% TX-100 (Thermo Fisher Scientific, BP151-100) in PBS, washed twice with PBS, and blocked for 1 h in PBS supplemented with 5% goat serum (Sigma, G9023) and 1% BSA (Thermo Fisher Scientific, BP1605-100). Samples were labeled with primary antibodies diluted in blocking solution for 1 hour at room temperature. Cells were washed 3 times for 5 min each in PBS, and then incubated in secondary antibody diluted in blocking solution for 1 hour at room temperature. When indicated in the Figure, Hoechst was included in the secondary antibody incubation at 0.5 µg/ml. Astrocyte samples were washed 3 times for 5 min each in PBS and kept in PBS for imaging. Neuron samples were washed 3 times for 5 min each in PBS, washed once in Milli-Q water, and mounted in ProLong Gold (Thermo Fisher Scientific/Molecular Probes, P36930).
Samples were imaged on a BioVision spinning disk confocal system consisting of a Yokagawa W1 spinning disk confocal, a Leica DMi8 inverted widefield microscope, and Photometrics Prime 95B sCMOS camera. Images were acquired with VisiView software using a 63X/1.4 NA Plan-Apochromat oil-immersion objective (with or without a 2X optical magnification) and solid-state 405, 488, 561, and 640 nm lasers for excitation. Z-stacks were obtained that spanned the entire depth of the cell at 0.2-µm sections. Images that would be quantitatively compared to each other used the same acquisition parameters across treatment conditions.
Live-cell imaging
Following treatments in the incubator, astrocytes and neurons were imaged immediately in HibE (Brain Bits, HE-Lf) imaging solution supplemented with 2% B-27 and 2 mM Glutamax, with continued presence of the relevant small molecule treatment. For Fig. S1, 0.5 µg/ml Hoechst was added to HibE imaging solution and incubated for 10 min at 37°C in 5% CO2 incubator prior to imaging. Coverslips with cortical neurons were imaged using a Chamlide CMB magnetic imaging chamber (BioVision Technologies). Images were acquired using the BioVision spinning disk confocal system for a maximum of 30 min. Z-stacks were obtained of the entire cell at 0.2-µm sections. Images that would be quantitatively compared to each other used the same acquisition parameters across treatment conditions.
For LysoTracker and Magic Red Cathepsin-B (MR-Cat B) labeling, astrocytes were treated with 100 nM LysoTracker Red or 1:500 dilution of Magic Red Cathepsin-B substrate (stock prepared according to manufacturer’s protocol) in the final 30 min of the indicated treatments (e.g. 24 h UPS inhibition, 4 h epoxomicin treatment, or 4 h Baf A1 treatment). For DQ-Red BSA and BSA-Alexa Fluor 647 experiments, astrocytes were treated with 25 μg/ml DQ-Red BSA or 25 μg/ml BSA-Alexa Fluor 647 in the final hour of 24 h UPS inhibition. For these experiments, 1.5 ml of the 2.0 ml treatment solution was removed and supplemented with LysoTracker Red, MR-Cat B, or BSA conjugates. The remaining volume on the cells was aspirated and replaced with the 1.5 ml treatment. Following incubation with the dyes and BSA conjugates, astrocytes were washed twice with glial media and imaged in a HibE imaging solution containing the appropriate drug concentration or volume of solvent control. Live-cell imaging was performed for a maximum of 20 min on the confocal microscope.
Immunoblotting
Following UPS inhibition, cells were washed in PBS (150 mm NaCl, 50 mm NaPO4, pH 7.4) and lysed with RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate [Thermo Fisher Scientific, BP349-100], 0.1% SDS [Thermo Fisher Scientific/Invitrogen, 15–553-027], 1X complete protease inhibitor mixture [Sigma, 11,697,498,001], 50 mM Tris-HCl, pH 7.4) for 30 min on ice. Following lysis, samples were centrifuged at 17,000 x g for 15 min at 4°C. Supernatants were analyzed by SDS-PAGE and transferred onto an Immobilon-P PVDF membrane. Immobilon-P PVDF membranes were blocked in 5% milk in TBS-Igepal block solution (2.7 mM KCl, 137 mM NaCl, 0.05% Igepal [Sigma, I3021], 24.8 mM Tris-HCl, pH 7.4) for 30 min at room temperature. Blots were then rocked overnight in primary antibody diluted in 5% milk in TBS-Igepal block solution at 4°C. The following day, membranes were washed 3 times for 20 min at room temperature in HRP wash buffer (150 mM NaCl, 0.1% BSA, 0.05% Igepal, 50 mm Tris-HCl, pH 8.0), and incubated for 45 min at room temperature in peroxidase-conjugated secondary antibody diluted in HRP wash buffer. Membranes were washed 3 times for 20 min at room temperature in the HRP wash buffer and immunoblots were developed using the SuperSignal West Pico Chemiluminescent Substrate for HRP conjugates (Thermo Fisher Scientific, PI34580).
Image analysis
Area occupied by LC3-positive puncta and SQSTM1-positive structures. Maximum projections of Z-stacks were generated in Fiji, and the entire astrocyte or neuronal soma was outlined and measured for total cross-sectional area. Ilastik was used to identify and segment LC3-positive puncta or SQSTM1-positive structures (includes puncta, fibrils, and aggresomes). Ilastik segmentations were imported into Fiji and the area of identified puncta was measured using “analyze particles”. Total area occupied by LC3-positive puncta or SQSTM1-positive structures (includes puncta, fibrils, and aggresomes) per cell was normalized to the corresponding cell area and plotted as a percentage. In (Figures 1F and 1I), to generate a mean value for LC3 puncta area per astrocyte, the total area occupied by GFP-LC3-positive puncta for a given cell was divided by the number of puncta measured in that cell. The mean values for LC3 puncta area for all astrocytes measured are plotted and reported as µm2. LC3-positive puncta measured by live-cell imaging of GFP-LC3 are designated as “GFP-LC3”, and LC3-positive puncta measured by immunostain of the GFP moiety of GFP-LC3 are designated as “GFP(LC3)”.
Colocalization between GFP(LC3) and SQSTM1. Colocalization of GFP(LC3) and SQSTM1 puncta was measured in Fiji by calculating the overlapping area between the Ilastik segmentations for GFP(LC3)-positive puncta and SQSTM1-positive structures (includes puncta, fibrils, and aggresomes) for each individual cell using the “and” function in the image calculator tool. This area was then divided by the total cell area of the corresponding cell and plotted as a percentage. In Fig. S5, the overlapping or colocalized area was also normalized to either the total cell area or the total LC3 puncta area for the corresponding cell and plotted as a percentage.
Cytosolic levels of GFP(LC3), SQSTM1, and ubiquitin. Maximum projections of Z-stacks were generated in Fiji. A segmented line was drawn in the cytoplasm extending from the tip of the longest astrocyte extension up to the nucleus, carefully avoiding puncta. The intensity of cytosolic GFP(LC3), SQSTM1, or ubiquitin was measured as the mean gray value along the line for each astrocyte. For reference, the mean gray value of the background signal outside the cell was consistently measured at ~110 in data from immunostain or live-cell imaging in either red or green channels.
Distribution of GFP(LC3)-positive puncta. The Ilastik segmentations of GFP(LC3)-positive puncta were used to get the area and X, Y coordinates of the centroid of each punctum using the Analyze Particles tool in FIJI. Nuclei were outlined to get the X, Y coordinates of the nuclear centroid using the Analyze Particles tool in FIJI. Distance between the centroid of each punctum and the centroid of the nucleus was calculated (Dp), and the distance between the furthest protrusion of each cell and the centroid of the nucleus was calculated (Dc). The relative position of each individual punctum was normalized to cell length by calculating Dp/Dc. Puncta distributed within 10% increments across the distance of the cell were pooled from three independent experiments to generate the total area occupied by GFP(LC3) puncta within each region of the cell. The area occupied by GFP(LC3) puncta in each region was divided by the total GFP(LC3) area measured across three independent experiments and expressed as a percentage.
Line Scan analysis. Colocalization of ubiquitin with SQSTM1 and GFP(LC3) (Figure 6), LAMP1 with SQSTM1 and GFP(LC3) (Figure 7), and GFP-Ub with mCherry-LC3 (Figure 8), and ubiquitin with SQSTM1 (Figure 9) was analyzed using line scans in FIJI. Using cells representative of the phenotype, a segmented line was drawn from the cell periphery to the nucleus, incorporating cytosol, puncta, and aggregates. The gray value of each pixel along the line was obtained using the Plot Profile function. The raw gray values were then normalized to the maximum gray value within each respective channel along that line and plotted as a function of distance across the line. Colocalized peaks measured with z-stacks were verified in single slices.
Quantification of lysosomal markers and BSA conjugates. Maximum projections of Z-stacks were generated in Fiji, and the entire astrocyte was outlined and measured for total cross-sectional area. Ilastik was used to identify and segment puncta positive for LysoTracker Red or the Magic Red Cathepsin B substrate. Ilastik segmentations were imported into Fiji and the area of identified puncta was measured using “analyze particles”. Total area occupied by puncta positive for LysoTracker Red or MR-Cat B was normalized to the corresponding cell area. We validated the use of z-stacks rather than single slice images for area analyses because z-stacks more accurately represented the totality of the organelle population. The regions of interest generated by the analyze particles tool that outline individual puncta were then overlaid onto the original maximum projection. A mean gray value across all the pixels selected as puncta positive for LysoTracker Red or MR-Cat B was determined to generate a single value for puncta intensity for each individual cell.
Total area occupied by BSA-Alexa Fluor 647-positive puncta was obtained as described above for LysoTracker and MR-Cat B. To quantitate the intensity of DQ-Red-BSA signal, maximum projections of Z-stacks were generated in FIJI, astrocytes were outlined, and the maximum gray value of the DQ-Red-BSA-positive puncta within each cell was measured. Astrocytes with any hot spots or saturated pixels were excluded from the analysis.
Quantification of UbG76V-GFP. Astrocytes with cytosolic UbG76V-GFP signal greater than background signal were counted in each imaging field and totaled for all fields acquired for a given experiment. This value was expressed as a percentage out of the total number of astrocytes as determined by Hoechst-positive nuclei in the acquired fields.
Quantification of immunoblotting. Protein levels were quantitated using the gel analyzer tool in Fiji. Boxes were used to designate the individual gel bands, and the integral of the intensity graph was taken as the value designating protein levels. To control for sample loading differences between lanes, values for LC3, SQSTM1, ubiquitin, CTSB, ATG5, p-ULK (Ser757), or total ULK1 (t-ULK1) were divided by values for GAPDH, TUBA/α-tubulin, or CANX loading controls from each corresponding lane. Values were then normalized relative to the relevant control sample and expressed as fold effect. In the case of ULK1, p-ULK:TUBA/α-tubulin was then divided by the respective total ULK:TUBA/α-tubulin to account for differences in total ULK1 levels.
Figure preparation. Images were adjusted for contrast and brightness in ImageJ Fiji. Images that are grayscale matched are denoted in the relevant figure legend. Figures were assembled using Adobe Illustrator.
Statistical analyses
All image measurements were obtained from the raw data. GraphPad Prism was used to plot graphs and perform statistical tests; statistical tests are denoted within each figure legend (ns, not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
Supplementary Material
Acknowledgments
We thank members of the Maday lab for advice with experiments and preparation of the manuscript.
Funding Statement
This work was supported by the grants from the McCabe Fund (UPenn), the University of Pennsylvania Alzheimer’s Disease Core Center, the Intellectual and Developmental Disabilities Research Center at the Children’s Hospital of Philadelphia and the University of Pennsylvania, and the Philadelphia Foundation to SM. and National Institute of Neurological Disorders and Stroke [R00NS082619]; National Institute of Neurological Disorders and Stroke [R01NS110716].
Disclosure statement
The authors declare no conflict of interest. .
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2084884
References
- [1].Winckler B, Faundez V, Maday S, et al. The endolysosomal system and proteostasis: from development to degeneration. J Neurosci. 2018. Oct 31;38(44):9364–9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harris JJ, Attwell D.. The energetics of CNS white matter. J Neurosci. 2012. Jan 4;32(1):356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Spalding KL, Bhardwaj RD, Buchholz BA, et al. Retrospective birth dating of cells in humans. Cell. 2005. Jul 15;122(1):133–143. [DOI] [PubMed] [Google Scholar]
- [4].Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010. Jan;119(1):7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chuang E, Hori AM, Hesketh CD, et al. Amyloid assembly and disassembly. J Cell Sci. 2018. Apr 13;131(8):jcs189928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020. Aug;21(8):439–458. [DOI] [PubMed] [Google Scholar]
- [7].Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. 2020. Jun 1;219(6):e202002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell. 2016. Dec 1;3(12):588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007. Aug 17;282(33):24131–24145. [DOI] [PubMed] [Google Scholar]
- [11].Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019. Oct 17;76(2):268–285. [DOI] [PubMed] [Google Scholar]
- [12].Kulkarni A, Chen J, Maday S. Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr Opin Neurobiol. 2018. Aug;51:29–36. [DOI] [PubMed] [Google Scholar]
- [13].Kulkarni VV, Maday S. Compartment-specific dynamics and functions of autophagy in neurons. Dev Neurobiol. 2018. Mar;78(3):298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yamamoto A, Yue Z. Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci. 2014;37:55–78. [DOI] [PubMed] [Google Scholar]
- [15].Nikoletopoulou V, Tavernarakis N. Regulation and roles of autophagy at synapses. Trends Cell Biol. 2018. Aug;28(8):646–661. [DOI] [PubMed] [Google Scholar]
- [16].Kim M, Sandford E, Gatica D, et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife. 2016. Jan 26;5:e12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Collier JJ, Guissart C, Olahova M, et al. Developmental consequences of defective ATG7-mediated autophagy in humans. N Engl J Med. 2021. Jun 24;384(25):2406–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Finkbeiner S. The autophagy lysosomal pathway and neurodegeneration. Cold Spring Harb Perspect Biol. 2020. Mar 2;12(3):a033993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maday S. Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res. 2016. Oct 15;1649(Pt B):143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017. Mar 8;93(5):1015–1034. [DOI] [PubMed] [Google Scholar]
- [21].Sung K, Jimenez-Sanchez M. Autophagy in astrocytes and its implications in neurodegeneration. J Mol Biol. 2020. Apr 3;432(8):2605–2621. [DOI] [PubMed] [Google Scholar]
- [22].Kulkarni A, Dong A, Kulkarni VV, et al. Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy. 2020. Sep;16(9):1651–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Maday S, Holzbaur EL. Compartment-specific regulation of autophagy in primary neurons. J Neurosci. 2016. Jun 1;36(22):5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bard JAM, Goodall EA, Greene ER, et al. Structure and function of the 26S proteasome. Annu Rev Biochem. 2018. Jun 20;87:697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Marshall RS, Vierstra RD. Dynamic regulation of the 26s proteasome: from synthesis to degradation. Front Mol Biosci. 2019;6:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Finley D, Chen X, Walters KJ. Gates, channels, and switches: elements of the proteasome machine. Trends Biochem Sci. 2016. Jan;41(1):77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hamilton AM, Oh WC, Vega-Ramirez H, et al. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 2012. Jun 21;74(6):1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Djakovic SN, Marquez-Lona EM, Jakawich SK, et al. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci. 2012. Apr 11;32(15):5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fonseca R, Vabulas RM, Hartl FU, et al. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006. Oct 19;52(2):239–245. [DOI] [PubMed] [Google Scholar]
- [30].Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008. Nov;9(11):826–838. [DOI] [PubMed] [Google Scholar]
- [31].Zheng Q, Huang T, Zhang L, et al. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front Aging Neurosci. 2016;8:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Thibaudeau TA, Anderson RT, Smith DM. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun. 2018. Mar 15;9(1):1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front Cell Dev Biol. 2018;6:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yin Z, Popelka H, Lei Y, et al. The roles of ubiquitin in mediating autophagy. Cells. 2020. Sep 2;9(9):2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Iwata A, Riley BE, Johnston JA, et al. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005. Dec 2;280(48):40282–40292. [DOI] [PubMed] [Google Scholar]
- [36].Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007. Jun 14;447(7146):859–863. [DOI] [PubMed] [Google Scholar]
- [37].Huang N, Erie C, Lu ML, et al. Aberrant subcellular localization of SQSTM1/p62 contributes to increased vulnerability to proteotoxic stress recovery in Huntington’s disease. Mol Cell Neurosci. 2018. Apr;88:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mitra S, Tsvetkov AS, Finkbeiner S. Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J Biol Chem. 2009. Feb 13;284(7):4398–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011. Jun 24;286(25):22426–22440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci. 2011. Oct 12;31(41):14508–14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Meng L, Mohan R, Kwok BH, et al. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A. 1999. Aug 31;96(18):10403–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dantuma NP, Lindsten K, Glas R, et al. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000. May;18(5):538–543. [DOI] [PubMed] [Google Scholar]
- [43].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000. Nov 1;19(21):5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004. Mar;15(3):1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Berg S, Kutra D, Kroeger T, et al. ilastik: interactive machine learning for (bio)image analysis. Nat Methods. 2019. Dec;16(12):1226–1232. [DOI] [PubMed] [Google Scholar]
- [46].Dantuma NP, Groothuis TA, Salomons FA, et al. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol. 2006. Apr 10;173(1):19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001. Aug;8(8):739–758. [DOI] [PubMed] [Google Scholar]
- [48].Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005. Nov 21;171(4):603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998. Dec 28;143(7):1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kageyama S, Sou YS, Uemura T, et al. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J Biol Chem. 2014. Sep 5;289(36):24944–24955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sha Z, Schnell HM, Ruoff K, et al. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J Cell Biol. 2018. May 7;217(5):1757–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001. Feb 19;152(4):657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. [DOI] [PubMed] [Google Scholar]
- [55].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. Feb;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sarraf SA, Shah HV, Kanfer G, et al. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. 2020. Dec 3;80(5):779–795 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Demishtein A, Fraiberg M, Berko D, et al. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy. 2017. Oct 3;13(10):1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wong ES, Tan JM, Soong WE, et al. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008. Aug 15;17(16):2570–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dragich JM, Kuwajima T, Hirose-Ikeda M, et al. Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife. 2016. Sep 20;5:e14810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gubas A, Dikic I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022. Jan;289(1):75–89. [DOI] [PubMed] [Google Scholar]
- [61].Milan G, Romanello V, Pescatore F, et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015. Apr 10;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Radhakrishnan SK, Lee CS, Young P, et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010. Apr 9;38(1):17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tydlacka S, Wang CE, Wang X, et al. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008. Dec 3;28(49):13285–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chua JP, De Calbiac H, Kabashi E, et al. Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy. 2021;31:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dong A, Kulkarni VV, Maday S. Methods for imaging autophagosome dynamics in primary Neurons. Methods Mol Biol. 2019;1880:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.