

Abstract
Background: Global or untargeted metabolomics is widely used to comprehensively investigate metabolic profiles under various pathophysiological conditions such as inflammations, infections, responses to exposures or interactions with microbial communities. However, biological interpretation of global metabolomics data remains a daunting task. Recent years have seen growing applications of pathway enrichment analysis based on putative annotations of liquid chromatography coupled with mass spectrometry (LC–MS) peaks for functional interpretation of LC–MS-based global metabolomics data. However, due to intricate peak-metabolite and metabolite-pathway relationships, considerable variations are observed among results obtained using different approaches. There is an urgent need to benchmark these approaches to inform the best practices. Results: We have conducted a benchmark study of common peak annotation approaches and pathway enrichment methods in current metabolomics studies. Representative approaches, including three peak annotation methods and four enrichment methods, were selected and benchmarked under different scenarios. Based on the results, we have provided a set of recommendations regarding peak annotation, ranking metrics and feature selection. The overall better performance was obtained for the mummichog approach. We have observed that a ~30% annotation rate is sufficient to achieve high recall (~90% based on mummichog), and using semi-annotated data improves functional interpretation. Based on the current platforms and enrichment methods, we further propose an identifiability index to indicate the possibility of a pathway being reliably identified. Finally, we evaluated all methods using 11 COVID-19 and 8 inflammatory bowel diseases (IBD) global metabolomics datasets.
Keywords: global metabolomics, LC-MS, peak annotation, pathway enrichment analysis, identifiability index
Introduction
The metabolome refers to the complete set of small molecules present in a biological sample and is closely related to an organism’s phenotype. Metabolomics is increasingly applied together with other omics to understand biological processes under various genetic, infectious or environmental influences [1–4]. Global or untargeted metabolomics aims to comprehensively study metabolome in an unbiased, high-throughput manner. Liquid chromatography coupled with mass spectrometry (LC–MS) is widely used for global metabolomics. However, the associated downstream data analysis remains a key bottleneck [5].
A typical LC–MS-based global metabolomics data analysis workflow starts with raw spectra processing. The step will generate a high-dimensional feature table containing abundance information for tens of thousands of LC–MS peaks across all samples. These peaks are characterized by their mass-to-charge ratios (m/z) and retention times (RT). A variety of statistical or machine learning methods can be directly applied to identify significant peaks or patterns of interest. However, further downstream analysis especially functional interpretation is not straightforward.
Pathway enrichment analysis is fundamental to integrate metabolomics data into biological contexts. Comprehensive databases, such as KEGG [6] or MetaCyc [7], contain manually curated pathways depicting well-structured functions or biological processes involving multiple biomolecules. Utilizing these resources, several enrichment methods, such as over-representation analysis (ORA) [8] and gene set enrichment analysis (GSEA) [9], which were initially developed for transcriptomics data analysis, have been adapted to perform enrichment analysis of data from targeted metabolomics [10, 11]. However, these methods cannot be directly applied to global metabolomics data, as pathways are defined by genes/proteins/metabolites, not by peaks. Reliably assigning peaks to specific metabolites based on their m/z values is difficult—a single m/z could potentially match multiple metabolites (redundancy), and one metabolite often generates multiple peaks (degeneracy). Using RT could narrow down the potential candidates but is not sufficient in general. To confidently pinpoint the underlying metabolite for a peak with a particular m/z, it is often necessary to conduct time-consuming wet-lab experiments such as MS/MS or using internal reference standards. In summary, performing accurate peaks annotation before enrichment analysis has significantly limited the throughput of global metabolomics.
One promising strategy is to shift the unit of analysis from individual compounds (which usually cannot be reliably identified from their very few MS peaks) to individual pathways (which can be more reliably identified based on many more peaks contributed by their collective members). In this conceptual framework, all input peaks are mapped to the predefined metabolic pathways or networks in a multiple-matching manner based on m/z and/or RT without knowing their true identities. All the putative annotations are submitted to the next stage for pathway enrichment analysis. Mummichog [12] is the first algorithm that leverages this concept and offers an efficient computational package to enable pathway activity prediction directly from high-resolution LC–MS peaks. The underlying argument is that if a list of significant peaks reflect pathway activities, those true hits should collectively identify those pathways, while falsely matched metabolites are distributed more randomly.
To narrow the gap between raw LC–MS spectra and functional insights, we have developed or adapted a series of enrichment analysis methods that are widely used by the metabolomics community through MetaboAnalyst [13–15]. Enrichment analysis methods are available for both targeted metabolomics (such as MSEA [16] and MetPA [17]) and global metabolomics (such as mummichog, also known as Peak Set Enrichment Analysis (PSEA) in MetaboAnalystR [18]). According to Google Analytics, MetaboAnalyst is accessed by ~2000 users/day, and ~30% of the traffic (~600 users/day) enters the functional analysis modules. Among them, around one-sixth or ~100 users/day perform peak set enrichment analysis. We have recently adapted GSEA to analyze global metabolomics data [14]. However, the enrichment results have shown considerable variations depending on the choices of methods and parameters. Multiple factors involved in peak annotation or enrichment analysis could have significant effects on the enrichment results. As most previous comparative studies were geared toward targeted metabolomics, there is a lack of general insights into the effectiveness of these different methods for global metabolomics.
To address this need, we have performed a comprehensive review of commonly used LC–MS peak annotation approaches and enrichment methods in current metabolomics studies. We further evaluated four representative enrichment methods and three annotation approaches under various scenarios. Based on the observations, we proposed an identifiability index for each pathway to inform whether it can be reliably identified using current platforms and enrichment methods. Finally, we evaluated all the methods on 11 COVID-19 and 8 inflammatory bowel diseases (IBD) global metabolomics datasets and demonstrated that using semi-annotated input can significantly improve biological interpretation. The overview of the workflow is shown in Figure 1.
Figure 1.
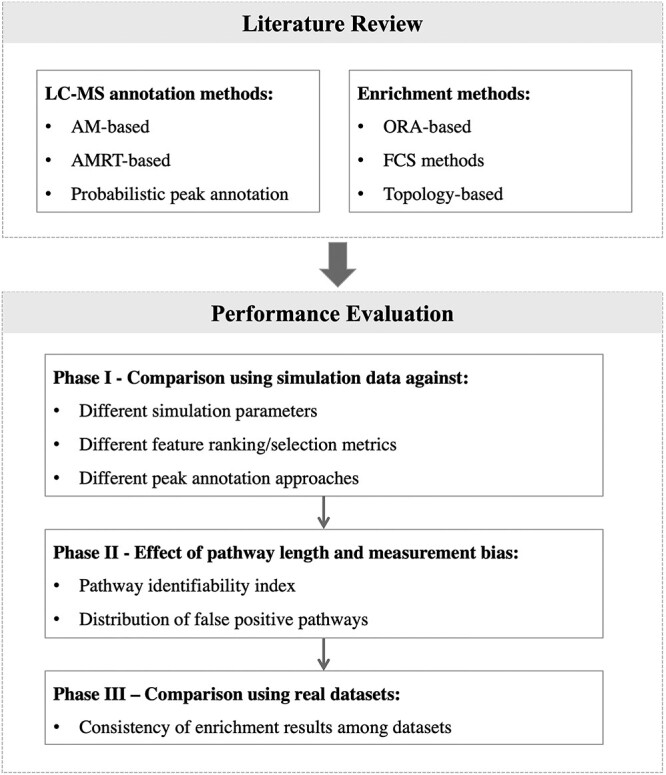
Literature review and study design. After a comprehensive literature review of approaches involved in peak annotation and enrichment analysis, selected methods were evaluated under different key parameters using both simulated and real datasets. AM: accurate mass; AMRT: accurate mass and retention time; ORA: over-representation analysis; FCS: functional class scoring.
LC–MS peak annotation strategies in global metabolomics
LC–MS global metabolomics of human samples can usually generate more than 10 000 peaks. However, only a small fraction of metabolic features can be annotated with high confidence [19]. Currently, the gold standard for metabolite identification is to match at least two physicochemical properties, such as accurate m/z value, RT and MS/MS (fragmentation pattern), of a measured feature against the authentic chemical standards measured through the same instrument following the same sample preparation protocols such as same chromatographic methods, ionization modes and collision energies [20–22]. While the chemical standards are limited by their commercial sources, it is impractical to generate all possible metabolites in the experiment samples on the same instruments. Different levels regarding current computational annotation strategies are proposed [23]. Here, we explore the common computational approaches for high-throughput LC–MS peak annotation without relying on comprehensive in-house spectra databases. In this context, three types of approaches are summarized for putative annotation in LC–MS data including (i) accurate mass (AM)-based annotation; (ii) accurate mass and retention time (AMRT)-based annotation and (iii) probabilistic peak annotation.
AM-based annotation
The wide accessibilities of high-resolution MS instruments, such as Orbitrap or quadrupole Time-of-Flight (qTOF), have significantly increased the precision of measured m/z values and improved the estimation of elemental composition [5]. There are two options for assigning putative metabolites to measured m/z values. For both options, the m/z values need to be converted to the neutral mass first. Next, we can either calculate the possible molecular formula(s) matching this neutral mass and search the formula(s) in the metabolomics database, or directly match the neutral mass to the accurate mass in chemical databases within a given mass error. For most medium-to-high resolving power instruments, the expected errors introduced during data acquisition in m/z measurements are <5 parts per million (ppm) [24]. Various open-access metabolomics databases are currently available for AM-based annotation [25]. Given the easy accessibility, AM-based annotation, ignoring the step of molecular formula conversion, has been successfully used for pathway analysis in global metabolomics data [12].
AMRT-based annotation
Due to the redundancy and degeneracy in LC–MS peaks, peak annotation purely based on AM will lead to a high number of false positives. To further improve peak annotation, RT is usually included to provide orthogonal information. RT describes the time from the injection of the sample to the time of compound elution, taking the apex of the peak that belongs to the specific molecular species [26]. Different from molecular properties such as m/z, RT is characteristic for a specific compound in a given analytical system involving chromatographic equipment, mobile and stationary phases, and separation conditions [27]. Thus, it is difficult to utilize experimental RT for the annotation of unknown peaks and for sharing information across laboratories. One strategy is to build the retention time index, which normalizes the deviations and allows for comparisons in different experimental conditions [27–29]. Others predict RT based on the quantitative structure–property relationships [30, 31]. In our current study, RT is directly used to cluster m/z values to reduce false positives of AM-based annotation when matching peaks to a pathway library, assuming that peaks of similar RT are more likely to belong to the same compounds.
Probabilistic peak annotation
In addition to m/z and RT, inter-peak relationships, either within spectra or based on biochemical transformations, can be leveraged to improve annotation. For instance, MetAssign [32] combines the information of m/z and RT with a quantitative model of inter-peak dependency structure to provide more robust annotations by means of sophisticated Bayesian statistics. Integrated Probabilistic Annotation (IPA) [33] enhanced the Bayesian annotation model by incorporating RT in the estimation of prior probabilities and considering the connectivity patterns among the peaks and adducts according to biochemical relationships. Network analysis has also been increasingly used in metabolomics peak annotation, as it intuitively capitalizes on peak–peak relationships to increase annotation scope and precision. Several popular network-based methods, such as GNPS [34] and MetDNA [35], require MS/MS data as input. A more recent algorithm, NetID [36], can directly work with LC–MS peak list. By leveraging an integer linear programming optimization algorithm, NetID optimizes a network of peak connections based on mass differences corresponding to the gain or loss of relevant chemical moieties while incorporating literature data on known metabolites and their retention times within specific metabolic models. NetID highlights the usage of all peak annotations simultaneously instead of sequentially to take full advantage of the entire available information to improve annotation. Thus, single annotations can be achieved by solving the global network optimization problem in a biological context. In this survey, we select NetID to illustrate the impact of probabilistic peak annotation on the functional interpretation of global metabolomics data.
Pathway enrichment analysis in global metabolomics
We have reviewed the available tools for pathway enrichment analysis in metabolomics data in the last 5 years, including web-based tools and R/Python packages. A summary of 18 current tools is listed in Supplementary Table S1. According to Khatri et al. [37], enrichment methods can be organized to three generations: (i) ORA-based methods, (ii) functional class scoring (FCS) methods, and (iii) topology (TP) based methods. ORA is the most widely used method among the tools in our survey. While mummichog is the only method directly supporting peak list as input for global metabolomics, all other methods require prior peak annotation. It is thus of great interest to explore the potential usage of these methods and to inform the community of the best practices in analyzing global metabolomics data. Therefore, we conducted a systematic comparison under the framework introduced by mummichog, namely putative peak annotation followed by enrichment analysis.
Four typical methods, including ORA (hypergeometric test), mummichog, GSEA and FELLA [38], were selected for further systematic evaluation. They cover the different method categories and represent the underlying concepts of each type. The input of all the selected methods is peak lists with differential statistics (such as P values, t-statistics or fold changes). We hereby briefly describe these methods. More details on these algorithms can be found in their original papers. The general features of each method are summarized in Table 1.
Table 1.
Main characteristics of the selected enrichment methods. Detailed explanations of these equations are provided in the Supplementary Text S1
| Method | Class | Statistics | P value assessment | Descriptions |
|---|---|---|---|---|
| ORA | ORA-based | Hypergeometric |
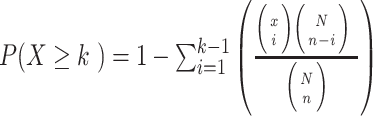
|
Features and pathways are treated equally; only significant features are used; arbitrary selection for the significant features |
| Mummichog | ORA-based | FET, EASE |
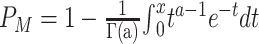
|
Same as above; using permutation to address the dependence assumption |
| GSEA | FCS | Kolmogorov–Smirnov-like statistic |
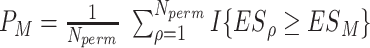
|
Small changes are included; features and pathways are treated equally; summarize the differential information to a rank list |
| FELLA | TP-based | Diffusion and PageRank algorithms |
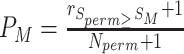
|
Combined topology information with ORA; built-in weighted network consisting of genes, enzymes, and metabolites; dependent on the annotated network relationships |
Over-representation analysis
ORA, also known as 2 × 2 table methods [39], divide the features based on whether they are significantly changed between different experimental conditions and whether they are members of a particular pathway M. The main idea is to determine if there is a greater overlap between differential features and M than expected by chance. The classical way to calculate the P-value for pathway M is using the hypergeometric test (same as the right-tailed Fisher’s exact test (FET) based on the hypergeometric distribution).
Mummichog
The mummichog algorithm [12] enhances the classical ORA in two ways: application to untargeted metabolomics based on putative identification of metabolites as previously explained, and a more robust P-value assessment. One of the most critical assumptions of the ORA methods is that the pathways can be treated independently. To enhance the robustness of this independence assumption, mummichog employs the null model by Berriz et al. [40], which estimates an adjusted P-value from the results of 1000 permutations. Instead of directly using empirical P-value, mummichog models the P-values from null distribution as a Gamma distribution based on the maximum likelihood estimation and calculates the cumulative distribution function. In addition, EASE [41], a more conservative version of ORA, was applied to increase the precision, which penalizes pathways with fewer hits by taking out one hit from each pathway. The framework designed by mummichog can also incorporate other enrichment methods to interpret global metabolomics data. For instance, MetaboAnalyst has extended it to include the GSEA approach, which can be easily achieved using the website and the R package.
Despite their extensive usage, ORA methods have several limitations [37]. Firstly, a subjective threshold is required, which could lead to variations depending on the chosen cut-offs. Secondly, only binary information between conditions is considered, and thus, the features are treated equally. Thirdly, the features and pathways are presumed to be independent. However, the equal status and independent assumption cannot be fulfilled in complex biological activities.
Gene set enrichment analysis
As a representative of the FCS methods, GSEA was designed to address the limitations of ORA in two aspects. It uses all features without the requirement of a pre-selected feature list and incorporates their effect sizes (such as t-scores) between conditions to indicate coordinated changes among individual features. The key idea is that in addition to significant features, minor and coordinated alterations of nonsignificant features can also contribute to pathway activities. GSEA is conducted in three steps. First, values from all the measurements are used to create a decreasing ranked list based on the differential statistics of the individual features. The enrichment score for each pathway is then calculated as the largest difference in a running-sum statistic corresponding to a weighted Kolmogorov–Smirnov (KS)-like statistics. Pathway significance is generated by permuting either the sample labels or feature labels [42].
Despite eliminating the need to preselect significant features, FSC still has some limitations. Firstly, summarizing complex biological information into a rank list may reduce the ability to detect real changes. Secondly, including weak signals may introduce noises and reduce the sensitivity in certain cases [43]. Thirdly, the network structures underlying pathways are not considered, which is similar to the ORA methods.
FELLA
TP-based methods address the interactions between pathways by taking topology information into account. While the development of TP-based methods keeps growing in genomics/transcriptomics, e.g. SPIA [44] and CePa [45], its application in metabolomics remains very limited. Recently, several studies have tried to combine ORA methods with TP information for metabolomics data enrichment analysis. For instance, FELLA [38] employed a subnetwork optimization strategy to determine the most affected subgraphs based on the input significant metabolites. FELLA retrieves pathways from KEGG as graph objects and generates scores for all the nodes through random walks to assess their importance relative to the metabolites in the input. The PageRank algorithm and heat diffusion model were used for calculating the scores. A further permutation step was conducted to estimate the null distribution of scores for each node. The node scores are subsequently normalized and ranked using their null distributions. The final P-value estimation was achieved either by the normalized score (z-score) or the empirical cumulative distribution function based on the null models.
Although TP-based methods are inherently limited by the incomplete knowledge on the pathways or networks, it has been reported to give better performance in detecting significant pathways over non-TP methods in certain cases. More investigations are needed to inform metabolomics-related pathway enrichment analysis.
Evaluation design
Overview of the evaluation process
We evaluated the performance and robustness of these selected methods under different scenarios based on the key parameters of each step (Figure 2). Our evaluation aims to assess the performance of each method under (i) different input conditions, e.g. magnitude of change (compound level), direction of change (compound level) and percentage of change (pathway level); (ii) different ranking metrics and significant feature selection and (iii) different parameters for peak annotation. The detailed parameters for each scenario are provided in Supplementary Table S2.
Figure 2.

Overview of the evaluation workflow. Three steps were involved in the evaluation of current enrichment methods. Both simulated and real datasets were used to provide comprehensive results. Different input abundance tables were submitted for t-tests and fold change analysis to rank the features. Input features were putatively annotated based on three methods. Four representative enrichment approaches were selected to identify significant pathways in different scenarios. The results were evaluated using multiple performance measures.
Datasets
Both simulated and real datasets were used to illustrate the performance of different methods.
Simulated datasets
The main advantage of using simulated data is that the features can be tuned with a fully known data generating process and ground truth. To maintain the characteristics of the real data as much as possible, we employed a strategy to generate simulated datasets by transforming multiple real datasets [46, 47]. The simulation was achieved in five steps, including background cleaning, random metabolite label assignment, signal simulation, m/z and RT label generating and sampling. Three datasets of different lengths and study types (see details in Supplementary Table S3) were selected as initial datasets for simulation. After the imputation of missing values, quantile normalization was used to remove the differences across the samples, followed by log transformation and centering. Thus, the original signals of individual dataset were erased, while the underlying distributions and inherent correlations among the features were preserved. The class labels were randomly swapped to nullify the covariances between the phenotypes. For the simulation, we consider both the percentage of the differential features within a dataset and a pathway. In particular, the percentage of differential features for each dataset was set to 5–25%, and a percentage of 30–100% features within a specific pathway was picked to be perturbed in different scenarios. We manipulated the signal x of a targeted metabolite by interfering sd with an index a termed the magnitude of change. To avoid the exponential increase in the original space without log transformation, we calculated the simulated value 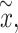 by
by
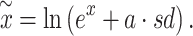 |
The direction of change for each compound can be either up or down according to different scenarios. An additional step is needed for global metabolomics data to access the peak labels. As the peaks cannot be fully annotated to metabolites, we first replaced the feature labels (m/z value of the peaks) with known compound identifiers and shuffled the labels to make them randomly correlated. This may not fulfill the methods requiring correlation information among features. However, those methods are hardly used in untargeted metabolomics and are not included in this study. After this step, we changed the metabolite ID back to its m/z value, and the retention time was estimated based on a deep-learning model by Xavier et al. [31]. A sampling procedure was performed to generate 1000 simulation datasets to allow more robust results.
Real datasets
Since the ground truth is unavailable for real metabolomics data, we chose to evaluate the consistency of enrichment analysis results using datasets from studies on the same phenotype. Global metabolomics datasets, including 11 datasets from COVID-19 studies and 8 datasets from IBD studies, were selected for evaluation. All the datasets were generated through LC–MS. The COVID-19 datasets were downloaded from COVID-19 Metabolomics Data Center in MetaboAnalyst, and the IBD dataset was downloaded from the Metabolomics Workbench (https://www.metabolomicsworkbench.org/) with the accession number of ST001000. For evaluation purposes, we focused on the comparison between the samples from patients diagnosed as COVID-19 positive or with Crohn’s disease (CD) versus the samples from healthy controls in each corresponding studies. More detailed descriptions are listed in Supplementary Tables S4 and S5. To obtain the input lists for enrichment analysis, t-tests were performed for all the datasets after log transformation.
Pathway library
Metabolic pathways of Homo sapiens from the KEGG database were used in our study. The KEGGREST package [48] was used to obtain the latest KEGG pathways. A recent study has shown that pathways of smaller size provide more precision information and are more meaningful for enrichment analyses [49]. High-level metabolic pathways at levels ‘A’ and ‘B’ based on BRITE hierarchy [50] were removed as they contain several hundred metabolites and do not provide specific functional insights. The remaining library consisted of 81 pathways, with sizes ranging from 5 to 85.
Result assessment
Following the evaluation metrics employed by Tarca et al. [51], we used recall (Rc, also known as sensitivity) and precision (Pc) in our simulation studies. For each method, several performance measures were calculated: true positives (TP), namely the significant pathways accessed and truly observed in the simulated dataset; false positives (FP), the significant pathways accessed but not observed; false negatives (FN), the significant pathways observed while not been accessed. Thus, Rc can be represented as TP / (TP + FN), and Pc can be calculated as TP/ (TP + FP). The overall performance was measured by F1 score, denoted by (2*Rc*Pc) / (Rc + Pc). We also evaluated the prioritization and reproducibility of the methods. Prioritization provides the ratio of N perturbed pathways during simulation that can be ranked in the top N pathways reported by each method.
To compare the consistency among the real datasets that study the same phenotype, we employed a score based on the Simpson index, which has been widely used to measure the stability of different methods. For each method, the consistency score is defined as the mean value of the Simpson index of the enumerated paired datasets tested given by
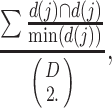
where d(i) represents the significant pathways accessed in the ith dataset and D is the total number of the datasets in the test.
Results
In this section, we first compared the performance of the selected methods using 1000 simulated datasets. The effect of exact metabolite annotation was surveyed among all the methods, followed by evaluating semi-annotated inputs. An identifiability index was proposed to evaluate the reliability of a pathway to be identified as significant by the current methods. We also investigated the potential of these pathways to be reported as false positives or hard to detect in real experiments. Finally, 11 COVID-19 and 8 IBD metabolomics datasets were used to assess the usefulness of each method on real complex untargeted metabolomics data.
Comparative results using simulation data
Effect of different simulation parameters
We first evaluated the influence of different parameters on the performance of the methods. A few notable trends are shown in Figure 3. The performance of all the methods (except GSEA) increased along with the increasing magnitude of changes as they focused on the significant differential features. The two groups can be totally separated when a = 2 based on PCA plots (Supplementary Figure S1). A turning point was observed when a = 2, followed by slight changes when a increased above 2 (Figure 3A). The performance of GSEA dropped obviously when a reached 10. A possible reason could be the enlarged signals of overlapping metabolites among the pathways, which prioritized the rank scores of irrelevant pathways leading to lower recall of the perturbed pathways.
Figure 3.

(A) Performance evaluations under different magnitude of changes at the compound level. (B) Performance evaluations under different percentages of differential features within pathways. (C) Performance evaluations with regard to the direction of changes at the compound level.
A consistent trend was observed for the effect of the percentage of change within pathways (Figure 3B). It is evident that the larger the percentage of differential features within pathways, the better outcomes were achieved by each method. Mummichog yielded high recall/sensitivity, reaching 65% at the percentage of change of 30%, while its precision significantly dropped when all the metabolites within a pathway becoming differential. In this case, the permutation step in mummichog led to very high recall of over 90%; it also introduced more false positives compared to the basic ORA. Both ORA and FELLA showed a sharp increase with increasing percentage of differential features within pathways, while the performance of GSEA was largely improved when more than 80% of metabolites within pathways were changed.
We also evaluated the responses of each method to the direction of change at the metabolite level (Figure 3C). The standard method of GSEA considers the up- and down-regulated features separately when calculating the pathway scores, thus informing the direction of change for the significant pathways accessed, while other methods only count the differential features/pathways as yes or no. The performance of GSEA decreased when the affected pathways contained both up- and down-regulated metabolites. However, this occurs in real experiments due to dynamic regulation of metabolic flux. Thus, calculating the effect of direction separately negatively impacts its performance within our evaluation schema. More studies are required to get a common definition for affected pathways in this context.
Effect of parameters for peak annotation
Many common enrichment methods are used for targeted metabolomics data analysis. Here we leveraged the mummichog peak annotation framework to other methods, including AM-based annotation (version 1), AMRT-based annotation (version 2) as well as the NetID method. The results are summarized in Figure 4A. The overall performance of all the methods was shown to be slightly improved by grouping m/z values by RT. However, the recall and precision of most methods were greatly increased using the NetID annotation, which is supposed to provide a single match for each peak with an annotation ratio of ~40% [36]. With annotation increase, mummichog achieved high recall (up to 92%), while the precision decreased.
Figure 4.

Performance evaluations based on: (A) peak annotation approaches, (B) ppm selection using AMRT- based annotation, and (C) ppm selection using NetID annotation.
The effect of mass tolerance varied between the annotation approaches. In general, 1 ppm and 3 ppm are recommended for AM and AMRT-based mapping to obtain better outcomes. For both methods, the recall started to decrease at 5 ppm and showed an apparent fall at 10 ppm (Figure 4B). The NetID approach showed similar performance when the tolerance increased from 5 to 10 ppm, and its recall started to decrease at 12 ppm (Figure 4C). The default distance tolerance for NetID is 10 ppm, which is useful for most methods. Specifically, mummichog was ranked at the top in terms of recall among the methods. The relatively lower F1 score was caused by the increasing false positives. Mummichog showed a similar trade-off between recall and precision since NetID could potentially increase both the true and false annotations.
We further investigated how accurate metabolite annotations could affect performance. The potential matched metabolites based AMRT were replaced by a certain percentage of real annotation. Most methods reached their performance plateau at around 30% (Figure 5), indicating that accurate biological interpretation can be achieved without requiring fully annotated metabolomics data. At this level, mummichog reached a recall ratio of >90%. The performance of GSEA was also significantly improved by a 30% accurate annotation, reaching ~60%. With the advances in the development of tools and methodologies for compound annotation, we envision that using a semi-annotated input would greatly enhance data interpretation. Further explanation is presented in the section based on the results obtained from the real datasets.
Figure 5.

Performance evaluations under different percentages of accurate peak annotation. Different percentages of real annotations were used to replace the putative annotations obtained based on AMRT method.
Effect of parameters for peak ranking and selecting differential peaks
For ranking peak lists, we focused on two widely used statistics: t-tests and fold changes (Figure 6A). Better performance was obtained for GSEA and ORA by leveraging the absolute value of fold changes. FELLA was the most robust method to the different metrics as its recall and precision remained at a similar level. Different from FELLA, the similarity of F1 scores obtained by mummichog was the result of a trade-off between rising recall and decreasing precision. GSEA performed better with absolute values, which is consistent with the observation of the direction effect in the previous section.
Figure 6.

Performance evaluations under different: (A) ranking metrics, and (B) cut-offs for significant feature selection.
For selecting significant or differential peaks from the ranked peak list, the original paper of mummichog recommended the number of differential peaks to be top 10% of the input list. We evaluated this parameter among the tested methods. The real significant signals were set between 5% and 25%. We confirmed that 10% selection is applicable for all the methods and can be used as the selection criteria for other ranking metrics such as fold changes (Figure 6B). We also investigated the effects of input peak list length (i.e. the number of original input peaks), and observed that longer input peak lists gave better results, which can be explained by the improved metabolome coverage by high-resolution MS.
The overall performance of each tested method is summarized in Table 2. Mummichog obtained a high recall of 0.85 while suffering from a relatively low precision. ORA showed a moderate performance with both recall and precision at around 50%, while the highest precision was observed for FELLA, up to 58%. GSEA showed the lowest overall performance, confirming the previous findings [52, 53].
Table 2.
Performance summary of different enrichment methods. The top performer in each category is highlighted in bold
| Recall | Precision | F1 score | Prioritization | Reproducibility | |
|---|---|---|---|---|---|
| FELLA | 0.41 ± 0.0158 | 0.58 ± 0.0121 | 0.48 ± 0.0109 | 0.44 ± 0.0195 | 0.36 ± 0.0181 |
| GSEA | 0.24 ± 0.0129 | 0.37 ± 0.0158 | 0.29 ± 0.0112 | 0.57 ± 0.0204 | 0.45 ± 0.0164 |
| Mummichog | 0.85 ± 0.0143 | 0.44 ± 0.0164 | 0.58 ± 0.0135 | 0.72 ± 0.0153 | 0.69 ± 0.0144 |
| ORA | 0.52 ± 0.0165 | 0.49 ± 0.0197 | 0.50 ± 0.0154 | 0.60 ± 0.0115 | 0.54 ± 0.0223 |
Pathway identifiability index
We further investigated whether different pathways have the same levels of identifiability when they are truly enriched across the tested methods. Figure 7 shows the detailed result regarding the distribution of pathways that can be correctly identified as significant during 1000 simulations. The best performance was achieved by mummichog, while FELLA showed the lowest power failing to identify more than half of the pathways. The patterns are similar among these methods against different pathways. Pathways of larger size are more likely to be identified with better reproducibility by all the methods. GSEA is highly sensitive to the pathway size, possibly due to the fact that all features are considered in the ranking score calculation. This may partially explain its low recall in metabolomics data analysis. The lowest outcome for all methods appears in the pathways of small size (containing less than 10 metabolites). Given this, we hypothesize that the reliability of a pathway to be identified by the current methods is correlated with its size. According to our observation and the previous study [54], overlaps among pathways also have a significant impact on the enrichment results. To describe the correlation, we propose an identifiability index. Firstly, a uniqueness score 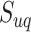 was defined for each pathway. For a pathway of the size of
was defined for each pathway. For a pathway of the size of 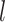 , we calculate its
, we calculate its 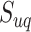 by
by
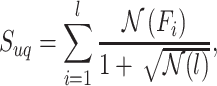 |
where 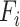 denotes the number of pathways containing the ith metabolite within the given pathway and
denotes the number of pathways containing the ith metabolite within the given pathway and 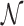 is the range normalization function. We then calculated the identifiability index by dividing the
is the range normalization function. We then calculated the identifiability index by dividing the 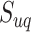 of each pathway by the maximum uniqueness score obtained among the evaluated pathways. As shown on the right side of the heatmap in Figure 7, the index generally represents the reliability of a given pathway being correctly identified by most current methods.
of each pathway by the maximum uniqueness score obtained among the evaluated pathways. As shown on the right side of the heatmap in Figure 7, the index generally represents the reliability of a given pathway being correctly identified by most current methods.
Figure 7.

Pathway identifiability across the tested methods. The heatmap shows the rate of each pathway to be correctly identified by different methods across 1000 permutations. The size indicates pathway length, and the index shows identifiability for each pathway.
Effect of metabolome coverage in pathway enrichment analysis
Since metabolites are not equally measured in global metabolomics, we further investigated whether the enrichment methods tend to report specific pathways as false positives across different experimental conditions. The three data tables collected from different studies and platforms were used to generate simulation datasets. All the background signals were removed, while no additional signals were added. The sample labels and original peak labels were permuted 1000 times. In this case, any significant pathways reported by any methods would be considered false positives. As shown in Supplementary Figure S2, mummichog reported the largest number of pathways as positives, while GSEA covered almost all the pathways but with a lower rate than mummichog. It is important to point out that most false positive pathways tend to be study specific rather than method specific. Several exceptions were observed. For instance, arachidonic acid metabolism and starch and sucrose metabolism were not reported by mummichog but were marked as false positive with a relatively high rate by the other three methods. On the other hand, tyrosine metabolism was reported by mummichog in every dataset with a very high ratio of around 50%, while the rate was much lower by other methods. These pathways are more likely to be false positives during functional interpretations. Similar observations can also be used to investigate if some pathways are never reported by using specific mass spectral platforms. As our study was limited to these three studies, further investigations are warranted to include more datasets from specific platforms.
Results from real datasets
We applied all the methods to 11 COVID-19 and 8 IBD global metabolomics datasets. Both the distance-based annotation of the input peak list (Figure 8A and Supplementary Figure S3A) and the semi-annotated metabolite/peak list (Figure 8B and Supplementary Figure S3B) were tested. The prior annotation was achieved through an enhanced version of NetID using OmicsNetR [55]. According to the heatmaps, an overall higher number of changed pathways were reported when using the semi-annotated input. We then calculated the consistency scores across the results obtained for each method. Prior annotation increased the consistency score by 9% on average, suggesting the additional pathways detected using semi-annotated data are likely to be real active pathways rather than false positives. For instance, tryptophan metabolism, which has been reported as a participant in COVID-19 process [56, 57], was reported by all the methods except for GSEA using the semi-annotated input. A similar case is retinol metabolism, a newfound player during COVID-19 process [58, 59]. Other pathways reported to be involved in COVID-19 with high consistency across the datasets include primary bile acid biosynthesis [36], steroid hormone biosynthesis [60], glycerolipid metabolism [61], caffeine metabolism [62] and vitamin B6 metabolism [63]. For the IBD datasets, the original study [64] showed that tryptophan metabolism genes were decreased in CD patients based on microbiome samples, while the metabolomics data indicated enrichment of primary bile acid biosynthesis. Here, tryptophan metabolism was reported by mummichog with high consistency using semi-annotated data; primary bile acid biosynthesis was detected by mummichog and ORA in both annotation approaches, while only captured by FELLA and GSEA using semi-annotated data.
Figure 8.
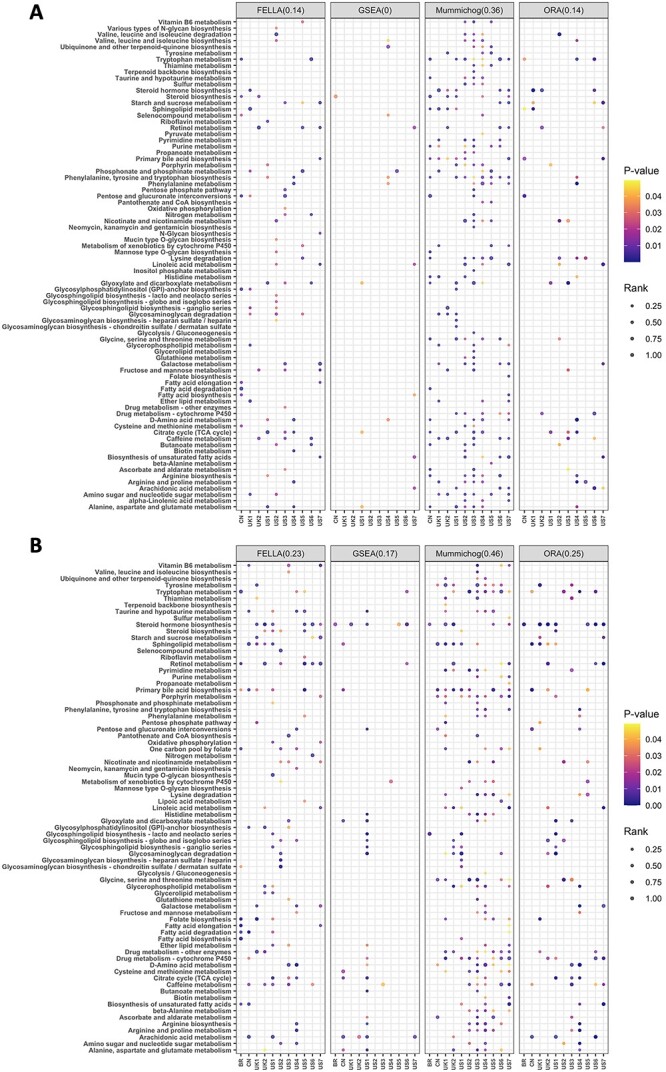
Consistency of enrichment results obtained using different enrichment methods obtained based on 11 COVID-19 datasets. For each method, its consistency score is indicated at the top inside the round brackets. (A) Results generated using peak list as input. (B) Results generated using the semi-annotated metabolite/peak list as input.
Overall, the highest consistency score was obtained for mummichog in both annotation approaches, but it risks a relatively high false positive ratio. Despite different patterns, FELLA and ORA showed a similar level of consistency, which was improved by using the semi-annotated data. However, a generally poor performance was observed for GSEA, consistent with our previous evaluations using simulated datasets.
Discussions and future directions
Global metabolomics has been widely used for biomarker discovery with applications to disease diagnosis, treatment monitoring as well as personalized medicine [65–67]. There is a growing interest in the application of global metabolomics to gain mechanistic and functional insights of diseases and biological processes [68, 69]. Pathway enrichment analysis plays a critical role in the biological interpretation of metabolomics data. Thus, a general guideline is both timely and necessary to inform the best practices of enrichment analysis for global metabolomics. In this manuscript, we have systematically reviewed three peak annotation approaches combined with four enrichment methods under various scenarios for global metabolomics.
A key challenge facing the design of a comparative study on pathway enrichment methods is that the underlying biological processes involved in the experimental conditions is far from fully understood. Therefore, we have no ground truth to compare the enrichment results, which makes it very difficult to evaluate performance across different methods. One solution is to leverage benchmark datasets. In gene enrichment analysis, some well-studied datasets can serve this purpose [45, 54, 70, 71]. Unfortunately, no such dataset has been made available for metabolomics in the current stage. Using simulated datasets can be a good alternative, which has been successfully employed in several studies [53, 72]. Another strategy is to evaluate the consistency among the real datasets measuring the same phenotype. Both strategies were employed in our current study.
Mummichog shows the highest recall/sensitivity in detecting significant pathways across all the scenarios tested at the sacrifice of precision. FELLA was found to have an edge on precision, indicating that incorporating topology information may help reduce false positives. GSEA, which is extensively used in gene expression profiling, was found to lack strength in metabolomics data with low recall and precision. This can be potentially explained by the relatively smaller size of metabolic pathways and their limited coverage by the current metabolomics platforms. The performance of GSEA can be enhanced by increasing the metabolite annotation and using the absolute values as ranking metrics.
In addition to the methods, the properties of pathway themselves can also greatly influence the enrichment results. For instance, overlaps among pathways have been found to significantly impact the enrichment results [54]. We proposed an identifiability index to indicate whether a pathway can be reliably reported as enriched.
According to our study, up to more than 90% of significant pathways can be captured by mummichog when accurate annotation arrives at ~30%, suggesting semi-annotated input without full annotation is sufficient for biological interpretation. Using NetID annotation to generate semi-annotated lists could enhance the performance of the enrichment methods, which was also confirmed using real datasets from COVID-19 and IBD studies based on better consistency scores across all methods. Multiple active pathways have been confirmed with high consistency, indicating the usage of semi-annotated data in finding new biological insights.
Our studies have some limitations. Firstly, only three datasets were selected as initial datasets for simulation which may lead to the potential bias towards the specific type of samples. More standardized benchmark datasets are necessary for future studies. Secondly, our simulation did not have a specific design to accommodate feature correlations, although none of the methods we have evaluated can deal with the correlation information. Moreover, some other factors, such as the choice of pathway databases, are also important in pathway analysis, which were not included in our study and need further investigations. Finally, some new methods leveraging machine learning, multi-omics integration for metabolite annotation and pathway enrichment were not considered in this study [73].
Key Points
Comprehensive review and benchmarking were performed to assess the effects of key parameters used in peak annotation approaches and pathway enrichment methods for LC–MS-based global metabolomics.
Mummichog combined with NetID was found to give the overall best performance.
Semi-annotation enables more accurate and consistent functional interpretation.
An identifiability index was proposed to indicate the probability of a pathway being reliably identified by different enrichment methods on current global metabolomics data.
Supplementary Material
Yao Lu is a PhD candidate at the Department of Microbiology and Immunology, McGill University. Her research interest focuses on functional annotation and integration of microbiome with metabolomics.
Zhiqiang Pang is a PhD candidate at the Institute of Parasitology, McGill University. His research project focuses on improving metabolomics raw data processing to gain deep biological insight on infectious diseases, cancer and host–microbiome interactions.
Jianguo Xia is an Associate Professor and Canada Research Chair at the Institute of Parasitology and the Department of Animal Science, McGill University. He is also an Associate Member of the Department of Microbiology and Immunology, McGill University. His research interest focuses on big data analytics in metabolomics, microbiomics and multi-omics.
Contributor Information
Yao Lu, Department of Microbiology and Immunology, McGill University, Quebec, Canada.
Zhiqiang Pang, Institute of Parasitology, McGill University, Quebec, Canada.
Jianguo Xia, Department of Microbiology and Immunology, McGill University, Quebec, Canada; Institute of Parasitology, McGill University, Quebec, Canada.
Funding
We thank Genome Canada, Génome Québec, US National Institutes of Health (U01 CA235493), Natural Sciences and Engineering Research Council of Canada (NSERC) and Canada Research Chairs (CRC) Program for funding support. Y. Lu is partially supported by a PhD scholarship from the China Scholarship Council.
References
- 1. Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol 2016;17:451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shen B, Yi X, Sun Y, et al. . Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 2020;182:59–72 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lotta LA, Pietzner M, Stewart ID, et al. . A cross-platform approach identifies genetic regulators of human metabolism and health. Nat Genet 2021;53:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buergel T, Steinfeldt J, Ruyoga G, et al. . Metabolomic profiles predict individual multidisease outcomes. Nat Med 2022;28:2309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chaleckis R, Meister I, Zhang P, et al. . Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr Opin Biotechnol 2019;55:44–50. [DOI] [PubMed] [Google Scholar]
- 6. Kanehisa M, Araki M, Goto S, et al. . KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008;36:D480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Caspi R, Billington R, Keseler IM, et al. . The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res 2020;48:D445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyle EI, Weng S, Gollub J, et al. . GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004;20:3710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subramanian A, Tamayo P, Mootha VK, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wieder C, Frainay C, Poupin N, et al. . Pathway analysis in metabolomics: Recommendations for the use of over-representation analysis. PLoS Comput Biol 2021;17:e1009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marco-Ramell A, Palau-Rodriguez M, Alay A, et al. . Evaluation and comparison of bioinformatic tools for the enrichment analysis of metabolomics data. BMC Bioinformatics 2018;19:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S, Park Y, Duraisingham S, et al. . Predicting network activity from high throughput metabolomics. PLoS Comput Biol 2013;9:e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pang Z, Chong J, Zhou G, et al. . MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res 2021;49:W388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pang Z, Zhou G, Ewald J, et al. . Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc 2022;17:1735–61. [DOI] [PubMed] [Google Scholar]
- 15. Chong J, Soufan O, Li C, et al. . MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 2018;46:W486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xia J, Wishart DS. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res 2010;38:W71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia J, Wishart DS. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010;26:2342–4. [DOI] [PubMed] [Google Scholar]
- 18. Chong J, Yamamoto M, Xia J. MetaboAnalystR 2.0: From Raw Spectra to Biological Insights. Metabolites 2019;9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Viant MR, Kurland IJ, Jones MR, et al. . How close are we to complete annotation of metabolomes? Curr Opin Chem Biol 2017;36:64–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sumner LW, Amberg A, Barrett D, et al. . Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007;3:211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Broeckling CD, Ganna A, Layer M, et al. . Enabling efficient and confident annotation of LC-MS metabolomics data through MS1 spectrum and time prediction. Anal Chem 2016;88:9226–34. [DOI] [PubMed] [Google Scholar]
- 22. Alden N, Krishnan S, Porokhin V, et al. . Biologically consistent annotation of metabolomics data. Anal Chem 2017;89:13097–104. [DOI] [PubMed] [Google Scholar]
- 23. Domingo-Almenara X, Montenegro-Burke JR, Benton HP, et al. . Annotation: a computational solution for streamlining metabolomics analysis. Anal Chem 2018;90:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nash WJ, Dunn WB. From mass to metabolite in human untargeted metabolomics: Recent advances in annotation of metabolites applying liquid chromatography-mass spectrometry data. TrAC Trends Anal Chem 2019;120:115324. [Google Scholar]
- 25. Johnson SR, Lange BM. Open-access metabolomics databases for natural product research: present capabilities and future potential. Front Bioeng Biotechnol 2015;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moldoveanu SC, David V. Chapter 2 - parameters that characterize HPLC analysis. In: Moldoveanu SC, David V (eds). Essentials in Modern HPLC Separations. Elsevier, Amsterdam, Netherlands, 2013, 53–83. [Google Scholar]
- 27. Stoffel R, Quilliam MA, Hardt N, et al. . N-Alkylpyridinium sulfonates for retention time indexing in reversed-phase-liquid chromatography-mass spectrometry-based metabolomics. Anal Bioanal Chem 2022;414:7387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kováts E. Gas-chromatographische Charakterisierung organischer Verbindungen. Teil 1: Retentionsindices aliphatischer halogenide, alkohole, aldehyde und ketone. Helv Chim Acta 1958;41:1915–32. [Google Scholar]
- 29. Kind T, Wohlgemuth G, Lee DY, et al. . FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal Chem 2009;81:10038–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cao M, Fraser K, Huege J, et al. . Predicting retention time in hydrophilic interaction liquid chromatography mass spectrometry and its use for peak annotation in metabolomics. Metabolomics 2015;11:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Domingo-Almenara X, Guijas C, Billings E, et al. . The METLIN small molecule dataset for machine learning-based retention time prediction. Nat Commun 2019;10:5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Daly R, Rogers S, Wandy J, et al. . MetAssign: probabilistic annotation of metabolites from LC-MS data using a Bayesian clustering approach. Bioinformatics 2014;30:2764–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Del Carratore F, Schmidt K, Vinaixa M, et al. . Integrated probabilistic annotation: a bayesian-based annotation method for metabolomic profiles integrating biochemical connections, isotope patterns, and adduct relationships. Anal Chem 2019;91:12799–807. [DOI] [PubMed] [Google Scholar]
- 34. Nothias LF, Petras D, Schmid R, et al. . Feature-based molecular networking in the GNPS analysis environment. Nat Methods 2020;17:905–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen X, Wang R, Xiong X, et al. . Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat Commun 2019;10:1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen L, Lu W, Wang L, et al. . Metabolite discovery through global annotation of untargeted metabolomics data. Nat Methods 2021;18:1377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol 2012;8:e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Picart-Armada S, Fernandez-Albert F, Vinaixa M, et al. . Null diffusion-based enrichment for metabolomics data. PLoS One 2017;12:e0189012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 2007;23:980–7. [DOI] [PubMed] [Google Scholar]
- 40. Berriz GF, King OD, Bryant B, et al. . Characterizing gene sets with FuncAssociate. Bioinformatics 2003;19:2502–4. [DOI] [PubMed] [Google Scholar]
- 41. Hosack DA, DennisG, Jr, Sherman BT, et al. . Identifying biological themes within lists of genes with EASE. Genome Biol 2003;4:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sergushichev AA. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016:060012.
- 43. Mubeen S, Tom Kodamullil A, Hofmann-Apitius M, et al. . On the influence of several factors on pathway enrichment analysis. Brief Bioinform 2022;23:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fang H, Li X, Zan X, et al. . Signaling pathway impact analysis by incorporating the importance and specificity of genes (SPIA-IS). Comput Biol Chem 2017;71:236–44. [DOI] [PubMed] [Google Scholar]
- 45. Ihnatova I, Popovici V, Budinska E. A critical comparison of topology-based pathway analysis methods. PLoS One 2018;13:e0191154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mathur R, Rotroff D, Ma J, et al. . Gene set analysis methods: a systematic comparison. BioData Min 2018;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma J, Shojaie A, Michailidis G. A comparative study of topology-based pathway enrichment analysis methods. BMC Bioinformatics 2019;20:546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. TDM B. KEGGREST: Client-side REST access to the Kyoto Encyclopedia of Genes and Genomes (KEGG). R package version 1.38.0, 2022. [Google Scholar]
- 49. Karp PD, Midford PE, Caspi R, et al. . Pathway size matters: the influence of pathway granularity on over-representation (enrichment analysis) statistics. BMC Genomics 2021;22:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kanehisa M, Goto S, Sato Y, et al. . KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012;40:D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tarca AL, Bhatti G, Romero R. A comparison of gene set analysis methods in terms of sensitivity, prioritization and specificity. PLoS One 2013;8:e79217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maleki F, Ovens K, McQuillan I, et al. . Size matters: how sample size affects the reproducibility and specificity of gene set analysis. Hum Genomics 2019;13:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McLuskey K, Wandy J, Vincent I, et al. . Ranking metabolite sets by their activity levels. Metabolites 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tarca AL, Draghici S, Bhatti G, et al. . Down-weighting overlapping genes improves gene set analysis. BMC Bioinformatics 2012;13:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou G, Pang Z, Lu Y, et al. . OmicsNet 2.0: a web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res 2022;50:W527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cai Y, Kim DJ, Takahashi T, et al. . Kynurenic acid may underlie sex-specific immune responses to COVID-19, Science Signaling2021;14:eabf8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eroğlu İ, Eroğlu BÇ, Güven GS. Altered tryptophan absorption and metabolism could underlie long-term symptoms in survivors of coronavirus disease 2019 (COVID-19). Nutrition 2021;90:111308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sarohan AR, Kizil M, Inkaya AC, et al. . A novel hypothesis for COVID-19 pathogenesis: Retinol depletion and retinoid signaling disorder. Cell Signal 2021;87:110121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sarohan AR, Akelma H, Arac E, et al. . Retinol depletion in COVID-19. Clin Nutr Open Sci 2022;43:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sezer S, Bal C, Kalem AK, et al. . COVID-19 patients with altered steroid hormone levels are more likely to have higher disease severity. Endocrine 2022;78:373–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Escarcega RD, Honarpisheh P, Colpo GD, et al. . Sex differences in global metabolomic profiles of COVID-19 patients. Cell Death Dis 2022;13:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Romero-Martinez BS, Montano LM, Solis-Chagoyan H, et al. . Possible beneficial actions of caffeine in SARS-CoV-2. Int J Mol Sci 2021;22:5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kumrungsee T, Zhang P, Chartkul M, et al. . Potential role of vitamin B6 in ameliorating the severity of COVID-19 and its complications. Front Nutr 2020;7:562051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Franzosa EA, Sirota-Madi A, Avila-Pacheco J, et al. . Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 2019;4:293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang A, Sun H, Wang X. Emerging role and recent applications of metabolomics biomarkers in obesity disease research. RSC Adv 2017;7:14966–73. [Google Scholar]
- 66. Nicholson JK, Holmes E, Kinross JM, et al. . Metabolic phenotyping in clinical and surgical environments. Nature 2012;491:384–92. [DOI] [PubMed] [Google Scholar]
- 67. Nicholls A, Theodoridis G, Wilson ID. Global metabolic profiling in health and disease. In: Global Metabolic Profiling: Clinical Applications. Future Science Ltd, London, United Kingdom, 2014, 2–5. [Google Scholar]
- 68. Gonzalez-Covarrubias V, Martinez-Martinez E, Del Bosque-Plata L. The potential of metabolomics in biomedical applications. Metabolites 2022;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rinschen MM, Ivanisevic J, Giera M, et al. . Identification of bioactive metabolites using activity metabolomics. Nat Rev Mol Cell Biol 2019;20:353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Geistlinger L, Csaba G, Santarelli M, et al. . Toward a gold standard for benchmarking gene set enrichment analysis. Brief Bioinform 2021;22:545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bayerlova M, Jung K, Kramer F, et al. . Comparative study on gene set and pathway topology-based enrichment methods. BMC Bioinformatics 2015;16:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rahmatallah Y, Emmert-Streib F, Glazko G. Gene set analysis approaches for RNA-seq data: performance evaluation and application guideline. Brief Bioinform 2016;17:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hosseini R, Hassanpour N, Liu LP, et al. . Pathway-activity likelihood analysis and metabolite annotation for untargeted metabolomics using probabilistic modeling. Metabolites 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.