

Abstract
The coronavirus disease of 2019 pandemic has catalyzed the rapid development of mRNA vaccines, whereas, how to optimize the mRNA sequence of exogenous gene such as severe acute respiratory syndrome coronavirus 2 spike to fit human cells remains a critical challenge. A new algorithm, iDRO (integrated deep-learning-based mRNA optimization), is developed to optimize multiple components of mRNA sequences based on given amino acid sequences of target protein. Considering the biological constraints, we divided iDRO into two steps: open reading frame (ORF) optimization and 5′ untranslated region (UTR) and 3′UTR generation. In ORF optimization, BiLSTM-CRF (bidirectional long-short-term memory with conditional random field) is employed to determine the codon for each amino acid. In UTR generation, RNA-Bart (bidirectional auto-regressive transformer) is proposed to output the corresponding UTR. The results show that the optimized sequences of exogenous genes acquired the pattern of human endogenous gene sequence. In experimental validation, the mRNA sequence optimized by our method, compared with conventional method, shows higher protein expression. To the best of our knowledge, this is the first study by introducing deep-learning methods to integrated mRNA sequence optimization, and these results may contribute to the development of mRNA therapeutics.
Keywords: mRNA vaccine optimization, sequence deep learning, transformer-based model
Introduction
The coronavirus disease of 2019 (COVID-19) pandemic has presented new challenges to individuals worldwide. Various vaccine platforms have been developed and the mRNA vaccine precedes other conventional vaccine platforms because of high potency, safe administration, rapid development potentials and cost-effective manufacturing. However, the variants continuously arise global wide and are challenging the efficacy of current vaccines. According to CDC’s report, during the Delta variant period (June–July 2021), adjusted vaccine effectiveness (VE) against infection among those fully vaccinated was 52.4% for Pfizer-BioNTech, and 50.6% for Moderna. (Data show at https://www.cdc.gov/mmwr/volumes/70/wr/mm7034e3.html.) More seriously, the omicron variant was first detected in southern Africa in late November 2021 and labeled a ‘variant of concern’ by the World Health Organization on 26th November 2021. According to a recent study, traditional dosing regimens of COVID-19 vaccines available in the United States do not produce antibodies capable of recognizing and neutralizing the Omicron variant, and an additional ‘booster’ dose of Moderna or Pfizer-BioNTech mRNA-based vaccine is needed to provide immunity against the Omicron variant of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2; [1]). The global data show the coronavirus pandemic is far away from over and thus, more variants are expectable and some of them may escape the immune response produced after vaccination. It raises concerns that how to keep the efficacy of existing mRNA vaccines on variants.
The fundamental structure of mRNA vaccine is lipid nanoparticle (LNP) encapsulated mRNA chain, which is composed of an open reading frame (ORF) region that encodes the protein, flanked by five-prime (5′) and three-prime (3′) untranslated region (UTR), and further stabilized by 7-methylgaunosine (m7G) 5′ cap and 3′ poly (A) tails, respectively (Figure 1A). The process of mRNA vaccine development is illustrated in Figure 1B.
Figure 1.

Schematic of immune responses elicited by COVID-19 vaccine. (A) Structure of mRNA. (B) Schematic illustration of mRNA vaccine production. (C) Schematic representation of mechanism of mRNA vaccines.
The immune responses elicited by COVID-19 vaccine are depicted in the schematic Figure 1C. Briefly, upon intramuscular vaccination (Figure 1C—step 1), mRNA encapsulated in LNP are taken up by antigen-presenting cells (APCs), such as dendritic cells (DCs). mRNA is directly translated into polypeptides which are processed by the proteasome system, leading to peptide presentation onto MHC-I on the cell surface (similarly as during a viral infection), and post-translationally modified to be folded into the protein which, depending on the mRNA design, can either be membrane-anchored or be secreted. Peptide presentation onto MHC-II may occur on APCs after protein uptake of extracellular proteins or of cell debris containing protein (Figure 1C—step 2). These APCs then traffic to the lymph nodes (Figure 1C—step 3) where they are able to prime CD4 and CD8 T lymphocytes (Figure 1C—steps 4 and 5). Antigen-primed CD4 T cells can differentiate into T follicular helper (Tfh) cells, which help to activate B cells in germinal center (GC) and production of antibody with high affinity (Figure 1C—step 6) [2, 3].
As the COVID-19 mRNA vaccine encoding spike protein needs to be translated to protein in human cells to function, increased translation efficiency will yield more target antigen protein and therefore elicit stronger immune protection, and this is even more critical for virus variants. According to CureVac’s report [4], the COVID-19 mRNA vaccine from CureVac was only 47% effective in a late-stage trial. One possible reason is CureVac used the dose of 12 μg, a lower dose than Moderna and Pfizer-BioNTech vaccines, to balance safety and efficacy. However, lower dose of CureVac’s vaccine translated into fewer antigens and elicited weaker immune response insufficient to protect recipient from SARS-CoV-2 variants. Thus, how to improve the translation efficiency of mRNA vaccine to yield enough antigens at given dose mRNA is critically important.
It is acknowledged that various organisms have their own pattern of mRNA sequence and this pattern has complex effect on translation efficiency. For example, in each organism there is a preference for certain codons (biased codons) over others (rare codons); therefore, synonymous codons occur with different frequencies, a phenomenon termed codon usage bias, which is observed across species [5]. The codon usage directly influences the translation efficiency and mRNA stability [6]. Meanwhile, the sequence and length of UTR also varies among different organism [7]. UTR can impact mRNA degradation rate and translation efficiency through interacting with RNA binding proteins [8]. Therefore, in mRNA-based heterologous expression systems, such as expressing virus antigen in human cells, how to optimize and convert the mRNA sequence pattern of virus gene to human is important [9]. Some direct and straightforward optimization strategies have been widely used but they all have shortcomings. For instance, directly replacing rare codons with biased codons will results in an imbalance of different tRNAs and eventually leads to the depletion of tRNA and termination of translation [10]. Use of the globin UTR represents a logical approach to improve mRNA stability as the globin mRNAs produce large amount of protein and have long half-life [11]. However, as the biology of the target ORF and the cellular context may influence overall efficacy of mRNA, using a single universal UTR to improve protein production from exogenously administered mRNA across different cells types and tissues may not be feasible [12]. Further methods have been reported to optimize codon usage [13, 14], 5′UTR [15, 16], 3′UTR [17], combination of 5′UTR/3′UTR [12]. Nevertheless, as the multiple components (5′UTR, codon, 3′UTR) work in coordination during translation, a gene specific integrated optimization method is needed.
To overcome the aforementioned issues, this paper represents a full-length mRNA optimizing algorithm, iDRO (integrated deep-learning-based mRNA optimization; Figure 2A). Based on the mRNA structure, our algorithm consists of two parts: ORF optimization and UTR generation. For ORF optimization, we employed BiLSTM-CRF (bidirectional long-short-term memory with conditional random field; [18]) to construct codon. Inspired by mBART (multilingual bidirectional and auto-regressive transformers; [2, 19]) in Natural Language Processing (NLP) research, we developed RNA-Bart for UTR generation. Together, we developed a novel pipeline for optimization of full mRNA sequence (including 5′UTR, ORF, 3′UTR) via deep-learning method. The experimental validation indicates that our pipeline can generate mRNA sequence with high translation efficiency. As a brief summary, the contribution of this paper includes: (i) This is the first work to optimize full-length mRNA sequence for human including 5′UTR, ORF and 3′UTR; (ii) We developed a novel RNA-Bart, a Transformer-based model, to generate UTRs that are similar to human genome and increase translation efficiency and (iii) The biological experiments showed that the optimized mRNA sequence generated by iDRO reaches higher protein expression than conventional optimization method, i.e. incorporating the UTRs of human globin.
Figure 2.

Integrated deep-learning-based mRNA optimization (iDRO) pipeline and model details. (A) iDRO pipeline. iDRO has two main stages (gray blocks), ORF optimization and UTR generation. (B) Network structure of iDRO. Arrows shows the information flow. BiLSTM and RNA-Bart are detailed in sub-figures C and D. (C) Structure of BiLSTM-CRF. (D) Structure of RNA-Bart.
The rest of paper is structured as follows. In section ‘Materials and methods’, we introduced the datasets, ‘Biological materials and biological methods’ used in this work. In section ‘Proposed computational approach’, we described the proposed model iDRO in details. In section ‘Results’, we showed the performance of the proposed model, minimum free energy (mfe) analysis, secondary structural analysis and experiment validation results. In section ‘Discussion’, we discussed the results and showed how to use our method to generate mRNA sequence for SARS-CoV-2 variants and the comparison with Pfizer-BioNTech vaccine BNT162b2 and Moderna vaccine mRNA-1273. Finally, we concluded this work and discussed the future directions in section ‘Conclusion and future work’.
Material and methods
Design and build high expression efficiency dataset
Based on natural facts, a basic and reasonable assumption is proposed [12, 20]: we assumed human gene is the most efficient sequence for translation in human cell, which can be considered as the ground truth. This assumption is supported by the facts: (i) different species have consistent and characteristic codon bias, and a conventional approach to improve exogenous gene expression is to substitute rare codons by frequent codon in CDS according to the genomic codon usage in a host organism [21, 22], (ii) the UTR of mRNA are obviously different among species [23, 24], and using 5′UTR and/or 3′UTR from human gene is standard approach to enhance translation efficiency of exogenous mRNA (e.g. COVID-19 vaccine from Moderna and Pfizer-BioNTech; [25]). Previous study has shown that deep learning can learn the codon rules in Escherichia coli, and then fulfil high protein expression with optimized codons [13]. In this study, we extended these results and assumed that these rules exist not only in E. coli but also in humans, not only in ORF but also in UTR.
To generate training dataset that covers high translation efficiency sequence, we used the NCBI human gene dataset and UCSC.hg19.knownGene [26]. UCSC.hg19.knownGene contains 63 691 samples, and each sample consists of 5′UTR, ORF and 3′UTR sequence. We chose the data whose UTR length was 50–500 bp and ORF length less than 2500 bp and obtained 17 029 training data. We also translated ORFs to amino acid sequences for ORF optimization training. In ORF optimization, the amino acid is the input, and the corresponding ORF is the ground truth. In UTR generation, ORF is the input, and the corresponding UTR is the ground truth.
Biological materials and methods
Chemicals and reagents
All chemicals and reagents were purchased from Thermo Fisher Scientifc, Inc. (Waltham, MA) unless otherwise specified.
Cell line
HEK-293 cells were purchased from ATCC and cultured in DMEM medium supplemented with 10% fetal bovine serum, 100-U/ml penicillin and 100-mg/ml streptomycin.
Plasmid construction
The Kozak sequence (5′-GCCACC-3′) was inserted before the start codon (ATG) of the optimized EGFP gene, and they together were flanked by 5′UTR and 3′UTR. In this study, best preprocessing iDRO sequence, only tokenized iDRO sequence, and human alpha globin 5′UTR/3′UTR [27] were used. The sequence necessary for the in vitro transcription of mRNA, including T7 promoter, 5′UTR, Kozak sequence, EGFP coding sequence and 3′UTR, were cloned into pUC57 vector by GeneScript (Piscataway, NJ) and transformed into DH5α competent E. coli by chemical transformation. The transformed E. coli was allowed to grow on LB broth plate with agar and 100 μg/ml−1 ampicillin. Individual colonies were inoculated and outgrown in LB broth liquid medium containing 100 μg/ml ampicillin overnight with vigorous shaking at 250 rpm. Plasmids were extracted using GeneJET Plasmid Miniprep kit. Concentration was measured on a NanoDrop 2000 Spectrophotometer. The region of interest in the plasmid from the T7 promoter to 3′ UTR was confirmed by Sanger Sequencing.
In vitro transcription of mRNA
The templates for in vitro transcription were generated by PCR amplification of the corresponding plasmids using a forward primer and a reverse primer containing 100T at the 5′ end. The PCR products were purified using GeneJET PCR Purification Kit. The in vitro transcription of mRNA was performed using mMESSAGE mMACHINE T7 ULTRA Transcription Kit and generated mRNA was purified using MEGAclear Transcription Clean-Up Kit. After measurement of concentration by a NanoDrop 2000 Spectrophotometer, all mRNAs were diluted to 50 ng/μl, aliquoted, and stored at −80°C for future use.
Transfection
Lipofectamine 3000 (Invitrogen, Carlsbad, CA) was used as the transfection reagent. Briefly, on day 0, HEK-293 cells were seeded at a density of 3 × 105 cells/well on 6-well plates at volumes of 3 ml with DMEM medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin. Cells were incubated at 37°C in the presence of 5% CO2 until the confluence reached 80%. On day 1, 3 μg of various mRNA was diluted into 125 μl Opti-MEM. Meanwhile, 9 μl of Lipofectamine 3000 was diluted into 125 μl of Opti-MEM. Then the diluted mRNA was added to the diluted Lipofectamine 3000. The mixture was incubated for 15 min prior to addition to the cells. Each of mRNA was tested in triplicate.
Flow cytometry
The transfected cells were analyzed by flow cytometry (BD Biosciences, San Jose, CA). After 24 h, the cells were digested with trypsin, washed twice with phosphate-buffered saline (PBS) and finally, resuspended in 1 ml of PBS. Ten thousand cells were collected from each group, and the green fluorescence signal was captured through a fluorescein isothiocyanate (FITC) channel.
Proposed computational approach iDRO
iDRO structure
The iDRO structure is shown in Figure 2B. The input is amino acid sequence, which is the candidate protein sequence (e.g. spike protein in SARS-CoV-2), and the output is the optimized mRNA sequence for human. There are two main components: BiLSTM-CRF and RNA-Bart. BiLSTM-CRF consists of three types of deep-learning layer (word2vec layer, BiLSTM layer and CRF layer), and aims to find the best codon arrangement for amino acid sequence. RNA-Bart is a transformer-based pretraining model, which contains tokenize layer, transformer layer and softmax layer. The amino acids are firstly input to BiLSTM-CRF to get the optimized ORF sequence. Then, the ORF is split into tokens and fed into RNA-Bart to generate 5′UTR and 3′UTR.
The hyperparameter setup is followed. As for BiLSTM-CRF, the hidden layer dimension was 200; the learning rate was 0.003; the batch size was 32 and the dropout rate was 0.5. As for RNA-Bart. The learning rate was 0.00005; layer number was 8; hidden layer dimension was 512. Next, we will describe the BiLSTM-CRF and RNA-Bart in details.
BiLSTM-CRF
Based on the injective relation between codon and amino acid, ORF optimization can be regarded as a NER (name entity recognition) task in NLP. The goal of NER is to recognize the category (people, location, etc.) for each word. In this task, we recognize the best codon for each amino acid. Here, we employ the BiLSTM-CRF network to optimize ORF. BiLSTM uses the context information of the input amino acid to predict the codon, and the CRF layer adjusts the codon choice to increase accuracy. Therefore, BiLSTM-CRF is suitable for the ORF optimization task because of the high accuracy of BiLSTM-CRF. The structure of BiLSTM-CRF is shown in Figure 2C. Each amino acid is first fed into a word2vec layer [28], which is pretrained with all amino acid sequences we collected. At this step, each amino acid is encoded to a word vector that carries some context information. We denote the input word vector as x; the timestamp as t; the hidden feature as h and the output as y. The value for hidden layer and output is computed as follows:
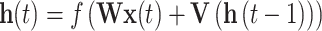 |
(1) |
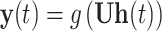 |
(2) |
where, W, V and U are learnable parameters. f(z) and g(z) are activation function. To improve the performance, CRF layer is attached to focus on sentence-level and ensures the predicted tags are correct. The codon box paradigm [13], shown in Supplementary Table 1 (Supplementary Data available online), is involved to reduce the tag class number and improve performance. We use cross-entropy loss to calculate the loss between output and ground truth.
RNA-Bart
The UTR generation can be regarded as a machine translation task, which aims to find the sentence that is most relevant to the input sentence. In our scenario, we aim to find the most relevant UTR sequence with given ORF. In this study, we developed a transformer-based pretraining model, RNA-Bart, to generate UTR. Transformer-based model can encode the long sequence precisely because it is fully based on attention mechanism [29]. The pretraining phase enables model to learn how to represent input sequences and serve for the downstream task [30]. Therefore, RNA-Bart can perform well in the machine translation task. The structure of RNA-Bart is shown in Figure 2D. It uses word embedding and position embedding to encode the input ORF token first, and then, the ORF sequence will be encoded to a vector with 512 dimensions and decoded to UTR. Similar to mBART, RNA-Bart consists of two training phases. In pretraining phase, we cover 15% tokens in UTR and ORF sequences and train an auto-regressive model with 512 max length to recover the masked tokens. Then, we keep the auto-regressive model and fine-tune it to machine translation model that can generate UTR sequences. Because of the relatively small dataset of RNA set, complex models such as mBART are prone to overfitting. In contrast, RNA-Bart is a light model that uses fewer layers and less parameters can avoid this issue. Meanwhile, some studies demonstrate that the fewer layers in transformer-based model would not affect accuracy [31, 32]. As for tokenizing, we use WordPiece model [33] with a 70 000 token vocabulary instead of sentence-piece model [34] like mBART.
We adopt tokenization, a common method in NLP, to reduce the length of input sequences. Tokenization greedily split mRNA sequence into several short sequences based on the frequency of these short sequences in corpus. For example, ‘ATGGACTTTC’ will be split into ‘ATGGAC’, ‘TTTC’ (two tokens only) instead of ‘A’, ‘T’, ‘G’, ‘G’, ‘A’, ‘C’, ‘T’, ‘T’, ‘T’ and ‘C’ (10 tokens) since ‘ATGGAC’ and ‘TTTC’ appear frequently in the training corpus.
Apart from tokenizing, there are two more preprocessing methods to increase the difference between ORF and UTR: adding special tokens and converting ORF into codon boxes. Adding special tokens refers to adding tokens that would not occur in ORF nor UTR before sequence. Detailed, we add '<ORF>' before ORF, '<5UTR>' before 5′UTR and '<3UTR>' before 3′UTR. As for converting ORF into codon boxes, the input sequence is the translated codon boxes instead of ORF. Because of these preprocessing methods, the difference between ORF and UTR is increased. Therefore, RNA-Bart can distinguish sequence regions and capture sequence features precisely. The codon box combined with the amino acid can determine a specific ORF, which ensures the most suitable UTR is generated.
Results
ORF optimization
Codons are not randomly selected among individuals [20], but follow some codon rules embedded in cellular environmental information. To accurately unravel the codon rules, we introduced BiLSTM-CRF. The input of BiLSTM-CRF is the amino acid sequence, and the output is the corresponding optimized ORF. In the training phase, we set the human’s ORF as the ground truth, because we assume that human’s ORF is the most optimal sequence for protein translation in human.
To test the performance of ORF optimization, we randomly split ORFs data into the training set, validation set, and test set as 8:1:1. The accuracy on the test data set and training dataset can reach 0.63, 0.71, respectively. This performance is comparable with that in bacterial cell [13]. In addition, we adjusted hidden layer dimension and learning rate of BiLSTM-CRF to test the robustness. The results are shown in Figure 3A. When the learning rate is 0.003 and the hidden layer dimension is 200, which means the model reaches the highest accuracy (0.63).
Figure 3.

Statistic results of iDRO. (A) Accuracy of different BiLSTM-CRF hyperparameter setup. Different groups represent different hidden layer dimension and colors represent different learning rate. The setup with 200 dimensions and 0.003 learning rate has the highest accuracy (B) Machine translation indicators for different preprocessing method. The combination of Codon Box and Special Tokens has the highest score for both Jaccard and Rouge. Jaccard and Rouge are commonly used to indicate the similarity between two sentences (C) Machine translation indicators of different learning rate. In both 5′UTR generation and 3′UTR generation, RNA-Bart with 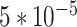 learning rate has the best performance.
learning rate has the best performance.
UTR generation
Our goal is to generate UTR sequence that mimic the pattern of human endogenous UTR. To test UTR optimization method, we randomly split data into the training set, validation set and test set as 8:1:1, and used cross validation to test RNA-Bart. The results are represented in Figure 3B. Jaccard score [35] and Rouge [36] are used as main index because they are commonly used indicators in machine translation to measure the similarity between two sequences. Jaccard is calculated as the size of the intersection divided by the size of the union of the sample sets. If two sequences are the same, the intersection will equal the union and Jaccard will be ‘1’. On the other hand, if two sequences are different, the intersection will be empty and Jaccard will be ‘0’. Rouge is calculated as the overlap of n-grams between the output and reference. N-gram is continuous N tokens. Similar output has more matched n-grams, as well the higher Rouge score. The higher Jaccard and Rouge score represent higher extend similarity of predicted sequence with human UTR sequence. We compared RNA-Bart with RNN (recurrent neural network), LSTM (long-short-term memory), BiLSTM (bi-direction long-short-term memory), BiLSTM with attention and Transformer. The results are shown in Table 1. RNA-Bart has the highest score (0.811 and 0.783), and significantly outperforms RNN (0.652 and 0.623), LSTM (0.704 and 0.692), BiLSTM (0.727 and 0.713). It also reaches higher score than Transformer (0.767 and 0.741). In addition, we compared the UTR sequence generated by different preprocessing methods. As Figure 3A shown, codon box combined with special tokens has the highest value for both 5′UTR (0.811 and 0.783) and 3′UTR (0.798 and 0.856), and are substantially better than the tokenize-only approach. The detailed results are shown in Supplementary Table 2 (Supplementary Data available online).
Table 1.
Jaccard and Rouge for different deep-learning method for generating 5′UTR
| Method | Jaccard | Rouge |
|---|---|---|
| RNN (recurrent neural network) | 0.652 | 0.623 |
| LSTM (long-short-term memory) | 0.704 | 0.692 |
| BiLSTM (Bi-direction long-short-term memory) | 0.727 | 0.713 |
| Transformer | 0.767 | 0.741 |
| RNA-Bart | 0.811 | 0.783 |
To further verify our model, we took EGFP as example and use miRanda [37] to predict miRNA (microRNA) binding site and secondary structure of our UTRs (sequences are shown in Supplementary Table 3, Supplementary Data available online) and the results are shown in Figure 4. As miRNA is known for negative regulating complementary mRNA by inducing translational repression and mRNA decay [38], the mRNA sequence that containing more miRNA binding sites may has lower stability. iDRO and globin UTR have comparable number of miRNA binding sites and both of them are much lower than control sequence (Figure 4A and Supplementary Table 4, Supplementary Data available online). In addition, based on the Nussinov algorithm and energy information, Zuker et al. proposed a minimum free energy (mfe) algorithm [39], which assumes that mRNA structure has a great relationship with energy. With the aid of this algorithm, we calculated the mfe and predicated the secondary structure of 5′UTR and 3′UTR with software RNAfold ([40]; Figure 4B).
Figure 4.

miRNA binding site, secondary structure and minimum free energy (mfe) (kcal/mol) for EGFP UTRs. (A) miRNA binding site. (B) Secondary structure for globin, control and iDRO UTRs. (C) Secondary structure and mfe (kcal/mol) for the top 5′UTR with the highest ribosome loading. (D) Secondary structure and mfe (kcal/mol) for 5′UTR from the top five genes with the largest fold changes of upregulated translation efficiency after LPS stimulation.
There is ample evidence that secondary structures with the 5′ and 3′UTR sequences mediate translational control [41]. We next analyzed the secondary structure of 5′ and 3′UTR. The iDRO-predicted 5′UTR has a more secondary structures than globin 5′UTR. The role of secondary structure on 5′UTR remains controversial. Although most highly structured 5′UTRs have an inhibitory effect on translation [42], there are some examples of translation enhancement by 5′UTR secondary structures such as Hsp70 [43, 44], surfactant protein A [45] and GLUT1 transporter [46, 47]. To confirm the high structure in 5′UTR is not always a negative regulator for translation, we generated 5′UTR using the deep-learning model established from polysome profiling of a library of 280 000 randomized 5′UTR [15]. We found that complex secondary structures exist in all top-ranked 5′UTR with the highest ribosome loading (Figure 4C). We also studied the 5′UTR in those genes that can upregulate translation levels rapidly in dendritic cells after lipopolysaccharide (LPS) stimulation using reported quantitative transcriptome and proteasome data [48, 49]. As results showed (Figure 4D), all the five genes (1433B, 2AAA, 3BP1, AL3A2 and AL9A1) with the largest fold change of translation upregulation after stimulation have highly structured 5′UTR. On the other hand, the secondary structure on 3′UTR is generally believed to aid mRNA translation and stability via interaction with trans-acting regulatory factors [41, 47]. Meanwhile, the 3′UTR sequence predicted by iDRO contains more complex secondary structure, which may improve the mRNA translation and stability (Figure 4B) compared with globin 3′UTR.
Experimental validation
We used the EGFP gene as an example to verify our method. EGFP amino acid sequence is input to iDRO. The combination (codon box plus Special tokens for 5′UTR and 3′UTR) was used to compare with the globin 5′UTR/3′UTR. In addition, to emphasize that preprocessing is necessary, the tokenize-only model was used as control sequence. The sequences of EGFP gene with optimized codon and 5′UTR/3′UTR used in this study were listed in Supplementary Table 3 (see Supplementary Data available online). The mRNAs were transfected into HEK 293 cells and translated into functional protein with high efficiency. The results of flow cytometry analysis showed that ≥90% cells expressed EGFP (Figure 5A). Next, we analyzed the geometric mean of EGFP fluorescence density of each group. The results showed that the EGFP fluorescence density of cells transfected with mRNA containing de novo designed 5′UTR and 3′UTR was 1.2-fold higher than the HBA 5′UTR and 3′UTR, which is widely used in mRNA synthesis [27]. Although the fluorescence density of cells transfected with mRNA containing control 5′UTR/3′UTR was half of HBA 5′UTR/3′UTR (Figure 5B).
Figure 5.

The flow cytometry analysis of mRNA transfected HEK-293 cells. (A) EGFP gene expression in mRNA transfected HEK-293 cells. (B) The relative geometric mean of EGFP fluorescence density in EGFP positive cells.
Discussion
Regarding the performance and robustness of iDRO, Figure 3A shows the cross-validation results of RNA-Bart. Both UTR and ORF are composed of ‘ATCG’ and the model can be confused. The ‘A’ in the UTR is not the same as the ‘A’ in the ORF. Without further preprocessing, the model may not be able to distinguish which area ‘A’ belongs to and leading to poor performance. On the contrary, special tokens directly indicate the domain, and the codon box changes the alphabet of ORF. These preprocessing methods help model identify areas and encode ‘ATCG’ correctly, resulting in better performance. In addition, we adjust hidden layer dimension and learning rate in a small range to test the robustness of iDRO. The results are shown in Figure 3B and C. Small-scale parameter adjustment has little effect on model performance, indicating that iDRO is robust to hyperparameters setup and has a good generalization.
Our approach takes advantage of self-attention mechanism, thus it is interesting to compare our approach with other attention features techniques such as genetic algorithm [50, 51], quantum-behaved multiverse optimization (QMVO; [52]) and BiLSTM with attention [53]. Genetic algorithm is a popular approach for optimization and feature selection, and QMVO is one of the bio-inspired algorithms based on multiverse theory in physics. Both of them can remove redundant information and may get a better data representation, whereas attention mechanism [53] can also increase model performance by emphasizing the connection importance in sequence. We compared our model iDRO with genetic algorithm, QMVO and BiLSTM with attention, and the results are presented in Table 2. iDRO (0.811 and 0.783) substantially outperforms genetic algorithm (0.719 and 0.701) and QMVO (0.734 and 0.713) since the token selection may introduce ambiguity into sequence. iDRO also significantly outperforms BiLSTM (0.753 and 0.736) because BiLSTM may not able to capture long-distance connections in sequences. The parameters for each approach were set as follows. First for genetic algorithm, we set cross rate as 0.5 mutation rate as 0.001, population size as 1000, epoch as 100; second for QMVO, we set min Wormhole existence probability as 0.2 and the max Wormhole existence probability as 1 and finally for BiLSTM with attention we set the hidden layer dimension as 200; the learning rate as 0.003; the batch size as 32 and the dropout rate as 0.5.
Table 2.
Jaccard and Rouge for feature selection for generating 5′UTR
| Method | Jaccard | Rouge |
|---|---|---|
| Genetic algorithm | 0.719 | 0.701 |
| QMVO (quantum-behaved multiverse optimization) | 0.734 | 0.713 |
| BiLSTM with attention | 0.753 | 0.736 |
| iDRO without feature selection | 0.811 | 0.783 |
In the experimental validation, we used the EGFP reporter gene which is a widely used strategy for mRNA sequence optimization [54]. As experiments results showed that mRNA sequence generated by iDRO yielded higher protein expression, we next used iDRO for SARS-CoV-2 spike protein (sequences shown in Supplementary Table 5, Supplementary Data available online). To be consistent with Pfizer-BioNTech vaccine BNT162b2 [55] and Moderna vaccine mRNA-1273 [56], in our optimization, the amino acids K986 and V987 were replaced with 2 prolines to stabilize the trimers of SARS-CoV-2 spike proteins. We analyzed miRNA binding site as well as secondary structure for mRNA-1273, BNT162b2 and iDRO UTRs. The results are shown in Figure 6A and Supplementary Table 6 (Supplementary Data available online). iDRO UTRs have fewer or comparable miRNA binding sites (4 sites in 5′UTR and 11 sites in 3′UTR), compared with mRNA-1273 (6 sites in 5′UTR and 10 sites in 3′UTR) and BNT162b2 (9 sites in 5′UTR and 11 sites in 3′UTR). The iDRO-generated 5′UTR and 3′UTR have lower mfe (−38.10 kcal/mol and −107.8 kcal/mol) compared with BNT162b2 (−9.70 and −37 kcal/mol) and mRNA-1273 (−4.62 and −86.40 kcal/mol) in Figure 6B, manifesting that iDRO-generated mRNA sequence may yield high level of antigen expression and elicit strong immune protection if used as vaccine. Compared with BNT162b and mRNA-1273, the iDRO 5′UTR forms long stem structure, composed of multiple G/C pairs (Figure 6B). The 5′UTR in mRNA-1273 is patented sequence de novo developed by Moderna (U.S. Patent US 10881730B2), and it only has a short 6 G/C pairs-formed stem structure. The 5′UTR of BNT162b2 incorporates the 5′UTR of human α-globin, and it has two short stem structures composed of G/C and A/U pairs. As mentioned in the EGFP mRNA optimization (Figure 4), contrary to conventional thinking, the stable secondary structure in iDRO 5′UTR does not necessarily imply low translation efficiency [57]. Another thing worth noting is all U nucleotides in the BNT162b and mRNA-1273 have been replaced with N1-methyl-pseudouridine (ψ) to reduce immune reaction towards mRNA and to increase protein production [55, 56], and thus the real secondary structure may be slightly different with the predicated results.
Figure 6.

miRNA binding site, secondary structure and minimum free energy (mfe) (kcal/mol) for SARS-CoV-2 spike protein. (A) miRNA binding site (B) Secondary structure and mfe (kcal/mol) for Pfizer-BioNTech vaccine BNT162b2, Moderna vaccine mRNA-1273 and iDRO UTRs. (C) Secondary structure and mfe (kcal/mol) for SARS-CoV-2 variants spike protein.
With the world starting another year in the pall of the pandemic, new SARS-CoV-2 variants have become a cause for concern for governments and epidemiologists around the world. The omicron variant is spreading at a rate not seen with previous variants and Moderna and Pfizer-BioNTech are working on the vaccine against the omicron variant. In this study, we also used iDRO to generate mRNA sequence for spike protein of alpha, beta, gamma, delta and omicron variants. The generated sequences are shown in Supplementary Table 7, Supplementary Data available online. The predicted secondary structure and minimum free energy for each UTRs are shown in Figure 6C. The Alpha and Beta variants have the same predicated UTR because they have similar spike proteins sequences.
For the long mRNA sequence data in this study, the long sequence dependency problem is one of the biggest obstacle for analysis. The long sequence dependency challenge is not only the high computation requirements but also the difficulty to learn the relationship between tokens. BiLSTM analyzes sequence data in forwarding order and reversing order. It uses forget and input gates to drop useless information and keep vital information. However, it still cannot fully address the long sequence dependency problem as it requires high computation resources. The transformer uses self-attention to analyze sequences, and it eliminates sequence dependency and allows full GPU acceleration. But the older version of Transformer cannot analyze the relationship between tokens well. To overcome above mentioned issues, we developed the integrated deep-learning model iDRO to optimize 5′UTR, codon usage and 3′UTR simultaneously that enables users to design the optimal mRNA sequence to enhance protein expression level, thus improving the efficacy of mRNA medicines. Through our pipeline, we can optimize the whole sequence of mRNA vaccines and handle various kinds of mRNA optimization.
Conclusion and future work
In this study, we developed an integrated mRNA sequence optimization method, iDRO. It can generate mRNA sequences that are similar to human genome and has high translation efficiency. The structural and MFE analysis suggested the predicted sequences are stable and may have better translation efficiency. Our biological experiments showed the sequence generated by iDRO has higher protein expression than the α-globin UTR. Although iDRO achieves good performance. This iDRO approach could be further improved, e.g. regarding to the transform from ORF to 5′ and 3′UTR, we might be able to get a good performance by considering multilingual model [58]. Contrastive learning [59], which helps model capture sequences features from different aspects, could be considered here to give a better representation of input data. Different pretraining tasks such as shuffled sentence [60] could also be recruited to improve the performance of our model. Finally, these strategies will be validated by biological experiments for various genes using multiple cell lines, as well as in vivo experiments. Eventually, our study will be widely applicable to all therapeutic applications of mRNA medicines, e.g. infectious disease, cancer and aging.
Tool availability
The software is freely available for academic use only upon request.
Key Points
mRNA vaccine optimization;
Sequence deep learning;
Transformer-based model.
Supplementary Material
Haoran Gong is a research assistant in West China Biomedical Big Data Center, West China Hospital, Sichuan University. His interests are sequence data analysis and bioinformatics.
Jianguo Wen is an assistant professor in the School of Biomedical Informatics at the University of Texas Health Science Center at Houston. His research interests include mRNA medicine and immunotherapies.
Ruihan Luo is a PhD student at West China Biomedical Big Data Center, West China Hospital,Sichuan University, China. Her main interests lie in bioinformatics, genomics and cancer immunology.
Yuzhou Feng is a PhD student at West China Biomedical Big Data Center, West China Hospital,Sichuan University, China. His interests are bioinformatics and genomics.
Jingjing Guo received her PhD degree in basic medicine in Shandong University, China. Her research interests are tumor single-cell transcriptome analysis, spatial transcriptome analysis and bioinformatics.
Hongguang Fu is a professor in University of Electronic Science and Technology of China. His research interests are symbolic computation, bioinformatics and cognitive intelligence.
Xiaobo Zhou received his PhD degree in applied mathematics from Beijing University, China, in 1998. His research interests are bioinformatics, systems biology, imaging informatics and clinical informatics.
Contributor Information
Haoran Gong, West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu 610041, China; Med-X Center for Informatics, Sichuan University, Chengdu 610041, China.
Jianguo Wen, Center for Computational Systems Medicine, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, Texas, 77030, USA; McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Ruihan Luo, West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu 610041, China.
Yuzhou Feng, West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu 610041, China.
JingJing Guo, West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu 610041, China; Med-X Center for Informatics, Sichuan University, Chengdu 610041, China.
Hongguang Fu, University of Electronic Science and Technology of China, Chengdu 611731, China.
Xiaobo Zhou, Center for Computational Systems Medicine, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, Texas, 77030, USA; McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA; School of Dentistry, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Funding
Gong, Luo and Feng were partially supported by Center of Excellence-International Collaboration Initiative Grant, West China Hospital, Sichuan University (no. 139170052) and 1·3·5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University (no. 2019HXFH022). Guo was supported by Sichuan Science and Technology Program, (no. 2022YFS0228). Fu was partially supported by National Natural Science Foundation of China (no. 61876034). Wen and Zhou were partially supported by NIH R01GM123037, U01AR069395, R01CA241930 and NSF 2217515.
Reference
- 1. Garcia-Beltran WF, Denis KJS, Hoelzemer A, et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 omicron variant. Cell 2021;184:2372–2383.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bettini E, Locci M. SARS-CoV-2 mRNA vaccines: immunological mechanism and beyond. Vaccine 2021;9:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cagigi A, Loré K. Immune responses induced by mRNA vaccination in mice, monkeys and humans. Vaccine 2021;9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Radford A, Narasimhan K, Salimans T, et al. Improving language understanding by generative pre-training. 2018;2018. [Google Scholar]
- 5. Grantham R, Gautier C, Gouy M, et al. Codon catalog usage and the genome hypothesis. Nucleic Acids Res 1980;8:r49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jia L, Qian S-B. Therapeutic mRNA engineering from head to tail. Acc Chem Res 2021;54:4272–82. [DOI] [PubMed] [Google Scholar]
- 7. Liu H, Yin J, Xiao M, et al. Characterization and evolution of 5′ and 3′ untranslated regions in eukaryotes. Gene 2012;507:106–11. [DOI] [PubMed] [Google Scholar]
- 8. Miao L, Zhang Y, Huang L. mRNA vaccine for cancer immunotherapy. Mol Cancer 2021;20:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanson G, Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol 2018;19:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Villalobos A, Ness JE, Gustafsson C, et al. Gene designer: a synthetic biology tool for constructing artificial DNA segments. BMC bioinformatics 2006;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ross J, Sullivan TD. Half-lives of beta and gamma globin messenger RNAs and of protein synthetic capacity in cultured human reticulocytes. Blood 1985;66:1149–54. [PubMed] [Google Scholar]
- 12. Asrani KH, Farelli JD, Stahley MR, et al. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol 2018;15:756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fu H, Liang Y, Zhong X, et al. Codon optimization with deep learning to enhance protein expression. Sci Rep 2020;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trösemeier J-H, Rudorf S, Loessner H, et al. Optimizing the dynamics of protein expression. Sci Rep 2019;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sample PJ, Wang B, Reid DW, et al. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol 2019;37:803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sultana N, Hadas Y, Sharkar MTK, et al. Optimization of 5′ untranslated region of modified mRNA for use in cardiac or hepatic ischemic injury. Mol Ther Methods Clin Dev 2020;17:622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niessen AGO, Poleganov MA, Rechner C, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3′ UTRs identified by cellular library screening. Mol Ther 2019;27:824–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang Z, Xu W, Yu K. Bidirectional LSTM-CRF models for sequence tagging arXiv preprint arXiv:1508.01991. 2015.
- 19. Liu Y, Gu J, Goyal N, et al. Multilingual denoising pre-training for neural machine translation. Trans Assoc Comput Linguist 2020;8:726–42. [Google Scholar]
- 20. Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet 2008;42:287–99. [DOI] [PubMed] [Google Scholar]
- 21. Quax TE, Claassens NJ, Söll D, et al. Codon bias as a means to fine-tune gene expression. Mol Cell 2015;59:149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Z, Dang Y, Zhou M, et al. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci 2016;113:E6117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leppek K, Das R, Barna M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 2018;19:158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayr C. Evolution and biological roles of alternative 3′ UTRs. Trends Cell Biol 2016;26:227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xia X. Detailed dissection and critical evaluation of the Pfizer/BioNTech and Moderna mRNA vaccines. Vaccine 2021;9:734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang T, Shi T, Zhang H, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol 2019;12:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhuang X, Qi Y, Wang M, et al. mRNA vaccines encoding the HA protein of influenza A H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccine 2020;8:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma L, Zhang Y. Using Word2Vec to process big text data. In: 2015 IEEE International Conference on Big Data (Big Data). New York: IEEE, 2015, 2895–7. [Google Scholar]
- 29. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. Adv Neural Inf Process Syst 2017;30. [Google Scholar]
- 30. Devlin J, Chang M-W, Lee K, et al. Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies 1:4171–86, https://aclanthology.org/N19-1423. [Google Scholar]
- 31. Jiao X, Yin Y, Shang L, et al. Tinybert: distilling bert for natural language understanding arXiv preprint arXiv:1909.10351. 2019.
- 32. Lan Z, Chen M, Goodman S, et al. Albert: a lite bert for self-supervised learning of language representations arXiv preprint arXiv:1909.11942. 2019.
- 33. Wu Y, Schuster M, Chen Z, et al. Google's neural machine translation system: bridging the gap between human and machine translation arXiv preprint arXiv:1609.08144. 2016.
- 34. Kudo T, Richardson J. Proceedings of the 2018 Conference on Empirical Methods in Natural Language Processing: System Demonstrations 66–71, https://aclanthology.org/D18-2012.
- 35. Real R, Vargas JM. The probabilistic basis of Jaccard's index of similarity. Syst Biol 1996;45:380–5. [Google Scholar]
- 36. Lin C-Y. Rouge: a package for automatic evaluation of summaries. In: Text Summarization Branches Out, Association for Computational Linguistics, 2004, 74–81, https://aclanthology.org/W04-1013.
- 37. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res 2019;47:D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iwakawa H-o, Tomari Y, Tomari Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol 2015;25:651–65. [DOI] [PubMed] [Google Scholar]
- 39. Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981;9:133–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gruber AR, Lorenz R, Bernhart SH, et al. The Vienna RNA websuite. Nucleic Acids Res 2008;36:W70–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adeli K. Translational control mechanisms in metabolic regulation: critical role of RNA binding proteins, microRNAs, and cytoplasmic RNA granules. Am J Physiol-Endocrinol Metabol 2011;301:E1051–64. [DOI] [PubMed] [Google Scholar]
- 42. Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005;361:13–37. [DOI] [PubMed] [Google Scholar]
- 43. Vivinus S, Baulande S, Zanten M, et al. An element within the 5′ untranslated region of human Hsp70 mRNA which acts as a general enhancer of mRNA translation. Eur J Biochem 2001;268:1908–17. [DOI] [PubMed] [Google Scholar]
- 44. Yueh YG, Yaworsky PJ, Kappen C. Herpes simplex virus transcriptional activator VP16 is detrimental to preimplantation development in mice. Mol Reprod Dev 2000;55:37–46. [DOI] [PubMed] [Google Scholar]
- 45. Wang G, Guo X, Floros J. Differences in the translation efficiency and mRNA stability mediated by 5′-UTR splice variants of human SP-A1 and SP-A2 genes. Am J Physiol 2005;289:L497–508. [DOI] [PubMed] [Google Scholar]
- 46. Boado RJ, Pardridge WM. The 5′-untranslated region of GLUT1 glucose transporter mRNA causes differential regulation of the translational rate in plant and animal systems. Comp Biochem Physiol Part B 1997;118:309–12. [DOI] [PubMed] [Google Scholar]
- 47. Boado RJ, Pardridge WM. Amplification of gene expression using both 5′-and 3′-untranslated regions of GLUT1 glucose transporter mRNA. Mol Brain Res 1999;63:371–4. [DOI] [PubMed] [Google Scholar]
- 48. Schinnerling K, García-González P, Aguillón JC. Gene expression profiling of human monocyte-derived dendritic cells–searching for molecular regulators of tolerogenicity. Front Immunol 2015;6:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Worah K, Mathan TS, Manh TPV, et al. Proteomics of human dendritic cell subsets reveals subset-specific surface markers and differential inflammasome function. Cell Rep 2016;16:2953–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anter AM, Moemen YS, Darwish A, et al. Multi-target QSAR modelling of chemo-genomic data analysis based on extreme learning machine. Knowl-Based Syst 2020;188:104977. [Google Scholar]
- 51. Anter AM, Abd Elaziz M, Zhang Z. Real-time epileptic seizure recognition using Bayesian genetic whale optimizer and adaptive machine learning. Future Gener Comput Syst 2022;127:426–34. [Google Scholar]
- 52. Anter AM, Elnashar HS, Zhang Z. QMVO-SCDL: a new regression model for fMRI pain decoding using quantum-behaved sparse dictionary learning. Knowl-Based Syst 2022;252:109323. [Google Scholar]
- 53. Liu G, Guo J. Bidirectional LSTM with attention mechanism and convolutional layer for text classification. Neurocomputing 2019;337:325–38. [Google Scholar]
- 54. Suknuntha K, Tao L, Brok-Volchanskaya V, et al. Optimization of synthetic mRNA for highly efficient translation and its application in the generation of endothelial and hematopoietic cells from human and primate pluripotent stem cells. Stem Cell Rev Rep 2018;14:525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vogel AB, Kanevsky I, Che Y, et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021;592:283–9. [DOI] [PubMed] [Google Scholar]
- 56. Corbett KS, Edwards DK, Leist SR, et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020;586:567–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Araujo PR, Yoon K, Ko D, et al. Before it gets started: regulating translation at the 5′ UTR. Comp Funct Genom 2012;2012:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang Y, Tran C, Li X, et al. Findings of the Association for Computational Linguistics ACL-IJCNLP 2021 3450–66.
- 59. Giorgi J, Nitski O, Wang B, et al. Declutr: deep contrastive learning for unsupervised textual representations arXiv preprint arXiv:2006.03659. 2020.
- 60. Cui Y, Yang Z, Liu T. PERT: pre-training BERT with permuted language model arXiv preprint arXiv:2203.06906. 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.