

Abstract
Cell–cell communications are vital for biological signalling and play important roles in complex diseases. Recent advances in single-cell spatial transcriptomics (SCST) technologies allow examining the spatial cell communication landscapes and hold the promise for disentangling the complex ligand–receptor (L–R) interactions across cells. However, due to frequent dropout events and noisy signals in SCST data, it is challenging and lack of effective and tailored methods to accurately infer cellular communications. Herein, to decipher the cell-to-cell communications from SCST profiles, we propose a novel adaptive graph model with attention mechanisms named spaCI. spaCI incorporates both spatial locations and gene expression profiles of cells to identify the active L–R signalling axis across neighbouring cells. Through benchmarking with currently available methods, spaCI shows superior performance on both simulation data and real SCST datasets. Furthermore, spaCI is able to identify the upstream transcriptional factors mediating the active L–R interactions. For biological insights, we have applied spaCI to the seqFISH+ data of mouse cortex and the NanoString CosMx Spatial Molecular Imager (SMI) data of non-small cell lung cancer samples. spaCI reveals the hidden L–R interactions from the sparse seqFISH+ data, meanwhile identifies the inconspicuous L–R interactions including THBS1−ITGB1 between fibroblast and tumours in NanoString CosMx SMI data. spaCI further reveals that SMAD3 plays an important role in regulating the crosstalk between fibroblasts and tumours, which contributes to the prognosis of lung cancer patients. Collectively, spaCI addresses the challenges in interrogating SCST data for gaining insights into the underlying cellular communications, thus facilitates the discoveries of disease mechanisms, effective biomarkers and therapeutic targets.
Keywords: single-cell spatial transcriptomics, spatial cell graph, adaptive graph model, triplet loss
Introduction
Cell–cell communications play important roles in cellular functions and tissue homeostasis [1]. These cellular communications are mediated by the active interactions of ligand–receptor (L–R). Cells with altered L–R signalling will affect their neighbouring cells and remodel their microenvironment. When cells do not interact or transfer messages properly, disease-related activities will happen. For example, tumour-associated macrophages may interact with tumour cells by deriving IL-6, which leads to the chemo-resistance of tumour cells [2]. Moreover, cancer-associated fibroblasts have been shown to release the CXCL12 chemokine in the pancreatic tumour microenvironment (TME), which interacts with CXCR4 and promotes tumour progression through growth-permissive modulation of the immune microenvironment [3]. Thus, identification and quantification of cell-to-cell communications are critical and fundamental for revealing the underlying mechanisms of complex diseases.
Recently, the breakthroughs in single-cell spatial transcriptomics (SCST) technologies [4–6] have allowed researchers to examine both the spatial and transcriptional landscapes of individual cells and are extremely useful for dissecting heterogeneity and cellular communications at unprecedented resolution. For example, seqFISH+ [7] can image mRNAs for 10 000 genes in single cells, which enables revealing subcellular mRNA patterns of spatially organized cells within the tissue. NanoString CosMx™ Spatial Molecular Imager (SMI) [8], Vizgen MERSCOPE [4] and 10× Genomics Xenium [9] are developed as a single-cell spatial solution to capture targeted transcripts in manually selected regions of interest [10], which enables to investigate morphologically intact tissues at unprecedented resolution. These new technologies hold the promise to spatially and functionally reveal complex architectures of tissues and to further our insights into intercellular interactions at unprecedented resolution.
Given the emerging spatial technologies, effective methods inferring L–R signalling axis and cell–cell communications from spatial transcriptomics (ST) data are still lacking. Currently, there are methods available to infer L–R interactions from single-cell RNA-seq (scRNA-seq) data. For example, iTALK infers the L–R interactions as those significantly differentially expressed among different cell groups [11]. CellPhoneDB v2.0 infers enriched L–R interactions between two cell groups based on their significant specificity [12]. CellChat focuses on the differentially over-expressed ligands and receptors to quantify the associations of L–R pairs modelled by the law of mass action [13]. Connectome [14] defines the weight of L–R interaction based on the product of cell type-wise normalized expression for ligand and receptor. Though these methods enable to infer L–R interactions from scRNA-seq data, their assumptions usually require that the average expression of ligand or receptor in a specific cell group is above its mean expression over the whole counts, which may cause false negatives due to the sparsity and dropout issues of SCST profiles [15]. More importantly, these methods do not consider the spatial cell locations, and thus may result in false positives due to the lack of physical adjacency for contact-based cell–cell interactions, autocrine or paracrine.
Therefore, to accurately reveal the landscape of cellular communications, we present a novel method, termed spaCI, to decipher cellular interactions in SCST data. spaCI incorporates both the spatial graphs of adjacent cells and their gene expressions to identify the L–R interactions. spaCI introduces the triplet loss that effectively avoids possible false positive or false negative interactions when detecting active L–R signalling pairs. The outperformance of spaCI is demonstrated using both simulation data and real ST data. More importantly, spaCI allows detecting the upstream transcriptional factors (TF) mediating the L–R signalling axis, which facilitates the understanding of the underlying molecular mechanisms of intercellular crosstalk.
Results
Overview of the spaCI method
A novel computational method, spaCI (Figure 1), is developed to infer cell–cell interactions from ST data. The ST data, especially the recently emerging SCST data, exhibits frequent dropout events and low data coverage. Such sparse and noisy data impedes the accurate identification of L–R interactions in complex tissues. Our method leverages the spatial relationships between cells as well as the intracellular gene–gene association patterns to overcome these challenges, as illustrated in Figure 1A. Limited by the ST data quality, the expression profile of ligand 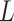 is compromised, thus the true relations between this ligand and its presumed receptor
is compromised, thus the true relations between this ligand and its presumed receptor 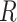 between two adjacent cells cannot be reliably determined. This challenge can be addressed by examining the gene expression patterns of those genes that are co-expressed with ligand
between two adjacent cells cannot be reliably determined. This challenge can be addressed by examining the gene expression patterns of those genes that are co-expressed with ligand 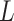 . The spaCI model utilizes the gene–gene interaction patterns, including both interactive pairs and non-interactive pairs, as well as the cell spatial graph to learn such a latent representation of genes, so that true L–R interactions, as well as co-expressed genes, are proximate to each other. The trained spaCI model thus is able to predict both L–R interactions and their upstream regulators such as transcription factors.
. The spaCI model utilizes the gene–gene interaction patterns, including both interactive pairs and non-interactive pairs, as well as the cell spatial graph to learn such a latent representation of genes, so that true L–R interactions, as well as co-expressed genes, are proximate to each other. The trained spaCI model thus is able to predict both L–R interactions and their upstream regulators such as transcription factors.
Figure 1.

Schematic overview of spaCI. (A) The spaCI method leverages the spatial relationships between cells as well as the expression data of all available genes to infer the L–R interactions. (B) In spaCI, genes are projected into the latent space through two components: a gene-based linear encoder as well as a cell-based attentive graph encoder. In this way, both gene expression patterns and spatial cell relations are incorporated into the latent space. Triplet loss is adopted in spaCI to assure a clear separation of interactive and non-interactive pairs.
The architecture of spaCI is illustrated in Figure 1B. Genes are projected into the latent space 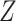 through two components: a gene-based linear encoder as well as a cell-based attentive graph encoder. Both gene expression patterns and spatial organization of the complex tissues are thus incorporated into the latent space. The model is then trained using triplet loss. Briefly, the interactive and non-interactive gene pairs are randomly assembled into positive-anchor-negative gene triplets. For each triplet, the latent representation of genes is learned so that the similarity between the positive pair is higher than that between the negative pairs with a margin of
through two components: a gene-based linear encoder as well as a cell-based attentive graph encoder. Both gene expression patterns and spatial organization of the complex tissues are thus incorporated into the latent space. The model is then trained using triplet loss. Briefly, the interactive and non-interactive gene pairs are randomly assembled into positive-anchor-negative gene triplets. For each triplet, the latent representation of genes is learned so that the similarity between the positive pair is higher than that between the negative pairs with a margin of 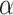 . Mathematically, the triplet loss defines a manifold in the latent space for each triplet. Thus, a group of co-expressed genes (such as the genes represented as nodes with different shades of blue colour) as well as the corresponding interactive L–R pairs (nodes in red colours) fall into a domain of arbitrary shape in the latent space (the larger grey domain). The margin
. Mathematically, the triplet loss defines a manifold in the latent space for each triplet. Thus, a group of co-expressed genes (such as the genes represented as nodes with different shades of blue colour) as well as the corresponding interactive L–R pairs (nodes in red colours) fall into a domain of arbitrary shape in the latent space (the larger grey domain). The margin 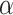 separates different gene interaction domains (such as the smaller grey domain formed by genes represented by green nodes). The triplet loss simplifies the optimization and simultaneously assures a clear separation of interactive and non-interactive cases. In this way, the expression data of all available genes are used to infer whether an L–R pair has interaction or not.
separates different gene interaction domains (such as the smaller grey domain formed by genes represented by green nodes). The triplet loss simplifies the optimization and simultaneously assures a clear separation of interactive and non-interactive cases. In this way, the expression data of all available genes are used to infer whether an L–R pair has interaction or not.
Through benchmarking with other methods on both simulation data and real data, we demonstrate that spaCI can effectively identify L–R interactions between two cell types as well as the cell types that communicate the most. To gain biological insights, we apply the spaCI to the single-cell spatial data from mouse cortex and lung cancer tissue. spaCI not only reveals L–R interactions that are not obvious in the original, sparse seqFISH+ data, but also identifies significant interactions between fibroblast and tumours in with strong L–R interactions such as the THBS1–ITGB1 interaction in lung cancer tissue. Moreover, spaCI is able to infer the upstream TF such as SMAD3 for such tumour-associated L–R interactions. Further analysis suggests that the intratumoural TF-receptor has strong predictive power in both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) patients. Overall, spaCI enables the interrogation of cell–cell communications in ST data and facilitates insights of the underlying disease mechanisms. Detailed explanations of spaCI are included in the Materials and Methods. The software for implementing spaCI is available at https://github.com/QSong-github/spaCI, with detailed manual and tutorials provided.
Evaluation of spaCI using simulation data
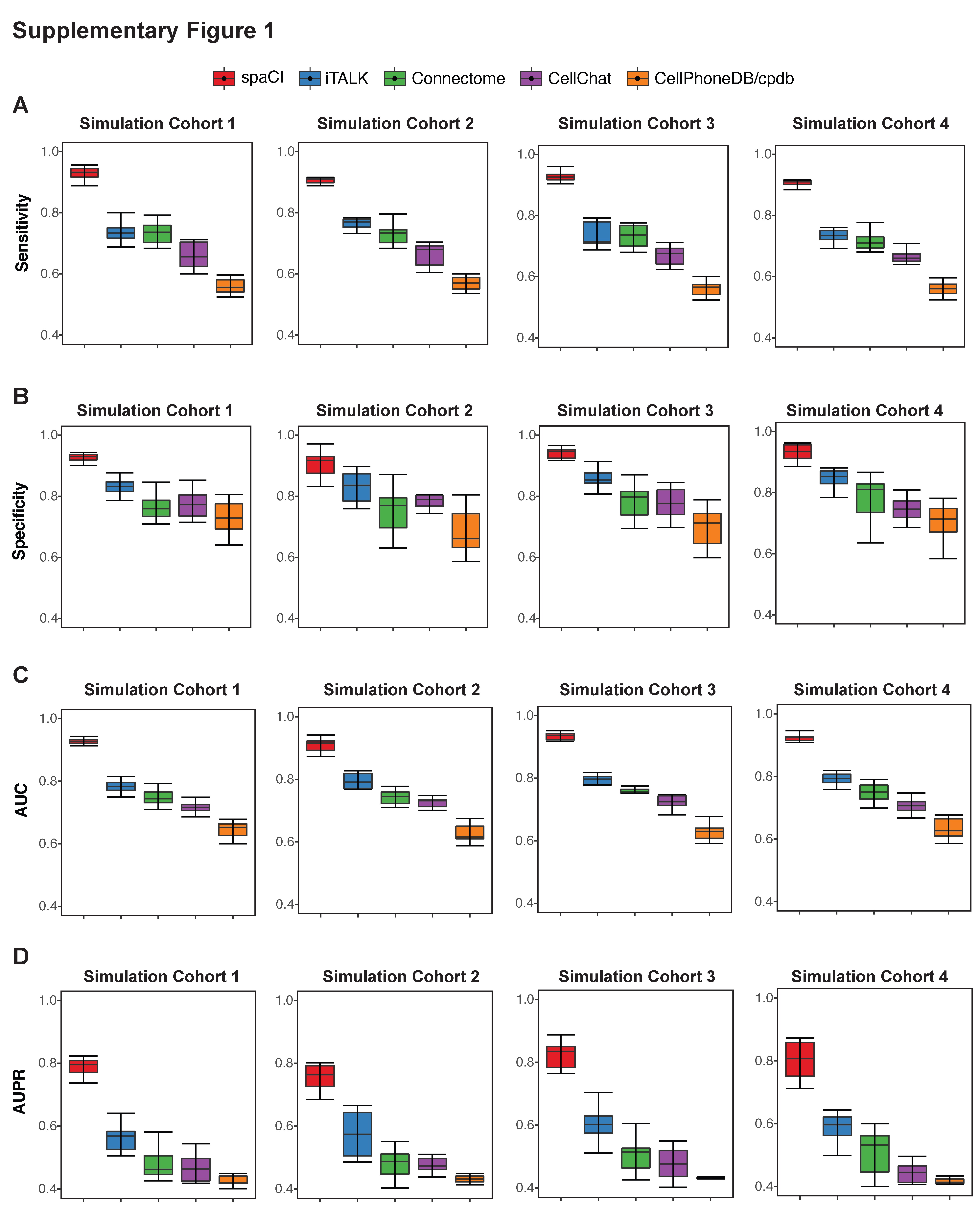
We first evaluated the performance of five different methods including spaCI, iTALK [11], CellPhoneDB [12] (i.e. cpdb), CellChat [13] and Connectome [14], based on the four cohorts of simulation data (see Section ‘Simulation data and performance evaluation’). Each simulation cohort consisted of 10 simulation data with both interaction pairs and non-interaction pairs generated by different parameters. F1 score was used to evaluate the performance of different methods in detecting accurate interaction pairs (Figure 2). The corresponding boxplots represented the F1 score of the identified interaction pairs by each method compared to the ground truth. Notably, spaCI accurately identified the true interaction pairs in four cohorts and demonstrated higher F1 (mean ± SE: 0.852 ± 0.014 for simulation cohort 1, 0.817 ± 0.05 for cohort 2, 0.859 ± 0.06 for cohort 3, 0.853 ± 0.03 for cohort 4). The other methods showed relatively lower F1 scores. For example, CellPhoneDB (mean ± SE: 0.453 ± 0.03) and CellChat (mean ± SE: 0.544 ± 0.03) showed lower F1 in simulation cohort 1. Relatively, Connectome (mean ± SE: 0.574 ± 0.04) had a comparable performance with CellChat (mean ± SE: 0.557 ± 0.02) in simulation cohort 2, while Connectome showed relatively higher F1 than CellChat in other simulation cohorts. Across the four simulation data cohorts, iTALK showed higher F1 than Connectome, but lower F1 scores than spaCI. In addition to the F1-score, metrics including sensitivity, specificity, AUC and AUPR (the area under the precision-recall curve) were also used for comprehensive evaluation of different methods (Supplementary Figure 1, Supplementary File). These results demonstrated the outperformance of spaCI in identifying accurate interaction L–R pairs based on the simulated data.
Figure 2.
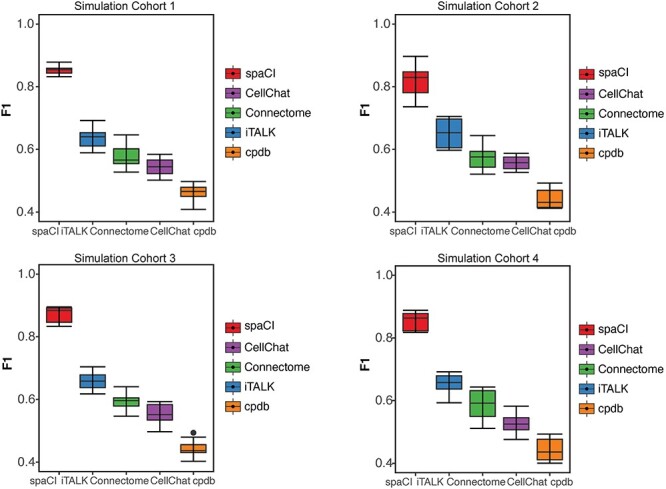
Evaluation of L–R pairs identified by different methods in simulation data. L–R pairs are predicted by five methods based on four simulation cohorts. Each cohort consists of 10 simulation data generated with different parameters. The corresponding boxplots represent the F1 score of the interaction pairs identified by each method.
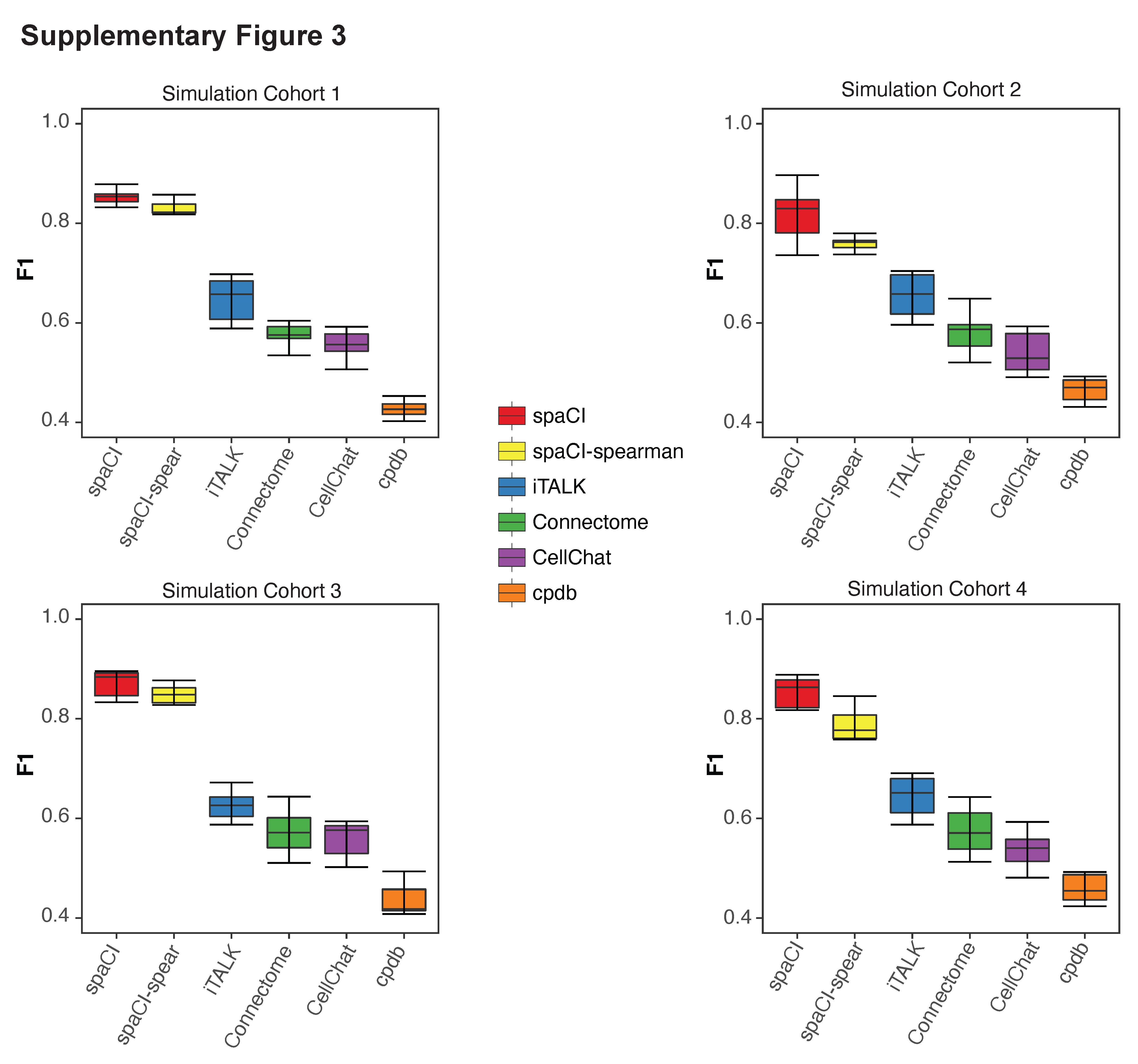
Moreover, to verify that spaCI also performed better in additional L–R interaction database, we performed benchmarking analysis using the LIANA CCC Consensus Resource (Supplementary Figure 2, Materials and Methods). We measured the performance of spaCI versus CellChat, Connectome, iTALK and cpdb based on four additional cohorts of simulation data generated from the Consensus Resource. Notably, spaCI identified true interaction pairs in four cohorts and demonstrated higher F1 (mean ± SE: 0.830 ± 0.022 for simulation cohort 1, 0.780 ± 0.012 for cohort 2, 0.847 ± 0.019 for cohort 3, 0.792 ± 0.034 for cohort 4) than other methods, including CellPhoneDB and CellChat. These results proved that the outperformance of spaCI did not depend on the selection of L–R database. In addition, spaCI also outperformed other methods when using Spearman association to generate gene triplets (Supplementary Figure 3).
Performance evaluation on ST data
To further demonstrate the performance of spaCI, we compared it with the other methods (iTALK, CellPhoneDB, CellChat and Connectome) on real ST data. For comparisons, we used 10 cropped regions of SCST data profiled from colon cancer sample using MERSCOPE (Figure 3A). We also included the SCST of lung cancer patients by NanoString CosMx (Figure 3B) for comparison. Details of these spatial datasets were provided in the Data availability section. To assess and quantify performance on these real spatial data, here we used evaluation metrics including the number of overlapped L–R interactions and the Jaccard index (see Section ‘Benchmarking methods and comparison measurements’).
Figure 3.

Evaluation of L–R pairs identified by different methods in SCST datasets. (A) Boxplots represent the number of L–R interactions shared by each method with the rest ones, based on the 10 regions of MERSCOPE colon tumour spatial data. (B) Boxplots represent the number of shared interaction pairs, based on the 20 FOVs of NanoString CosMx data. (C) Heatmap shows the Jaccard index of L–R interactions between any two methods. The filled colour is proportional to the Jaccard index. The average Jaccard index between a given method and the rest of the methods is shown in the bar plot above the heatmap.
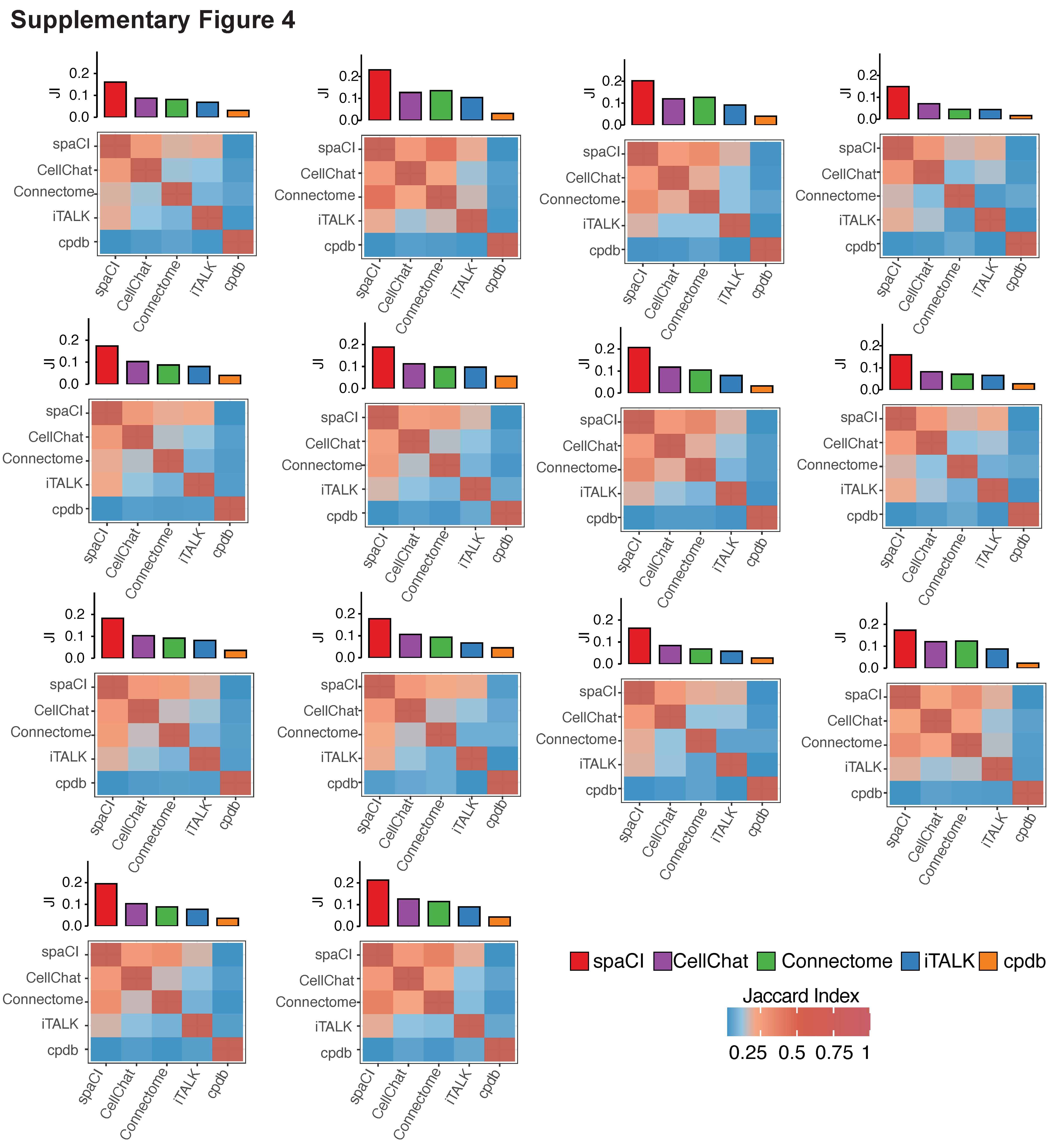
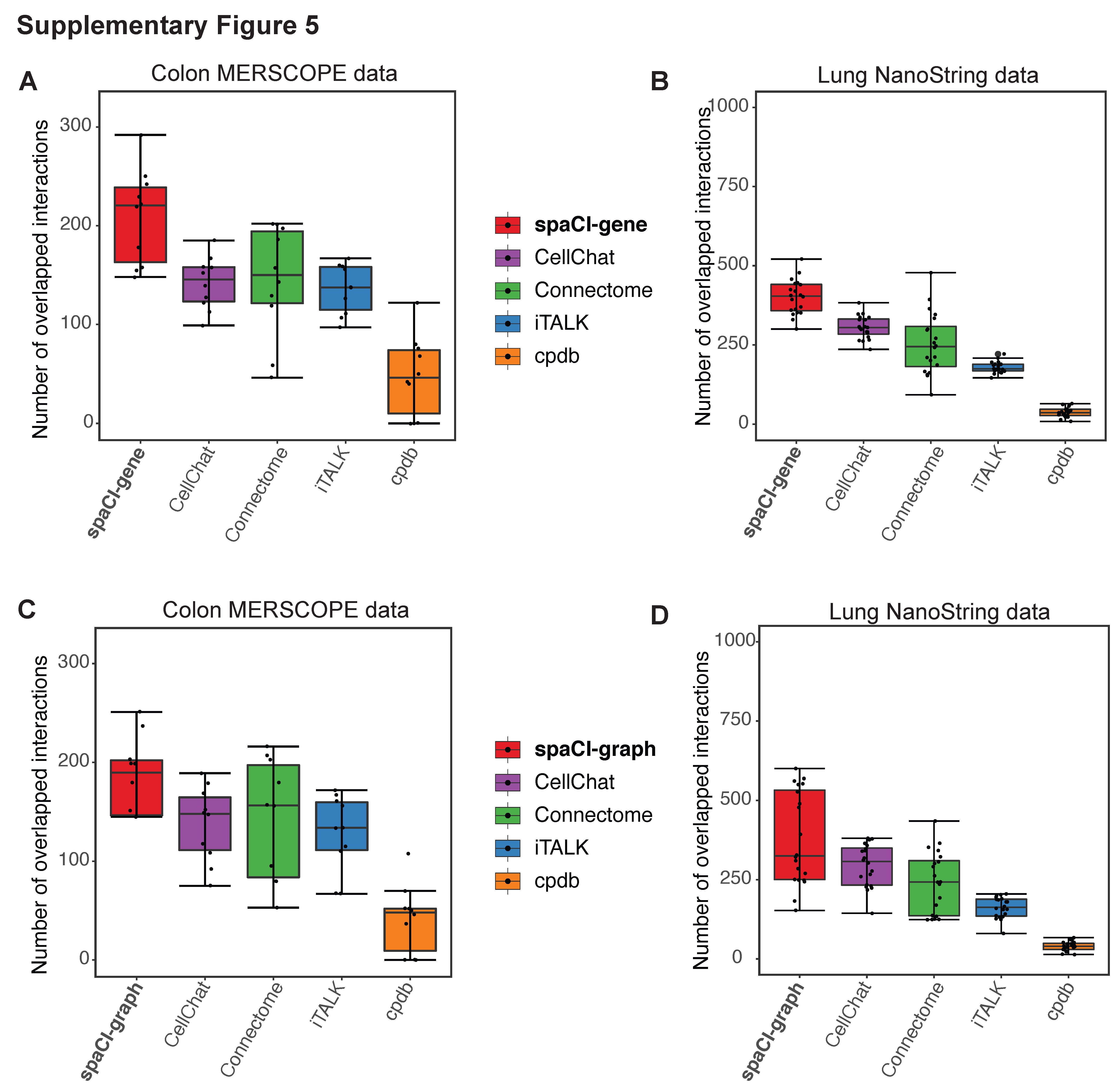
Based on the Vizgen’s MERSCOPE data from colon cancer sample (Figure 3A), spaCI identified the most overlapped L–R interactions (mean = 236.2) with the rest of the methods, especially higher than CellChat (mean = 151.3) and iTALK (mean = 141.9). CellPhoneDB (mean = 49.1) presented the least number of interactions in common with other methods. Connectome showed a relatively higher number of overlapped interactions (mean = 154.1) but was still lower than spaCI. For the other single-cell spatial data from lung tumours (Figure 3B), spaCI consistently identified the largest number of L–R interactions (mean = 462.9) shared with the rest of the methods. Connectome (mean = 283.8) presented less overlapped L–R interactions in common with other methods, but it was higher than CellPhoneDB (mean = 40.7) and iTALK (mean = 185.1). CellChat also presented lower performance than spaCI (mean = 328.5). In addition, we also calculated the Jaccard index of each method based on the 20 Field Of View (FOVs) (Figure 3C, Supplementary Figure 4). The Jaccard index of L–R pairs between any two methods was shown in the heatmap. The average Jaccard index between a given method and the rest of the methods was shown in the bar plot above the heatmap. Notably, spaCI presented the highest average rank based on the Jaccard index of L–R pairs. Moreover, considering the hybrid architecture of spaCI model, we also evaluated spaCI’s performance with ablation study (Supplementary Figure 5, Supplementary File). Collectively, these comparison results demonstrated that spaCI achieved superior performance on real spatial datasets and proved to identify accurate L–R interactions.
Application of spaCI to the seqFISH+ data of mouse cortex
To gain biological insights, spaCI was applied to the seqFISH+ mouse cortex data [7], which consisted of 10 000 genes and 12 different cell types. Figure 4A showed the spatial location of different cells and the UMAP plot of different cell types, with the edges linking neighbouring cells. Based on this data, spaCI identified 30 L–R interactions, while these L–R pairs presented poor associations in their raw gene expressions. Further interrogation of the embeddings and raw data of L–R pairs revealed the reasons why these L–R interactions were hidden in their expressions. For example, the embeddings of Nrg1 and Erbb3 were observed with strong associations in the latent space (Figure 4B). In contrast, both Nrg1 and Erbb3 presented extensive zeros in their expressions, with only a small portion of expressed cells observed, which explained the low correlation (Pearson correlation = 0.18) of Nrg1−Erbb3 in raw data. The other L−R pair, Fgf14−Fgfr2, was also observed with associations in latent space but not in raw gene expressions. These results demonstrated that spaCI revealed the L–R interactions which otherwise could be hidden due to data sparsity. Moreover, cells expressing Nrg1−Erbb3 and Fgf14−Fgfr2 presented prevalent adjacent cellular interactions (Figure 4C). The contour plots indicated that most cellular interactions of Nrg1−Erbb3 and Fgf14−Fgfr2 occurred in the spatial regions of L5 eNeuron and L4 eNeuron cells.
Figure 4.

spaCI reveals L–R interactions in the seqFISH+ data of mouse cortex. (A) The spatial plot of cells profiled from mouse cortex data by seqFISH+. UMAP plot of the profiled cells with edges linking adjacent ones. (B) The scatter plot shows the embeddings and the scaled expression values of identified L–R interactions, including Nrg1−Erbb3 and Fgf14−Fgfr2. (C) The left panel shows the spatial plots of cells expressing Nrg1−Erbb3 and Fgf14−Fgfr2. The right panel shows the contour plots of two L–R interactions. (D) Heatmap shows the identified L–R interaction strength across different cell types. (E) Network diagram of the summarized communications between different cell types. The edge width represents the communication strength.
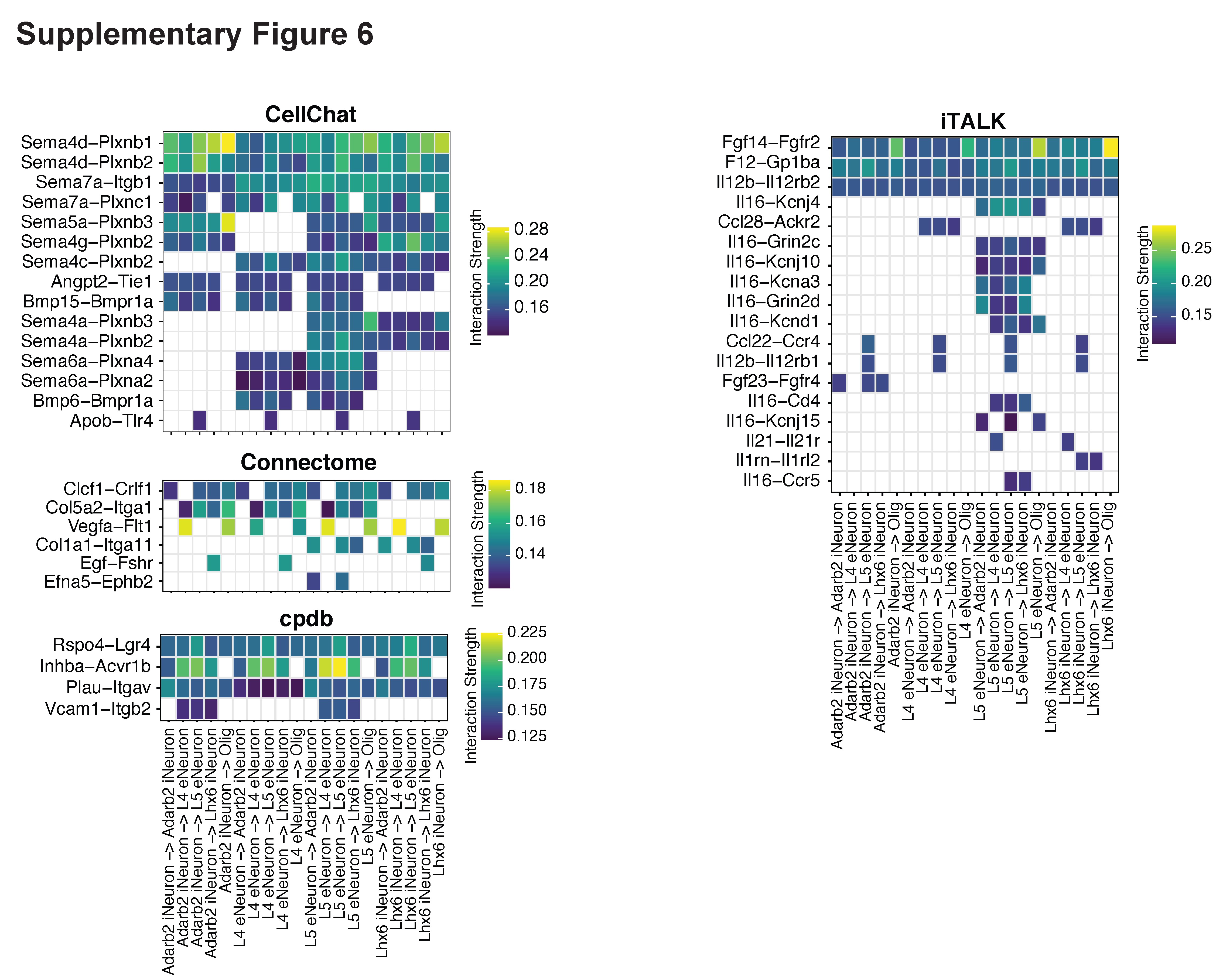
With the identified L–R pairs by spaCI, we further interrogated the L−R interaction strength across different cell types (Figure 4D, Materials and Methods). Specifically, L5 eNeuron expressed Nrg1 showed strong communication strength with the oligodendrocytes (Olig)−expressed Erbb3. The Nrg1−Erbb3 also presented strong communications between Lhx6 iNeuron and Olig. The Fgf14−Fgfr2 interaction was observed prevalently from L5 eNeuron, Lhx6 iNeuron and Adarb2 iNeuron, to Olig. To visualize the communications between different cell types, a summary network showed the cell-to-cell crosstalk, with the edge width indicating the interaction strength between different cell types (Figure 4E). Of note, we observed strong interactions between Lhx6 iNeuron and Olig, as well as L5 eNeuron and Olig, suggesting that oligodendrocytes might play a central role in the cellular crosstalk of brain. The benchmarking methods were also applied to this cortex data, which revealed prevalent inflammatory L–R pairs that were not common in healthy cortex tissues (Supplementary Figure 6, Supplementary File).
Applying spaCI to the SCST data of lung cancer patients
We then applied spaCI to the NanoString CosMx™ SMI dataset [8] with 20 FOVs profiled from a lung cancer patient, with a total of 81 236 cells and 18 different cell types (Figure 5A). The cell–cell communications identified by spaCI among different cell types were shown in the summary chord diagram (Figure 5B). The chord width between two cell types indicated their interaction strength. Of note, we observed strong interactions from fibroblast, endothelial cells and CD4 T memory cells to tumours, as well as intra-tumour interactions. To quantitively measure the interactions that occurred for each cell type, we used a scatter plot to visualize the involved number and strength of L–R interactions (Figure 5C), where the x-axis represents the number of interactions, and the y-axis represents the interaction strength. This scatter plot pinpointed fibroblasts and tumours as the most communicated cell types in cellular crosstalk.
Figure 5.

spaCI reveals strong interactions from fibroblasts to tumours in the lung cancer microenvironment. (A) The spatial plot of 81 236 cells from the 20 FOVs of NanoString CosMx SMI dataset. (B) Summary chord diagram of the identified cell–cell communication network. The chord width is proportional to the interaction strength across different cell types. (C) The scatter plot shows the involved number of L–R interactions and the interaction strength for each cell type. To keep the x-axis and y-axis at the same scale, we divide the value, i.e. the number of interactions (x-axis) and interaction strength (y-axis), by their maximums respectively. (D) The spatial plot of cells within the FOV comprising abundant fibroblast and tumours. (E) Summary chord diagram of the cell–cell communication network of major cell types. The chord width is proportional to the interaction strength. (F) Heatmap shows the identified L–R interactions between major cell types and tumour cells. (G) Spatial plots of the expressions of identified L–R interaction pairs.
To investigate if the globally dominant crosstalk also existed locally, we scrutinized a specific FOV with abundant fibroblast and tumour cells (Figure 5D). Based on this FOV, spaCI identified the cellular communications among major cell types (Figure 5E). Consistent with the cell–cell interactions identified globally (Figure 5B), we also observed strong interactions from fibroblast to tumour cells. The involved L–R pairs that contributed to the fibroblast-tumour crosstalk were presented (Figure 5F), along with the L–R interactions from other major cell types to tumour cells. Different colours represented the interaction strength. Specifically, fibroblast expressed DCN (ligand) showed strong interactions with tumour-expressed mesenchymal-epithelial transition (MET) (receptor). DCN has been reported to interact antagonistically with the MET factor (c-MET) and significantly influences angiogenesis [16–19]. Other L–R pairs including THBS1/THBS2−CD47 [20, 21], THBS1/THBS2−ITGB1 [22, 23], DCN−EGFR [24, 25], COL5A2−DDR1 [26], COL11A1−DDR1 [27, 28] also contributed to the commutations between fibroblasts and tumours. Of note, strong intra-tumour communications were also observed through COL18A1−ITGA3 [29] interaction. Moreover, the spatial expressions of ligand DCN were located adjacent to the receptor MET (Figure 5G). Similarly, adjacent expressions of the interaction pairs COL18A1−ITGA3 and THBS1−ITGB1 were also observed. In contrast, the interaction pairs identified by benchmarking methods were lack of spatial adjacency (Supplementary Figure 7, Supplementary File). These results confirmed the necessity of utilizing spatial adjacent graphs in spaCI to identify L–R interactions and the effectiveness of spaCI in detecting physical interactions in ST data.
spaCI identifies the upstream regulators mediating the cellular crosstalk
Interrogation of cellular crosstalk without elucidating the upstream mediators was not sufficient to fully understand the underlying mechanisms. Given the merit of our model, spaCI enabled us to predict not only the L–R interactions, but also the interactions of any gene pairs. Therefore, spaCI was able to infer the potential upstream TF of each ligand or receptor, to facilitate the discovery of L–R signalling activities. To achieve this, we used the hTFtarget database [30] with TF–target regulations specific to lung tissue, to identify the potential TFs of tumour receptors. In this way, spaCI revealed the upstream regulators of receptors within tumour cells that mediated the L–R interactions between fibroblast and tumour cells (Figure 6A). The Sankey diagram presented the identified L–R–TF signalling axis, with the source (fibroblast) expressing ligands, and the receiver (tumour) expressing receptors that were mediated by upstream TFs in tumour cells. Different colours represented different TF-mediated L–R interactions. From the Sankey plot, we observed prevalent interactions between THBS1/THBS2 and ITGB1 [31], with upstream TFs including SMAD3, JUN and EST1. These identified upstream TF were also confirmed through the motif enrichment analysis (Supplementary File).
Figure 6.

spaCI reveals the prognostic upstream TF mediating the L–R interactions. (A) spaCI predicts the upstream transcription factors of active receptors in tumour cells. (B) Survival analysis of SMAD3−ITGB1 expression in terms of overall survival using the lung cancer patient samples (LUAD, LUSC) from TCGA. (C) Survival analysis of SMAD3−ITGB1 expression with the clinicopathologic factor of different stages. (D) Comparison of the expression levels of SMAD3−ITGB1 at different stages in LUAD and LUSC patients, respectively.
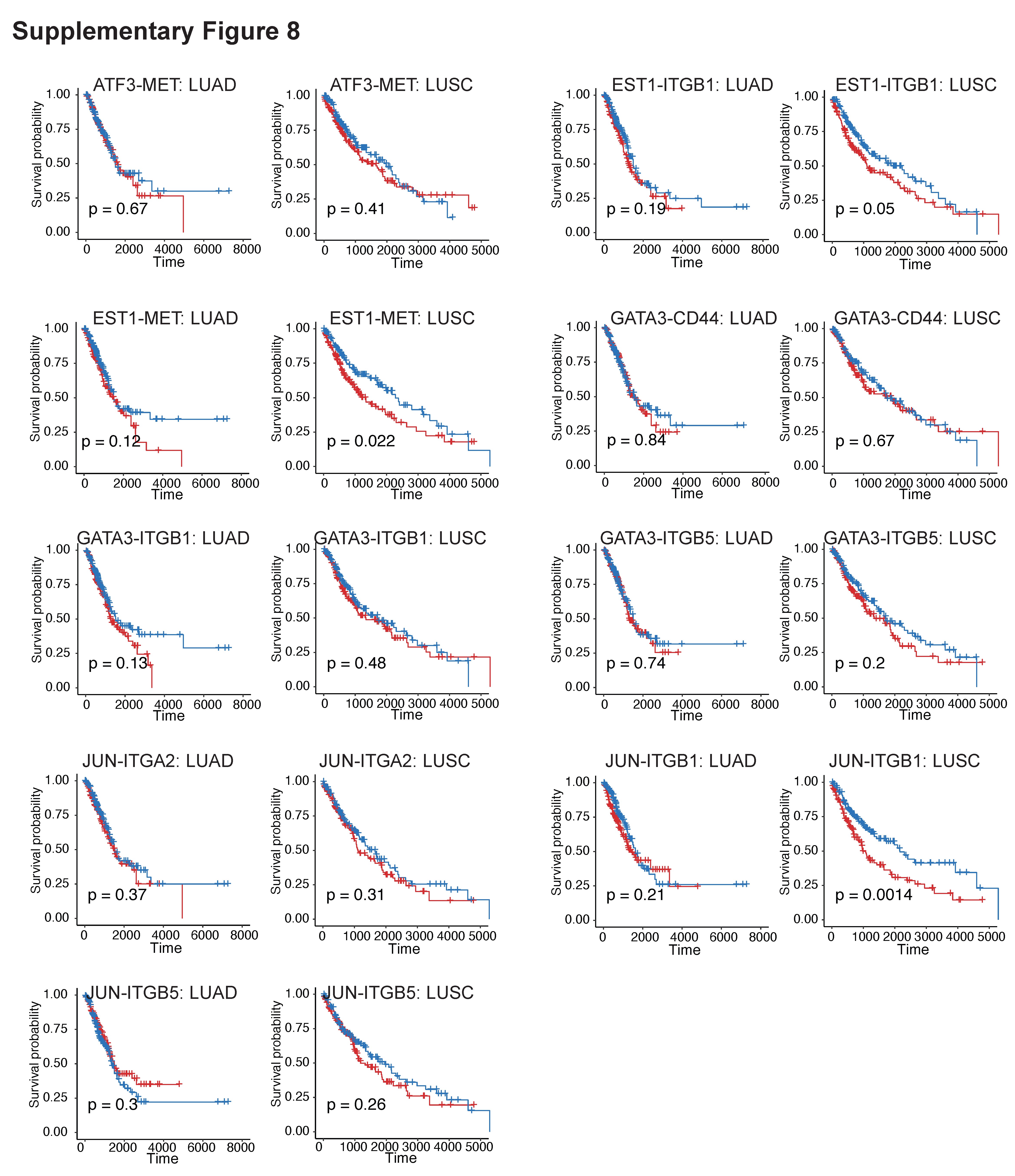
Such TF-mediated interaction axis may play important role in lung cancer prognosis. To evaluate the prognostic effects of these TF-mediated receptors (TF-R pair) in lung cancer patients, we used Kaplan–Meier survival analysis with the log-rank test to estimate the relationship between the TF-R expression and the overall survival (Figure 6B). Among the identified TF-R pairs, patients with high SMAD3−ITGB1 expression had significantly poor overall survival in both LUAD (P-value = 0.005) and LUSC (P-value = 0.05) patients in TCGA. However, other TF-mediated receptors (TF-R pair) did not present significant prognosis value in both LUAD and LUSC patients (Supplementary Figure 8). Moreover, patients with high expressions of SMAD3−ITGB1 also presented poor overall survival in advanced stage for both LUAD (P-value = 0.0001) and LUSC (P-value = 0.035) patients (Figure 6C). Consistently, the SMAD3−ITGB1 axis showed higher expression in the advanced stage in LUAD patients (P-value = 0.04), rather than in LUSC patients (Figure 6D).
Discussion
Cell–cell communications generally involve L–R interactions, which are vital for various biological signalling and disease pathogenesis. The recent advance of spatial technologies has effectively uncovered the spatial cellular heterogeneity within complex tissues, facilitating the systematic investigation of cell–cell communications at unprecedented resolution. However, effective methods of identifying cell–cell communication in ST, especially SCST data, are still lacking. Herein, we have proposed a novel adaptive graph attention model, i.e. spaCI, to detect the L–R interactions and reveal cellular crosstalk in ST profiles. spaCI is developed as a graph-based deep learning model, which has been shown effectively in spatial data analysis [32]. Through the comprehensive benchmarking, spaCI proves to achieve superior performance using both simulation data and real spatial datasets.
spaCI is not only able to reveal the L–R interactions, but also identify the upstream regulators mediating the active L–R signalling axis. In this work, we identified the THBS1 (ligand)−ITGB1(receptor) interaction between fibroblasts and tumour cells. spaCI further identified SMAD3 as an important regulator of ITGB1 within tumour cells, which would affect its interaction with fibroblasts. Moreover, spaCI can also be used to identify the upstream TF of THBS1, which provides an additional intervening strategy of targeting the upstream TF of ligand genes to inhibit the interactions between fibroblast and tumours, thus could further impede tumour progression. Collectively, spaCI not only reveals the interactions between ligands and receptors, but also the relations between TFs and ligands, as well as TFs and receptors, extending the interrogation of cellular dynamics from intercellular to intracellular. Therefore, spaCI is anticipated to provide novel biological hypotheses and facilitate insights into the underlying mechanisms.
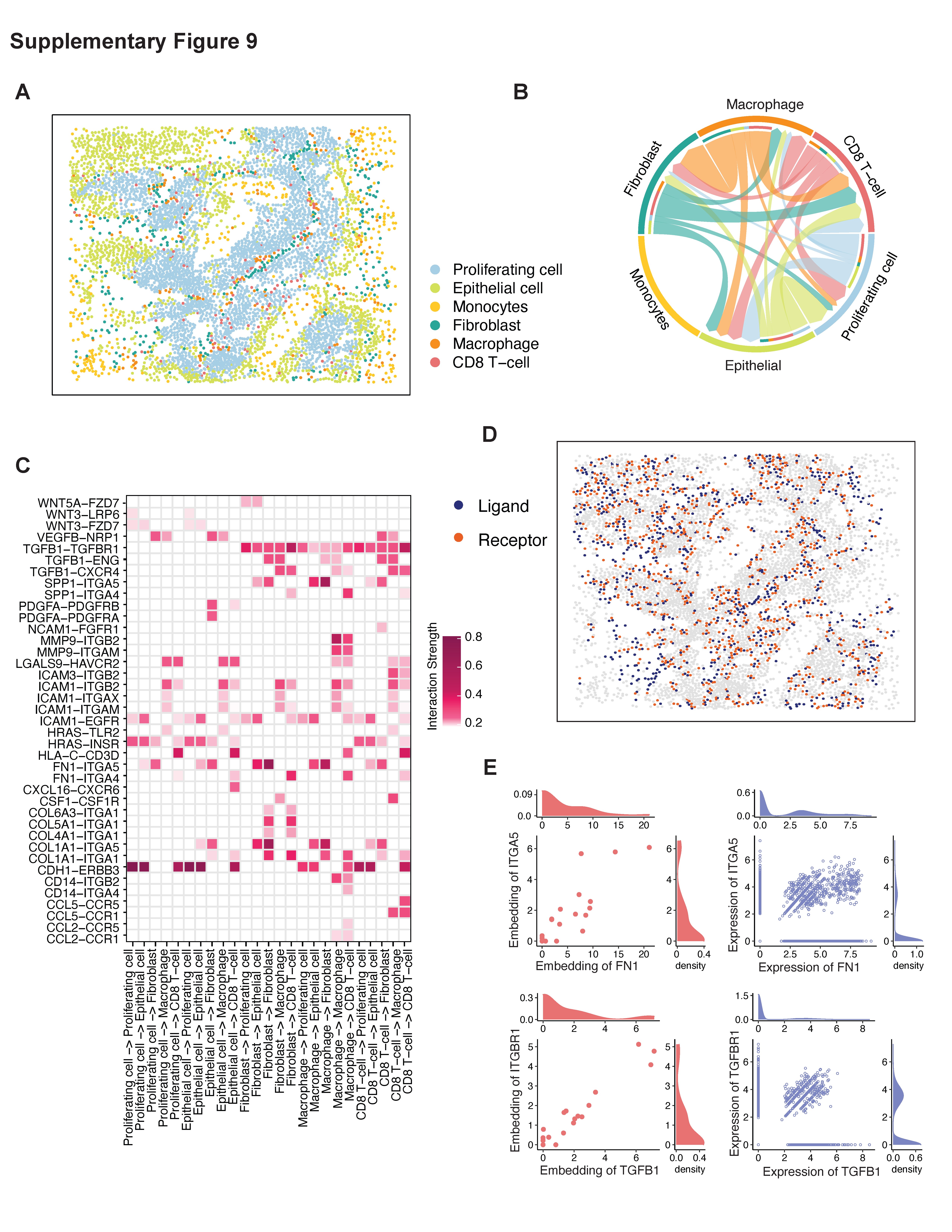
Currently, immunotherapy works as a promising way to treat and cure lung cancer [33]. Though the recent use of immune checkpoint inhibitors has demonstrated significant clinical efficacy in the treatment of lung cancer patients, 40–60% of patients do not achieve a significant therapeutic response [34–36]. Thus, there remains a need for predictive biomarkers to determine in advance those with the most potential to benefit from immune checkpoint blockade, as well as the potential targets to overcome the therapeutic resistance to immunotherapy. The immuno-oncology therapeutic effects are often mediated by multifaceted interactions between different cell types within the TME [37]. In addition to lung cancer, we also applied spaCI to the SCST data of colon patient sample (Supplementary Figure 9) and melanoma sample (Supplementary Figure 10) and revealed hidden L–R interactions and upstream regulators. Therefore, we anticipate that the L–R pairs identified by spaCI hold the promise to discriminate patients’ responses to immunotherapy.
Despite the advantages of spaCI, there are several aspects that spaCI can be improved. First, evaluating the predicted L–R interactions without ground truth poses a significant challenge. With experimentally validated L–R pairs in public resources, systematic and fair evaluations of different methods will be performed. Second, with the rapid development of spatial multi-omics technologies [38], spaCI will be further improved by incorporating new layers of omics data. Though the current version of spaCI enables the predictions of L–R interactions based on ST data, spaCI can be further improved by utilizing new data types, such as spatial proteomic profiles [39], to predict other types of molecular interaction data such as protein–protein interactions and genetic interactions. Moreover, as spaCI uses the cell adjacent graph based on the spatial distance between cells, incorporating the topological structure of spatial profiles using cell–cell similarity graph [40] and affinity graph [41] will gain more insights into the spatial gene expression pattern, which may further improve the model performance.
Materials and methods
spaCI model
Collection of L–R pairs
For real spatial data, we first assemble a collection of L–R pairs for predictions from International Union of Pharmacology [42], Connectome [43], FANTOM5 [43], HPRD [44], Human Plasma Membrane Receptome [45] and Database of Ligand−Receptor Partners [46], which encompasses 815 ligands, 780 receptors and 3398 reliable L–R pairs. This collection is used for candidate L–R interactions in spaCI. Moreover, the LIANA CCC Consensus Resource [47], the L–R reference dataset from OmniPath collection [48] is also used to evaluation the performance of spaCI.
Gene triplet set for model training
Pearson association is used to measure the associations of any gene pairs. The topmost associated gene pairs will be used as the ‘interaction’ pairs, denoted as 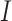 , which refers to a list of gene pairs that have strongly related expressions. The topmost un-associated gene pairs will be selected as the ‘non-interaction’ pairs, denoted as
, which refers to a list of gene pairs that have strongly related expressions. The topmost un-associated gene pairs will be selected as the ‘non-interaction’ pairs, denoted as 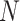 , referring to the gene pairs without relations. Here the correlation threshold
, referring to the gene pairs without relations. Here the correlation threshold 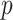 is set as 1% by default, which is learnable in spaCI. spaCI also provides alternative spearman correlation for identifying gene pairs. The two lists of interaction and non-interaction pairs are then used to construct the gene triplets [49] (Figure 1A), i.e. (
is set as 1% by default, which is learnable in spaCI. spaCI also provides alternative spearman correlation for identifying gene pairs. The two lists of interaction and non-interaction pairs are then used to construct the gene triplets [49] (Figure 1A), i.e. (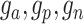 ), where
), where 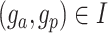 ,
, 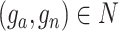 , for training the spaCI model. Here
, for training the spaCI model. Here 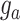 represents the ‘anchor’,
represents the ‘anchor’, 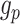 refers to the ‘positive’, and
refers to the ‘positive’, and 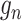 refers to the ‘negative’. In this way, the well-trained spaCI enables us to predict the interactions of our curated collection of L–R pairs.
refers to the ‘negative’. In this way, the well-trained spaCI enables us to predict the interactions of our curated collection of L–R pairs.
Spatial graph construction
We hypothesize that spatially adjacent cells should have a better chance of cell–cell communications than distant cells, especially for contact-based cell–cell interactions and paracrine signalling. Thus, for 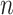 cells, the spatial graph, represented by its adjacent matrix
cells, the spatial graph, represented by its adjacent matrix 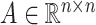 , is constructed according to cell locations (Figure 1A) using
, is constructed according to cell locations (Figure 1A) using 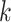 -nearest neighbours. For two cells
-nearest neighbours. For two cells 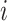 and
and 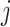 , we have
, we have 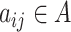 , and
, and 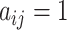 if the two cells are adjacent, otherwise
if the two cells are adjacent, otherwise 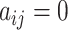 . Here the number of nearest neighbours
. Here the number of nearest neighbours 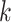 for constructing cell adjacency graph by default is set as 5, which is learnable in spaCI.
for constructing cell adjacency graph by default is set as 5, which is learnable in spaCI.
Model architecture
As shown in Figure 1B, the spatial gene expression data is denoted as 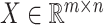 , where
, where 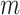 represents the number of genes and
represents the number of genes and 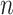 refers to the number of cells respectively. spaCI converts the gene expression features into the latent representation of genes through three major components:
refers to the number of cells respectively. spaCI converts the gene expression features into the latent representation of genes through three major components:
(i) Gene-based embedding 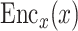 . The original gene expression matrix
. The original gene expression matrix 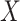 can be represented as a list of gene-based samples, that is,
can be represented as a list of gene-based samples, that is, 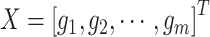 , with the
, with the 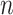 -dimensional vector
-dimensional vector 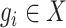 representing the expressions of gene
representing the expressions of gene 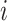 across the
across the 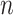 cells. Thus, genes in
cells. Thus, genes in 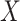 are projected to the latent space through a series hidden linear layers for gene-based embedding, with the
are projected to the latent space through a series hidden linear layers for gene-based embedding, with the 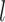 th layer as:
th layer as:
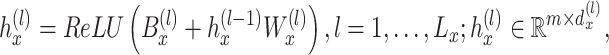 |
where 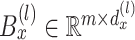 is a bias parameter,
is a bias parameter, 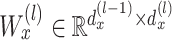 is the weight matrix in the
is the weight matrix in the 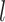 th layer to transform the embedding,
th layer to transform the embedding, 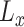 is the total depth of layers,
is the total depth of layers, 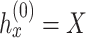 ,
, 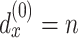 and the obtained embedding
and the obtained embedding 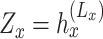 ;
;
(ii) Spatially guided embedding 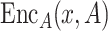 . We further incorporate the spatial graph of cell adjacency for an additional embedding
. We further incorporate the spatial graph of cell adjacency for an additional embedding 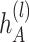 by
by
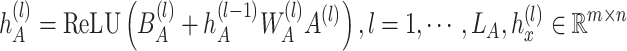 |
where 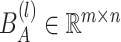 is a bias parameter,
is a bias parameter, 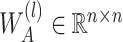 is the weight matrix in the
is the weight matrix in the 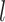 th layer to transform the embedding,
th layer to transform the embedding, 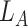 is the depth of graph attentive layers. Specifically, the
is the depth of graph attentive layers. Specifically, the 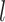 th adaptive adjacent matrix
th adaptive adjacent matrix 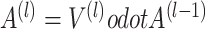 learns weighted edges for the spatial graph through an attention mechanism, with
learns weighted edges for the spatial graph through an attention mechanism, with 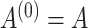 and
and  is the element-wise multiplication. To model the cell–cell relations, we leverage a learnable layer-independent attentive matrix
is the element-wise multiplication. To model the cell–cell relations, we leverage a learnable layer-independent attentive matrix 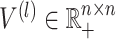 . In this way,
. In this way, 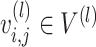 captures the importance of between two neighbour cells
captures the importance of between two neighbour cells 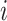 and
and 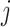 . For non-neighbour cells, since for
. For non-neighbour cells, since for 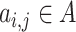 ,
, 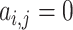 , we always have
, we always have 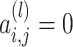 for
for 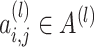 . We evaluate
. We evaluate 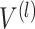 from previous embeddings in
from previous embeddings in 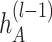 as:
as:
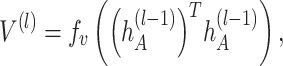 |
where 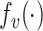 is a two-layer fully connected neural network with
is a two-layer fully connected neural network with 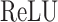 and sigmoid activation function in the hidden and output layers, respectively. Note that the attentive matrix
and sigmoid activation function in the hidden and output layers, respectively. Note that the attentive matrix 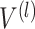 is an element wisely multiplied to the adaptive adjacency matrix
is an element wisely multiplied to the adaptive adjacency matrix 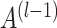 , so the value of each element
, so the value of each element 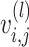 serves as an attention score between the
serves as an attention score between the 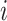 th and
th and 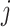 th cell and the message of edges with higher prediction scores would give a higher weight to propagate. The obtained embedding is
th cell and the message of edges with higher prediction scores would give a higher weight to propagate. The obtained embedding is 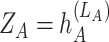 .
.
(iii) Hybrid embedding 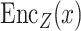 . With the two major embeddings
. With the two major embeddings 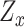 and
and 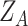 considering both gene expressions and spatial cell locations, we combine them and map the raw gene expression feature into the final latent representation via a standard Multilayer Perceptron (MLP):
considering both gene expressions and spatial cell locations, we combine them and map the raw gene expression feature into the final latent representation via a standard Multilayer Perceptron (MLP):
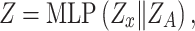 |
where 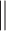 is the concatenation operator,
is the concatenation operator, 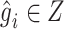 is the latent representation of the
is the latent representation of the 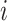 -th gene.
-th gene.
Loss function
With the latent embedding of genes within triplet list 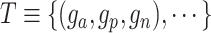 , we apply a modified version of triplet loss [49] as the objective function:
, we apply a modified version of triplet loss [49] as the objective function:
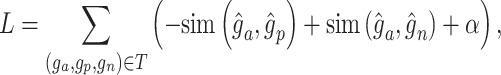 |
where 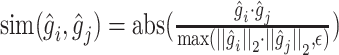 refers to the cosine similarity between two latently represented genes, and
refers to the cosine similarity between two latently represented genes, and 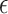 is a small value to avoid division by zero,
is a small value to avoid division by zero, 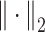 is
is 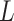 -2 norm, and
-2 norm, and 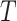 is the triplet list. The rationale is that the similarity between an anchor gene and its positively interacting gene should be significantly higher than its non-interacting genes. To avoid a trivial solution,
is the triplet list. The rationale is that the similarity between an anchor gene and its positively interacting gene should be significantly higher than its non-interacting genes. To avoid a trivial solution, 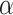 serves as a margin and enforces a significant difference between the similarity in interaction pairs and non-interaction pairs. Cosine similarity is used to measure the relationship of two genes in the latent space to ensure the gene pairs have associated latent embeddings [49].
serves as a margin and enforces a significant difference between the similarity in interaction pairs and non-interaction pairs. Cosine similarity is used to measure the relationship of two genes in the latent space to ensure the gene pairs have associated latent embeddings [49].
Prediction of L–R interactions
For each L–R pair, its expression across cells, 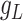 and
and 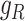 , are projected onto the latent space as
, are projected onto the latent space as 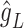 and
and 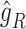 , respectively, by the trained spaCI model. The L–R pair is considered as interactive if:
, respectively, by the trained spaCI model. The L–R pair is considered as interactive if:
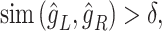 |
where 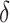 is an empirical parameter and is determined by the model training procedure.
is an empirical parameter and is determined by the model training procedure.
Parameter tuning
The essential parameters in the spaCI model are the triplet loss margin 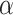 , the number of nearest neighbours
, the number of nearest neighbours 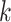 for constructing cell adjacency graph, and the correlation threshold
for constructing cell adjacency graph, and the correlation threshold 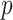 for constructing triplets. spaCI provides a detailed grid search approach for hyperparameter tuning, with
for constructing triplets. spaCI provides a detailed grid search approach for hyperparameter tuning, with 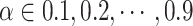 ,
, 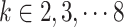 , ,and
, ,and 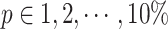 . The model performance is evaluated by the F1 score on the validation set.
. The model performance is evaluated by the F1 score on the validation set.
Simulation data and performance evaluation
To evaluate the performance of spaCI, we generate the simulation data with the ground truth of L–R interactions across different cell types. First, we simulate the gene expressions of cells with multiple cell types. Briefly, for any cell 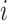 from the same cell type
from the same cell type 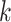 , the expression of gene
, the expression of gene 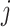 is sampled from the same negative binomial (NB) distribution, i.e.
is sampled from the same negative binomial (NB) distribution, i.e. 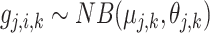 , where
, where 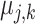 and
and 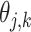 are the mean and dispersion of the NB distribution, respectively, and the probability function is:
are the mean and dispersion of the NB distribution, respectively, and the probability function is: 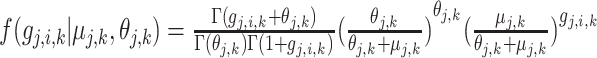 , with
, with 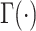 denoting the gamma function. Expressions in different cell types are generated using different NB distribution parameters [50].
denoting the gamma function. Expressions in different cell types are generated using different NB distribution parameters [50].
For the L–R interactions, we consider two scenarios of L–R interactions: those that demonstrate strong associations, and those that demonstrate similar expression levels. Since both scenarios can be intracellular or intercellular, for simplicity, we only consider the cases of intracellular interactions within the same cell type and intercellular interactions across two different cell types.
Scenario 1): Associated expression patterns for L–R interactions. In such cases, associated expressions of ligand and receptor involved in cellular interactions may demonstrate similar expression patterns but may be of different expression levels. That is, the expression of ligand 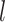 is generated for a group of cells,
is generated for a group of cells, 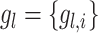 as previously described. Then the expression of its interacted receptor
as previously described. Then the expression of its interacted receptor 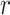 ,
, 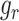 , is generated using the linear combination of
, is generated using the linear combination of 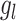 and the a strong correlation
and the a strong correlation 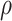 between
between 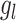 and
and 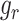 , i.e. for cell
, i.e. for cell 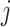 ,
, 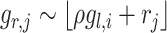 , where
, where 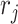 is a random noise following normal distribution.
is a random noise following normal distribution.
Scenario 2): Similar expression levels for L–R interactions. In such cases, ligands and receptors demonstrate similar expression levels. For an L–R pair, we first generate the expression 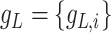 of a specific ligand
of a specific ligand 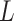 over a group of cells by the NB distribution as previously described. Then the expression of its paired receptor
over a group of cells by the NB distribution as previously described. Then the expression of its paired receptor 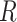 in another group of cells is generated with a small parameter
in another group of cells is generated with a small parameter 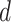 , i.e. for cell
, i.e. for cell 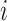 ,
, 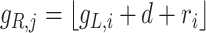 , where
, where 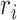 is a random noise following normal distribution, and
is a random noise following normal distribution, and 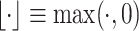 converts the native simulated expression values to 0s. Thus, we generate the expressions of L–R pairs with similar but different overall expression levels.
converts the native simulated expression values to 0s. Thus, we generate the expressions of L–R pairs with similar but different overall expression levels.
Based on the above scenarios, we generate four simulation cohorts. Each cohort is composed of 10 simulated data with different parameters. For each data, total 3000 genes for 4000 cells from five cell types are simulated. For each cohort, the L–R interactions are simulated as the following: Cohort (1) For each simulation data, we generate 250 associated L–R pairs using Scenario 1, with 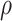 randomly selected from 0.5 to 1. We assign these 250 L–R pairs to the same cell types for the detection of intracellular interactions. Cohort (2) Similar to Cohort 1, but we assign the 250 L–R pairs to different cell types for the detection of intercellular interactions. Cohort (3) For each simulation data, we generate 250 associated L–R pairs using Scenario 2, with
randomly selected from 0.5 to 1. We assign these 250 L–R pairs to the same cell types for the detection of intracellular interactions. Cohort (2) Similar to Cohort 1, but we assign the 250 L–R pairs to different cell types for the detection of intercellular interactions. Cohort (3) For each simulation data, we generate 250 associated L–R pairs using Scenario 2, with 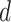 values randomly sampled from the uniform distribution [0, 1.5], to simulate L–R interactions within the same cell type. Cohort (4) Similar to Cohort 3, but we assign the 250 L–R pairs to different cell types for detection of intercellular interactions. The same number of non-interactive L–R pairs are simulated in a similar way for each cohort, with
values randomly sampled from the uniform distribution [0, 1.5], to simulate L–R interactions within the same cell type. Cohort (4) Similar to Cohort 3, but we assign the 250 L–R pairs to different cell types for detection of intercellular interactions. The same number of non-interactive L–R pairs are simulated in a similar way for each cohort, with 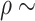 [0, 0.2] or
[0, 0.2] or 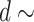 [5, 10], respectively.
[5, 10], respectively.
Benchmarking methods and comparison measurements
To evaluate the performance, we compare spaCI with four methods that are designed for predicting L–R interactions, including iTALK [11], CellPhoneDB [12], CellChat [13] and Connectome [14]. Specifically, when training the spaCI model, we use the strongly associated gene pairs as training set to predict the interactions of L–R pairs. iTALK is evaluated using the default criteria, including filtering out genes detected in less than three cells and low-quality cells with less than 200 expressed genes, and using mean count to identify the highly expressed ligands and receptors. For CellPhoneDB, as recommended by the tutorial, interactions with ligands and receptors expressed in at least 10% of the cells are considered. CellChat is performed with the default settings of 1000 permutations, following their provided tutorial for cell–cell communication. Connectome is also evaluated with the default settings, with the differentially expressed genes filtered by P-value less than 0.05.
For simulation data with ground truth, we use the F1 score to evaluate the performance of different methods in identifying true positive and true negative interaction pairs. For real spatial data, since there is no ground truth for evaluation, we use concordance analysis of identified L–R pairs among different methods to evaluate model performance. The sensitivity of a method is evaluated by the overlapped number of identified L–R pairs between this method and the rest of the methods. The specificity of a method is measured by the Jaccard indexes against other methods. Specifically, the Jaccard index is defined as the number of overlapped L–R pairs divided by the total number of identified L–R pairs by different methods. That is 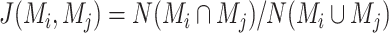 , where
, where 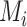 and
and 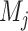 represent the L–R pairs by different methods
represent the L–R pairs by different methods 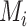 and
and 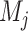 , based on the intersection and union of the identified L–R pairs.
, based on the intersection and union of the identified L–R pairs.
Characterization of L–R interaction strength
With the identified L–R interactions, we further characterize the interaction strength among different types of cells. For an L–R pair, we define its interaction strength as the multiplication of their average expression values, where the top and bottom 10% expressions of the ligand and the receptor are ignored [13]. The interaction strength of all identified L–R pairs is then summarized as the interaction strength between two cell types. Thus, the higher value of the interaction strength, the stronger the two cell types interact. The definition of interaction strength in terms of L–R pairs and cell types allows revealing the major active L–R signalling and their contribution to highly communicated cell types.
Key Points
We have developed a novel adaptive graph model with designed triplet loss function named spaCI, to incorporate both spatial locations and gene expressions of cells for revealing the active L–R signalling axis across neighbouring cells.
spaCI is developed tailored for SCST and provided available as a ready-to-use open-source software, which demonstrates high accuracy and robust performance over existing methods.
spaCI is able to identify the upstream TF mediating the identified L–R interactions, which allows gaining further insights into the underlying cellular communications, the discoveries of disease mechanisms and effective biomarkers.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ziyang Tang is a PhD candidate in the Department of Computer and Information Technology, Purdue University, NC, USA. His research focuses on developing novel artificial intelligence methods in interdisciplinary science.
Tonglin Zhang is an associate professor in the Department of Statistics, Purdue University, NC, USA. His research focuses on developing novel statistical model for interdisciplinary research.
Baijian Yang is a professor in the Department of Computer and Information Technology, Purdue University, NC, USA. His research focuses on computer and information technology.
Jing Su is an assistant professor in the Department of Biostatistics and Health Data Science, Indiana University School of Medicine, IN, USA. His research focuses on graph artificial intelligence and machine learning in biomedical informatics and precision health.
Qianqian Song is an assistant professor in the Center for Cancer Genomics and Precision Oncology, Wake Forest School of Medicine, NC, USA. Her research focuses on machine learning and deep learning in bioinformatics and precision oncology.
Contributor Information
Ziyang Tang, Department of Computer and Information Technology, Purdue University, Indiana, USA.
Tonglin Zhang, Department of Statistics, Purdue University, Indiana, USA.
Baijian Yang, Department of Computer and Information Technology, Purdue University, Indiana, USA.
Jing Su, Department of Biostatistics and Health Data Science, Indiana University School of Medicine, Indiana, USA.
Qianqian Song, Center for Cancer Genomics and Precision Oncology, Wake Forest Baptist Comprehensive Cancer Center, Atrium Health Wake Forest Baptist, Winston Salem, NC, USA; Department of Cancer Biology, Wake Forest School of Medicine, Winston Salem, NC, USA.
Data availability
SeqFISH+ mouse cortex data: The seqFISH+ dataset was obtained from the Giotto [51] repository (https://github.com/RubD/spatial-datasets/tree/master/data/2019_seqfish_plus_SScortex). We used the gene count matrix of the mouse cortex dataset, with FOVs ranging from 0 to 4, which covers 10 000 genes and 523 cells. NanoString CosMx SMI data: This single-cell spatial dataset contains 20 FOVs, which are profiled by the CosMx SMI on Formalin-Fixed Paraffin-Embedded (FFPE) samples of non-small-cell lung cancer (NSCLC) tissue [8]. The dataset (Lung 13) is available from https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/. Vizgen MERSCOPE datasets: We used the spatial profiles of colon and melanoma tumour from Vizgen MERFISH FFPE Human Immuno-oncology datasets, which are accessible from https://info.vizgen.com/merscope-ffpe-solution. The colon dataset (Colon cancer 2) contains a MERFISH measurement of a 500 gene panel and 817 588 cells, of which the cropped region (dim x: 11 250–13 750 and dim y: 4000–5000) was in the application of spaCI. Ten cropped regions were identified for performance evaluation. The melanoma dataset (Melanoma 2) contains a MERFISH measurement of a 500 gene panel and 207 869 cells, where the cropped region (dim x: 5000–6000 and dim y: 5000–6000) was used in this study.
Code availability
The spaCI method is provided as an open-source python package in GitHub https://github.com/QSong-github/spaCI, with detailed manual and tutorials.
Funding
Comprehensive Cancer Center of Wake Forest University Health Sciences (Bioinformatics Shared Resources under the NCI Cancer Center Support Grant P30CA012197 to Q.S.); Indiana University Precision Health Initiative (to J.S.); National Cancer Institute (Indiana University Melvin and Bren Simon Comprehensive Cancer Center Support Grant P30CA082709 to J.S.); National Library of Medicine of the National Institutes of Health (award number R01LM013771 to J.S.).
References
- 1. Armingol E, Officer A, Harismendy O, et al. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet 2021;22(2):71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iriki T, Ohnishi K, Fujiwara Y, et al. The cell-cell interaction between tumor-associated macrophages and small cell lung cancer cells is involved in tumor progression via STAT3 activation. Lung Cancer 2017;106:22–32. [DOI] [PubMed] [Google Scholar]
- 3. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci 2013;110(50):20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu J, Tran V, VNP V, et al. Comparative analysis of MERFISH spatial transcriptomics with bulk and single-cell RNA sequencing. 2022. bioRxiv 2022.03.04.483068; 10.1101/2022.03.04.483068. [DOI] [PMC free article] [PubMed]
- 5. Moffitt JR, Lundberg E, Heyn H. The emerging landscape of spatial profiling technologies. Nat Rev Genet 2022;23:741–59. 10.1038/s41576-022-00515-3. [DOI] [PubMed] [Google Scholar]
- 6. Moses L, Pachter L. Museum of spatial transcriptomics. Nat Methods 2022;19(5):534–46. 10.1038/s41592-022-01409-2. [DOI] [PubMed] [Google Scholar]
- 7. Eng C-HL, Lawson M, Zhu Q, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019;568(7751):235–9. 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. He S, Bhatt R, Brown C, et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol 2022. Advance online publication. 10.1038/s41587-022-01483-z. [DOI] [PubMed]
- 9. Janesick A, Shelansky R, Gottscho A, et al. High resolution mapping of the breast cancer tumor microenvironment using integrated single cell, spatial and in situ analysis of FFPE tissue. 2022. bioRxiv 2022.10.06.510405; 10.1101/2022.10.06.510405. [DOI] [PMC free article] [PubMed]
- 10. Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26(3):317–25. [DOI] [PubMed] [Google Scholar]
- 11. Wang Y, Wang R, Zhang S, et al. iTALK: an R package to characterize and illustrate intercellular communication. bioRxiv. 2019;507871. 10.1101/507871. [DOI]
- 12. Efremova M, Vento-Tormo M, Teichmann SA, et al. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc 2020;15(4):1484–506. [DOI] [PubMed] [Google Scholar]
- 13. Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun 2021;12(1):1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raredon MSB, Yang J, Garritano J, et al. Computation and visualization of cell–cell signaling topologies in single-cell systems data using connectome. Sci Rep 2022;12(1):4187. 10.1038/s41598-022-07959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qiu P. Embracing the dropouts in single-cell RNA-seq analysis. Nat Commun 2020;11(1):1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khan GA, Girish GV, Lala N, et al. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol 2011;25(8):1431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang W, Ge Y, Cheng Q, et al. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018;9(4):5480–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daum R, Visser D, Wild C, et al. Fibronectin adsorption on electrospun synthetic vascular grafts attracts endothelial progenitor cells and promotes endothelialization in dynamic in vitro culture. Cell 2020;9(3):778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Järveläinen H, Sainio A, Wight TN. Pivotal role for decorin in angiogenesis. Matrix Biol 2015;43:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daubon T, Léon C, Clarke K, et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat Commun 2019;10(1):1146. 10.1038/s41467-019-08480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaur S, Bronson SM, Pal-Nath D, et al. Functions of thrombospondin-1 in the tumor microenvironment. Int J Mol Sci 2021;22(9):4570. Epub 2021/05/01. 10.3390/ijms22094570. PubMed PMID: 33925464; PMCID: PMC8123789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Terrinoni M, Holmgren J, Lebens M, et al. Proteomic analysis of cholera toxin adjuvant-stimulated human monocytes identifies Thrombospondin-1 and integrin-β1 as strongly upregulated molecules involved in adjuvant activity. Sci Rep 2019;9(1):2812. 10.1038/s41598-019-38726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joost S, Jacob T, Sun X, et al. Single-cell Transcriptomics of traced epidermal and hair follicle stem cells reveals rapid adaptations during wound healing. Cell Rep 2018;25(3):585–97.e7. 10.1016/j.celrep.2018.09.059. [DOI] [PubMed] [Google Scholar]
- 24. Santra M, Reed CC, Iozzo RV. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J Biol Chem 2002;277(38):35671–81. Epub 2002/07/10. 10.1074/jbc.M205317200. PubMed PMID: 12105206. [DOI] [PubMed] [Google Scholar]
- 25. Csordás G, Santra M, Reed CC, et al. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J Biol Chem 2000;275(42):32879–87. Epub 2000/07/27. 10.1074/jbc.M005609200. PubMed PMID: 10913155. [DOI] [PubMed] [Google Scholar]
- 26. Vogel W, Gish GD, Alves F, et al. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell 1997;1(1):13–23. [DOI] [PubMed] [Google Scholar]
- 27. Lai SL, Tan ML, Hollows RJ, et al. Collagen induces a more proliferative, migratory and chemoresistant phenotype in head and neck cancer via DDR1. Cancer 2019;11(11):1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ling Lai S, Leng Tan M, Hollows RJ, et al. Collagen induces a more proliferative, migratory and chemoresistant phenotype in head and neck cancer via DDR1. Cancers (Basel) 2019;11(11):1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang B, Xu C, Liu J, et al. Nidogen-1 expression is associated with overall survival and temozolomide sensitivity in low-grade glioma patients. Aging (Albany NY) 2021;13(6):9085–107. Epub 2021/03/19. 10.18632/aging.202789. PubMed PMID: 33735110; PMCID: PMC8034893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Q, Liu W, Zhang H-M, et al. hTFtarget: a comprehensive database for regulations of human transcription factors and their targets. Genom Proteomics Bioinform 2020;18(2):120–8. 10.1016/j.gpb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calzada MJ, Annis DS, Zeng B, et al. Identification of novel beta1 integrin binding sites in the type 1 and type 2 repeats of thrombospondin-1. J Biol Chem 2004;279(40):41734–43. Epub 2004/08/05. 10.1074/jbc.M406267200. PubMed PMID: 15292271. [DOI] [PubMed] [Google Scholar]
- 32. Song Q, Su J. DSTG: deconvoluting spatial transcriptomics data through graph-based artificial intelligence. Brief Bioinform 2021;22(5):bbaa414. 10.1093/bib/bbaa414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med 2016;375:1823–33. [DOI] [PubMed] [Google Scholar]
- 34. Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018;378(24):2288–301. [DOI] [PubMed] [Google Scholar]
- 35. Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. N Engl J Med 2018;379(21):2040–51. [DOI] [PubMed] [Google Scholar]
- 36. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med 2018;378(22):2078–92. [DOI] [PubMed] [Google Scholar]
- 37. Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adap,,tive, and acquired resistance to cancer immunotherapy. Cell 2017;168(4):707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perkel JM. Single-cell analysis enters the multiomics age. Nature 2021;595(7868):614–6. [Google Scholar]
- 39. Marx V. A dream of single-cell proteomics. Nat Methods 2019;16(9):809–12. Epub 2019/08/14. 10.1038/s41592-019-0540-6. PubMed PMID: 31406385. [DOI] [PubMed] [Google Scholar]
- 40. Wu W, Zhang W, Ma X. Network-based integrative analysis of single-cell transcriptomic and epigenomic data for cell types. Brief Bioinform 2022;23(2):bbab546. 10.1093/bib/bbab546. [DOI] [PubMed] [Google Scholar]
- 41. Wang H, Ma X. Learning deep features and topological structure of cells for clustering of scRNA-sequencing data. Brief Bioinform 2022;23(3):bbac068. Epub 2022/03/19. 10.1093/bib/bbac068. PubMed PMID: 35302164. [DOI] [PubMed] [Google Scholar]
- 42. Sharman JL, Benson HE, Pawson AJ, et al. IUPHAR-DB: updated database content and new features. Nucleic Acids Res 2013;41(D1):D1083–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramilowski JA, Goldberg T, Harshbarger J, et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun 2015;6:7866. Epub 2015/07/23. 10.1038/ncomms8866. PubMed PMID: 26198319; PMCID: PMC4525178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prasad TK, Kandasamy K, Pandey A. Human protein reference database and human proteinpedia as discovery tools for systems biology. Methods Mol Biol Clifton, N.J., 2009;577:67–79.. 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 45. Ben-Shlomo I, Yu Hsu S, Rauch R, et al. Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci STKE 2003;2003(187):re9-re. [DOI] [PubMed] [Google Scholar]
- 46. Graeber TG, Eisenberg D. Bioinformatic identification of potential autocrine signaling loops in cancers from gene expression profiles. Nat Genet 2001;29(3):295–300. [DOI] [PubMed] [Google Scholar]
- 47. Dimitrov D, Türei D, Garrido-Rodriguez M, et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat Commun 2022;13(1):3224. 10.1038/s41467-022-30755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turei D, Valdeolivas A, Gul L, et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol Syst Biol 2021;17(3):e9923.Epub 2021/03/23. 10.15252/msb.20209923. PubMed PMID: 33749993; PMCID: PMC7983032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schroff F, Kalenichenko D, Philbin J. (eds). Facenet: a unified embedding for face recognition and clustering. 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). 2015, pp. 815–23. 10.1109/CVPR.2015.7298682. [DOI]
- 50. Song Q, Zhu X, Jin L, et al. SMGR: a joint statistical method for integrative analysis of single-cell multi-omics data. NAR genomics and bioinformatics 2022;4(3):lqac056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dries R, Zhu Q, Dong R, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol 2021;22:78. 10.1186/s13059-021-02286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SeqFISH+ mouse cortex data: The seqFISH+ dataset was obtained from the Giotto [51] repository (https://github.com/RubD/spatial-datasets/tree/master/data/2019_seqfish_plus_SScortex). We used the gene count matrix of the mouse cortex dataset, with FOVs ranging from 0 to 4, which covers 10 000 genes and 523 cells. NanoString CosMx SMI data: This single-cell spatial dataset contains 20 FOVs, which are profiled by the CosMx SMI on Formalin-Fixed Paraffin-Embedded (FFPE) samples of non-small-cell lung cancer (NSCLC) tissue [8]. The dataset (Lung 13) is available from https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/. Vizgen MERSCOPE datasets: We used the spatial profiles of colon and melanoma tumour from Vizgen MERFISH FFPE Human Immuno-oncology datasets, which are accessible from https://info.vizgen.com/merscope-ffpe-solution. The colon dataset (Colon cancer 2) contains a MERFISH measurement of a 500 gene panel and 817 588 cells, of which the cropped region (dim x: 11 250–13 750 and dim y: 4000–5000) was in the application of spaCI. Ten cropped regions were identified for performance evaluation. The melanoma dataset (Melanoma 2) contains a MERFISH measurement of a 500 gene panel and 207 869 cells, where the cropped region (dim x: 5000–6000 and dim y: 5000–6000) was used in this study.