

Abstract
We draw from the assumption that similarities between pathogens at both pathogen protein and host protein level, may provide the appropriate framework to identify and rank candidate drugs to be used against a specific pathogen. Vir2Drug is a drug repurposing tool that uses network-based approaches to identify and rank candidate drugs for a specific pathogen, combining information obtained from: (a) ranked pathogen-to-pathogen networks based on protein similarities between pathogens, (b) taxonomy distance between pathogens and (c) drugs targeting specific pathogen’s and host proteins. The underlying pathogen networks are used to screen drugs by means of specific methodologies that account for either the host or pathogen’s protein targets. Vir2Drug is a useful and yet informative tool for drug repurposing against known or unknown pathogens especially in periods where the emergence for repurposed drugs plays significant role in handling viral outbreaks, until reaching a vaccine. The web tool is available at: https://bioinformatics.cing.ac.cy/vir2drug, https://vir2drug.cing-big.hpcf.cyi.ac.cy
Keywords: drug repurposing, pathogen networks, protein interactions
Introduction
The advent of omics technologies has provided a wide spectrum of knowledge related to pathogens, leading to a series of available repositories that hold information ranging from molecular, functional and morphological, up to various phenotypic data. However, the way in which pathogens are functionally linked within an overall network of existing pathogens is largely unexplored. Indeed, despite the plurality on pathogen-based information as well as the plurality of drug repurposing tools, the feedback obtained through the SARS-COV-2 trajectory towards repurposing drugs against COVID-19 symptoms, in time of emergence, was quite disappointing [1], emerging the need for drug repurposing tools that employ alternative approaches. Systemic advancements in the field have been recently focused on network-based approaches [2–4], indicatively using methodologies that draw from disease-based networks [5], drug–target networks [6], artificial-intelligence-based methods [7–9] or even combinations of them [10]. Characteristically, Ma et al. [11] proposed a pioneer network-based computational method for drug repurposing, providing 483 microbiota–disease associations, suggesting that: microbe-based inference of novel drug indications represents a new approach for drug repurposing and deserves further large-scale studies. Herein, the present work draws from the assumption that similarities between pathogens at protein level, may provide an appropriate framework to identify and rank candidate drugs to be used against the viral infection of a specific pathogen, even if the pathogen is a new uncategorized one without a yet identified proteome. Specifically, we propose the Vir2Drug: a drug repurposing web tool that uses network-based approaches to identify and rank candidate drugs for a specific pathogen, combining information obtained from: (a) ranked pathogen-to-pathogen (P2P) networks based on protein similarities between pathogens at host and/or pathogen’s proteome level, (b) taxonomy distance between pathogens and (c) drugs targeting specific host and/or pathogen proteins. Vir2Drug allows the creation of P2P similarity networks based either at host or pathogen level, where the edges are scored by means of specific equations depending on whether the pathogen under study is categorized or not. P2P networks are used to screen through seven available methodologies that account for either the host or pathogen’s protein targets and/or the taxonomic positioning of the pathogen with respect to others, while the drug ranking is performed by means of eight proposed available equations.
Material and methods
The Vir2Drug methodologies towards identifying candidate drugs, come with a stepwise frontend web interface that consists of the mainframe and a help page, all written using HTML, PHP and JavaScript language environments. The backend interface has been written in R language environment, holding all the proposed methodologies and equations that serve the overall pipeline. The proposed tool is available online through the webpage of the Bioinformatics Department, at the Cyprus Institute of Neurology and Genetics (CING) (https://bioinformatics.cing.ac.cy/vir2drug). Vir2Drug is also served by a Docker space at the High-Performance Computer Facility of the Cyprus Institute (https://vir2drug.cing-big.hpcf.cyi.ac.cy). Figure 1 depicts the overall pipeline, towards obtaining the final drug list for a specific pathogen under study.
Figure 1.

Overall pipeline showing the required components towards obtaining the final drug list through the obtained P2P network.
The overall pipeline draws from a pathogen-based repository developed in the context of this work, and provides three individual steps designed to guide the user until the end of the workflow process. These read as follows: (a) importing candidate proteins/genes related (or not) to a specific pathogen, (b) creating ranked P2P networks based on protein similarities between proteins at host and/or pathogen’s proteome and/or taxonomic level and (c) performing a series of drug screening and scoring methodologies towards obtaining the candidate drug list for a specific set of proteins under study. In the following sections, we describe in detail the main components and methodologies used for the implementation of the proposed tool.
The pathogen repository
A pathogen-based repository has been developed by collecting and combining all the pathogens’ proteins as well as their host–protein interactions from all organisms found in IntAct [12], Phisto [13] and VirHostNET [14] data repositories. The underlying pathogens were verified by means of the NCBI taxonomy IDs [15], and further filtered for non-pathogenic organisms like metazoan and plants, using a custom written script in R. The final list of pathogens included viruses, bacteria and known human parasites from eukaryotes, forming a data repository of 625 pathogens in total. The ‘taxize’ R [16] package has been used to construct the taxonomy tree and calculate the taxonomy distances for each pair of pathogens included in the above repository. The drug general information along with their target proteins have been obtained from the DrugBank [17] repository. The above data are regularly updated and serve as the main pathogen repository where all the available services and methodologies draw from.
Creating P2P networks
By definition, the term Pathogen Networks employed in this work refers to two types of P2P networks: (a) P2P networks based on host proteins reported as pathogen–protein interactors, and (b) P2P networks based on pathogen’s proteins, accordingly. Depending on the network type, the node size represents the number of either host or pathogen’s proteins, and the edge connection refers to whether there is a number of common host interactors or pathogen’s proteins in-between two pathogens. An R pipeline has been developed that allows the creation of P2P networks based on specific proteins of interest. The latter networks have been further enriched with an additional edge score (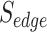 ), according to the following equation:
), according to the following equation:
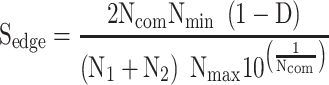 |
(1) |
where 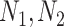 : are the number of host proteins interacting with the two pathogens forming a single edge.
: are the number of host proteins interacting with the two pathogens forming a single edge. 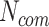 : is the number of common host proteins that the two pathogens interact with.
: is the number of common host proteins that the two pathogens interact with. 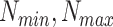 : are the maximum and minimum estimations of
: are the maximum and minimum estimations of 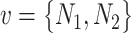 vector.
vector. 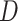 : is the taxonomy distance between two pathogens. Equation (1) aims to bring drugs from pathogens that are strongly connected with the pathogen understudy, based on the assumption that those drugs may also be candidate against the infection symptoms of the pathogen understudy. This is in line with the following critical assumptions.
: is the taxonomy distance between two pathogens. Equation (1) aims to bring drugs from pathogens that are strongly connected with the pathogen understudy, based on the assumption that those drugs may also be candidate against the infection symptoms of the pathogen understudy. This is in line with the following critical assumptions.
Assumption 1: the relation between two pathogens can be approximated by the taxonomy distance D in-between them, as well as by their observed commonality (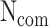 ) mainly at host protein and in some cases at pathogen’s protein level. The combination of these two metrics aims to get a better approximation by means of two different approaches of estimating the similarity between two pathogens. For unclassified pathogens, Equation (1) comes without the (1-D) parameter.
) mainly at host protein and in some cases at pathogen’s protein level. The combination of these two metrics aims to get a better approximation by means of two different approaches of estimating the similarity between two pathogens. For unclassified pathogens, Equation (1) comes without the (1-D) parameter.
Assumption 2: the significance of a connection between two pathogens should also be approximated by the number of proteins (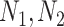 ) each pathogen interacts with. Specifically, we consider that an edge formed by two pathogens with relatively similar number of protein interactions
) each pathogen interacts with. Specifically, we consider that an edge formed by two pathogens with relatively similar number of protein interactions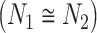 is more significant than an edge where
is more significant than an edge where 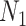 is much different than
is much different than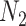 . In addition, a limited number of a pathogen’s host–protein interactions may be a result of a limited knowledge for the specific pathogen. This in turn may affect the overall score, since commonalities at protein interaction level may not be an optimum representative approximation. Thus, we further used the factor
. In addition, a limited number of a pathogen’s host–protein interactions may be a result of a limited knowledge for the specific pathogen. This in turn may affect the overall score, since commonalities at protein interaction level may not be an optimum representative approximation. Thus, we further used the factor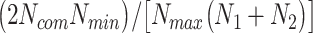 , followed by an overall commonality penalty of (
, followed by an overall commonality penalty of (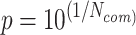 ), aiming to keep a balance between the number of common protein interactions
), aiming to keep a balance between the number of common protein interactions 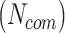 and the total number of protein interactions per pathogen (
and the total number of protein interactions per pathogen (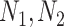 ). Figure 2 depicts an example of a P2P network showing the Escherichia coli STRAIN CFT073 pathogen sharing host–protein interactions with 16 additional pathogens. The node size represents the number of host proteins related to a specific pathogen, and the edge size represents the edge score obtained by means of Equation (1).
). Figure 2 depicts an example of a P2P network showing the Escherichia coli STRAIN CFT073 pathogen sharing host–protein interactions with 16 additional pathogens. The node size represents the number of host proteins related to a specific pathogen, and the edge size represents the edge score obtained by means of Equation (1).
Figure 2.
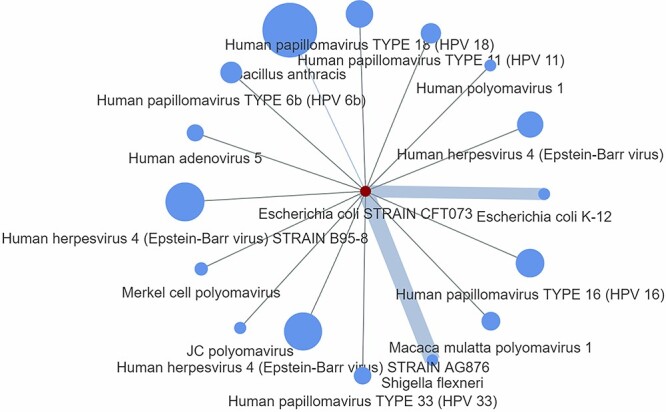
A P2P network showing the E. coli STRAIN CFT073 pathogen (depicted with red colour) along with its first neighbours. The node size represents the number of host proteins related to a specific pathogen, and the edge size represents the edge score obtained by means of Equation (1).
It is observed that the candidate pathogen has two strong connections with E. coli K-12 and Shigella flexneri pathogens, sharing the P16070 host protein, namely the CD44 antigen. Indeed, it has been stated that members of the genus Shigella share common characteristics with members of the genus Escherichia and the genetic relatedness clearly suggests that they are a subtype of E. coli [18]. The association between E. coli STRAIN CFT073 and E. coli K-12 has been also investigated in Ref. [19]. Although these three pathogens interact with the same host protein, the edge score between E. coli STRAIN CFT073 and S. flexneri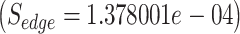 has been found larger than the edge score in between E. coli STRAIN CFT073 and E. coli K-12, which was found to be
has been found larger than the edge score in between E. coli STRAIN CFT073 and E. coli K-12, which was found to be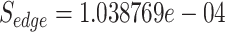 . This is due to the fact that the E. coli K-12 interacts with more host proteins (40 in total), and according to Equation (1), it has been considered as less significant edge compared with S. flexneri, which interacts with 30 in total host proteins. However, Equation (1) by no means suggests any biological truth but just simply a statistical score that aims to characterize in network terms the significance of a specific edge. Thus, in the following sections we further propose additional equations and methodologies in the prospect to create protein-based scenarios adequate to represent specific biological conditions related to the pathogen under study. For those that do not want to use Equation (1) to score their network edges, Vir2Drug provides two alternative scores: (1) the number of common proteins
. This is due to the fact that the E. coli K-12 interacts with more host proteins (40 in total), and according to Equation (1), it has been considered as less significant edge compared with S. flexneri, which interacts with 30 in total host proteins. However, Equation (1) by no means suggests any biological truth but just simply a statistical score that aims to characterize in network terms the significance of a specific edge. Thus, in the following sections we further propose additional equations and methodologies in the prospect to create protein-based scenarios adequate to represent specific biological conditions related to the pathogen under study. For those that do not want to use Equation (1) to score their network edges, Vir2Drug provides two alternative scores: (1) the number of common proteins 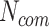 in between two pathogens, and (2) the taxonomic distance
in between two pathogens, and (2) the taxonomic distance 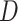 in between them.
in between them.
The drug screening intermediate process based on P2P network
The drug screening process is an intermediate process that aims to screen drugs based on the edge properties of the P2P network described in previous section, and to further provide a separate drug list for each edge in the network. Drug screening is performed by means of two diverse approaches: (1) the PPI network-based drug screening process that comes with six diverse methodologies that draw from the PPI information of the pathogens involved in the P2P network, and (2) the taxonomy-based drug screening process that is based on the taxonomic distance in-between two pathogens that form a specific edge. For a detailed description on those approaches please refer to the supplementary document.
The drug scoring intermediate process
Each drug contained in the lists obtained from the drug screening process is further ranked by means of specific equation that draws from the proteins included in each pair of pathogens that form the specific edge. This is an initial ranking process that aims to score a deriving drug by means of the edge and the node content of the P2P network. Due to the plethora of drug information that could be obtained from the targeted proteins contained in the two pathogens that form an edge, we propose eight diverse equations for this type of ranking, each one using a different set of parameters. For a detailed description on those methodologies and their parameters please refer to the supplementary document.
Obtaining the final drug list
In order to provide the final candidate drug list for the pathogen under study, we developed two generalized equations: one for the PPI network-based and one for the taxonomic-based methodologies, accordingly. These read as follows:
PPI network-based drug scoring
The underlying methodology draws from the following assumptions.
Assumption 1: a drug that appears very often during the screening process of the P2P network is more likely to be a candidate drug.
Assumption 2: a drug that targets mostly the protein sets of the pathogen under study is more likely to be significant, rather than a drug that targets mostly the X-pathogen’s proteins.
Assumption 3: drugs that derive from highly ranked edges of the P2P network are considered more significant than those deriving from low ranked edges.
Taking in to account the above-mentioned assumptions, the final equation reads as follows:
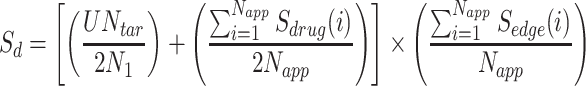 |
(2) |
where 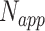 is the number of times a specific drug appeared through the drug screening process,
is the number of times a specific drug appeared through the drug screening process, 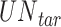 is the number of unique targets appeared in the scan process,
is the number of unique targets appeared in the scan process, 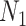 is the number of host proteins involved in the pathogen under study and
is the number of host proteins involved in the pathogen under study and 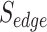 ,
, 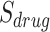 are the estimated edge and drug scores obtained from Equations (1) and those described in the supplementary file (Equation S1–S7).
are the estimated edge and drug scores obtained from Equations (1) and those described in the supplementary file (Equation S1–S7).
Taxonomic-based drug scoring
Herein, the final drug score is calculated by means of the minimum distance of organisms affected by the same drug, the range 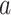 of its activity spectrum and the diversity in its inter-taxon distances. The scoring equation reads as follows:
of its activity spectrum and the diversity in its inter-taxon distances. The scoring equation reads as follows:
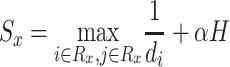 |
(3) |
where 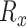 is the set of n pathogens with known protein targets for drug
is the set of n pathogens with known protein targets for drug 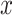 ,
, 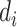 is the taxonomy distance of the
is the taxonomy distance of the 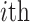 pathogen targeted by drug
pathogen targeted by drug 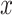 and parameter
and parameter 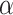 is the maximum difference of distances
is the maximum difference of distances 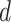 across the pathogens in set
across the pathogens in set 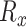 , which can be written as:
, which can be written as:
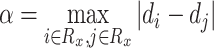 |
(4) |
Finally, the Shannon diversity of distances 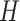 can be expressed as:
can be expressed as:
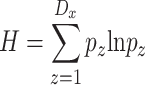 |
(5) |
where 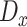 is the set of discrete distance values observed in
is the set of discrete distance values observed in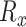 , whereas
, whereas 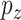 is the ratio of the distance
is the ratio of the distance 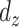 frequency over the total number (n) of measured distances in
frequency over the total number (n) of measured distances in 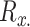
Results
We recall that a part of Vir2Drug methodology has been already applied, with noteworthy results in Ref. [4], where the authors used Equations (1)–(3) and (S1 described in supplementary file) to identify drugs that could be used against COVID-19 symptoms. Some of these drugs are already in experimental state or granted an FDA Emergency Use Authorization. Herein, in order to assess the efficiency, the adaptability and the quality of the output of Vir2Drug, we launched three real-life scenario-based research inquiries and we assessed the predictive performance of the system based on its output. The inquiries were based on three scenarios that cover three forms of anticipated user needs: (a) interested in drugs against a known virus, (b) interested in drugs against a newly discovered human protein target that is involved in the infectivity of a known virus and (c) interested in drugs with a known action against a human protein target that may have antiviral properties against any virus. We created a scenario for each of these three needs and after we selected the appropriate options from the provided tools to set up our analysis framework, we run a Vir2Drug inquiry for each scenario and assessed the output. In the following subsections we describe these inquiries in detail.
Scenario 1
Herein, the user is a virologist studying HIV-1 and is interested in a list of candidate drugs that could be tested in the lab for potential antiviral efficacy against HIV-1, independently of mechanism of action. To run this inquiry, we selected the options of system tools as shown in Table 1 and set up the analysis framework using 904 relevant host proteins and 65 relevant pathogen proteins, accordingly.
Table 1.
Input parameters and output information generated for the implementation of Scenario 1 described in the text
| Scenario 1 analysis framework | ||||||||
|---|---|---|---|---|---|---|---|---|
| Step1 | Step 2 | Step 3 | ||||||
| Input | Pathogen selection | Proteins used | Reference network | NET scoring methodology | Edges to keep | Drug screening methodology | Drug scoring methodology | Drugs to keep |
| HIV-1 | All available | Based on host protein | TaxScore Equation (1) |
10 | 5a | 4PH-fold & 8-folda | 30 | |
| Output | Identified proteins | Pathogen connections identified | Total top-scored candidate drugs | |||||
| Host | Pathogen | 10 | 120 b | |||||
| 904 | 65 | |||||||
One of the five combinations of options used in this scenario to generate four lists of top-scored drugs (Common/4PH-Fold; V1-Prot/4PH-Fold; V2-Prot/8-fold; V2-Host).
Total number of candidate drugs from all four lists.
The generated P2P network obtained from Step 2 involved 377 ranked connections with other pathogens, of which 10 top-rated of them were strictly kept and considered as the most significant ones. Figure 3 depicts the outcome of this process showing strong protein commonality with other strains of the HIV1 virus; a score obtained by means of Equation (1), namely the taxScore equation.
Figure 3.
P2P network generated as an output from Step 2. The node size represents the number of host proteins related to a specific pathogen, and the edge size represents the edge score obtained by means of Equation (1), namely the taxScore equation.
We further used multiple screening and scoring methodology combinations (Step 3) to generate four separate scored drug lists, ranked by score. To assess the reliability of the generated Vir2Drug lists of candidate drugs, we positively identified drugs that are currently FDA-approved for HIV-1 infection in the four generated top-30 drug lists (https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines). More precisely, Vir2Drug identified as top-30 ranked the 13 out of 14 drugs approved drugs in all three major classes (nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors and protease inhibitors), with the exception of etravirine. The top-two ranked drugs in the main generated list were lamivudine and tenofovir, which are both part of the recommended first-line anti-retroviral (ARV) regimen in treatment-naive patients (https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv/what-start-initial-combination-regimens-antiretroviral-naive). In fact, all the first-line ARV drugs in the NIH guidelines are included in the four top-30 generated lists (i.e. abacavir, emtricitabine). Interestingly, none of the integrase stand transfer inhibitors (i.e. dolutegravir, cabotegravir) or fusion inhibitors (i.e. enfuvirtide) were included in any of the four top-30 ranked lists using the options of tools described. We therefore conclude that the Vir2Drug lists produced under the studied scenario offer high reliability. However, since the studied scenario was created with the aim to identify new candidates against HIV, we also looked more closely at the top-ranked drugs in the list that were not classified as HIV-approved in order to scrutinize the quality of the output. From the four lists produced, we selected drugs that were either experimental/investigational or approved for other conditions in order to fulfil the above aim. From the 120 candidate drugs included in the four top-30 ranked generated lists, we identified seven drugs that covered the selection criteria and were being in the top-10 of one of the four generated lists. In the following, we present a summary of the two most highly ranked drugs of these seven candidate drugs (myristic acid and memantine), as representative examples of the quality examination analysis performed to all seven drugs.
Memantine [an N-methyl-D-aspartate (NMDA) antagonist approved against Alzheimer’s disease] was reported by Vir2Drug in the COMMON list (drugs that target proteins that are common/shared between HIV-1 and other pathogens; identified from the generated pathogen network). This drug has been highly scored in this list due to the physical association of its known NMDA-receptor target with protein PLPCb4 (phospholipase C beta 4; code Q15147), which is an enzyme that catalyzes the formation of DAG and IP3 from PIP2. The NMDA receptors and phospholipase C are known to have important functional interactions [20, 21]. There are reports that HIV-1 envelope glycoproteins gp120 and gp160 directly impair the CD3/TcR-mediated activation of phospholipase C via CD4 in uninfected T cells [22] and that the HIV-1 regulatory protein Tat induces the release of calcium and TNF-alpha production, an effect mediated by the activation of phospholipase C [23]. Moreover, Vir2Drug reported that another protein, Platelet-activating factor receptor (code: P25105), which also has an association with the NMDA receptor since its activation results in the activation of platelet-activating factor [24], plays a crucial step in HIV-1 neuropathogenesis [25]. Based on the above associations, memantine has been shown to be a particularly interesting candidate against HIV-1. Indeed, studies in the early 90s identified of memantine prevented HIV-induced neuronal damage [26] and later studies confirmed that memantine plays a therapeutic role in HIV-associated cognitive impairment [27, 28] and neuropathy [29]. Nevertheless, to the best of our knowledge, no studies, clinical or in vitro, have been performed to study any direct effect of memantine against HIV-1 viability and infectivity. All the above confirm the relevance of memantine in AIDS pathophysiology and HIV-1 pathology and represents a positive-quality output in our assessment as a potential candidate for further experimentation against HIV-1 and AIDS.
Another drug that Vir2Drug identified and ranked highly in our seven-drug selection list is myristic acid (a saturated fatty acid used by cells as a lipid anchor of certain proteins/receptors on the biomembrane). Myristoylation is a lipidation modification process where a myristoyl group from myristic acid is linked to an N-terminal glycine residue of a target protein. Myristoylation is a very important process for HIV-1, since it relies on it to successfully package its genome, assemble and mature into a new infectious particle. HIV accessory proteins, such as Nef, are myristoylated and are essential for AIDS progression [30]. Some myristic acid analogues have been tested for their anti-HIV efficacy [31] and some have been found to have significant cytopathic effect against HIV (i.e. 12-thioethyldodecanoic acid). However, there is not much depth in research around this class of drugs against HIV and further investigation is needed to see whether these agents can be clinically applicable against AIDS. Nevertheless, the above evidence represents another positive-quality output in our assessment of the generated list on this Scenario.
Scenario 2
Herein, the user is a molecular biologist who is interested in candidate drugs against the human protein exportin-1 or XPO1 (UniProtKB code: O14980), which has been recently found to react with influenza-A virus, along with other viruses, in the infected human cells, in order to help viral particles mature into ready-to-infect viruses. Therefore, XPO1 is presented as a novel antiviral target. To run this inquiry, similarly to Scenario 1, we selected the options of system tools as shown in Table 2 and set up the analysis framework accordingly.
Table 2.
Table shows the input (tools and options used) and output information generated by Vir2Drug
| Scenario 2 analysis framework | ||||||||
|---|---|---|---|---|---|---|---|---|
| Step1 | Step 2 | Step 3 | ||||||
| Input | Pathogen selection | Proteins used | Reference network | NET scoring methodology | Edges to keep | Drug screening methodology | Drug scoring methodology | Drugs to keep |
| Influenza A virus (H7N9) | XPO1 (O14980) | Based on host proteins | TaxScore Equation (1) |
10 | 3a | 4PH-fold | 30 | |
| Output | Identified proteins | Pathogen connections identified | Total top-scored candidate drugs | |||||
| Host | Pathogen | 10 | 35 b | |||||
| 1 | 1 | |||||||
One of the five combinations of options used in this scenario to generate three lists of top-scored drugs (Common/4PH-Fold; V2-Host/4PH-Fold; V2-Prot/4PH-Fold).
Total number of candidate drugs from all three lists.
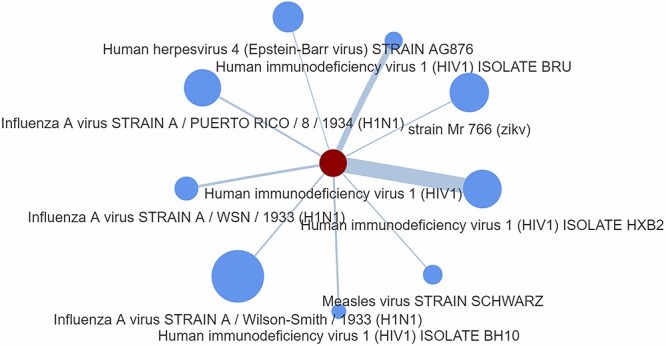
In this scenario, we are interested in drugs that can potentially target XPO1 and affect the influenza A virus pathology. For this, we create a P2P network based on common host proteins between pathogens in the prospect to identify drugs that affect these linked pathogens, which may considered as potential candidates. Figure 4 depicts the outcome of this process showing strong protein commonality with the ‘Murine minute virus strain MVM Prototype’ pathogen; a score obtained by means of Equation (1), namely the taxScore equation.
Figure 4.
P2P network generated as an output from Step 2. The node size represents the number of host proteins related to a specific pathogen, and the edge size represents the edge score obtained by means of Equation (1), namely the taxScore equation.
As in Scenario 1, we used options depicted in Step 3, to generate three separate ranked scored drug lists. The COMMON list (common proteins between pathogens) and the V1-HOST list (target pathogen’s host protein) options produced the same only one drug (selinexor; approved drug). This challenge was expected, since there is only one target host protein in our scenario and selinexor is the only first-in-class selective inhibitor of nuclear export (SINE), an XPO1-antagonist, which is currently used in the clinic against multiple myeloma as it causes the accumulation of tumour suppressor proteins in the cell nucleus (https://www.ema.europa.eu/en/medicines/human/EPAR/nexpovio). Vir2Drug has correctly identified this compound as a positively candidate drug against the specified target. We then continued to use the V2-HOST list (using the host proteins from all pathogens in the linked network) and the V2-PROT list (using the pathogen proteins from all pathogens in the linked network). The former produced 30 drugs of which 24 were approved drugs, whereas the latter produced only four compounds, all experimental. Selinexor has been included in the two out of three drug lists generated by Vir2Drug as number one in these lists (COMMON and V2-HOST), reflecting a positive reliable hit in this test. Nevertheless, since the scenario presented the challenge of looking at drug candidates against a pathogen using a host target, which had been under-researched in terms of therapeutic applicability, we had to focus our quality assessment on the approved drugs generated by the V2-HOST list. However, by list definition, these were drugs that exploit host targets other than XPO1 and therefore it was difficult to provide an accurate quality assessment of these drugs as potential SINE compounds, since these drugs may have an indirect effect on XPO1 and/or influenza-A by unknown mechanisms. For example, one of the drugs included in the generated V2-HOST list was digoxin (a cardiac glycoside used for treating heart failure), which has been recently shown to inhibit cytokine production in influenza-A infections [32]. The experimental SINE compound, called KPT-185, has been shown to inhibit cytokine production [33] and the replication of HIV [20], a virus that is included in the pathogen network of our scenario. It is, therefore, possible that digoxin may exert its anti-cytokine action against influenza-A virus through the inhibition of XPO1, but data to support this hypothesis are lacking. Therefore, the above info provides a positive assessment for the inclusion of digoxin in the generated list of our inquiry. However, similar interconnections have not been established for other 24 approved drugs after a limited investigation of the literature.
Scenario 3
Herein, the user is a pharmacologist who is interested in identifying candidate drugs against the human protein Cyclin Dependent Kinase 1 (CDK1), which is known to be involved in viral transcription and replication in different viruses. For this inquiry, which does not look at any specific pathogen, we selected the options of system tools as shown in Table 3 and set up the analysis framework for finding potential novel CDK1 antagonists with a wide-range antiviral efficacy.
Table 3.
Table shows the input (tools and options used) and output information generated by the Vir2Drugtool
| Scenario 3 analysis framework | ||||||||
|---|---|---|---|---|---|---|---|---|
| Step1 | Step 2 | Step 3 | ||||||
| Input | Pathogen selection | Proteins used | Reference network | NET scoring methodology | Edges to keep | Drug screening methodology | Drug scoring methodology | Drugs to keep |
| Unspecified | CDK1 (P06493) | Based on host proteins | GenScore Equation (1) |
0 (unlimited) | 3a | 4PH-fold | 30 | |
| Output | Identified proteins | Pathogen connections identified | Total top-scored candidate drugs | |||||
| Host | Pathogen | 16 | 35 b | |||||
| 1 | 1 | |||||||
These options are one of the five combinations of options used in this scenario to generate two lists of top-scored drugs (V1-Host/4PH-Fold; V2-Host/4PH-Fold).
Total number of candidate drugs from all two lists.
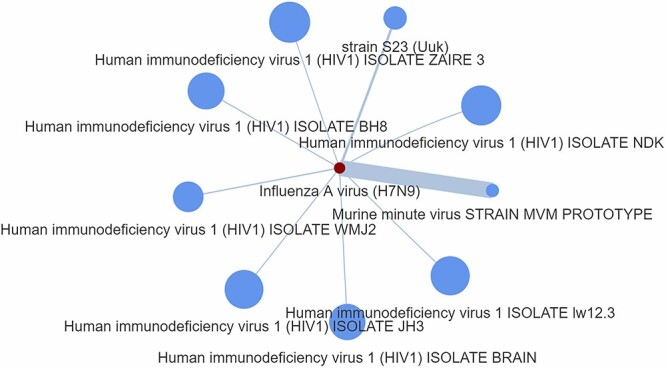
It should be stressed that in Step 1 we used five different protein codes accounted for CDK1 or for novel alterations of CDK1 complexes (P06493, P06493-1, P06493-2, A0A024QZP7, A0A087WZZ9), which were available in the database. In Step 2, we opted for zero ‘number of edges to keep’ in creating a host-based P2P network, which would allow to produce its maximum size independent of connection strength and therefore without excluding any connections from the network. This is a straight forward process, which is not based on specific pathogen but takes advantage of a hypothetical P2P network that may be used for drug screening and scoring, accordingly. Figure 5 depicts the outcome of this process showing strong commonality between a hypothetically unknown pathogen that involves the CDK1 and the ‘Human adenovirus 2’ pathogen. Since the pathogen and its proteins are unknown, the edge scores have been obtained by means of Equation (1), namely the genScore, which comes without the (1-D) parameter.
Figure 5.
P2P network generated as an output from Step 2. The node size represents the number of host proteins related to a specific pathogen, and the edge size represents the edge score obtained by means of the generalized Equation (1), namely the genScore, which comes without the (1-D) parameter.
Unlike previous scenarios, in Step 3 in this inquiry we used one screening and scoring methodology combination to generate one ranked scored drug list; the V2-HOST list (using the host proteins from all pathogens in the linked network). This is because this inquiry had no targeted pathogen but rather a host protein. The V2-HOST list uses the generated pathogen network, produced based on the association of the pathogens in the network with the target host protein. Assessing the reliability of the drug list, the approved drug fostamatinib (a tyrosine kinase inhibitor used in treatment of chronic immune thrombocytopenia) was ranked first in the generated list. Fostamatinib is known to have a CDK1 inhibitor activity and negatively regulates G2/M transition by down-regulating CDK1 kinase activity [34]. As SarsCov2 is included in the 16-pathogen network generated by Vir2Drug for our inquiry, we note that fostamatinib has been proposed by a number of repurposing studies to be efficacious against COVID-19 pathology [35, 36] and recently it has been shown to reduce serious adverse effects and risk of death by 50% in hospitalized COVID-19 patients in a Phase II study [37]. However, its effects have not been linked to mechanisms other than its CDK1 activity. The second ranked drug in the generated list was the experimental drug alvocidib (also known as flavopiridol; a semi-synthetic flavone plant-extract), which was the first CDK inhibitor to reach human clinical trials in patients with non-small cell lung cancer, in combination with paclitaxel [38]. Flavopiridol is a non-specific CDK inhibitor, and therefore has inhibitory activity to other CDKs apart from CDK1 [39]. More interestingly, flavopiridol has been suggested to have an antiviral effect against influenza-A virus [40] and HIV-1 [41] among others. In the generated list, there are drugs that are particularly interesting. One of these is the investigational drug seliciclib (or else roscovitine), which has been recently identified as a non-selective CDK inhibitor [42]; however, there are no studies available to date about the potential antiviral efficacy of this agent. Another interesting compound in the generated list was the experimental drug alsterpaullone; a 20-year old CDK1 and CDK2 inhibitor [43] that has been shown to possess a good molecular efficacy against HIV-1 replication [44]. A number of investigational and experimental kinase inhibitors, which include the ones mentioned here and a few more included in our generated V2-HOST list, have been characterized as underexplored of their antiviral efficacy and clinical applicability to date [45]. As reflected in the above examples, there is a positive verdict on the assessment of the reliability and quality of the generated list in this inquiry.
Discussion
Vir2Drug provides a flexible framework for drug repurposing, which integrates pathogen-based knowledge into a single network and prioritizes protein commonalities to be used for drug screening and scoring for further interpretation. To author’s knowledge, Vir2Drug is the only available web-framework for drug repurposing, which is based on pathogen similarities, rather than other computational approaches [46]. A significant novelty of Vir2Drug is the ability to identify candidate drugs for unknown pathogens by simply obtaining similarities with existing pathogens. Evaluation by means of three well-designed and successive scenarios, provided positive feedback on the significance the reliability and the quality of obtained results, as well as the flexibility to screen and score drugs based of different assumptions. Vir2Drug puts significant contribution to the understanding of pathogen relationships, while at the same time offers a pipeline that fills a significant gap between pathogen protein interactions and drug identification.
Key Points
Vir2Drug is a web-based drug repurposing tool that uses network-based approaches to identify and rank candidate drugs for a specific pathogen.
It draws from the assumption that similarities between pathogens at protein level, may provide the appropriate framework to identify and rank candidate drugs.
The tool uses ranked pathogen-to-pathogen networks based on protein commonalities, by means of four edge-scoring methodologies.
The tool further provides eight methods for drug screening and eight methods for drug scoring as well allowing the user to perform combinations of them.
Innovation of the tool is the ability to work on both known and unknown pathogens especially in periods where the emergence for repurposed drugs plays significant role in handling viral outbreaks.
Supplementary Material
Acknowledgement
None declared.
Dr George Minadakis is an associate scientist at the Department of Bioinformatics at The Cyprus Institute of Neurology and Genetics. He holds a BSc in Biomedical Engineering, a Master’s degree in Data Communication Systems and a PhD in Electronic & Computer Engineering.
Dr Marios Tomazou is an associate scientist at the Neurogenetics Group at The Cyprus Institute of Neurology and Genetics. He holds a BSc in Biology, a Master’s degree and a PhD in the field of Systems and Synthetic Biology.
Dr Nikolas Dietis is an Assistant Professor of Pharmacology at the Medical School, University of Cyprus. He holds a BSc in Neuroscience & Pharmacology, a Master’s degree in Applied Biosciences (Neuropharmacology) and a PhD in Pharmacology.
Prof. George M. Spyrou is a Bioinformatics European Research Area Chair Holder and the Head of the Bioinformatics Department at the Cyprus Institute of Neurology and Genetics. He holds a BSc in Physics, MSc in Medical Physic, MSc in Bioinformatics and a PhD in Medical Physics.
Contributor Information
George Minadakis, Bioinformatics Department, The Cyprus Institute of Neurology & Genetics, 6 Iroon Avenue, 2371 Ayios Dometios, Nicosia, Cyprus | PO Box 23462, 1683 Nicosia, Cyprus; The Cyprus School of Molecular Medicine, 6 Iroon Avenue, 2371 Ayios Dometios, PO Box 23462, 1683 Nicosia, Cyprus.
Marios Tomazou, Bioinformatics Department, The Cyprus Institute of Neurology & Genetics, 6 Iroon Avenue, 2371 Ayios Dometios, Nicosia, Cyprus | PO Box 23462, 1683 Nicosia, Cyprus; The Cyprus School of Molecular Medicine, 6 Iroon Avenue, 2371 Ayios Dometios, PO Box 23462, 1683 Nicosia, Cyprus.
Nikolas Dietis, Medical School, University of Cyprus, Nicosia 1678, Cyprus.
George M Spyrou, Bioinformatics Department, The Cyprus Institute of Neurology & Genetics, 6 Iroon Avenue, 2371 Ayios Dometios, Nicosia, Cyprus | PO Box 23462, 1683 Nicosia, Cyprus; The Cyprus School of Molecular Medicine, 6 Iroon Avenue, 2371 Ayios Dometios, PO Box 23462, 1683 Nicosia, Cyprus.
Data Availability
Vir2Drug is available online through the following links: (https://bioinformatics.cing.ac.cy/vir2drug, https://vir2drug.cing-big.hpcf.cyi.ac.cy)
Funding
No funding scheme used for this work.
References
- 1. Ameratunga R, Woon S-T, Steele R, et al. Perspective: the nose and the stomach play a critical role in the NZACE2-Pātari*(modified ACE2) drug treatment project of SARS-CoV-2 infection. Expert Rev Clin Immunol 2021;17:553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karaman B, Sippl W. Computational drug repurposing: current trends. Curr Med Chem 2019;26:5389–409. [DOI] [PubMed] [Google Scholar]
- 3. Karatzas E, Minadakis G, Kolios G, et al. A web tool for ranking candidate drugs against a selected disease based on a combination of functional and structural criteria. Comput Struct Biotechnol J 2019;17:939–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomazou M, Bourdakou MM, Minadakis G, et al. Multi-omics data integration and network-based analysis drives a multiplex drug repurposing approach to a shortlist of candidate drugs against COVID-19. Brief Bioinform 2021;22:bbab114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiang AP, Butte AJ. Systematic evaluation of drug–disease relationships to identify leads for novel drug uses. Clin Pharmacol Ther 2009;86:507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rakshit H, Chatterjee P, Roy D. A bidirectional drug repositioning approach for Parkinson's disease through network-based inference. Biochem Biophys Res Commun 2015;457:280–7. [DOI] [PubMed] [Google Scholar]
- 7. Bai Q, Tan S, Xu T, et al. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 2020;22:bbaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu S, Bai Q, Li L, et al. Drug repositioning in drug discovery of T2DM and repositioning potential of antidiabetic agents. Comput Struct Biotechnol J 2022;20:2839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bai Q, Liu S, Tian Y, et al. Application advances of deep learning methods for de novo drug design and molecular dynamics simulation. Wiley Interdiscip Rev Comput Mol Sci 2022;12:e1581. [Google Scholar]
- 10. Liu H, Song Y, Guan J, et al. Inferring new indications for approved drugs via random walk on drug-disease heterogenous networks. BMC Bioinform 2016;17:269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma W, Zhang L, Zeng P, et al. An analysis of human microbe–disease associations. Brief Bioinform 2017;18:85–97. [DOI] [PubMed] [Google Scholar]
- 12. Kerrien S, Aranda B, Breuza L, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res 2012;40:D841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Durmuş Tekir S, Çakır T, Ardıç E, et al. PHISTO: pathogen–host interaction search tool. Bioinformatics 2013;29:1357–8. [DOI] [PubMed] [Google Scholar]
- 14. Guirimand T, Delmotte S, Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res 2015;43:D583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Federhen S. The NCBI taxonomy database. Nucleic Acids Res 2012;40:D136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chamberlain S, Szoecs E, Foster Z, et al. Taxize: taxonomic search and retrieval in R. F1000 Research, 2013;2:191. 10.12688/f1000research.2-191.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006;34:D668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jennison AV, Verma NK. Shigella flexneri infection: pathogenesis and vaccine development. FEMS Microbiol Rev 2004;28:43–58. [DOI] [PubMed] [Google Scholar]
- 19. Welch RA, Burland V, Plunkett G, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci 2002;99:17020–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao Z, Jaiswal MK, Deng PY, et al. Requirement of phospholipase C and protein kinase C in cholecystokinin-mediated facilitation of NMDA channel function and anxiety-like behavior. Hippocampus 2012;22:1438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horne EA, Dell'Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci 2007;27:3523–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cefai D, Debre P, Kaczorek M, et al. Human immunodeficiency virus-1 glycoproteins gp120 and gp160 specifically inhibit the CD3/T cell-antigen receptor phosphoinositide transduction pathway. J Clin Invest 1990;86:2117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mayne M, Holden CP, Nath A, et al. Release of calcium from inositol 1, 4, 5-trisphosphate receptor-regulated stores by HIV-1 Tat regulates TNF-α production in human macrophages. J Immunol 2000;164:6538–42. [DOI] [PubMed] [Google Scholar]
- 24. Brady ST, Siegel GJ, Albers RW, et al. Basic Neurochemistry: Principles of Molecular, Cellular, and Medical Neurobiology. Academic Press, 2012;i–iii1. [Google Scholar]
- 25. Perry SW, Hamilton JA, Tjoelker LW, et al. Platelet-activating factor receptor activation: an initiator step in HIV-1 neuropathogenesis. J Biol Chem 1998;273:17660–4. [DOI] [PubMed] [Google Scholar]
- 26. Lipton SA. Memantine prevents HIV coat protein-induced neuronal injury in vitro. Neurology 1992;42:1403–3. [DOI] [PubMed] [Google Scholar]
- 27. Schifitto G, Navia BA, Yiannoutsos CT, et al. Memantine and HIV-associated cognitive impairment: a neuropsychological and proton magnetic resonance spectroscopy study. AIDS 2007;21:1877–86. [DOI] [PubMed] [Google Scholar]
- 28. Zhao Y, Navia BA, Marra CM, et al. Memantine for AIDS dementia complex: open-label report of ACTG 301. HIV Clin Trials 2010;11:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schifitto G, Yiannoutsos CT, Simpson DM, et al. A placebo-controlled study of memantine for the treatment of human immunodeficiency virus–associated sensory neuropathy. J Neurovirol 2006;12:328–31. [DOI] [PubMed] [Google Scholar]
- 30. Morgan CR, Miglionico BV, Engen JR. Effects of HIV-1 Nef on human N-myristoyltransferase 1. Biochemistry 2011;50:3394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parang K, Wiebe LI, Knaus EE, et al. In vitro antiviral activities of myristic acid analogs against human immunodeficiency and hepatitis B viruses. Antiviral Res 1997;34:75–90. [DOI] [PubMed] [Google Scholar]
- 32. Pollard BS, Blanco JC, Pollard JR. Classical drug digitoxin inhibits influenza cytokine storm, with implications for COVID-19 therapy. In Vivo 2020;34:3723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood J Am Soc Hematol 2012;120:4621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rolf MG, Curwen JO, Veldman-Jones M, et al. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol Res Perspect 2015;3:e00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saha S, Halder AK, Bandyopadhyay SS, et al. Drug repurposing for COVID-19 using computational screening: is Fostamatinib/R406 a potential candidate? Methods 2022;203:564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kost-Alimova M, Sidhom E-H, Satyam A, et al. A high-content screen for mucin-1-reducing compounds identifies fostamatinib as a candidate for rapid repurposing for acute lung injury. Cell Rep Med 2020;1:100137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strich JR, Tian X, Samour M, et al. Fostamatinib for the treatment of hospitalized adults with coronavirus disease 2019: a randomized trial. Clin Infect Dis 2021;75:e491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Avendaño C, Menéndez J. Chapter 10 - Drugs That Inhibit Signaling Pathways for Tumor Cell Growth and Proliferation: Kinase Inhibitors. Medicinal Chemistry of Anticancer Drugs (Second Edition) 2015;391–491. [Google Scholar]
- 39. Chen R, Keating MJ, Gandhi V, et al. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood 2005;106:2513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perwitasari O, Yan X, O'Donnell J, et al. Repurposing kinase inhibitors as antiviral agents to control influenza A virus replication. Assay Drug Dev Technol 2015;13:638–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ali A, Ghosh A, Nathans RS, et al. Identification of flavopiridol analogues that selectively inhibit positive transcription elongation factor (P-TEFb) and block HIV-1 replication. Chembiochem 2009;10:2072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta P, Narayanan S, Yang D-H. Chapter 9 - CDK inhibitors as sensitizing agents for cancer chemotherapy. In: Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy. Academic Press, 2019;4:125–49. [Google Scholar]
- 43. Soni DV, Jacobberger JW. Inhibition of cdk1 by alsterpaullone and thioflavopiridol correlates with increased transit time from mid G2 through prophase. Cell Cycle 2004;3:347–55. [PubMed] [Google Scholar]
- 44. Guendel I, Agbottah ET, Kehn-Hall K, et al. Inhibition of human immunodeficiency virus type-1 by cdk inhibitors. AIDS Res Therapy 2010;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. García-Cárceles J, Caballero E, Gil C, et al. Kinase inhibitors as underexplored antiviral agents. J Med Chem 2021;65:935–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Savva K, Zachariou M, Oulas A, et al. Chapter 4 - Computational Drug Repurposing for Neurodegenerative Diseases. In Silico Drug Des. Academic Press. 2019;85–118. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Vir2Drug is available online through the following links: (https://bioinformatics.cing.ac.cy/vir2drug, https://vir2drug.cing-big.hpcf.cyi.ac.cy)