

Abstract
A method for the enantioselective, Lewis base-catalyzed sulfenofunctionalization of cyclic and (Z)-alkenes is reported. The intermediate thiiranium ion generated in the presence of a selenophosphoramide catalyst is intercepted by a variety of nucleophiles. A diverse array of inter- and intramolecular functionalizations proceed in high yield and good to high enantioselectivity (86:14–98:2 er). Prior experimental and computational studies indicated such enantiotopic face discrimination to be poor; however, the results disclosed herein remediate the previous findings. Control experiments were performed to investigate the different behavior of (Z)-alkenes and their more established (E)-counterparts.
Keywords: Lewis base, thiiranium, sulfenium, sulfenofunctionalization, organocatalysis
Graphical Abstract

INTRODUCTION
Functionalization of unactivated olefins via enantioselective group transfer is a powerful strategy for generating molecular complexity from simple building blocks. Among them, a variety of epoxidations,1 aziridinations,2 halo-,3 sulfeno-,4 and selenofunctionalizations5 have been reported. The methods of greatest utility have demonstrated excellent substrate recognition of various double-bond substitution patterns to functionalize alkenes with high levels of diastereo- and enantiocontrol. However, such processes are rare, and the catalysts often require engineered substrates bearing coordinating groups to achieve high enantioselectivity.
Sulfenium ion transfer to unactivated alkenes by enantioselective thiiranium ion formation, pioneered in these laboratories and expanded upon by other groups, has been successfully applied for the functionalization of unactivated olefins.4 Enantioenriched thiiranium intermediates generated from mono-, di-, and trisubstituted alkenes can be captured by a variety of nucleophiles including oxygen,6 nitrogen,7 and carbon8 moieties, acting in both an intra- and intermolecular fashion (Scheme 1).
Scheme 1.

Generalized Asymmetric Sulfenofunctionalizations of Alkenes
Despite great advances in recent years, reports of enantioselective sulfenylations of both (Z)- and (E)-configured 1,2-disubstituted alkenes using one catalyst are extremely rare. For example, Shi’s sulfenoamination protocol catalyzed by chiral phosphoric acids is well suited for alkyl-substituted (Z)-amino-alkenes to form corresponding pyrrolidines and piperidines with high enantiomeric ratios (89:11–93:7 er), whereas moderate enantioselectivities were obtained when (E)-substituted substrates were employed (72:28–85:15 er).7b In 2014, Zhao reported the enantioselective construction of trifluoromethylthiolated azaheterocycles in the presence of an indane-based, bifunctional, chiral selenide catalyst.7d High levels of enantiocontrol were achieved using alkyl- and aryl-substituted (E)-alkenes; however, no product was observed when a (Z)-alkene was employed, which was attributed to inherent reactivity and steric hindrance in the alkene.9 It is clearly shown in Figure 1 that regardless of the trajectory for sulfenium ion transfer to the alkene, cis-alkenes necessarily have unavoidable steric repulsions rendering a higher energy transition state.
Figure 1.
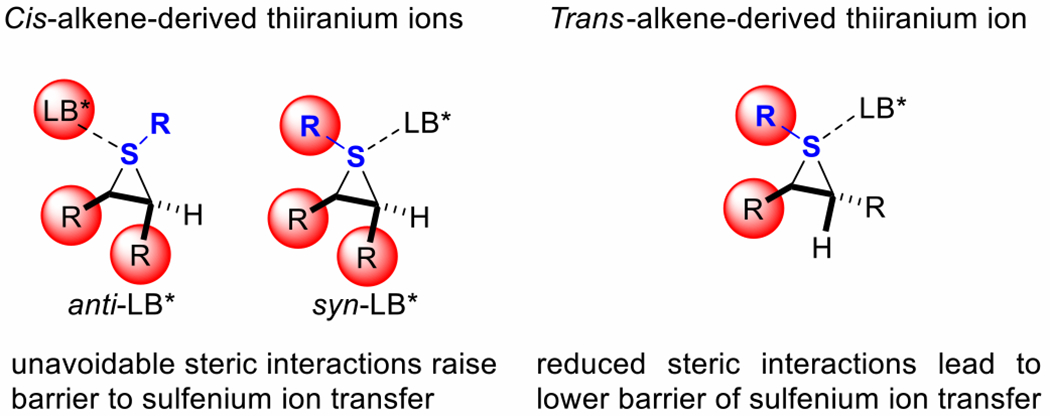
Steric interactions of thiiranium ions.
Indeed, our own efforts to engage (Z)-configured 1,2-disubstituted substrates have been plagued by reduced reactivity and moderate or poor enantioselectivity. For example, in the seminal report from these laboratories on the enantioselective sulfenylation of simple alkenes, the enantiocontrol with substrate (E)-3 (96:4 er) was far superior to the corresponding Z-analogue (54:46 er) (Scheme 2A).6a Later in 2014, another attempt to employ aryl-substituted (Z)-6 in an intramolecular sulfenoamination resulted in reduced reactivity (68% yield), poor endo/exo-selectivity, and weak enantioinduction (63:37 er) (Scheme 2B).7a Several additional reports from these laboratories have shown a similar trend. These failures were explained owing to the topology of the BINAM-derived selenophosphoramide Lewis base catalyst and the corresponding active species using a diagonally symmetric quadrant model of occupied and unoccupied space.10 In addition, the presence of the bulky Lewis base makes both thiiranium ion intermediates stemming from the (Z)-alkene (syn and anti) more sterically hindered than its (E)-counterpart, disfavoring reactions with (Z)-alkenes.10b,11
Scheme 2.

Sulfenofunctionalization of 1,2-Disubstituted (E)- vs (Z)-Alkenes
Hence, engaging both (Z)- and (E)-alkenes for sulfeno-functionalization reactions in a highly enantioselective fashion using a single catalyst remains a challenge and is addressed in this report.
RESULTS AND DISCUSSION
To investigate the reactivity of (Z)-alkenes, a general substrate that rules out the influence of coordinating groups (e.g., −OH or −NHR) on enantioselectivity was selected for the model reaction. On the basis of our recent report on the intermolecular sulfenoamination of alkenes,12 the combination of (Z)-β-methylstyrene 9 and 4-bromoaniline was well suited for the catalyst survey (Table 1). All reactions were performed in 1,1,1,3,3,3-hexafluoroisopropyl alcohol (HFIP) using 2,6-diisopropylphenyl sulfenylating agent 10, which affords higher enantioselectivities compared to its less bulky analogues.10
Table 1.

|
All reactions performed on a 0.05 mmol scale.
Yields determined by 1H NMR spectroscopic analysis using 1,1,1,2-tetrachloroethane as an internal standard.
Enantiomeric ratio determined by CSP-HPLC analysis.
ND = not determined.
Orienting experiments to identify (Z)-alkene-selective Lewis bases began with bisimidazoline-derived selenophosphoramides, which previously showed promising enantioselectivity in bromocycloetherification.13 However, in this case, very low reactivity was observed using catalyst 12, with most of the starting materials returned unreacted. Because tetrahydrothiophene is a privileged Lewis base for sulfenium ion transfer, a chiral analogue was examined. Inspired by the desymmetrization of alkenoic diols with chiral tetrahydrothiophene 1314 on 2,2-disubstituted alkenes, it was synthesized and tested in its efficiency on the model reaction. Unfortunately, Lewis base 13 afforded no enantioinduction and product formation was very slow. Aggarwal’s isothiocineole 1415 applied in sulfur-ylide-mediated epoxidations and aziridinations resulted in good reactivity (76% yield); however, minimal levels of enantiocontrol were obtained (54:46 er). Dithiin 15 is reported by Pasquato and co-workers to generate sulfonium salts from MeSCl and SbCl5 followed by stoichiometric delivery of the sulfenium ion to (E)-3-hexene with good selectivity.16 However, catalyst 15 was insoluble in HFIP and resulted in no reactivity. Good yield but moderate levels of enantioselectivity were observed using BINAS-based disulfide 1617 (66:34 er). BINOL-derived bifunctional Lewis base 17 bearing a free hydroxy group inspired by Shirakawa for applications in enantioselective bromolactonizations18 afforded high conversion to the desired product, albeit in a racemic fashion.
Owing to the lack of significant enantioinduction, it was decided to benchmark these results against catalysts previously developed in this group. Selenophosphoramidate (R)-18 and selenophosphoramide (R)-2b were subjected to the model reaction conditions. Whereas (R)-18 afforded promising enantioselectivity (89:11 er), much to our surprise, using BINAM-derived (R)-2b, sulfenoamination product 11a was isolated in 90% yield and 97:3 er. Importantly, comparable yield (80%) and selectivity (98:2 er) were obtained for the sulfenoamination of (E)-β-methylstyrene with 4-bromoaniline under similar reaction conditions.12 The high enantioselectivity observed contradicted what has been established over the last 10 years, namely, (Z)-alkenes were not well recognized by these catalyst scaffolds.5a,6a With this unexpected result in hand, the generality of catalyst (R)-2b using a variety of (Z)-alkenes in both inter- and intramolecular sulfenofunctionalization reactions was investigated using 4-chloroaniline as the model nucleophile (Table 2).
Table 2.

|
All reactions performed on a 1.00 mmol scale.
Yield of isolated, analytically pure product.
Diastereomeric ratio determined by 1H NMR spectroscopic analysis of the crude reaction mixture. Unless otherwise noted, the dr of the products is >99:1.
Enantiomeric ratio determined by CSP-HPLC analysis.
Enantiomeric ratio determined on the dinitrobenzoyl derivative.
Obtained as a mixture of constitutional isomers.
The influence of steric bulk on the sulfenium ion transfer and the electronic effects were explored by changing the substituents near the alkene fragment. No profound steric effect was observed in the presence of an ortho-methyl group such that the corresponding sulfenyl amine 11b was formed in good yield and enantiocontrol. In the case of an electron-withdrawing CF3-group, the reactivity was maintained, and desired product 11c was isolated in synthetically useful yield (78%) and enantioselectivity (98:2 er). Having a methoxy group in the 4-position resulted in the formation of 11d as a diastereomeric mixture (64:36 dr) favoring the formation of the anti-product. Both diastereoisomers were obtained with high enantiocontrol (96:4 and 98:2 er). This reaction likely proceeded through an open carbocation intermediate stabilized by the strongly electron-donating nature of the methoxy group. Preferential formation of anti-11d is attributed to relieving A1,3-strain of the methyl group with the aromatic ring prior to interception by the nucleophile. In addition, (Z)-β-ethylstyrene reacted with exceptional yield and enantioselectivity (11e), while (Z)-β-isopropylstyrene had a precipitous drop in conversion but retained satisfactorily high enantiocontrol (11f).
Cyclic alkenes were also explored since the challenge of enantioselective sulfenium ion transfer to an internal double bond is well known.7b Gratifyingly, the reaction of indene with 4-chloroaniline gave 11g in excellent yield and enantioselectivity (Table 2). It is worth noting that the indane-based chiral bifunctional sulfide catalyst, developed by Zhao and co-workers for asymmetric sulfenofunctionalization,6c–f,7c,d,8a,b can be accessed through this process in a single synthetic step. 1,2-Dihydronaphthalene behaved similarly to indene to afford near-quantitative conversion to highly enantioenriched product 11h. 2H-Chromene analogues represent a privileged class of substrates for medicinal chemistry19 and chromene derivative 11i was formed as a single diastereomer albeit with reduced enantioselectivity (86:14 er). The exact reason for the stereochemical erosion using this substrate is yet to be determined.
The scope of the reaction was extended by surveying the nucleophilic partners, for which (Z)-β-methylstyrene was chosen as the model alkene (Table 2). It is important to note that a judicious selection of nucleophiles allowed the suppression of a competitive opening of the thiiranium ion by HFIP.20 In general, to avoid the oxysulfenylated product arising from solvent capture, the amine nucleophile must not be basic enough to deprotonate HFIP, thus rendering it inactive, but nucleophilic enough to perform the desired transformation. In addition to anilines, the reactivity of secondary and primary amines was examined. Morpholine and piperazine are abundantly used as substituents or scaffolds in drugs and medicinal chemistry owing to their positive impact on the ADME properties.21 Accordingly, products 11k and 11l were obtained in good yield (~80%) and with consistently high enantioselectivity (95:5 and 94:6 er, respectively). Although tosylamide could not outcompete HFIP, the more acidic triflamide was a competent nucleophile to furnish 11m in 69% yield and 90:10 er. To establish the absolute stereochemical course for the reactions of (Z)-alkenes, the full stereostructure of 11m was determined by X-ray crystallographic analysis and was shown to be R,R.22 This outcome comports with the observed stereochemical pathway for (E)-alkenes using the same catalyst. The scope was further extended to primary amines and other anilines, with benzyl-amine providing 11n in good yield and enantioselectivity, whereas p-anisidine (11o) and 2-fluoroaniline (11p) afford the sulfenoamination products in near-quantitative yields. Next, oxygen nucleophiles were explored. 4-Nitrophenol afforded highly enantioenriched product 11q in 96% yield. Incorporation of oxygen using TMSOAc resulted in the desired product formation (11r) with good enantiocontrol (90:10 er). Finally, benzoic acid was also competent leading to the formation of benzoate 11s in 96% yield and 93:7 er. It is notable that both carboxylate nucleophiles suffered from poor selectivity between the α and β-positions of the styrene. It is posited that the initial attack of the nucleophile proceeded kinetically at the α-position but reformation of the thiiranium-ion with concomitant ejection of the nucleophile followed by recapture allowed an equilibration event to happen during the course of the reaction.
The reaction scope was further evaluated by probing (Z)-alkenes of different chain lengths bearing various nucleophilic moieties that can engage in intramolecular sulfenofunctionalizations and deliver useful heterocyclic motifs (Table 3). Phenyl-substituted alcohol 19a afforded corresponding ether 20a in 63% yield and 95:5 er. In addition, pyrrolidine 20b was obtained in a highly enantioenriched form (entry 2). As expected, lactonization of 4-methoxyphenyl-substituted carboxylic acid 19c proceeded through an open carbocation, which resulted in a mixture of diastereomers (20c), albeit in an improved ratio (80:20 dr) compared to the 4-methoxy-substituted substrate in the intermolecular sulfenofunctionalization (64:36 dr, Table 2). Both isomers were obtained in excellent yields and enantioselectivities (entry 3). Naphthyl-substituted lactone 20d derived from acid 19d was furnished with high efficiency (entry 4). Pyran 20e was formed as the exclusive product with no loss of enantioinduction. Finally, amide 19f affords oxazoline 20f with exclusive exo-selectivity albeit with extended reaction times owing to the reduced nucleophilicity of the double bond and perhaps solvation of the amide oxygen nucleophile. Starting material 19f remained after 48 h.
Table 3.

|
All reactions performed on a 1.00 mmol scale.
Yield of isolated, analytically pure product.
Diastereomeric ratio determined by 1H NMR spectroscopic analysis of the crude reaction mixture. Unless otherwise noted, the dr of the products is >99:1.
Enantiomeric ratio determined by CSP-HPLC analysis.
Enantiomeric ratio determined for major diastereomer.
Enantiomeric ratio determined on the corresponding sulfone.
Notably, all cyclizations of Z-alkenes led to the predominant formation of the kinetic exo-product as expected unless that product would be a four-membered ring.23 The rationale for this phenomenon is depicted in Scheme 3A whereby the endo transition state for (Z)-alkenes necessarily has a destabilizing 1,3-diaxial interaction that is not observed in the exo-transition state. A head-to-head comparison is provided to demonstrate this effect on two nearly identical substrates, which vary only by alkene geometry (Scheme 3B). The trans-alkene affords the endo-cyclization product (pyran) exclusively both as the thermodynamic and the kinetic product. This sharply contrasts the cis-alkene that affords roughly a 4:1 mixture of the kinetic furan product as the major component. In light of the putative 1,3-diaxial interaction that favors exo-cyclization of cis-alkenes, a tertiary alcohol bearing a geminal dimethyl group was synthesized and subjected to standard reaction conditions (Scheme 3C) to examine how an increase in diaxial strain may improve endo/exo-selectivity. Exclusive formation of the furan provides further evidence for the role of nonbonding interactions governing the transition state, which leads to either five- or six-membered ring formation.
Scheme 3. Rationale for exo-Selective Cyclization of (Z)-Alkenesa.

aAll reactions performed on a 0.1 mmol scale. Yields in parentheses determined by 1H NMR analysis of the crude reaction mixture using 1,1,2,2-tetrachloroethane as the internal standard. *Enantiomeric ratio determined on the corresponding sulfone.
The unexpected effectiveness of the BINAM-derived selenophosphoramide catalyst in enantiotopic face discrimination of (Z)-alkenes motivated a study to better understand the basis for its selectivity. Two key changes have been implemented since the inception of these research programs: the first is changing to a bulkier sulfenylating agent, which affords better selectivity, and the second is changing the solvent from CH2Cl2 to HFIP. To further examine how these factors may have conspired to afford such unexpected results, a series of studies were performed modulating the solvent and substrate. Accordingly, the (E)-isomer of 19a was prepared and subjected to cyclization conditions in both HFIP and CH2Cl2. As expected, trans-20a was produced with excellent enantioselectivity irrespective of solvent (Table 4). Furthermore, the same cyclization with (Z)-19a in dichloromethane afforded similar results as with HFIP. These preliminary findings suggest that solvent plays little role in the enantiodetermining step nor offers viable racemization pathways.10b Furthermore, the significant increase in enantioinduction between the smaller –SPh sulfenylating agent 1 versus the bulkier agent 10 is noteworthy. For styryl alkenes, sulfenylating agent 1 afforded a 63:37 er with an amine nucleophile (Scheme 2b), whereas sulfenylating agent 10 afforded greater than 95:5 er for all cyclization products (Tables 3 and 4).
Table 4.

| ||||
|---|---|---|---|---|
| Entry | Substrate | Conditions | Product | e.r. |
| 1 |

|
HFIP | trans-20a | 97:3 |
|
|
|
|||
| 2 | CH2Cl2 MsOH (0.4 equiv) |
trans-20a | 97:3 | |
|
| ||||
| 3 |

|
HFIP | cis-20a | 98:2 |
|
|
|
|||
| 4 | CH2Cl2 MsOH (0.4 equiv) |
cis-20a | 97:3 | |
All reactions performed on a 0.10 mmol scale.
Enantiomeric ratio determined by CSP-HPLC analysis.
The focus was then directed toward the nature of the alkene and differentiation between similar methylene substituents. Substrate (E)-3 was synthesized and cyclized to give a mixture of furan and pyran constitutional isomers but with consistently high enantioselectivity (Table 5). Curiously, a different product distribution was obtained with the more strongly ionizing MsOH component through a thermodynamic equilibration process to afford greater amounts of trans-21b than with HFIP. However, when (Z)-3 was cyclized, only weak enantioinduction was observed in either solvent albeit with slightly higher levels in HFIP. Taken together, these results indicate that (R)-2b is capable of discriminating both (Z)- and (E)-styrenes as well as (E)-dialkyl-alkenes but not (Z)-dialkyl-alkenes. The bulkier sulfenylating agent was instrumental in achieving high selectivities compared to previously employed N-phenylthiophthalimide 1. In view of the result shown in Scheme 2A for the cyclization of 3, it is clear that the combination of selenophosphoramide catalyst 2b and sulfenylating agent 10 confers a moderate increase in enantiotopic face discrimination. Similar reactivity trends for constitutional selectivity remain constant, however, namely, exo-cyclization of (Z)-alkenes is preferred in the absence of any activating groups.
Table 5.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conditions | Selectivity (a:b) | Product | e.r. |
| 1 |

|
HFIP | 1.3:1 | anti-21a + trans-21b | (6)c = 98:2 (5) = 98:2 |
|
|
|
||||
| 2 | CH2Cl2 MsOH (0.4 equiv) |
1:2 | anti-21a + trans-21b | (6)c = 98:2 (5) = 98:2 |
|
|
| |||||
| 3 |

|
HFIP | >20:1 | syn-21a + cis-21b | 71:29d |
|
|
|
||||
| 4 | CH2Cl2 MsOH (0.4 equiv) |
>20:1 | syn-21a + cis-21b | 67:33d | |
All reactions performed on a 0.10 mmol scale.
Enantiomeric ratio determined by CSP-HPLC analysis.
Enantiomeric ratio determined on corresponding sulfone.
Enantiomeric ratio determined for syn-21a.
Control studies were carried out next to demonstrate the difference in reactivity between (Z)- and (E)-alkenes for sulfenium ion capture (Scheme 4). A competition experiment between (Z)- and (E)-β-methylstyrene using one equivalent of sulfenylating agent 10 and 4-bromoaniline was performed. After only 30 min, full consumption of 4-bromoaniline was observed.1H NMR analysis of the crude reaction mixture revealed a 72:28 ratio of the anti/syn products, demonstrating that (E)-β-methylstyrene reacted about 2.5 times faster. This result is consistent with the analysis of steric hindrance in the corresponding (Z)-alkene-derived thiiranium ion intermediate discussed above.
Scheme 4.

Competition Experiment between (E)- and (Z)-β-Methylstyrene
CONCLUSIONS
In conclusion, enantioselective sulfenofunctionalization of (Z)- and cyclic alkenes has been described. Despite the challenge of the reduced reactivity and steric congestion of these substrates, the BINAM-derived selenophosphoramide catalyst was able to engage a variety of substrates in inter- and intramolecular sulfenofunctionalization. Comparable levels of reactivity and selectivity were achieved as previously demonstrated for more established (E)-analogues. The development of a diverse scope as well as a better understanding of the difference between (E)- and (Z)-configured double bonds has been accomplished.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to the National Institutes of Health (GM R35 127010) for generous financial support. They also thank the UIUC SCS support facilities (X-ray, microanalysis, mass spectrometry, and NMR spectroscopy) for their assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c01232.
Experimental procedures and characterization data for all new compounds along with copies of NMR spectra and CSP-HPLC chromatograms (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.2c01232
The authors declare no competing financial interest.
Contributor Information
Anastassia Matviitsuk, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States.
Jesse Lee Panger, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States.
Scott E. Denmark, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States
REFERENCES
- (1).For a review on organocatalytic, enantioselective epoxidation of alkenes see:; (a) Zhu Y; Wang Q; Cornwall RG; Shi Y Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem. Rev 2014, 114, 8199–8256. [DOI] [PubMed] [Google Scholar]; For relevant book chapters on epoxidation see:; (b) Katsuki T; Martin VS Asymmetric Epoxidation of Allylic Alcohols: The Katuski-Sharpless Epoxidation Reaction. Org. React 1996, 48, 1–208, Chapter 1. [Google Scholar]; Katsuki T Catalytic Asymmetric Synthesis, 2nd ed.; Ojima I, Ed.; Wiley-VCH, 2000, Chapter 6B. [Google Scholar]
- (2).For relevant book chapters on aziridination see:; (a) Muchalski H; Johnston JN Science of Synthesis, de Vries JG, Ed.; Georg Thieme Velag KG, 2011; Vol. 1, pp 155–184. [Google Scholar]; (b) Mossner C; Bolm C Transition Metals for Organic Synthesis, 2nd ed.; Beller M; Bolm C, Eds.; Wiley-WCH Verlag: Weinheim, 2004; pp 389–402. [Google Scholar]
- (3).For a review on catalytic, asymmetric halofunctionalization of alkenes see:; Denmark SE; Kuester WE; Burk MT Catalytic, Asymmetric Halofunctionalization of Alkenes–A Critical Perspective. Angew. Chem., Int. Ed 2012, 51, 10938–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For a review on catalytic, asymmetric sulfenofunctionalizations of alkenes see:; Matviitsuk A; Panger JL; Denmark SE Catalytic, Enantioselective Sulfenofunctionalization of Alkenes: Development and Recent Advances. Angew. Chem., Int. Ed 2020, 59, 19796–19819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Denmark SE; Collins WR Lewis Base Activation of Lewis Acids: Development of a Lewis Base Catalyzed Selenolactonization. Org. Lett 2007, 9, 3801–3804. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE; Kalyani D; Collins WR Preparative and Mechanistic Studies toward the Rational Development of Catalytic, Enantioselective, Selenoetherification Reactions. J. Am. Chem. Soc 2010, 132, 15752–15765. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wei Q; Wang Y-Y; Du Y-L; Gong L-Z Organocatalytic Asymmetric Selenofunctionalization of Tryptamine for the Synthesis of Hexahydropyrrolo[2,3-b]indole Derivatives. Beilstein J. Org. Chem 2013, 9, 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang H; Lin S; Jacobsen EN Enantioselective Selenocyclization via Dynamic Kinetic Resolution of Seleniranium Ions by Hydrogen-Bond Donor Catalysts. J. Am. Chem. Soc 2014, 136, 16485–16488. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Niu W; Yeung Y-Y Catalytic and Highly Enantioselective Selenolactonization. Org. Lett 2015, 17, 1660–1663. [DOI] [PubMed] [Google Scholar]; (f) See JY; Yang H; Zhao Y; Wong MW; Ke Z; Yeung Y-Y Desymmetrizing Enantio- and Diastereoselective Selenoetherification through Supramolecular Catalysis. ACS Catal. 2018, 8, 850–858. [Google Scholar]
- (6).(a) Denmark SE; Kornfilt DJP; Vogler T Catalytic Asymmetric Thiofunctionalization of Unactivated Alkenes. J. Am. Chem. Soc 2011, 133, 15308–15311. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guan H; Wang H; Huang D; Shi Y Enantioselective Oxysulfenylation and Oxy-selenenylation of Olefins Catalyzed by Chiral Brønsted Acids. Tetrahedron 2012, 68, 2728–2735. [Google Scholar]; (c) Liu X; An R; Zhang X; Luo J; Zhao X Enantioselective Trifluoromethylthiolating Lactonization Catalyzed by and Indane-Based Chiral Sulfide. Angew. Chem., Int. Ed 2016, 55, 5846–5850. [DOI] [PubMed] [Google Scholar]; (d) Liu X; Liang Y; Ji J; Luo J; Zhao X Chiral Selenide-Catalyzed Enantioselective Allylic Reaction and Intermolecular Difunctionalization of Alkenes: Efficient Construction of C-SCF3 Stereogenic Molecules. J. Am. Chem. Soc 2018, 140, 4782–4786. [DOI] [PubMed] [Google Scholar]; (e) Liang Y; Zhao X Enantioselective Construction of Chiral Sulfides via Catalytic Electrophilic Azidothiolation and Oxythiolation of N-Allyl Sulfonamides. ACS Catal 2019, 9, 6896–6902. [Google Scholar]; (f) Qin T; Jiang Q; Ji J; Luo J; Zhao X Chiral Selenide-Catalyzed Enantioselective Synthesis of Trifluoromethylthiolated 2,5-Disubstituted Oxazolines. Org. Biomol. Chem 2019, 17, 1763–1766. [DOI] [PubMed] [Google Scholar]; (g) Luo H-Y; Xie Y-Y; Song X-F; Dong J-W; Zhu D; Chen Z-M Lewis Base-Catalyzed Asymmetric Sulfenylation of Alkenes: Construction of Sulfenylated Lactones and Application to the Formal Syntheses of (−)-Nicotlactone B and (−)-Galbacin. Chem. Commun 2019, 55, 9367–9370. [DOI] [PubMed] [Google Scholar]; (h) Luo H-Y; Dong J-W; Xie YY; Song X-F; Zhu D; Ding T; Liu Y; Chen Z-M Lewis Base/Brønsted Acid Co-Catalyzed Asymmetric Thiolation of Alkenes with Acid-Controlled Divergent Regioselectivity. Chem.-Eur. J 2019, 25, 15411–15418. [DOI] [PubMed] [Google Scholar]; (i) Matviitsuk A; Denmark SE Enantio- and Diastereoselective, Lewis Base-Catalyzed, Cascade Sulfenoacetalization of Alkenyl Aldehydes. Angew. Chem., Int. Ed 2019, 58, 12486–12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Denmark SE; Chi HM Lewis Base Catalyzed, Enantioselective, Intramolecular Sulfenoamination of Olefins. J. Am. Chem. Soc 2014, 136, 8915–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li L; Li Z; Huang D; Wang H; Shi Y Chiral Phosphoric Acid Catalyzed Enantioselective Sulfamination of Amino-Alkenes. RSC Adv. 2013, 3, 4523–4525. [Google Scholar]; (c) Luo J; Liu X; Zhao X Development of Chalcogenide Catalysts towards Trifluoromethylthiolation. Synlett 2017, 28, 397–401. [Google Scholar]; (d) Luo J; Liu Y; Zhao X Chiral Selenide-Catalyzed Enantioselective Construction of Saturated Trifluoromethylthiolated Azaheterocycles. Org. Lett 2017, 19, 3434–3437. [DOI] [PubMed] [Google Scholar]
- (8).(a) Luo J; Cao Q; Cao X; Zhao X Selenide-Catalyzed Enantioselective Synthesis of Trifluoromethylthiolated Tetrahydronaphthalenes by Merging Desymmetrization and Trifluoromethylthiolation. Nat. Commun 2018, 9, No. 527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang Y; Ji J; Zhang X; Jiang Q; Luo J; Zhao X Enantioselective Construction of Axially Chiral Amino Sulfide Vinyl Arenes by Chiral Sulfide-Catalyzed Electrophilic Carbothiolation of Alkynes. Angew. Chem., Int. Ed 2020, 59, 4959–4964. [DOI] [PubMed] [Google Scholar]; (c) Wang J-J; Yang H; Gou B-B; Zhou L; Chen J Enantioselective Organocatalytic Sulfenylation of β-Naphthols. J. Org. Chem 2018, 83, 4730–4738. [DOI] [PubMed] [Google Scholar]; (d) Song X-F; Ye A-H; Xie Y-Y; Dong J-W; Chen C; Zhang Y; Chen Z-M Lewis-Acid-Mediated Thiocyano Semipinacol Rearrangement of Allylic Alcohols for Construction of α-Quaternary Center β-Thiocyano Carbonyls. Org. Lett 2019, 21, 9550–9554. [DOI] [PubMed] [Google Scholar]; (e) Xie Y-Y; Chen Z-M; Luo HY; Shao H; Tu Y-Q; Bao X; Cao R-F; Zhang S-Y; Tian J-M Lewis Base/Brønsted Acid Co-Catalyzed Enantioselective Sulfenylation Semipinacol Rearrangement of Di- and Trisubstituted Allylic Alcohols. Angew. Chem., Int. Ed 2019, 58, 12491–12496. [DOI] [PubMed] [Google Scholar]
- (9).Ashtekar KD; Marzijarani NS; Jaganathan A; Holmes D; Jackson JE; Borhan B A New Tool to Guide Halofunctionalization Reactions: The Halenium Affinity (HalA) Scale. J. Am. Chem. Soc 2014, 136, 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Denmark SE; Hartmann E; Kornfilt DJP; Wang H Mechanistic, Crystallographic, and Computational Studies on the Catalytic, Enantioselective Sulfenofunctionalization of Alkenes. Nat. Chem 2014, 6, 1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hartmann E; Denmark SE Structural, Mechanistic, Spectroscopic, and Preparative Studies on the Lewis Base Catalyzed, Enantioselective Sulfenofunctionalization of Alkenes. Helv. Chim. Acta 2017, 100, No. e1700158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Huisgen R Kinetics and Mechanism of 1,3-Dipolar Cyclo-additions. Angew. Chem., Int. Ed 1963, 2, 633–645. [Google Scholar]
- (12).Roth A; Denmark SE Enantioselective, Lewis Base-Catalyzed, Intermolecular Sulfenoamination of Alkenes. J. Am. Chem. Soc 2019, 141, 13767–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Böse D; Denmark SE Investigating the Enantiodetermining Step of a Chiral Lewis Base Catalyzed Bromocycloetherification of Privileged Alkenes. Synlett 2018, 29, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ke Z; Tan CK; Chen F; Yeung Y-Y Catalytic Asymmetric Bromoetherification and Desymmetrization of Olefinic 1,3-Diols with C2-Symmetric Sulfides. J. Am. Chem. Soc 2014, 136, 5627–5630. [DOI] [PubMed] [Google Scholar]
- (15).Illa O; Namutebi M; Saha C; Ostovar M; Chen CC; Haddow MF; Nocquet-Thibault S; Lusi M; McGarrigle EM; Aggarwal VK Practical and Highly Selective Sulfur Ylide-Mediated Asymmetric Epoxidations and Aziridinations Using a Cheap and Readily Available Chiral Sulfide: Extensive Studies to Map Out Scope, Limitations, and Rationalization of Diastereo- and Enantioselectivities. J. Am. Chem. Soc 2013, 135, 11951–11966. [DOI] [PubMed] [Google Scholar]
- (16).Lucchini V; Modena G; Pasquato L Enantiopure Thiosulfonium Salts in Asymmetric Synthesis. Face Selectivity in Electrophilic Additions to Unfunctionalised Olefins. J. Chem. Soc., Chem. Commun 1994, 1565–1566. [Google Scholar]
- (17).Fabbri D; Delogu G; De Lucci O Preparation of Enantiomerically Pure 1,1′-Binaphthalene-2,2′-diol and 1,1′-Binaphthalene-2,2′-dithiol. J. Org. Chem 1993, 58, 1748–1750. [Google Scholar]
- (18).Okada M; Kaneko K; Yamanaka M; Shirakawa S BINOL-derived Bifunctional Sulfide Catalysts for Asymmetric Synthesis of 3,3-Disubstituted Phthalides via Bromolactonization. Org. Biomol. Chem 2019, 17, 3747–3751. [DOI] [PubMed] [Google Scholar]
- (19).(a) Weston AH In the Pharmacology of Antihypertensive Therapeutics: Canter D; Mulrow PJ Eds.; Springer-Verlag: New York, 1989; 643. [Google Scholar]; (b) Ashwood VA; Buckingham RE; Cassidy F; Evans JM; Raruk EA; Hamilton TC; Nash DJ; Stemp G; Willcocks K Synthesis and Antihypertensive Activity of 4-(Cyclic Amido)-2H-1-Benzopyrans. J. Med. Chem 1986, 29, 2194–2201. [DOI] [PubMed] [Google Scholar]
- (20).Nucleophiles that have a pKaH higher than the pKa of HFIP (9.3) favor capture of the thiiranium ion intermediate by HFIP due to its partial or full deprotonation. In addition, stoichiometric amount of alcohols could not outcompete HFIP used in solvent quantities.
- (21).Al-Ghorbani M; Bushra BA; Zabiulla BBA; Mamatha SV; Khanum SA Piperazine and Morpholine: Synthetic Preview and Pharmaceutical Applications. Res. J. Pharm. Technol 2015, 8, 611–628. [Google Scholar]
- (22).CCDC 2020665 contains the crystallographic data for compound 11m. These data can be obtained free of charge from the Cambridge Crystallographic Data Center (www.ccdc.cam.ac.uk).
- (23).(a) Burk MT; Denmark SE Enantioselective Bromocycloetherification by Lewis Base/Chiral Brønsted Acid Cooperative Catalysis. Org. Lett 2012, 14, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ting PCj Bartlett, P. A. Stereocontrolled Synthesis of Trans-2,5-Disubstituted Tetrahydrofurans. J. Am. Chem. Soc 1984, 106, 2668–2671. [Google Scholar]; (c) Parker KA; O’Fee R Halonium-Initiated Cyclization of Allylic Urethanes: Stereo— and Regioselectivity in Functionalizing the Olefinic Bond. J. Am. Chem. Soc 1983, 105, 655–656. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.