

Abstract
Background
Metabolic syndrome is characterized by insulin resistance, which impairs intracellular signaling pathways and endothelial NO bioactivity, leading to cardiovascular complications. Extracellular signal‐regulated kinase (ERK) is a major component of insulin signaling cascades that can be activated by many vasoactive peptides, hormones, and cytokines that are elevated in metabolic syndrome. The aim of this study was to clarify the role of endothelial ERK2 in vivo on NO bioactivity and insulin resistance in a mouse model of metabolic syndrome.
Methods and Results
Control and endothelial‐specific ERK2 knockout mice were fed a high‐fat/high‐sucrose diet (HFHSD) for 24 weeks. Systolic blood pressure, endothelial function, and glucose metabolism were investigated. Systolic blood pressure was lowered with increased NO products and decreased thromboxane A2/prostanoid (TP) products in HFHSD‐fed ERK2 knockout mice, and Nω‐nitro‐l‐arginine methyl ester (L‐NAME) increased it to the levels observed in HFHSD‐fed controls. Acetylcholine‐induced relaxation of aortic rings was increased, and aortic superoxide level was lowered in HFHSD‐fed ERK2 knockout mice. S18886, an antagonist of the TP receptor, improved endothelial function and decreased superoxide level only in the rings from HFHSD‐fed controls. Glucose intolerance and the impaired insulin sensitivity were blunted in HFHSD‐fed ERK2 knockout mice without changes in body weight. In vivo, S18886 improved endothelial dysfunction, systolic blood pressure, fasting serum glucose and insulin levels, and suppressed nonalcoholic fatty liver disease scores only in HFHSD‐fed controls.
Conclusions
Endothelial ERK2 increased superoxide level and decreased NO bioactivity, resulting in the deterioration of endothelial function, insulin resistance, and steatohepatitis, which were improved by a TP receptor antagonist, in a mouse model of metabolic syndrome.
Keywords: endothelium, extracellular signal‐regulated kinase 2, hypertension, insulin resistance, metabolic syndrome
Subject Categories: Endothelium/Vascular Type/Nitric Oxide, Oxidant Stress, Vascular Biology, Hypertension
Nonstandard Abbreviations and Acronyms
- EE2KO
endothelial‐specific extracellular signal‐regulated kinase 2 knockout
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular signal‐regulated kinase
- ET1
endothelin 1
- HFHSD
high‐fat/high‐sucrose diet
- HOMA‐IR
homeostatic model assessment of insulin resistance
- ipGTT
intraperitoneal glucose tolerance test
- ITT
insulin tolerance test
- L‐NAME
Nω‐nitro‐l‐arginine methyl ester
- MEK
mitogen‐activated protein kinase kinase
- MetS
metabolic syndrome
- ND
normal diet
- PI3K
phosphatidylinositol‐3 kinase
- SBP
systolic blood pressure
- TP
thromboxane A2/prostanoid receptor
- TXA2
thromboxane A2
Clinical Perspective
What Is New?
This is the first study to clarify the role of endothelial ERK2 (extracellular signal‐regulated kinase 2) on systolic blood pressure and insulin resistance in a mouse model of metabolic syndrome in vivo.
What Are the Clinical Implications?
The present study indicates that a TP (thromboxane A2/prostanoid receptor) antagonist could be a therapeutic target for metabolic syndrome, which increases long‐term risk of cardiovascular events and death.
Metabolic syndrome (MetS) and type II diabetes are obesity‐associated diseases and well‐known risk factors for cardiovascular disease characterized by endothelial dysfunction and insulin resistance. 1 , 2 , 3 , 4 The elevation of blood pressure in MetS was associated with reduced NO bioactivity and increased production of superoxide, leading to endothelial dysfunction. 5 , 6 MetS induces steatohepatitis, which is closely associated with insulin resistance and also regarded as a risk for cardiovascular diseases. 7 The mechanical relationship between elevated systolic blood pressure (SBP) with endothelial dysfunction and insulin resistance has not yet been fully clarified, and there are no specific treatments targeted to vascular insulin resistance in MetS. 8
We have previously reported that insulin resistance in the MetS‐type II diabetes model influences the feed‐forward cycle, which exacerbates steatosis and endothelial function. 9 Although insulin receptor substrate/phosphatidylinositol‐3 kinase (PI3K)/protein kinase B (AKT) pathway supports NO biogenesis, resulting in vasorelaxation, the rat sarcoma (Ras)/rapidly accelerated fibrosarcoma (Raf)/mitogen‐activated protein kinase (MEK)/extracellular signal‐regulated kinase (ERK) pathway produces vasoconstrictive factors such as endothelin 1 (ET1), prostanoids, and superoxide in the endothelium. 10 , 11 , 12 In MetS with insulin resistance, the PI3K/AKT/endothelial nitric oxide synthase (eNOS)/NO pathway is impaired, and the Ras/Raf/MEK/ERK pathway is activated by inflammation and reactive oxygen species, 13 , 14 resulting in vasoconstraction. 15 , 16 , 17 This might be, in part, the mechanism that obesity/insulin resistance increases blood pressure. 10 , 18 , 19 Moreover, the activation of ERK2, with various agonists or cytokines, induces the production of vasocontraction factors in cultured endothelial cells. 11 , 12 , 20 However, the role of endothelial ERK2 in MetS in vivo remains to be clarified.
In this study, we generated endothelial‐specific ERK2 knockout (EE2KO) mice to clarify if the endothelial ERK2 was related to endothelial dysfunction and insulin resistance in vivo in a mouse model of MetS induced by feeding with a high‐fat/high‐sucrose diet (HFHSD). 9 , 21
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals, Genotyping, and Diets
A Cre‐lox P strategy was used to generate EE2KO mice. ERK2‐floxed mice were created as described previously, and they were backcrossed with C57BL/6J mice for 10 generations. 22 Transgenic mice expressing Cre recombinase under the control of the Tie2 promoter (Tie2‐Cre; Jackson Laboratory, Bar Harbor, ME), in which Cre‐recombinase activity was confined to endothelial cells, were used to drive recombination. The ERK2‐floxed mice were crossed with Tie2 promoter‐Cre mice, which had been maintained on the same C57BL/6J background. The resultant Tie2‐Cre(+/−);ERK2(lox/lox) mice (EE2KO) were viable and fertile with a normal appearance. The littermate controls used in this study were Tie2‐Cre(−/−);ERK2(lox/lox) mice (Control). Genotyping for the ERK2‐floxed allele and the presence of Cre was performed by polymerase chain reaction analysis using genomic DNA isolated from the tail tip. 9
The animals were housed in a temperature‐controlled room at 23±1 °C on a 12‐hour light–dark cycle. Male EE2KO and Control littermates were fed either a normal diet (ND) (CE‐7; CLEA Japan, Tokyo, Japan) or HFHSD (F2HFHSD with 28.3% of calories from carbohydrates, 54.5% from fat, and 17.2% from protein; Oriental Yeast, Tokyo, Japan) (Table 1) ad libitum from 6 weeks of age for 24 weeks. Body weight was recorded continuously every 2 weeks. In this study, mainly HFHSD‐fed Control (Control‐HFHSD) and HFHSD‐fed EE2KO (EE2KO‐HFHSD) mice were compared.
Table 1.
Composition of the ND and HFHSD Groups
| ND (CE‐7) | HFHSD (F2HFHSD) | |||
|---|---|---|---|---|
| g/100 g | kcal, % | g/100 g | kcal, % | |
| Total calories | 343 kcal | 481 kcal | ||
| Protein | 17.7 g | 20.6 | 20.7 g | 17.2 |
| Fat | 3.8 g | 10 | 29.1 g | 54.5 |
| Carbohydrate | 59.4 g | 69.4 | 34.0 g | 28.3 |
Ingredients: casein 25%, cellulose 5%, a‐corn starch 14.869%, sucrose 20%, vitamin mix (American Institute of Nutrition [AIN]‐93) 1%, mineral mix (AIN‐93G) 3.5%, beef tallow 14%, choline bitartrate 0.25%, tert‐butylhydroquinone 0.006%, lard 14%, soybean oil 2%, L‐cysteine 0.375%. Data represent mean±SEM. (Controls on ND, n=11; EE2KO on ND, n=9; controls on HFHSD, n=11; EE2KO on HFHSD, n=9). EE2KO indicates endothelial‐specific extracellular signal‐regulated kinase 2 knockout; HFHSD, high‐fat/high‐sucrose diet; and ND, normal diet. *P<0.05 vs corresponding ND. **P<0.01 vs corresponding ND. † P<0.05 vs controls with HFHSD. ‡ P<0.01 vs controls with HFHSD.
All experiments were conducted according to the institutional ethical guidelines for animal experiments and the safety guidelines for gene manipulation experiments of the National Defense Medical College, Japan, consistent with recommendations of the American Heart Association statement. The experiments were approved by the Committee for Animal Research of the National Defense Medical College, Japan (protocol number 13022).
Tissue Preparation and Histology
At the end of the diet, mice were anesthetized and euthanized with cervical dislocation using isoflurane inhalation (4%–5%), and an adequate level of anesthesia was confirmed by loss of pedal reflex. Blood was collected by puncture at the apex of the heart, and organs were perfused with Krebs‐Ringer bicarbonate solution (118.3 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3, 5.5 mmol/L D‐glucose). The liver and aorta were fixed in 4% paraformaldehyde for 24 hours, embedded in paraffin, and sectioned. Liver samples were stained with hematoxylin and eosin and Masson's trichrome. ERK2 expression in the aortic endothelium was visualized by immunohistochemistry with an anti‐ERK2 antibody (Abcam, Cambridge, United Kingdom).
Western Blotting of Tissues
Tissues (brain, heart, liver, kidneys, skeletal muscle, epididymal fat, and aorta) were collected and homogenized with homogenization buffer (20 mmol/L Tris–HCl [pH 7.4], 150 mmol/L NaCl, 1 mmol/L Na2EDTA, 1 mmol/L EGTA, 1% NP‐40, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L monoglycerophosphate, 1 mmol/L Na2VO4) containing 1 mmol/L phenylmethylsulfonyl fluoride and protease inhibitor cocktail. The homogenates were centrifuged at 13000g for 20 minutes at 4 °C, and supernatants were collected. Protein concentrations were measured by using the Bradford assay with bovine serum albumin as a standard. 23 Protein lysates were resolved by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes at 30 V for 2 hours at 4 °C and immunoblotted with primary antibodies to ERK1/2 (Cell Signaling Technology, Danvers, MA).
Preparation of Aortic Rings and Isometric Tension Measurement in an Organ Chamber
Isometric tension was measured as previously described. 24 The thoracic aorta was quickly removed and dissected from adhering connective tissue. Aortas from mice were cut into 3‐mm rings, with special care taken to preserve the endothelium, and mounted in organ baths filled with Krebs‐Ringer bicarbonate solution (118.3 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3, 5.5 mmol/L D‐glucose) aerated with 95% O2 and 5% CO2 at 37 °C. The rings of the aorta were attached to a force transducer, and isometric tension was recorded. The rings were primed with 30 mmol/L KCl and then precontracted with 10−5.5 mol/L L‐phenylephrine, producing a submaximal contraction. After the plateau was attained, the rings were exposed to the cumulative addition of 10−9 to 10−5 mol/L acetylcholine or 10−9 to 10−5 mol/L sodium nitroprusside. Changes in the tension of the aortic rings were measured.
Semiquantitative Assessment of Vascular Superoxide Level
Superoxide level in the aortic rings was assessed with dihydroethidium staining (Thermo Fisher Scientific, Waltham, MA), as previously described. 25 Aortic rings were snap‐frozen with liquid nitrogen and embedded in optimal cutting temperature medium (Sakura Finetek Japan, Tokyo, Japan). The frozen samples were cut immediately into 10‐μm‐thick sections and mounted on glass slides. The samples were incubated at room temperature for 30 minutes with 2.0×10−6 mol/L dihydroethidium and protected from light. Images were obtained with a BZ‐X710 microscope (Keyence, Osaka, Japan) with an excitation wavelength of 540 nm and an emission wavelength of 605 nm. The fluorescence intensity of dihydroethidium staining was measured semiquantitatively using software (Keyence).
Investigation of the Upstream and Downstream Components of the Ras/Raf/MEK/ERK Signaling Cascade
Aortic rings were incubated for 30 minutes with 1 μmol/L BQ123 (Adipogen Life Sciences, San Diego, CA) as an antagonist of the ET1 receptor, 1 μmol/L S18886 (terutroban; Cayman Chemical, Ann Arbor, MI) as an antagonist of the TXA2 (thromboxane A2)/TP (thromboxane A2/prostanoid receptor), and 30 μmol/L U0126 (Promega, Madison, WI) as an inhibitor of MEK. 26 Immediately after that, measurement of isometric tension and dihydroethidium staining of the aorta were performed.
Measurement of 11‐Dehydro Thromboxane B2 in Urine
Urine was collected from 30‐week‐old HFHSD‐fed mice for 24 hours. Because TXA2 is unstable, 11‐dehydro thromboxane B2 was analyzed as a metabolite of thromboxane B2 using an ELISA kit (Cayman Chemical).
Measurements of SBP, Heart Rate, and Serum NO2 − and NO3 − Levels
SBP and heart rate were measured by the tail‐cuff method (MK‐2000; Muromachi Kikai, Tokyo, Japan) without anesthesia at the end of the special diets. EE2KO‐HFHSD and Control‐HFHSD mice were injected intraperitoneally with 100 mg/kg body weight L‐NAME (Nω‐nitro‐l‐arginine methyl ester) (Cayman Chemical) as a NO synthase inhibitor for 7 days. SBP was measured before L‐NAME injection and 7 days after of injections. NO2 − and NO3 − levels were measured after fasting for 12 hours using an assay kit (Dojindo, Kumamoto, Japan).
Measurement of Metabolites
Blood glucose levels were determined using a blood glucose monitoring system (FreeStyle; Nipro, Osaka, Japan). Serum insulin levels were measured using an ELISA kit (Fujifilm Wako Shibayagi, Gunma, Japan). The homeostatic model assessment of insulin resistance (HOMA‐IR) was calculated by multiplying fasting serum glucose levels by insulin levels. The serum levels of alanine aminotransferase were measured by an enzymatic assay (Fujifilm Wako Pure Chemical Industries, Osaka, Japan). Intraperitoneal glucose tolerance tests (ipGTTs) were performed after fasting for 6 hours, whereas insulin tolerance tests (ITTs) were performed after fasting for 4 hours; the mice were injected intraperitoneally with 3 g/kg body weight D‐glucose or 0.5 U/kg body weight human regular insulin (Humulin R; Eli Lilly, Indianapolis, IN), respectively. Vein blood was collected at 0, 15, 30, 60, and 120 minutes after glucose injection for the ipGTTs and at 0, 15, 30, 45, 60, and 120 minutes after insulin injection for the ITTs. Blood insulin levels were measured at 0, 15, and 30 minutes after glucose injection.
Change of Endothelial Function, Glucose Metabolism, and Steatohepatitis by Oral Intake of S18886
EE2KO‐HFHSD and Control‐HFHSD mice were administered 5 mg/kg per day S18886, which was added to their drinking water for 6 weeks according to a previous report. 26 After the S18886 administration period ended, SBP, heart rate, and acetylcholine‐induced relaxation were evaluated as measures of endothelial function. Body weight, fasting serum glucose, and insulin were measured, and HOMA‐IR was calculated for glucose metabolism. Moreover, hematoxylin and eosin staining and Masson's trichrome staining of the liver were performed and evaluated according to nonalcoholic fatty liver disease activity scores, a commonly used histology scoring system for nonalcoholic fatty liver disease activity, and the fibrosis score for steatohepatitis. The evaluation items of nonalcoholic fatty liver disease activity scores are steatosis, lobular inflammation, and ballooning, and generates a score between 0 and 8. Liver fibrosis was evaluated with a range of 0 to 4 using Masson's trichrome staining. 27
Preparation of Lentivirus Expressing shRNA Targeting Human ERK2 ( MAPK1 ) mRNA
Homo sapien MAPK1 (gene ID 5594) mRNA (NM_002745) coding sequence and 3′‐noncoding sequences were used for the selection of shRNA. An in‐house computer program identified a highly specific sequence (5′‐gaccagctgaaccacatt‐3′) that recognizes all of the variants of MAPK1 (NM_002745 and NM_138957) with at least a 4‐nucleotide mismatch to nontarget sequences. The methods for preparing a human U6‐driven shRNA‐expressing entry vector, lentivirus plasmid, and production of lentivirus were previously described. 28 The nontargeting control shRNA lentivirus was also prepared in the same fashion.
Western Blotting of Human Umbilical Vein Endothelial Cells Treated With shRNA
Human umbilical vein endothelial cells (HUVECs) were seeded onto culture plates at 30% confluence. A lentivirus containing shRNA for ERK suppression or control was added. After 24 hours, human umbilical vein endothelial cells with the target shRNA were selected by 0.5 μg/mL blasticidin (InvivoGen, San Diego, CA). The culture was seeded to the next passage. When the cells were 80% confluent, an insulin stimulation test was performed. Insulin (100 μmol/L) (Sigma‐Aldrich, St. Louis, MO) was added to the cells after 6 hours incubation with 100 μmol/L palmitate acid (Sigma‐Aldrich) and 50 mmol/L L‐carnitine (Sigma‐Aldrich). After 30 minutes, the cultured cells were collected with a buffer (20 mmol/L Tris–HCl, pH 7.4, 150 mmol/L NaCl, 1 mmol/L Na2EDTA, 1 mmol/L EGTA, 1% NP‐40, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L monoglycerophosphate, 1 mmol/L Na2VO4) containing 1 mmol/L phenylmethylsulfonyl fluoride and protease inhibitor cocktail. The mixtures were centrifuged at 13 000g for 20 minutes at 4 °C, and the supernatants were collected. Protein concentrations were measured by using the Bradford assay, with bovine serum albumin as a standard. 20 Protein lysates were resolved by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes at 30 V for 2 hours at 4 °C and immunoblotted with primary antibodies to phosphorylated‐ERK1/2 (T202/Y204), ERK1/2, phosphorylated‐AKT (S473), AKT, phosphorylated‐eNOS (S1177) (Cell Signaling Technology), eNOS (Becton, Dickinson and Company, Franklin Lakes, NJ), and GAPDH (Cell Signaling Technology).
Statistical Analysis
The results are shown as mean±SEM. The Kolmogorov‐Smirnov test was used to test normality. Data for body weight, ipGTTs, ITTs, and vascular relaxation were analyzed by 2‐way ANOVA with repeated measures followed by the post hoc Bonferroni correction for multiple comparisons. Between‐group differences were compared by Student t test and 1‐way ANOVA, as appropriate. All statistical analyses were performed with GraphPad Prism software version 7 (GraphPad Software, La Jolla, CA). Differences were considered significant for P<0.05.
RESULTS
Phenotypical Changes in EE2KO With ND
ERK2 expression was assessed by immunohistochemical staining in the endothelium of aortas from Control and EE2KO mice (Figure 1A). ERK2 expression was predominantly decreased in the endothelium of EE2KO mice, whereas ERK2 expression was preserved in the aortic endothelium of Control mice. ERK2 protein expressions in other organs were not different between Control and EE2KO mice, as assessed by Western blotting (Figure 1B). We also checked that ERK2 expression is well preserved in macrophages and bone marrow in EE2KO mice (Figure 2). Control and EE2KO mice were fed either an ND or HFHSD for 24 weeks (Figure 1C). The body weight of Control and EE2KO mice on ND for 24 weeks was similar (Figure 1D and 3A). SBP was slightly lowered, and the serum NO2 − and NO3 − levels were increased in EE2KO mice with ND, although acetylcholine‐induced relaxation was not changed (Figure 3B through 3D). The fasting glucose, insulin levels, and HOMA‐IR were not changed in EE2KO mice with ND (Table 2).
Figure 1. Efficiency of ERK2 deletion and characteristics of EE2KO mice.
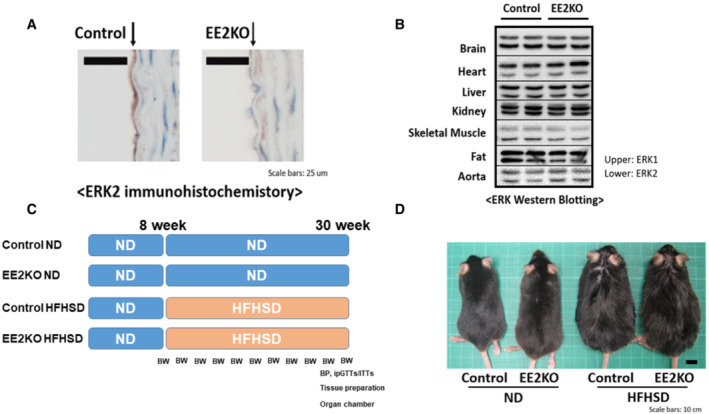
A, Immunohistochemistry of the aorta with an anti‐ERK2 antibody. Scale bar, 25 μm. B, Western blotting of tissues with an anti‐ERK antibody. C, Study protocols. D, Photographs of 30‐week‐old ND‐ or HFHSD‐fed control and EE2KO mice. Scale bar, 10 mm. BW and BP are assessment of body weight and blood pressure. BP indicates blood pressure; BW, body weight; EE2KO, endothelial‐specific extracellular signal‐regulated kinase 2 knockout; ERK, extracellular signal‐regulated kinase; HFHSD, high‐fat/high‐sucrose diet; ipGTTs, intraperitoneal glucose tolerance tests; ITT, insulin tolerance tests; and ND, normal diet.
Figure 2. ERK2 expression levels of macrophage from peritoneal cavity and bone marrow between control and EE2KO.
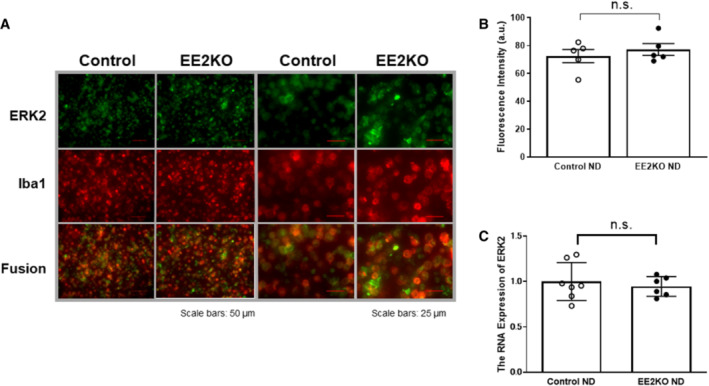
A, Double immunohistochemistry of macrophage from peritoneal cavity with anti‐ERK2 antibody (green) and anti‐ionized calcium‐binding adapter molecule‐1 (Iba1) antibody (red). Scale bar is 50 μm (left) and 25 μm (right). B, Fluorescence intensity of ERK2 in macrophage (n=5 per each group). C, The RNA expression of ERK2 in bone marrow (Control‐ND, n=7; EE2KO‐ND, n=6). Error bars represent SEM. Between‐group differences were compared by Student t test. a.u. indicates arbitrary unit; EE2KO, endothelial‐specific extracellular signal‐regulated kinase 2 knockout; ERK, extracellular signal‐regulated kinase; ND, normal diet; and n.s., not significant.
Figure 3. Characteristics of Control and EE2KO mice in ND.
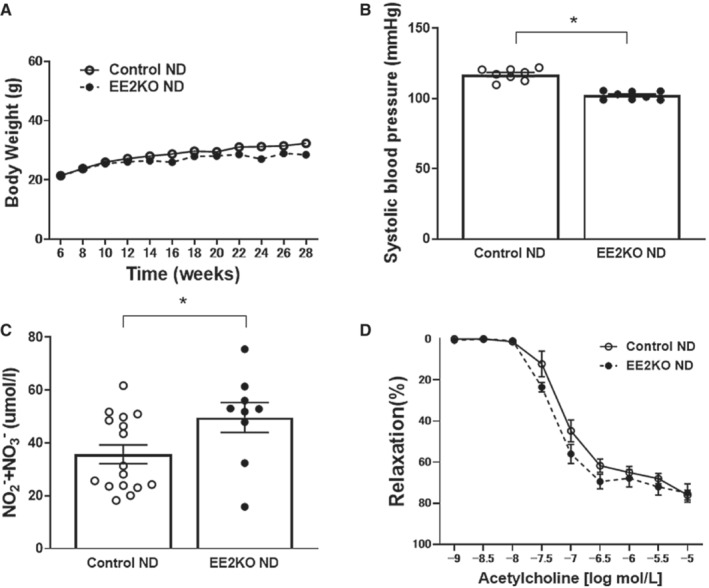
A, Body weight of ND‐fed Control and EE2KO mice. B, SBP of ND‐fed Control and EE2KO mice (n=8 per group). C, Serum levels of NO2 − and NO3 − of ND‐fed Control and EE2KO mice (Control‐ND, n=16; EE2KO‐ND, n=9). D, Endothelium‐dependent relaxation (Controls‐ND, n=8; EE2KO‐ND, n=7). Error bars represent SEM. Data for body weight and endothelium‐dependent relaxation were analyzed by 2‐way ANOVA with repeated measures (A and D). Between‐group differences were compared by Student t test (B and C). *P<0.05 (compared with Control‐ND mice). EE2KO indicates endothelial‐specific extracellular signal‐regulated kinase 2 knockout; ND, normal diet; and SBP, systolic blood pressure.
Table 2.
Serum Glucose and Insulin in ND‐ or HFHSD‐Fed Control and EE2KO Mice
| ND | HFHSD | |||
| Control | EE2KO | Control | EE2KO | |
| Serum glucose (mg/dL) | 90.6±3.3 | 85.8±6.7 | 136.1±4.8 | 109.3±6.1 |
| Serum insulin (ng/mL) | 0.44±0.11 | 0.123±0.02 | 2.23±0.36 | 1.11±0.22 |
| HOMA‐IR | 25.9±5.64 | 7.86±1.31 | 240.9±48.4 | 108.4±20.9 |
EE2KO indicates endothelial‐specific extracellular signal‐regulated kinase 2 knockout; HFHSD, high‐fat/high‐sucrose diet; HOMA‐IR, homeostatic model assessment of insulin resistance; and ND, normal diet.
NO Production Is Elevated and SBP Is Lowered in EE2KO‐HFHSD Mice
The body weight of Control and EE2KO mice on HFHSD for 24 weeks was similar (Figure 1D and 4A). SBP was lower in EE2KO‐HFHSD mice compared with Control‐HFHSD mice without changes in heart rate (Figure 4B). Because the bioavailability of NO in the endothelium is an important regulator of blood pressure, 29 the changes in SBP were observed with an administration of L‐NAME, an NO synthase inhibitor. SBP was significantly lower in EE2KO‐HFHSD than in Control‐HFHSD mice before L‐NAME injection. After L‐NAME injection, the increase in SBP was more prominent in EE2KO‐HFHSD mice, and the difference in SBP between both groups was abolished (Figure 4C). Next, the serum levels of NO2 − and NO3 −, which are catabolic products of NO, were measured. The serum levels of NO2 − and NO3 − were higher in EE2KO‐HFHSD than in Control‐HFHSD mice (Figure 4D). These data suggested that NO production was increased, resulting in lower SBP in EE2KO‐HFHSD mice.
Figure 4. The lowering SBP in EE2KO‐HFHSD mice with decreased TXB2 production and increased NO production.
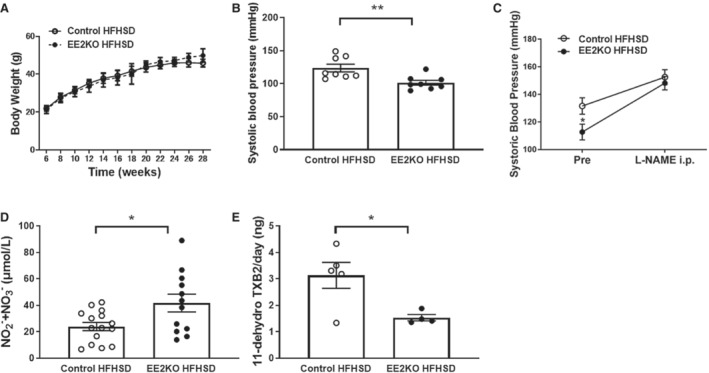
A, Body weight of Control‐HFHSD and EE2KO‐HFHSD mice. B, SBP of Control‐HFHSD and EE2KO‐HFHSD mice (n=8 per group). C, Serial change of SBP of Control‐HFHSD and EE2KO‐HFHSD mice treated with L‐NAME (n=7 per group). D, Serum levels of NO2 − and NO3 − of Control‐HFHSD and EE2KO mice (Control‐HFHSD, n=15; EE2KO‐HFHSD, n=12). E, 11‐dehydro thromboxane B2, which is a metabolic product of TXA2, in urine in Control‐HFHSD and EE2KO‐HFHSD mice (Control‐HFHSD, n=5; EE2KO‐HFHSD, n=4). Error bars represent SEM. Data for body weight were analyzed by 2‐way ANOVA with repeated measures (A). Between‐group differences were compared by Student t test (B–E). *P<0.05, **P<0.01 (compared with Control‐HFHSD mice). 11‐dehydro TXB2 indicates 11‐dehydro thromboxane B2; EE2KO, endothelial‐specific extracellular signal‐regulated kinase 2 knockout; HFHSD, high‐fat/high‐sucrose diet; L‐NAME, Nω‐nitro‐l‐arginine methyl ester; SBP, systolic blood pressure; TXA2, thromboxane A2; and TXB2, thromboxane B2.
11‐Dehydro Thromboxane B2 Levels Are Lower in EE2KO‐HFHSD Than in Control‐HFHSD Mice
The levels of 11‐dehydro thromboxane B2, which is a catabolite of TXA2, were examined in urine. 11‐dehydro thromboxane B2 levels were lower in EE2KO‐HFHSD than in Control‐HFHSD mice (Figure 4 E).
Endothelial Function Is Increased in EE2KO‐HFHSD Mice
Because NO from eNOS, which is largely produced by the vascular endothelium, could be increased in EE2KO‐HFHSD mice, we next assessed endothelium‐dependent, acetylcholine‐induced relaxation with isometric tension measurement of aortic rings. Acetylcholine‐induced relaxation was increased in EE2KO‐HFHSD compared with Control‐HFHSD mice (Figure 5A). Sodium nitroprusside‐induced (endothelium‐independent) relaxation was similar between the 2 groups (Figure 5B).
Figure 5. The increased endothelium‐dependent relaxation with lowering superoxide level in HFHSD‐fed EE2KO mice.
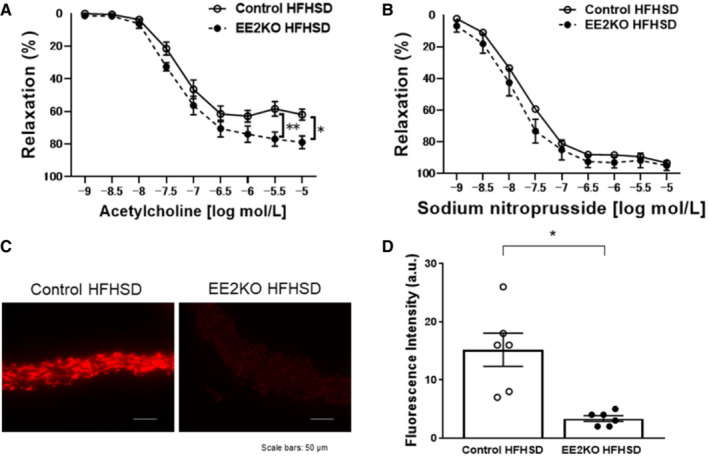
Vascular relaxation of aortic rings with acetylcholine (A) and sodium nitroprusside (B) (Control‐HFHSD, n=10; EE2KO‐HFHSD, n=7). Dihydroethidium staining of aortic rings (C) and quantification of dihydroethidium fluorescence intensity (D) in Control‐HFHSD and EE2KO‐HFHSD mice (Control‐HFHSD, n=6; EE2KO‐HFHSD, n=3). Scale bar, 50 μm. Error bars represent SEM. Vascular relaxation was analyzed by 2‐way ANOVA with repeated measures followed by the post hoc Bonferroni correction for multiple comparisons (A and B). Between‐group difference was compared by Student t test (D). *P<0.05, **P<0.01 (compared with Control‐HFHSD mice). a.u. indicates arbitrary unit; EE2KO, endothelial‐specific extracellular signal‐regulated kinase 2 knockout; and HFHSD, high‐fat/high‐sucrose diet.
Increases in Vascular Oxidative Stress With the HFHSD Are Blunted in EE2KO Mice
There were no histological changes in the aorta of Control‐HFHSD and EE2KO‐HFHSD mice (data not shown). Dihydroethidium staining was used to evaluate superoxide levels in the aorta, because superoxide levels reduce NO bioactivity because of their rapid interaction and the production of peroxynitrite, which induces eNOS uncoupling and impairs NO production from eNOS. 30 , 31 Aortic rings from Control‐HFHSD mice increased fluorescence intensity but not in EE2KO‐HFHSD mice (Figure 5C and 5D).
To further investigate the possible pathways by which ERK2 increases reactive oxygen species (ROS) production, we performed dihydroethidium staining of aortic rings from Control‐HFHSD mice with 1% cholesterol using carbonylcyanide m‐chlorophenylhydrazone (protonophore mitochondrial uncoupler), L‐NAME, tempol (superoxide dismutase), apocynin (NADPH‐oxidase inhibitor), rotenone (inhibitor of complex I of the mitochondrial electron transport chain), and oxypurinol (xanthine oxidase inhibitor). The fluorescence intensity was decreased in the aorta following treatment with carbonylcyanide m‐chlorophenylhydrazone, L‐NAME, and tempol, suggesting mitochondria and eNOS were major sources (Figure 6).
Figure 6. The fluorescence intensity in the aorta of HFHSD‐fed mice in dihydroethidium staining.
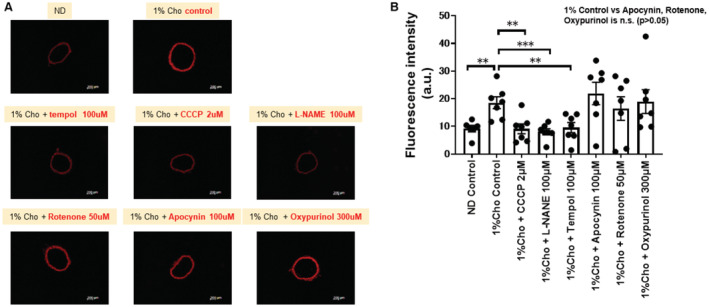
In this study we used the Control‐HFHSD with 1% cholesterol to clarify the pharmacological effects. Representative dihydroethidium staining of aortic rings (A) and quantification of dihydroethidium fluorescence intensity (B). Between‐group differences were compared by 1‐way ANOVA. **P<0.01, ***P<0.001. a.u. indicates arbitrary unit; CCCP, carbonylcyanide m‐chlorophenyl‐hydrazone; Cho, cholesterol; HFHSD, high‐fat/high‐sucrose diet; L‐NAME, Nω‐nitro‐l‐arginine methyl ester; and n.s., not significant.
TP Receptor Could Be the Downstream of ERK2 to Induce Endothelial Dysfunction With Superoxide Level
Because ERK2 induced endothelial dysfunction via superoxide level, we assessed the role of the upstream and downstream components of ERK2 in the regulation of endothelial function. MEK is known as the upstream of ERK2, whereas TXA2 and ET1 are endothelium‐derived vasoconstrictive peptides potentially located downstream of ERK2. 11 , 20 , 32 Thus, we tested the endothelial function of aortic rings with preincubations of either BQ123 (ET1 receptor antagonist), S18886 (TP receptor antagonist), or U0126 (MEK inhibitor) for 30 minutes. There were no changes of the relaxations in Control‐HFHSD and EE2KO‐HFHSD mice treated with BQ123. However, U0126 and S18886 significantly increased acetylcholine‐induced relaxation in the aorta in Control‐HFHSD mice but not in EE2KO‐HFHSD mice (Figure 7A and 7B). Next, changes in superoxide level were examined in aortas treated with or without the same agents for 30 minutes. In Control‐HFHSD but not EE2KO‐HFHSD mice, fluorescence intensity was decreased in the aorta following treatment with S18886 and U0126, whereas BQ123 had no effect (Figure 7C through 7E).
Figure 7. The increased endothelium‐dependent relaxation and the decreased superoxide level in Control‐HFHSD but not in EE2KO‐HFHSD mice with S18886.
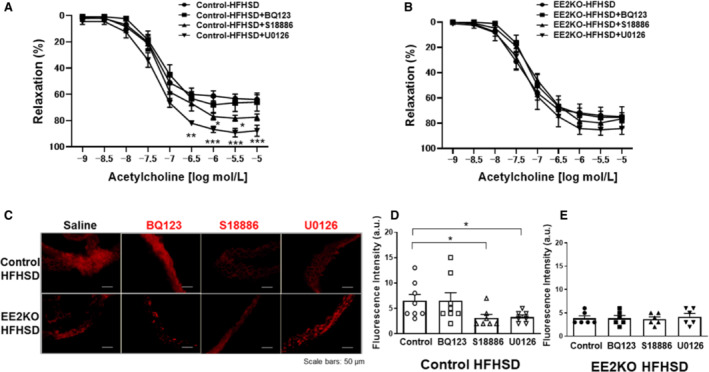
Vascular relaxation of aortic rings of HFHSD‐fed control mice (A) and EE2KO (B) mice treated with BQ123, S18886, and U0126 (Control‐HFHSD, n=9; EE2KO‐HFHSD, n=7) and BQ123 (A and B) (Control‐HFHSD, n=9; EE2KO‐HFHSD, n=7), S18886 (A and B) (Control‐HFHSD, n=9; EE2KO‐HFHSD, n=7) and U0126 (A and B) (Control‐HFHSD, n=9; EE2KO‐HFHSD, n=10). Error bars represent SEM. *P<0.05, **P<0.01, ***P<0.001 (compared with controls). Dihydroethidium staining of aortic rings with BQ123, S18886, and U0126 (C) and quantification of dihydroethidium fluorescence intensity of Control‐HFHSD (D) (saline, n=8; BQ123, n=8; S18886, n=7; U0126, n=7) and EE2KO‐HFHSD (E) (n=6 per group) mice. Scale bar, 50 μm. Error bars represent SEM. Vascular relaxation was analyzed by 2‐way ANOVA with repeated measures followed by the post hoc Bonferroni correction for multiple comparisons (A and B). Between‐group differences were compared by 1‐way ANOVA (D and E). *P<0.05, **P<0.01, ***P<0.001. a.u. indicates arbitrary unit; EE2KO, endothelial‐specific extracellular signal‐regulated kinase 2 knockout; and HFHSD, high‐fat/high‐sucrose diet.
Serum Fasting Glucose and Insulin Levels and HOMA‐IR Are Lowered, and Glucose Tolerance and Insulin Sensitivity Are Mitigated in EE2KO‐HFHSD
Because endothelial function is closely related to insulin resistance, the levels of blood glucose and insulin for glucose metabolism were measured. 33 The fasting glucose and insulin levels were lower in EE2KO‐HFHSD than Control‐HFHSD mice. HOMA‐IR was also lowered in EE2KO‐HFHSD mice to ≈50% of the value observed in Control‐HFHSD mice (Table 2).
Because insulin resistance in EE2KO‐HFHSD mice could be mitigated by fasting, ipGTTs and ITTs were used to further evaluate insulin resistance and sensitivity. In ipGTTs, the increases in glucose levels were significantly blunted in EE2KO‐HFHSD mice at 15 and 60 minutes after glucose injection, and areas under the curve were lower compared with Control‐HFHSD mice (Figure 8A and 8B). Serum insulin levels were almost the same between both groups (Figure 8C). In ITTs, insulin lowered glucose levels more prominently in EE2KO‐HFHSD than in Control‐HFHSD mice at 60 and 120 minutes after insulin injection (Figure 8D). Areas under the curve of ITTs were lower in EE2KO‐HFHSD mice compared with Control‐HFHSD mice (Figure 8E).
Figure 8. The improved glucose tolerance and insulin sensitivity in HFHSD‐fed EE2KO mice.
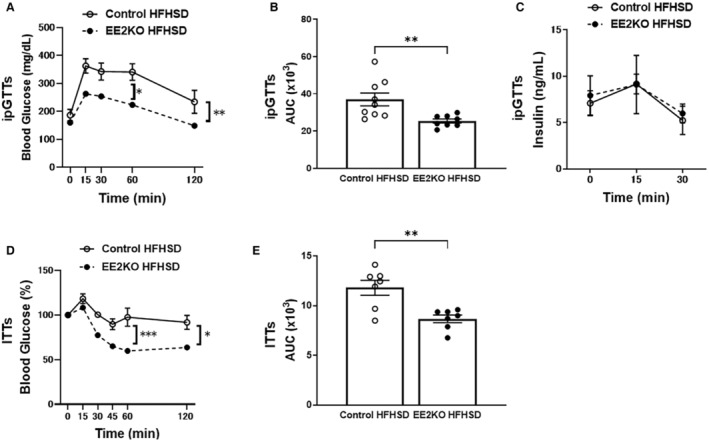
ipGTTs for 30‐week‐old HFHSD‐fed control and EE2KO mice. A, Glucose levels. B, AUC for ipGTTs. C, Insulin levels (Control‐HFHSD, n=10; EE2KO‐HFHSD, n=10). D, ITTs for 30‐week‐old HFHSD‐fed control and EE2KO mice (Control‐HFHSD, n=7; EE2KO‐HFHSD, n=7). E, AUC for ITTs. ipGTTs and ITTs results are expressed as mean blood glucose or insulin concentration±SEM and mean percentage of basal blood glucose concentration±SEM, respectively. Error bars represent SEM. Data for ipGTTs and ITTs were analyzed by 2‐way ANOVA with repeated measures followed by the post hoc Bonferroni correction for multiple comparisons (A, C, and D). Between‐group differences were compared by Student t test (B and E). *P<0.05, **P<0.01, ***P<0.001 (compared with Control‐HFHSD mice). AUC indicates areas under the curve; EE2KO, endothelial‐specific extracellular signal‐regulated kinase 2 knockout; HFHSD, high‐fat/high‐sucrose diet; ipGTTs, intraperitoneal glucose tolerance tests; and ITTs, insulin tolerance tests.
Oral Intake of S18886 Improves Endothelial Function, Glucose Metabolism, and Steatohepatitis With HFHSD
The study with isometric tension measurements ex vivo indicated that the TP receptor was involved in the ERK2 signaling cascade for endothelial dysfunction. The improvements of glucose metabolism and insulin resistance were also associated with the improved endothelial function by the deletion of ERK2. Therefore, endothelial function, glucose metabolism, and steatohepatitis, which was a major character for insulin resistance, were examined following long‐term in vivo inhibition of the TP receptor with S18886. EE2KO‐HFHSD and Control‐HFHSD mice were treated with 5 mg/kg per day S18886 added to the drinking water for 6 weeks. 26 Oral administration of S18886 did not change body weight in either group (Figure 9A). The administration of S18886 increased acetylcholine‐induced relaxation in aortic rings from Control‐HFHSD mice but not from EE2KO‐HFHSD mice (Figure 9B). The administration of S18886 lowered the SBP in Control‐HFHSD mice but not in EE2KO‐HFHSD mice (Figure 9C). For glucose metabolism, the administration of S18886 did not change the insulin levels in both groups; however, fasting serum glucose levels were decreased in both groups. The administration of S18886 decreased HOMA‐IR only in Control‐HFHSD mice (Figure 9D through 9F), suggesting marked improvement of insulin sensitivity. Finally, steatohepatitis was assessed in HFHSD‐fed mice because it is closely associated with insulin resistance in obesity. The administration of S18886 markedly improved steatohepatitis in Control‐HFHSD mice, as indicated by smaller lipid droplets, lowered nonalcoholic fatty liver disease activity scores, and improved fibrosis observed using Masson's trichrome staining in the liver (Figure 10).
Figure 9. The lowered SBP, the increased endothelium‐dependent relaxation, and the improved insulin resistance with S18886 in Control‐HFHSD mice but not in EE2KO‐HFHSD mice.
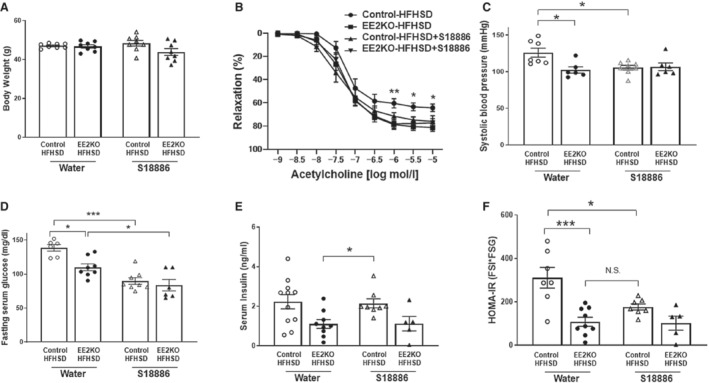
A, Body weight. B, Vascular relaxation with acetylcholine (n=6 per group). C, Systolic blood pressure (Control without S18886, n=7; EE2KO without S18886, n=8; Control with S18886, n=8; EE2KO with S18886, n=6). D, Fasting serum glucose levels. E, Serum insulin levels. F, HOMA‐IR of control and EE2KO mice with or without oral administration of S18886 (Control without S18886, n=6; EE2KO without S18886, n=8; Control with S18886, n=8; EE2KO with S18886, n=6). Error bars represent SEM. Between‐group differences were compared by 1‐way ANOVA (A, C, and D through F). *P<0.05, ***P<0.001. Vascular relaxation was analyzed by 2‐way ANOVA with repeated measures followed by the post hoc Bonferroni correction for multiple comparisons to compare with Control‐HFHSD mice (B). ***P<0.001 (compared between Control‐HFHFS and Control‐HFHSD‐S18886 mice). EE2KO indicates endothelial‐specific extracellular signal‐regulated kinase 2 knockout; HFHSD, high‐fat/high‐sucrose diet; and HOMA‐IR, homeostatic model assessment of insulin resistance.
Figure 10. The improved steatohepatitis with S18886 in Control‐HFHSD but not in EE2KO‐HFHSD mice.
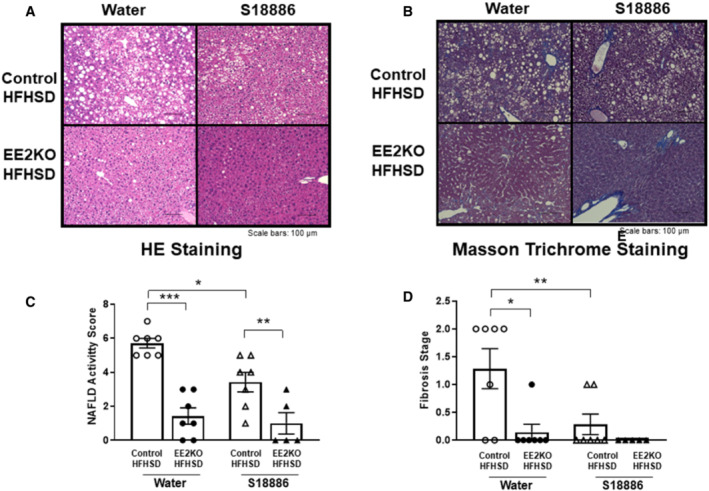
HE staining (A) and Masson's trichrome staining (B) of liver sections from Control‐HFHSD and EE2KO‐HFHSD mice with or without oral administration of S18886. Scale bars, 100 μm. NAFLD activity score (C) (Control without S18886, n=7; EE2KO without S18886, n=7; controls with S18886, n=7; EE2KO with S18886, n=5) and fibrosis score (D) of Control‐HFHSD and EE2KO‐HFHSD mice with or without oral administration of S18886. Scale bar, 100 μm. Error bars represent SEM. Between‐group differences were compared by 1‐way ANOVA (B and D).*P<0.05, **P<0.01, ***P<0.001. EE2KO indicates endothelial‐specific extracellular signal‐regulated kinase 2 knockout; HE, hematoxylin and eosin; HFHSD, high‐fat/high‐sucrose diet; and NAFLD, nonalcoholic fatty liver disease.
Phosphorylation of AKT and eNOS With Insulin Was Increased in HUVECs Treated With Lentivirus for ERK2 Suppression After 6 Hours Incubation With Palmitic Acid
Palmitic acid could induce insulin resistance in endothelial cells. We tested the insulin‐induced phosphorylation for AKT/eNOS with insulin after the 6 hours incubation of palmitic acid with or without the downregulation of ERK2 with shRNA. Insulin induced the phosphorylation of AKT/eNOS after an incubation of palmitic acid was increased by the downregulation of ERK2 with shRNA (Figure 11).
Figure 11. Phosphorylation of AKT and eNOS is increased in HUVECs treated with lentivirus for ERK2 suppression and incubated with palmitic acid.
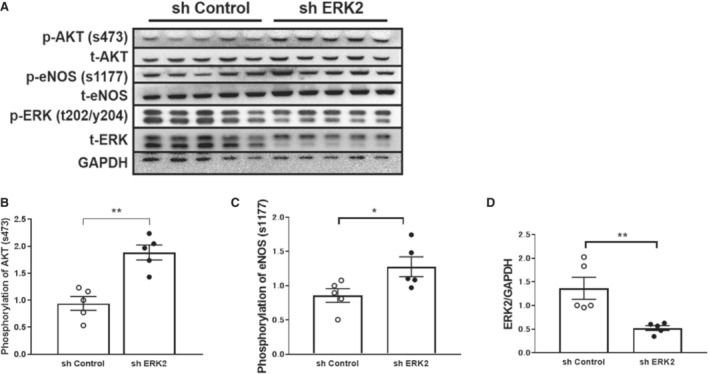
A, Western blotting of HUVECs treated with lentivirus for ERK2 suppression and incubated with palmitic acid. B, Relative levels of AKT phosphorylation. C, Relative levels of eNOS phosphorylation. D, Relative levels of ERK2 expression (n=5 per group). Error bars represent SEM. Between‐group differences were compared by Student t test (B through D). *P<0.05, **P<0.01 (compared with sh Control). AKT indicates protein kinase B; eNOS, endothelial nitric oxide synthase; ERK2, extracellular signal‐regulated kinase 2; HUVECs, human umbilical vein endothelial cells; p‐AKT, phosphorylation of AKT; p‐eNOS, phosphorylation of eNOS; p‐ERK2, phosphorylation of ERK2; t‐AKT, total AKT; t‐eNOS, total eNOS; t‐ERK2, total ERK2; and sh, small hairpin RNA.
DISCUSSION
The Effect of ERK2 Deficiency and TP Receptor Antagonist on Oxidative Stress, NO Bioactivity, and Endothelial Function
This is the first study to clarify the role of endothelial ERK2 in a mouse model of MetS in vivo. To investigate the role of endothelial ERK2, EE2KO mice were created and fed HFHSD. Because Tie2‐Cre promoter might affect the expression in blood cells, 34 we confirmed no changes in ERK2 expression on bone marrow and peritoneal macrophages (Figure 2). EE2KO‐HFHSD mice had decreased SBP, endothelial superoxide level, and improved endothelial function as assessed by acetylcholine‐induced relaxation in aorta. These data suggested that endothelial ERK2 increased production of superoxide and induced endothelial dysfunction with the decreased bioactivity of NO, resulting in the elevation of SBP in a mouse model of MetS.
Vascular tonus is regulated by the balance between endothelium‐derived relaxation factors and endothelium‐derived contraction factors such as ET1 and TXA2, which can be released via the Ras/Raf/MEK/ERK pathway in the endothelium. 11 , 20 In addition, oxidative stress decreases NO bioactivity in the vessel wall through multiple mechanisms, 35 , 36 , 37 and inactivation of NO by superoxide is augmented in various vascular diseases. 38 , 39 Zhang et al reported that the activation of the TP receptor increased superoxide and peroxynitrite levels, resulting in eNOS uncoupling in the endothelium. 40 Meanwhile, aortic ERK was reported to induce the production of superoxide and regulate spontaneous contractile tone. 41 Therefore, we tested the inhibitors of the receptors for endothelium‐derived contraction factors (BQ123, an ET1 receptor antagonist, and S18886, a TP receptor antagonist) or a MEK inhibitor (U0126) on the endothelial function and the superoxide level of aorta from HFHSD‐fed mice. Administration of S18886 and U0126 decreased superoxide level and improved endothelium‐dependent relaxation in aortas from Control‐HFHSD mice but not in EE2KO‐HFHSD mice, and there was no change for BQ123 in HFHSD‐fed control mice. These data suggested that MEK/ERK2 released prostanoids and activates the TP receptor to increase superoxide and endothelial dysfunction in MetS. Meanwhile, some reports showed that another substance induced endothelial dysfunction in MetS. 42 , 43 The superoxide level of aortas from Control‐HFHSD mice was decreased with carbonylcyanide m‐chlorophenylhydrazone and L‐NAME (Figure 6), suggesting the source of superoxide could be mitochondria and NOS. EE2KO‐HFHSD mice had decreased blood pressure together with a decrease in the urinary levels of TXA2 metabolites and an increase in the serum levels of NO metabolites. Moreover, L‐NAME, a NOS inhibitor, increased the blood pressure of EE2KO‐HFHSD mice to a similar level as in Control‐HFHSD mice with L‐NAME. The balance between NO and TP receptor could be responsible for vascular oxidative stress, NO bioactivity, and blood pressure in our MetS model.
Insulin signaling cascades in the endothelium consist of 2 major pathways, namely, PI3K/AKT/eNOS and Ras/Raf/MEK/ERK. In the healthy state, PI3K/AKT/eNOS is predominant for insulin signaling and decrease of vascular tonus. 30 , 44 PI3K/AKT/eNOS and Ras/Raf/MEK/ERK can be balanced, and the latter pathway could be enhanced in insulin resistance. ERK can phosphorylate and impair insulin receptor substrate‐1 and inhibits PI3K/AKT/eNOS pathway, and the mechanism has been proposed for endothelial dysfunction in insulin resistance. 15 , 17 , 45 , 46 The deletion of ERK2 with shRNA increased phosphorylation of AKT and eNOS in HUVEC with palmitic acid supporting the notion (Figure 11). The activation of the Ras/Raf/MEK/ERK pathway also increased the release of prostanoids and the activation of TP receptors, which could compete with NO signaling resulting in vasoconstriction. Because S18886 improved endothelial function in aortic rings, we tested if administration of S18886 for a longer period in vivo improved endothelial dysfunction and insulin resistance in both groups. As we considered, either the deletion of ERK2 or administrations of S18886 in vivo improved endothelial function of the aorta and decreased SBP without changing body weight in Control‐HFHSD mice but not in EE2KO‐HFHSD mice. The data indicated that the ERK2/TP receptors with HFHSD impaired vascular NO bioactivity in vivo.
Improvement of Insulin Resistance and Steatosis With ERK2 Deficiency or TP Receptor Antagonist
Our data also indicated that the endothelial ERK2/TP pathway could contribute to glucose metabolism with insulin resistance in MetS. EE2KO‐HFHSD mice had lower serum glucose, insulin and HOMA‐IR, and improved glucose tolerance and insulin tolerance tests compared with Control‐HFHSD mice, indicating an improvement of insulin resistance without changing body weight. Moreover, in histological studies, EE2KO‐HFHSD mice ameliorated steatohepatitis, a major complication of insulin resistance. Moreover, administration of S18886 in vivo also decreased fasting glucose levels and HOMA‐IR as well as ameliorating steatohepatitis. S18886 had no additional effects on EE2KO‐HFHSD, suggesting endothelial ERK2 was upstream for the activation of TP receptors to improve the glucose metabolism and steatohepatitis in our MetS model. The data suggested the pathological interactions among endothelial dysfunction, glucose intolerance, and steatohepatitis in insulin resistance with HFHSD.
Previous studies by our and other groups have demonstrated a strong relationship between insulin resistance and steatohepatitis, and MetS is bidirectionally linked with steatohepatitis as a consequence of the inflammatory and metabolic processes characterizing this condition. 47 We recently published the data indicating steatohepatitis with insulin resistance induced global metabolic remodeling including fatty acid and arginine metabolism which could affect NO bioactivity. 48 Because the deletion of endothelial ERK2 improved endothelial NO bioactivity, insulin sensitivity, and glucose metabolism in HFHSD‐fed mice, there might be a causal relationship between endothelial dysfunctions and steatohepatitis. Endothelial dysfunction in skeletal arterial or hepatic circulation might affect the exchange of glucose between blood and tissues. Previous reports suggested that the endothelial function of the hepatic microvasculature is impaired in nonalcoholic fatty liver disease model rats, leading to the reduction of oxygenation and modulation of hepatic microcirculatory perfusion by NO. 49 , 50 , 51 Hypertension with endothelial dysfunction is associated with insulin resistance, and hyperinsulinemia could promote steatohepatitis with lipid synthesis. Our data suggested that the inhibition of the endothelial ERK2/TP receptor might improve not only vascular NO bioactivity but also glucose metabolism and steatohepatitis independent of body weight. In previous studies S18886 inhibits the development of atherosclerosis in apolipoprotein E deletion and low‐density lipoprotein–receptor deletion mice. 26 , 52 Aspirin inhibits the production of TXA2; however, no reports have shown that aspirin improves insulin resistance, endothelial function, or steatohepatitis in MetS models. Because not only TXA2 but also other prostanoids, such as prostaglandin H2, can activate the TP receptor, antagonists of the TP receptor may be a better target than inhibiting TXA2 synthesis. In another report, S18886 was shown to improve portal pressure in cirrhosis, resulting in decreased hepatic vascular resistance, suggesting that hepatic endothelial dysfunction is also associated with the TP receptor. 53 From these data, the endothelial MEK/ERK/TP receptor pathway could be a novel therapeutic target for steatohepatitis in patients with MetS.
The present study had several limitations that should be addressed in future studies. Although ERK1 and ERK2 may have the similar structure and targets, they have distinct roles in each organ, the role of ERK1 in MetS in each organ has not been clarified. Systemic ERK1 knockout mice are healthy; however, systemic ERK2 deletion is lethal. Previously, the metabolism in ERK1 knockout mice with HFHSD was reported. ERK1 knockout mice with HFHSD revealed less body weight with improving glucose intolerance and steatohepatitis. In this study, ERK1 knockout mice in adipocyte played a significant role for maturing adipocytes. In our study we first reported that endothelial ERK2 played a role in endothelial function and glucose metabolism in MetS. Further studies are required for the difference roles in endothelial ERK1 and ERK2 in MetS.
Next, we assessed endothelial function only in the aorta, not in microvessels in skeletal muscle or the hepatic microcirculatory system. The balance between endothelium‐derived relaxation factors and endothelium‐derived contraction factors released with insulin could be important for the microcirculatory system.
Sources of Funding
This work was supported by a Grant‐in‐Aid for Scientific Research (B) (18H02815) and Scientific Research (C) (numbers 18K08120, 17K09565, and 17K09596) and a Grant‐in‐Aid from the Ministry of Defense.
Disclosures
None.
Acknowledgments
The authors thank the members of our animal institute for their assistance with animal care and the Central Research Laboratory of the National Defense Medical College for assistance with pathology.
Some of the results of this study were presented at the 90th American Heart Association's Scientific Sessions 2017, Anaheim, California; the 82nd Annual Meeting of the Japanese Circulation Society, Osaka, Japan; the 19th Annual Scientific Meeting of the NO Society of Japan, Kurume, Japan (Young Investigator Award); European Society of Cardiology Congress 2019, Paris, France; and the 3rd Japanese Circulation Society Council Forum on Basic Cardiovascular Research (Young Investigator Award).
For Sources of Funding and Disclosures, see page 15.
REFERENCES
- 1. Malik S, Wong ND, Franklin SS, Kamath TV, L'Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E [DOI] [PubMed] [Google Scholar]
- 2. Reaven G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin North Am. 2004;33:283–303. doi: 10.1016/j.ecl.2004.03.002 [DOI] [PubMed] [Google Scholar]
- 3. Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, Taskinen MR, Groop L. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689. doi: 10.2337/diacare.24.4.683 [DOI] [PubMed] [Google Scholar]
- 4. Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, Salonen JT. The metabolic syndrome and total and cardiovascular disease mortality in middle‐aged men. JAMA. 2002;288:2709–2716. doi: 10.1001/jama.288.21.2709 [DOI] [PubMed] [Google Scholar]
- 5. Vallance P, Collier J, Moncada S. Effects of endothelium‐derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/S0140-6736(89)91013-1 [DOI] [PubMed] [Google Scholar]
- 6. Pierdomenico S, Lapenna D, Di Tommaso R, Di Carlo S, Caldarella M, Neri M, Mezzetti A, Cuccurullo F. Prognostic relevance of metabolic syndrome in hypertensive patients at low‐to‐medium risk. Am J Hypertens. 2007;20:1291–1296. doi: 10.1016/j.amjhyper.2007.06.011 [DOI] [PubMed] [Google Scholar]
- 7. Giovanni T, Luciano Z, Lorenzo B, Felice P, Giancarlo F. Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes. 2005;54:3541–3546. doi: 10.2337/diabetes.54.12.3541 [DOI] [PubMed] [Google Scholar]
- 8. Mancia G, Bombelli M, Corrao G, Facchetti R, Madotto F, Giannattasio C, Trevano F, Grassi G, Zanchetti A, Sega R. Metabolic syndrome in the Pressioni Arteriose Monitorate E Loro Associazioni (PAMELA) study: daily life blood pressure, cardiac damage, and prognosis. Hypertension. 2007;49:40–47. doi: 10.1161/01.HYP.0000251933.22091.24 [DOI] [PubMed] [Google Scholar]
- 9. Kujiraoka T, Satoh Y, Ayaori M, Shiraishi Y, Arai‐Nakaya Y, Hakuno D, Yada H, Kuwada N, Endo S, Isoda K, et al. Hepatic extracellular signal‐regulated kinase 2 suppresses endoplasmic reticulum stress and protects from oxidative stress and endothelial dysfunction. J Am Heart Assoc. 2013;2:e000361. doi: 10.1161/JAHA.113.000361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rask‐Madsen C, Kahn CR. Tissue‐specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2012;32:2052–2059. doi: 10.1161/ATVBAHA.111.241919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gluais P, Paysant J, Badier‐Commander C, Verbeuren T, Vanhoutte PM, Feletou M. In SHR aorta, calcium ionophore A‐23187 releases prostacyclin and thromboxane A2 as endothelium‐derived contracting factors. Am J Physiol Heart Circ Physiol. 2006;291:H2255–H2264. doi: 10.1152/ajpheart.01115.2005 [DOI] [PubMed] [Google Scholar]
- 12. Feletou M, Verbeuren TJ, Vanhoutte PM. Endothelium‐dependent contractions in SHR: a tale of prostanoid TP and IP receptors. Br J Pharmacol. 2009;156:563–574. doi: 10.1111/j.1476-5381.2008.00060.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clavreul N, Adachi T, Pimental DR, Ido Y, Schoneich C, Cohen RA. S‐glutathiolation by peroxynitrite of p21ras at cysteine‐118 mediates its direct activation and downstream signaling in endothelial cells. FASB J. 2006;20:518–520. doi: 10.1096/fj.05-4875fje [DOI] [PubMed] [Google Scholar]
- 14. Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. s‐glutathiolation of Ras mediates redox‐sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200 [DOI] [PubMed] [Google Scholar]
- 15. Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate‐1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ Res. 2004;94:1211–1218. doi: 10.1161/01.RES.0000126501.34994.96 [DOI] [PubMed] [Google Scholar]
- 16. Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–457. doi: 10.1172/JCI5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sesti G, Federici M, Hribal M, Lauro D, Sbraccia P, Lauro R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 2001;15:2099–2111. doi: 10.1096/fj.01-0009rev [DOI] [PubMed] [Google Scholar]
- 18. David Vicent JI, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL, Kahn CR. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest. 2003;111:1373–1380. doi: 10.1172/JCI200315211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rong Li HZ, Wang W, Wang X, Huang Y, Huang C, Gao F. Vascular insulin resistance in prehypertensive rats: role of PI3‐kinase/Akt/eNOS signaling. Eur J Pharmacol. 2010;628:140–147. doi: 10.1016/j.ejphar.2009.11.038 [DOI] [PubMed] [Google Scholar]
- 20. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0 [DOI] [PubMed] [Google Scholar]
- 21. Vial G, Chauvin MA, Bendridi N, Durand A, Meugnier E, Madec AM, Bernoud‐Hubac N, Pais de Barros JP, Fontaine É, Acquaviva C, et al. Imeglimin normalizes glucose tolerance and insulin sensitivity and improves mitochondrial function in liver of a high‐fat, high‐sucrose diet mice model. Diabetes. 2015;64:2254–2264. doi: 10.2337/db14-1220 [DOI] [PubMed] [Google Scholar]
- 22. Satoh Y, Endo S, Nakata T, Kobayashi Y, Yamada K, Ikeda T, Takeuchi A, Hiramoto T, Watanabe Y, Kazama T. ERK2 contributes to the control of social behaviors in mice. J Neurosci. 2011;31:11953–11967. doi: 10.1523/JNEUROSCI.2349-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999 [DOI] [PubMed] [Google Scholar]
- 24. Dong YF, Liu L, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, Nako H, Ogawa H, Kim‐Mitsuyama S. Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes. Diabetologia. 2010;53:180–191. doi: 10.1007/s00125-009-1575-5 [DOI] [PubMed] [Google Scholar]
- 25. Miller FJ, Gutterman D, Rios C, Heistad D, Davidson B. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.RES.82.12.1298 [DOI] [PubMed] [Google Scholar]
- 26. Zuccollo A, Shi C, Mastroianni R, Maitland‐Toolan KA, Weisbrod RM, Zang M, Xu S, Jiang B, Oliver‐Krasinski JM, Cayatte AJ, et al. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circulation. 2005;112:3001–3008. doi: 10.1161/CIRCULATIONAHA.105.581892 [DOI] [PubMed] [Google Scholar]
- 27. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp‐Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701 [DOI] [PubMed] [Google Scholar]
- 28. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP‐activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hutchinson PJ, Palmer RM, Moncada S. Comparative pharmacology of EDRF and nitric oxide on vascular strips. Eur J Pharmacol. 1987;141:445–451. doi: 10.1016/0014-2999(87)90563-2 [DOI] [PubMed] [Google Scholar]
- 30. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532 [DOI] [PubMed] [Google Scholar]
- 31. Zou M‐H, Shi C, Cohen RA. Oxidation of the zinc‐thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–826. doi: 10.1172/jci0214442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease ‐ a 30th anniversary update. Acta Physiol (Oxf). 2017;219:22–96. doi: 10.1111/apha.12646 [DOI] [PubMed] [Google Scholar]
- 33. Vincent MACL, Lindner JR, Klibanov AL, Clark MG, Rattigan S, Barrett EJ. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes. 2004;53:1418–1423. doi: 10.2337/diabetes.53.6.1418 [DOI] [PubMed] [Google Scholar]
- 34. Tang YHA, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48(9):563–567. doi: 10.1002/dvg.20654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shimokawa H, Flavahan NA, Vanhoutte PM. Loss of endothelial pertussis toxin‐sensitive G protein function in atherosclerotic porcine coronary arteries. Circulation. 1991;83:652–660. doi: 10.1161/01.cir.83.2.652 [DOI] [PubMed] [Google Scholar]
- 36. Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium‐derived relaxing factor. Am J Physiol. 1986;250:H822–H827. doi: 10.1152/ajpheart.1986.250.5.H822 [DOI] [PubMed] [Google Scholar]
- 37. Masaki N, Ido Y, Yamada T, Yamashita Y, Toya T, Takase B, Hamburg NM, Adachi T. Endothelial insulin resistance of freshly isolated arterial endothelial cells from radial sheaths in patients with suspected coronary artery disease. J Am Heart Assoc. 2019;8:e010816. doi: 10.1161/JAHA.118.010816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sugamura K, Keaney JJ. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gokce N, Keaney JF Jr, Hunter LM, Watkins MT, Menzoian JO, Vita JA. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: a prospective study. Circulation. 2002;105:1567–1572. doi: 10.1161/01.cir.0000012543.55874.47 [DOI] [PubMed] [Google Scholar]
- 40. Zhang M, Song P, Xu J, Zou MH. Activation of NAD(P)H oxidases by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2011;31:125–132. doi: 10.1161/ATVBAHA.110.207712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding L, Chapman A, Boyd R, Wang H. ERK activation contributes to regulation of spontaneous contractile tone via superoxide anion in isolated rat aorta of angiotensin II‐induced hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H2997–H3005. doi: 10.1152/ajpheart.00388.2006 [DOI] [PubMed] [Google Scholar]
- 42. Lynch CM, Kinzenbaw DA, Chen X, Zhan S, Mezzetti E, Filosa J, Ergul A, Faulkner JL, Faraci FM, Didion SP. Nox2‐derived superoxide contributes to cerebral vascular dysfunction in diet‐induced obesity. Stroke. 2013;44:3195–3201. doi: 10.1161/STROKEAHA.113.001366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Du J, Fan LM, Mai A, Li JM. Crucial roles of Nox2‐derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle‐aged mice. Br J Pharmacol. 2013;170:1064–1077. doi: 10.1111/bph.12336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aisaka K, Gross SS, Griffith OW, Levi R. NG‐methylarginine, an inhibitor of endothelium‐derived nitric oxide synthesis, is a potent pressor agent in the Guinea pig: does nitric oxide regulate blood pressure in vivo? Biochem Biophys Res Commun. 1989;160:881–886. doi: 10.1016/0006-291x(89)92517-5 [DOI] [PubMed] [Google Scholar]
- 45. Goodyear L, Giorgino F, Sherman L, Carey J, Smith R, Dohm G. Insulin receptor phosphorylation, insulin receptor substrate‐1 phosphorylation, and phosphatidylinositol 3‐kinase activity are decreased in intact skeletal muscle strips from obese subjects. J Clin Invest. 1995;95:2195–2204. doi: 10.1172/JCI117909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vanhoutte PM, Boulanger CM. Endothelium‐dependent responses in hypertension. Hypertens Res. 1995;18:87–98. doi: 10.1291/hypres.18.87 [DOI] [PubMed] [Google Scholar]
- 47. Yki‐Järvinen H. Non‐alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2:901–910. doi: 10.1016/S2213-8587(14)70032-4 [DOI] [PubMed] [Google Scholar]
- 48. Kujiraoka T, Kagami K, Kimura T, Ishinoda Y, Shiraishi Y, Ido Y, Endo S, Satoh Y, Adachi T. Metabolic remodeling with hepatosteatosis induced vascular oxidative stress in hepatic ERK2 deficiency mice with high fat diets. Int J Mol Sci. 2022;23:8521. doi: 10.3390/ijms23158521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pasarin M, Abraldes J, Liguori E, Kok B, La Mura V. Intrahepatic vascular changes in non‐alcoholic fatty liver disease: potential role of insulin‐resistance and endothelial dysfunction. World J Gastroenterol. 2017;23:6777–6787. doi: 10.3748/wjg.v23.i37.6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ijaz S, Yang W, Winslet MC, Seifalian AM. The role of nitric oxide in the modulation of hepatic microcirculation and tissue oxygenation in an experimental model of hepatic steatosis. Microvasc Res. 2005;70:129–136. doi: 10.1016/j.mvr.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 51. Selzner M, Rudiger HA, Sindram D, Madden J, Clavien PA. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000;32:1280–1288. doi: 10.1053/jhep.2000.20528 [DOI] [PubMed] [Google Scholar]
- 52. Worth N, Berry C, Thomas A, Campbell J. S18886, a selective TP receptor antagonist, inhibits development of atherosclerosis in rabbits. Atherosclerosis. 2005;183:65–73. doi: 10.1016/j.atherosclerosis.2005.02.034 [DOI] [PubMed] [Google Scholar]
- 53. Rosado E, Rodriguez‐Vilarrupla A, Gracia‐Sancho J, Tripathi D, Garcia‐Caldero H, Bosch J, Garcia‐Pagan J. Terutroban, a TP‐receptor antagonist, reduces portal pressure in cirrhotic rats. Hepatology. 2013;58:1424–1435. doi: 10.1002/hep.26520 [DOI] [PubMed] [Google Scholar]