

Abstract
Autoimmune pulmonary alveolar proteinosis (PAP) is a rare disease characterized by myeloid cell dysfunction, abnormal pulmonary surfactant accumulation, and innate immune deficiency. It has a prevalence of 7–10 per million; occurs in individuals of all races, geographic regions, sex, and socioeconomic status; and accounts for 90% of all patients with PAP syndrome. The most common presentation is dyspnea of insidious onset with or without cough, production of scant white and frothy sputum, and diffuse radiographic infiltrates in a previously healthy adult, but it can also occur in children as young as 3 years. Digital clubbing, fever, and hemoptysis are not typical, and the latter two indicate that intercurrent infection may be present. Low prevalence and nonspecific clinical, radiological, and laboratory findings commonly lead to misdiagnosis as pneumonia and substantially delay an accurate diagnosis. The clinical course, although variable, usually includes progressive hypoxemic respiratory insufficiency and, in some patients, secondary infections, pulmonary fibrosis, respiratory failure, and death. Two decades of research have raised autoimmune PAP from obscurity to a paradigm of molecular pathogenesis-based diagnostic and therapeutic development. Pathogenesis is driven by GM-CSF (granulocyte/macrophage colony–stimulating factor) autoantibodies, which are present at high concentrations in blood and tissues and form the basis of an accurate, commercially available diagnostic blood test with sensitivity and specificity of 100%. Although whole-lung lavage remains the first-line therapy, inhaled GM-CSF is a promising pharmacotherapeutic approach demonstrated in well-controlled trials to be safe, well tolerated, and efficacious. Research has established GM-CSF as a pulmonary regulatory molecule critical to surfactant homeostasis, alveolar stability, lung function, and host defense.
Keywords: granulocyte/macrophage colony–stimulating factor, autoantibodies, alveolar macrophages, surfactant, BAL
Contents
Epidemiology
-
Pathogenesis
Historical Perspective
Lung Abnormalities
Surfactant Abnormalities
GM-CSF Signaling Disruption
Macrophage Abnormalities
Neutrophil Abnormalities
Other Cell Abnormalities
-
Natural History
Presentation
Clinical Course
Secondary Infections
Pulmonary Fibrosis
-
Evaluation and Diagnosis
Pulmonary Function Tests
Radiological Evaluation
Bronchoscopy Findings
GM-CSF Autoantibody Test
GM-CSF Signaling Tests
Other Laboratory Tests
Genetic Tests
Lung Biopsy
Diagnosis
-
Therapy
Therapeutic Lung Lavage
GM-CSF Augmentation
Other Treatment Approaches
Conclusions
Autoimmune pulmonary alveolar proteinosis (PAP) is a rare disease characterized by progressive accumulation of surfactant in pulmonary alveoli and resulting hypoxemia (Figure 1), disruption of GM-CSF (granulocyte/macrophage colony–stimulating factor) signaling, alveolar macrophage and neutrophil dysfunction, innate immunodeficiency, and in some individuals serious secondary infections, pulmonary fibrosis, respiratory failure, and death (1–6). It belongs to a heterogeneous group of distinct PAP-causing diseases defined by a shared cardinal feature, the abnormal accumulation of pulmonary alveolar surfactant; the constellation of surfactant accumulation and associated symptoms and signs is referred to as PAP syndrome prior to disease-specific diagnosis (7).
Figure 1.

Schematic depicting the anatomy and biology of the alveolus in health (left) and autoimmune pulmonary alveolar proteinosis (PAP) (right). Normally, alveolar structure and function are maintained by a surfactant layer sufficiently thick (about one to three molecules) to reduce surface tension and thin enough to permit adequate diffusion of oxygen across the alveolar wall. Homeostasis is maintained by balanced secretion of surfactant by alveolar type II cells and clearance by type II cells and by alveolar macrophages, which require GM-CSF (granulocyte/macrophage colony–stimulating factor) to export cholesterol normally. In autoimmune PAP, autoantibodies bind and block GM-CSF from activating receptors on alveolar macrophages (and other cells, e.g., neutrophils). Because surfactant uptake by alveolar macrophages is GM-CSF independent and export of surfactant-derived cholesterol is GM-CSF dependent, cholesterol accumulates within the cytoplasm. Alveolar macrophages esterify and sequester cholesterol within intracytoplasmic vesicles (as a cell-protective mechanism), resulting in foamy-appearing cells with secondary impairment of multiple macrophage functions, including phagocytosis and surfactant clearance. Over time, alveoli fill with insoluble surfactant sediment, cellular debris, cholesterol crystals, and several cytokines (indicated; see text). A thickened surfactant layer and surfactant-filled alveoli contribute to reduced oxygen delivery, resulting in hypoxemia, dyspnea, and in severe cases polycythemia, respiratory failure, and death. Pulmonary fibrosis (not shown) also occurs in some patients. See text for further details. MCP-1 = monocyte chemoattractant protein-1; M-CSF = macrophage colony–stimulating factor.
Historically, the reporting of PAP-causing diseases was challenged by their overlapping radiographic appearance; histopathology; pathophysiology; symptomatology; clinical presentation in men, women, children, and infants; and lack of disease-specific diagnostic tests. Nevertheless, they are usefully grouped mechanistically as disorders of surfactant clearance or surfactant production (7). The clearance disorders can be further divided into those caused by disruption of GM-CSF signaling (primary PAP) or by an underlying condition that reduces either the number and/or the functions of alveolar macrophages (secondary PAP). The disorders of surfactant production constitute a group of genetic diseases resulting in either insufficient or dysfunctional surfactant (congenital PAP or pulmonary surfactant metabolic dysfunction disorders). Other systems of nomenclature have also been proposed (3, 8).
Advances over the past two decades have improved our understanding of autoimmune PAP, led to novel methods for its diagnosis and treatment, and, importantly, defined the critical role pulmonary GM-CSF plays in alveolar macrophage specification, pulmonary surfactant homeostasis, alveolar structure, lung function, host defense, and inflammatory and autoimmune diseases. Consequently, in this review we focus on autoimmune PAP.
Epidemiology
A national PAP registry study in Japan identified 248 adult patients with PAP syndrome, of whom 223 (89.9%) had autoimmune PAP (on the basis of GM-CSF autoantibody testing; see below), 24 (9.7%) had secondary PAP, and 1 (0.4%) had unclassified PAP; pediatric cases were not counted (9). The incidence and prevalence of autoimmune PAP were estimated at 0.49 ± 0.13 and 6.7 per million, respectively. A study conducted in the United States on the basis of comprehensive health insurance claims data for 15 million covered individuals estimated the prevalence of PAP syndrome to be 6.87 per million in the general population, similar to the Japanese estimate (10). Notwithstanding agreement of these estimates, 69 (31%) of the Japanese patients were identified through mandatory health screening and were asymptomatic at diagnosis, suggesting that symptom-based ascertainment may underestimate detection. Indeed, a recent study using a Poisson-based analysis of annual incidence data returned values of 1.65 and 26.6 per million for the incidence and prevalence of autoimmune PAP, respectively (11). Analysis of 1,045 different patients reported in several large studies confirmed that autoimmune PAP accounts for 90% of cases (3, 9, 12–14). Age and smoking are risk factors (3). Although occupational inhalation exposure contributes to the development of secondary PAP, its contribution to autoimmune PAP is less clear (15). Several studies have sought a genetic predisposition to the development of autoimmune PAP (16, 17); one evaluating 198 patients with autoimmune PAP and 395 control subjects of Japanese ancestry identified two major histocompatibility complex alleles as independent risk factors: HLA-DRB1*08:03 (odds ratio, 5.2; P = 4.8 × 10−12) and HLA-DPβ1 (odds ratio, 0.28; P = 3.7 × 10−7) (17). In summary, autoimmune PAP accounts for about 90% of all cases of PAP syndrome; occurs in men, women, and children of all ethnic backgrounds and geographic locations, independent of sex, race, and socioeconomic status; and has an estimated prevalence of 7–10 (possibly up to 26) per million in the general population.
Pathogenesis
Historical Perspective
PAP was first recognized as a disorder of pulmonary surfactant accumulation in the mid-1950s (1), shortly after surfactant was discovered (18). Subsequently, the observation of enlarged, foamy, lipid-laden alveolar macrophages with reduced survival, chemotaxis, adhesion, and microbial killing suggested that the alveolar microenvironment was abnormal (19). The concept that an abnormal protein may mediate pathogenesis was supported by reports that lung lavage from patients with PAP blocked the proliferation of monocytes (20) and contained a 40-kD macrophage function–inhibiting protein (21) and that whole-lung lavage (WLL) therapy improved alveolar macrophage functions (22). The observation that PAP spontaneously develops in gene-knockout mice deficient in GM-CSF serendipitously implicated the absence of GM-CSF signaling in the pathogenesis of PAP (23, 24). Subsequently, the “inhibitory factor” in PAP lung lavage (and serum) was found to be polyclonal immunoglobulin that bound and neutralized GM-CSF (25, 26). Subsequent studies characterized these antibodies and established their role in what is considered an autoimmune disease specifically targeting GM-CSF (2, 27–30).
Lung Abnormalities
The cardinal histopathological feature of autoimmune PAP on light microscopy of a lung biopsy sample is near complete filling of alveoli with acellular, eosinophilic, periodic acid–Schiff–positive sediment (1, 3, 7). Interstitial B- and T-cell lymphocyte infiltration is also typically present but is somewhat variable (1, 31, 32). Importantly, the alveolar architecture is usually well preserved except when pulmonary fibrosis is also present (7), which may occur late in the clinical course. Electron microscopic examination reveals the presence of surfactant sediment and foamy alveolar macrophages (33, 34).
Surfactant Abnormalities
Normally, pulmonary surfactant is composed of approximately 80% polar lipids, 10% neutral lipids, and 10% proteins (35). The polar lipid fraction is composed of ∼75% phosphatidylcholine and ∼10% phosphatidylglycerol by mass and lesser amounts of other species (35); half of the phosphatidylcholine is unsaturated. The neutral lipid fraction is composed of ∼90% cholesterol and lesser amounts of mono-, di-, and triacylglycerol and free fatty acids, mainly palmitate (36). The protein fraction includes four surfactant protein (SP) species, SP-A, SP-B, SP-C, and SP-D (37, 38). Anatomically, surfactant normally comprises a multilaminar film on the alveolar wall thick enough to reduce the surface tension on the alveolar wall and prevent alveolar collapse and thin enough to permit sufficient diffusion of gas (39). SP-B and SP-C are lipophilic and contribute to the surface-active biophysical properties of surfactant, while SP-A and SP-D are hydrophilic and contribute to lung host defense (40–43). The cholesterol content determines surfactant layer fluidity (36, 44, 45), which is regulated dynamically, for example, during extremes of atmospheric temperature in reptiles (46), during hibernation (47), and during exercise in humans (48). Surfactant homeostasis is maintained by tightly balanced secretion of surfactant lipids and proteins from type 2 alveolar epithelial cells and clearance in roughly equal proportions both by type 2 cells (through recycling and catabolism) and by alveolar macrophages (through catabolism of phospholipids and proteins and export of cholesterol) (49, 50).
In autoimmune PAP, alveolar surfactant accumulation is due to an increase in polar lipids and an even greater increase in nonpolar lipids, primarily cholesterol and esterified cholesterol (51, 52). Early studies demonstrating preservation of the relative proportions of various phospholipid species normally present in surfactant (53, 54) led to a widely held belief that surfactant composition was essentially normal in PAP except for uncleared cytokines and cellular debris (55). However, simultaneous evaluation of polar and neutral lipids revealed that cholesterol was increased relatively more than phospholipid in autoimmune PAP (51, 52). Quantitative lipidomic analysis on the basis of mass spectrometry confirmed this increase in cholesterol-to-phospholipid ratio in autoimmune PAP and identified some other changes in lipid composition in autoimmune PAP (56). The cholesterol-to-phospholipid ratio in surfactant is also increased in several other PAP-causing diseases (56) (B. Trapnell, unpublished results) and may represent a common biochemical manifestation caused by the disruption of macrophage-mediated cholesterol clearance by different mechanisms.
GM-CSF Signaling Disruption
GM-CSF is a 23-kD dimeric glycoprotein cytokine identified in the 1970s, cloned in 1984 (57), and studied intensely because of its ability to stimulate the proliferation and differentiation of neutrophilic and monocyte/macrophage lineage colonies from hematopoietic cells in vitro (58–60). GM-CSF is present in most if not all mammalian species (59); expressed by a variety of cell types, including lung epithelial cells (60, 61); and widely recognized for its role in inflammation, autoimmunity, and host defense (62, 63). Human and nonhuman primate GM-CSF are highly homologous (95%) and functionally interchangeable, whereas human and mouse GM-CSF share little homology (55%) and are not immunologically cross-reactive or functionally interchangeable (64–67). Despite structural differences, GM-CSF regulates myeloid cell functions in a strikingly similar fashion in all three species (27–29), highlighting evolutionary conservation and the importance of GM-CSF–regulated functions. Pulmonary GM-CSF stimulates the survival, proliferation, differentiation, and functions of alveolar macrophages (68–71) and in mice is essential for establishing this population at birth (72, 73), for maintaining it by self-renewal throughout normal adult life (74), and for determining population size through a reciprocal feedback loop (75). Notwithstanding, physiological concentrations of GM-CSF are low and often undetectable (76–79).
The pleiotropic, dose-dependent effects of GM-CSF are mediated by heterodimeric cell-surface receptors composed of a low-affinity GM-CSF–binding α chain (CDw116), an affinity-enhancing β chain (CD131) (both without known enzymatic activity [80]), and the β chain–associated Janus-activating kinase 2 (JAK2), which has tyrosine phosphorylation activity (81). Ligand binding causes the assembly of GM-CSF/α/β/JAK2 dodecameric (and higher order) complexes, proximity-dependent reciprocal phosphorylation and activation of JAK2, phosphorylation of the α (82) and β (83, 84) chains, and activation of multiple intracellular signaling pathways (85–88). Signaling via the β chain is mediated by phosphorylation of either a serine or a tyrosine residue in a cytoplasmic tail motif, which functions as a molecular switch converting an analog input (GM-CSF extracellular concentration) into a binary output (signaling via phospho-Ser585 or phospho-Tyr577) (89). Phosphorylation of Ser585 occurs in GM-CSF dose–dependent, unimodal fashion with a peak at 1 pM, while phosphorylation of Tyr577 occurs as a GM-CSF dose–dependent, exponential rise to a maximum starting at 1 pM and plateauing at ∼100 pM (89). Low concentrations of GM-CSF initiate signaling via Ser585 with recruitment and phosphorylation-mediated activation of the transcription factor 14-3-3 and are associated with alveolar macrophage survival and differentiation (69, 89). High concentrations initiate signaling via Tyr577 with recruitment and phosphorylation-mediated activation of extracellular signal–regulated kinase and STAT5 (signal transducer and activator of transcription 5) and are associated with survival, proliferation, and activation (86, 89). GM-CSF receptors are expressed on alveolar macrophages and a number of other cells, including monocytes, granulocytes, lymphocytes, and alveolar epithelial cells. The β chain is also common to IL-3 and IL-5 receptors, which may operate by a similar mechanism (86, 90–92).
Mice deficient in either GM-CSF or the α or β subunit of its receptor (Csf2KO, Csf2raKO, or Csf2rbKO mice, respectively) spontaneously develop PAP similar to autoimmune PAP regarding physiological, radiological, histopathological, biochemical, and immunological manifestations (23, 24, 93, 94). These mice are also susceptible to a broad range of microbial pathogens (23, 71, 95–99) and have increased infection-related mortality (3, 27, 95). PAP is reversed in Csf2KO mice by expression of GM-CSF in the lungs (100–102), in Csf2rbKO mice by transplantation of normal bone marrow (103, 104), and in Csf2rbKO and Csf2raKO mice by pulmonary macrophage transplantation (75, 105). GM-CSF is present at very low or undetectable (but nonzero) concentrations in the lungs normally (69); however, its absence causes PAP, and constitutive high concentrations cause marked accumulation of alveolar macrophages and a phenotype resembling desquamative interstitial pneumonitis (106). Polycythemia develops over time in the absence of GM-CSF signaling (75).
These results identify alveolar macrophages as the cellular site of pathogenesis in models of experimental PAP and identify pulmonary GM-CSF as a low-abundance, albeit critical, regulator of surfactant homeostasis, alveolar structure, lung function, and host defense.
In 1999, Nakata’s group observed GM-CSF–specific autoantibodies in patients with acquired (idiopathic) PAP but not in patients with secondary PAP or other lung diseases or in healthy people (25, 26), a finding confirmed in subsequent studies (2, 3, 9, 107, 108). The GM-CSF autoantibodies were polyclonal, were composed predominantly of IgG1 and IgG2, were highly specific for GM-CSF, bound epitopes throughout the molecule with high affinity (KD = ∼20 pM) exceeding that of the GM-CSF receptor α-chain binding (KD = ∼1–10 nM), and neutralized GM-CSF at concentrations thousands of times higher than physiological concentrations of GM-CSF (26, 107, 109). Yet, GM-CSF autoantibody concentrations do not correlate with lung disease severity in patients with PAP (108). Furthermore, GM-CSF autoantibodies were later found to be ubiquitous in healthy people, albeit at concentrations far lower than in patients with PAP (109), and were also present in pharmaceutical immunoglobulin prepared from healthy individuals (30, 110).
A “Koch’s postulates” approach was used to demonstrate that a high concentration of GM-CSF autoantibodies mediated the pathogenesis of PAP (2, 27–29). Results showed that GM-CSF autoantibodies were present at high concentrations only in patients with (then) idiopathic PAP (2), could be isolated in pure form from these patients (27), reproduced the cardinal features of PAP after injection into healthy nonhuman primates (28), and had the same functional capacity after recovery directly from these patients or from passively immunized nonhuman primates (29). These data provided strong evidence that GM-CSF autoantibodies were in fact the pathogenic driver of idiopathic PAP, which is now recognized as “autoimmune PAP” (30).
The lack of correlation between disease severity and serum GM-CSF autoantibody concentrations in patients with PAP was reconciled by the identification of a critical threshold of GM-CSF autoantibodies (5 μg/ml serum) required for an increased risk of autoimmune PAP (108, 109). Values above this threshold are strongly associated with the development of autoimmune PAP in humans (109) and passively immunized nonhuman primates (29); below this threshold, GM-CSF signaling is reduced in proportion to the concentration of GM-CSF autoantibodies in humans (109) and passively immunized nonhuman primates (29). Furthermore, the discovery of neutralizing GM-CSF autoantibodies in patients with PAP also provided an explanation for the absence of a GM-CSF–stimulated increased in peripheral blood neutrophils (111), which is normally seen in healthy people (60).
Although the etiology of the increased GM-CSF autoantibody concentrations in patients with autoimmune PAP remains unknown, detailed studies of patient-derived monoclonal GM-CSF autoantibody clones revealed that they use multiple immunoglobulin V genes and target at least multiple nonoverlapping epitopes, suggesting that GM-CSF itself, and not a cross-reactive, pathogen-related B-cell epitope, is responsible (112).
Macrophage Abnormalities
In patients with autoimmune PAP and mice deficient in GM-CSF signaling, alveolar macrophages are abnormally large and foamy, have reduced viability, and have impaired functions, including reduced expression of multiple receptors, proinflammatory cytokine responses, phagocytosis, microbial killing, cell adhesion, and clearance of surfactant and debris from the alveolar surface (2, 19, 53, 70, 71, 113–116). In Csf2KO mice, all these defects can be corrected by restoring GM-CSF expression specifically in the lungs (100–102). Pharmacologic administration of inhaled GM-CSF also corrects alveolar macrophage abnormalities in patients with autoimmune PAP (32, 114).
Without GM-CSF, human and murine alveolar macrophages internalize and process surfactant-derived phospholipids and proteins but are unable to export surfactant-derived cholesterol normally (51, 52). Consequently, cytoplasmic cholesterol accumulates and is esterified and sequestered in intracytoplasmic lipid droplets (as a cellular protective mechanism) resulting in the characteristic foamy appearance and enhanced oil red O staining (7). GM-CSF–deficient bone marrow–derived macrophages have abnormalities similar to those of alveolar macrophages except without the foamy appearance, which can be induced by exposure to cholesterol-containing surfactant but not cholesterol-free surfactant (51). GM-CSF withdrawal slows the clearance of cholesterol-containing surfactant (the half-life increases from 1.75 to 21 h) but does not affect the clearance of cholesterol-free surfactant (51). As lipid droplets accumulate, the cells become increasingly disabled and unable to clear additional surfactant.
Cholesterol export by macrophages is mediated by several pathways, including membrane transporters (ABCG1 [ATP-binding cassette subfamily G member 1], ABCA1 [ATP-binding cassette subfamily A member 1], SRB1 [scavenger receptor class B type 1]), fat-binding protein carriers (ApoE [apolipoprotein E]), and cholesterol-modifying enzymes (sterol-27-hydroxylase) (117–121). Despite this redundancy, several observations suggest that ABCG1 is critical for cholesterol export in alveolar macrophages. First, ABCG1 is abundant in normal alveolar macrophages but reduced in alveolar macrophages from patients with autoimmune PAP and GM-CSF signaling–deficient mice (119). Second, ABCG1-deficient mice develop a severe form of PAP lung disease (122). Third, GM-CSF stimulates Abcg1 gene expression and cholesterol export in macrophages in a reversible, rheostatic, and dose-dependent fashion (51).
GM-CSF signaling requires the master macrophage transcription factor PU.1 (purine box binding protein 1) to regulate the differentiation and functions of human and mouse alveolar macrophages (69, 114). Numerous impaired functions of GM-CSF–deficient macrophages, including surfactant clearance, can be restored by vector-mediated, forced expression of PU.1 in the absence of GM-CSF (69). The lipid metabolism–related transcription factor PPARγ (peroxisome proliferator–activated receptor γ) is also required for specification of alveolar macrophages but not for development of macrophages in the peritoneum, liver, brain, heart, kidneys, intestine, and fat (123). Both transcription factors regulate ABCG1 gene expression and are required for macrophage-mediated surfactant clearance and export of cholesterol from macrophages (51, 52, 124–126). Conditional disruption of Pparγ gene expression in mice simultaneously reduces expression of Abcg1, causes cholesterol accumulation in alveolar macrophages, and causes the development of PAP (125). Nevertheless, although GM-CSF is required for differentiation of alveolar macrophages, the specific intracellular signaling pathways involved are incompletely defined, and how their disruption impairs alveolar macrophage functions in PAP is poorly understood.
Neutrophil Abnormalities
Neutrophils from patients with autoimmune PAP and GM-CSF–deficient mice have normal morphology, ultrastructure, and phenotypic marker expression, suggesting that they are fully differentiated; however, they have impaired host defense functions, including phagocytosis, adhesion, oxidative burst capacity, and microbial killing (27). Primates injected with patient-derived GM-CSF autoantibodies also have morphologically normal but functionally impaired neutrophils (29). In vitro exposure of normal human neutrophils to GM-CSF autoantibodies also causes functional impairment (27). Studies in humans, nonhuman primates, and mice indicate that GM-CSF constitutively regulates the basal functional capacity of neutrophils in vivo in rheostatic fashion (27). Functionally impaired neutrophils contribute to innate immune deficiency and provide an explanation for the increased infection risk observed in humans and mice with PAP caused by disruption of GM-CSF signaling (27).
Other Cell Abnormalities
Interstitial lymphoid hyperplasia is a histopathological feature of PAP caused by disruption of GM-CSF signaling in humans and mice (1, 31, 32). Although increased pulmonary concentrations of MCP-1 (monocyte chemoattractant protein-1) may contribute to pulmonary lymphocyte accumulation, the precise mechanism(s) responsible have not been defined. Type II alveolar epithelial cells normally express GM-CSF receptors and respond to pulmonary overexpression of GM-CSF by proliferating and becoming hyperplastic (127). However, no major abnormalities of alveolar epithelial cells have been reported in humans or mice with PAP caused by disruption of GM-CSF signaling.
Natural History
Presentation
Autoimmune PAP usually presents as dyspnea of insidious onset between the third and fifth decades of life, although it can present in children as young as 3 years and adults as old as 90 years (7, 9, 13). Most patients describe exertional dyspnea that progresses to resting dyspnea over months to years. Some patients also present with throat clearing, chest pain, chest congestion, or production of scant whitish sputum (Figure 2A). Fever and hemoptysis are uncommon unless secondary infection is also present (7). In the Japanese national registry, 31% of patients were asymptomatic at diagnosis and were identified incidentally by mandatory health screening (9). Generally, the physical examination is unremarkable but can identify crackles or even cyanosis in severe cases. Digital clubbing is not a feature of autoimmune PAP.
Figure 2.
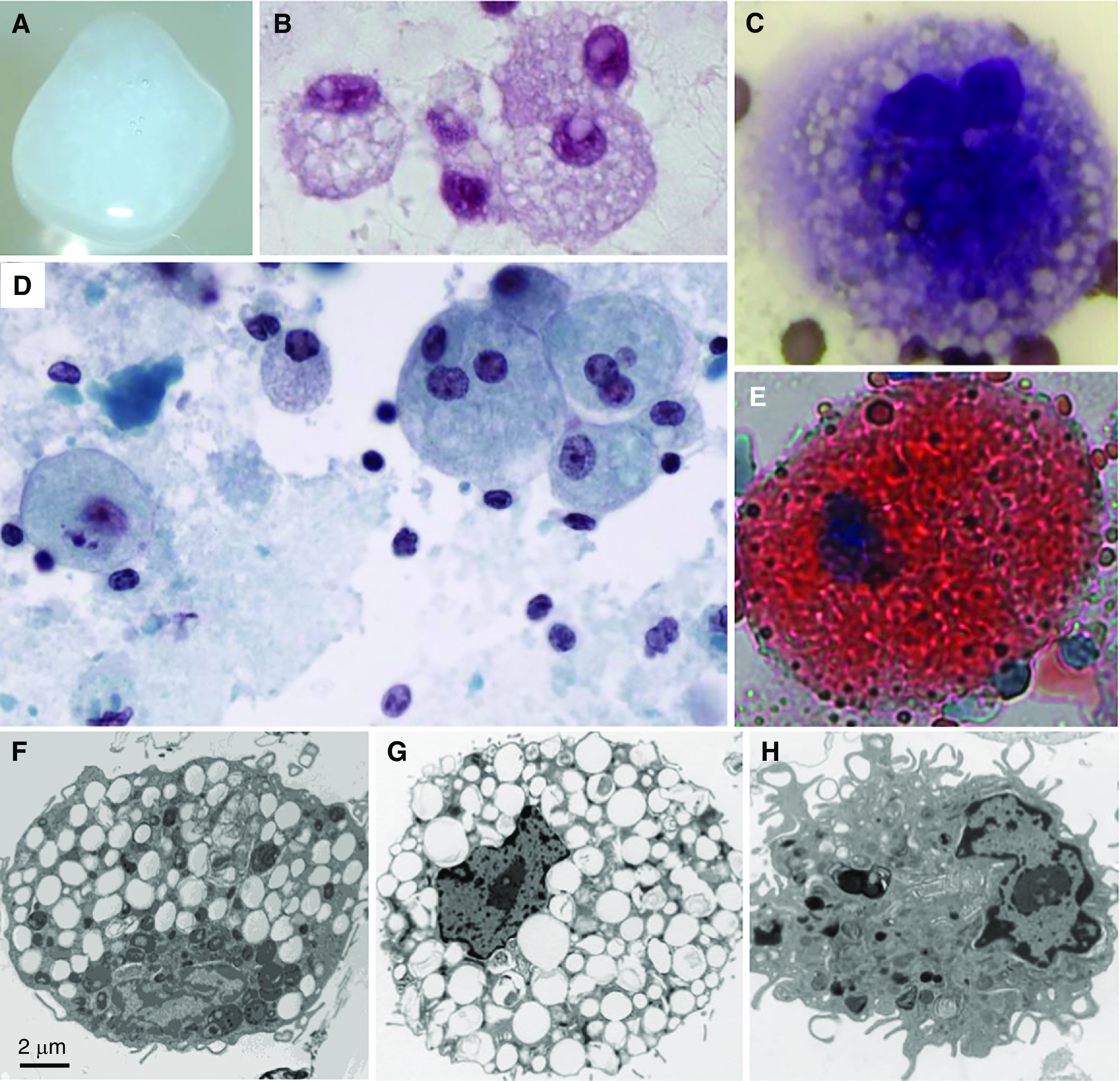
Sputum, sputum cytology, and alveolar macrophage ultrastructure in autoimmune pulmonary alveolar proteinosis (PAP). (A) Gross appearance of freshly expectorated sputum from a patient with autoimmune PAP. (B–E) Microscopic appearance of sputum cytology after staining with periodic acid–Schiff reagent (B); Diff-Quick, showing a foamy alveolar macrophage (C); Papanicolaou reagent (D); or oil red O, showing an oil red O–positive alveolar macrophage (E). (F) Alveolar macrophage obtained by BAL from an 18-year-old man with autoimmune PAP several months after lung transplantation performed as therapy for pulmonary fibrosis. Note the numerous, single membrane–delimited, intracytoplasmic lipid droplets. Scale bar, 2 μm. (G and H) Uranyl acetate staining. Alveolar macrophages obtained from a nonhuman primate passively immunized with highly purified, human GM-CSF (granulocyte/macrophage colony–stimulating factor) autoantibodies derived from patients with autoimmune PAP, showing intracytoplasmic lipid droplets (G), or a saline-injected primate, showing a normal macrophage appearance (H).
Clinical Course
No longitudinal studies on the natural history of PAP have been reported, but several large cross-sectional cohorts have provided some information (3, 9, 12–14). The clinical course of autoimmune PAP comprises three patterns: stable disease, progressive deterioration, and spontaneous resolution (2). In the largest study reported to date, a meta-analysis of 343 patients with PAP revealed survival rates of 78% at 2 years, 75% at 5 years, and 68% at 10 years; more than 80% of deaths occurred within 12 months of diagnosis, and respiratory failure was the most common cause of death, followed by uncontrolled infection (3). In a separate cross-sectional cohort of 223 patients with autoimmune PAP, no deaths occurred over the 5-year period of the study (9). Spontaneous alleviation of autoimmune PAP appears to occur; indeed it was reported in the initial description of PAP syndrome (1), and in the largest meta-analysis to date, 7.9% of cases were reported to have experienced spontaneous resolution (3). In contemporary studies from cohorts in Japan, China, Germany, and Italy, it is estimated that spontaneous resolution may occur in 5–7% of patients (9, 12–14).
Secondary Infections
Autoimmune PAP is associated with an increased risk of serious infections, consistent with the observed impaired innate immune functions of myeloid cells, which include reduced phagocytosis, microbial killing, proinflammatory signaling, and others (27, 69, 95, 109, 128). Causative organisms include community-acquired and opportunistic microbial pathogens (4, 27). Commonly identified opportunistic pathogens include Nocardia spp., Mycobacteria, and Aspergillus, and resulting infections are associated with a poor prognosis and increased mortality (129). Secondary infections are relatively common, account for 18–20% of deaths attributable to PAP, and occur at presentation or during the clinical course, at intra- or extrapulmonary sites, consistent with the systemic innate immunodeficiency caused by disruption of GM-CSF signaling (3, 27).
Pulmonary Fibrosis
Clinically significant pulmonary fibrosis can occur in autoimmune PAP but is among the least well-defined manifestations of the disease (4, 7, 130, 131). In one recent study, pulmonary fibrosis was reported in 9 (∼20%) of 44 patients with PAP syndrome and was associated with a poorer outcome (132). Of the 33 patients in the series with autoimmune PAP, 9 (∼27%) initially had minimal radiological features of fibrosis but subsequently developed traction bronchiectasis, and computed tomography (CT) findings worsened over time (132). The pathogenic mechanism driving fibrosis in autoimmune PAP is not known but has been speculated to result from WLL or long-term exposure to higher-than-normal oxygen concentrations from supplemental oxygen therapy (133, 134). Another possibility suggested by murine studies is that reduction in GM-CSF stimulated expression of prostaglandin E2, a potent antifibrotic eicosanoid (135). Although pulmonary fibrosis can occur and may be infrequent in autoimmune PAP, it can be a prominent feature of secondary PAP caused by toxic inhalation of silica and other dusts (13, 136–139) and is a major component of the pathology in congenital PAP caused by surfactant gene mutations (41, 140–142).
Evaluation and Diagnosis
Pulmonary Function Tests
Results of spirometry are normal in many patients with autoimmune PAP, limiting its diagnostic and prognostic utility. Lung volumes can be normal in patients with mild disease or may show a restrictive ventilatory pattern of impairment with lung volumes reduced in proportion to disease severity, especially in more advanced disease (3, 128). DlCO is typically reduced in proportion to disease severity in most patients with autoimmune PAP and provides a useful means to follow changes in disease severity over time (3, 9). A 6-minute-walk test can identify exercise-induced decline in SpO2 (peripheral blood oxygen saturation) but is hindered by variation in patient effort and test performance; in mild cases, results do not clearly identify an abnormality. Arterial blood gas analysis is generally effective in identifying a decrease in PaO2, an increase in alveolar–arterial difference in oxygen concentration (a-aDo2), and an increase in arterial shunt fraction; PaCO2 is often unaffected unless respiration is severely compromised (143). Increases in a-aDo2 and exercise-induced reduction in PaO2 correlate with decreases in DlCO, providing useful measures of disease severity and the need for treatment in patients with autoimmune PAP (3, 143).
Radiological Evaluation
Chest radiography in patients with autoimmune PAP (Figures 3A and 3B) typically reveals nonspecific, diffuse, patchy, bilateral air space disease similar in appearance to that of pulmonary edema but without the other radiographic findings of left heart failure (144, 145). Other patterns occur, including mixed alveolar, interstitial, or nodular infiltrates and focal or asymmetrical abnormalities (1, 7, 146).
Figure 3.

Appearance of the chest radiograph and chest computed tomography scan in autoimmune pulmonary alveolar proteinosis (PAP). (A and B) Posterior–anterior (A) and lateral (B) chest radiographs of a 19-year-old woman with autoimmune PAP showing diffuse ground-glass opacification of the lung parenchyma. (C–F) Representative images from computed tomography of the chest showing the diversity of radiographic findings in autoimmune PAP in a 45-year-old man (C), a 58-year-old woman (D), a 15-year-old girl (E), and an 18-year-old man (F). (C) Image showing ground-glass opacification involving some but not all secondary lobules resulting in a distinctive “geographic” pattern. Also, note the disproportionate involvement of the left and right lung parenchyma. (D) Image showing a distinctive pattern of interlobular septal thickening superimposed on ground-glass opacification, often referred to as “crazy paving.” Also, note the sharply demarcated differences in the degree of involvement between adjacent lung lobes. (E) Image revealing a homogeneous pattern of crazy paving throughout all regions of the lung parenchyma. (F) Image revealing extensive pulmonary fibrosis with parenchymal distortion from traction bronchiectasis. This patient underwent bilateral lung transplantation for pulmonary fibrosis and respiratory failure shortly after this image was obtained.
High-resolution CT (HRCT) of the chest without contrast (Figures 3C–3F) reveals a pattern of ground-glass opacification resulting in polygonal shapes with sharp-angled borders and superimposed interlobular septal thickening referred to as “crazy paving” (145, 147). Although characteristic, this pattern is not diagnostic of autoimmune PAP and can also be seen in other PAP-causing diseases, cardiogenic pulmonary edema, hypersensitivity pneumonitis, Pneumocystis jirovecii pneumonia, lymphangitic carcinomatosis, and acute lung injury/acute respiratory distress syndrome (147). HRCT findings do not distinguish autoimmune PAP from other PAP-causing diseases (148) but do correlate with disease severity as measured by the degree of hypoxemia (145).
Bronchoscopy Findings
Bronchoscopic examination of the airways is unremarkable in autoimmune PAP, while BAL fluid has a distinctive, opaque, milky-white/yellow appearance in nonsmokers and a tan/brown appearance in smokers and becomes pink/red if procedural bleeding occurs (3, 7, 32). A layer of sediment forms in the BAL when left standing, the height of which reflects the amount of alveolar sediment recovered and can reflect disease severity (2). BAL cytology reveals copious extracellular debris; enlarged, foamy-appearing alveolar macrophages (Figures 2B–2D); lymphocytes; fragmented cells in various stages of decay; and isolated nuclei (3, 7, 32). Neutrophils and other inflammatory cells may also be present and prominent if infection is also present. Periodic acid–Schiff staining identifies basophilic, acellular globules (149) and enlarged, foamy macrophages (Figure 2B), which also stain strongly with oil red O (Figure 2E) because of the accumulated intracytoplasmic lipid droplets (3, 9, 150, 151). Differential cytometry is hindered by the copious amounts of sediment and fragility of the enlarged, foamy macrophages, and findings can be normal or demonstrate increased numbers of CD4+ and CD8+ lymphocytes and uncleared neutrophils (152). Electron microscopy demonstrates that the acellular globules comprise tubular myelin and lamellar structures characteristic of pulmonary surfactant and that alveolar macrophages are enlarged and foamy because of the accumulation of intracytoplasmic lamellar bodies and numerous, small, single membrane–delimited lipid droplets (Figure 2F). This morphology can be induced in nonhuman primates by injection of GM-CSF autoantibodies derived from patients with PAP (Figures 2G and 2H) (28, 29). Biochemical analysis reveals that surfactant lipids recovered by BAL contain increased phospholipids, proteins, and cholesterol; a markedly increased cholesterol-to-phospholipid ratio; and increased amounts of macrophage colony–stimulating factor and MCP-1 (4, 56, 153, 154).
GM-CSF Autoantibody Test
GM-CSF autoantibodies are markedly increased in patients with autoimmune PAP (2, 155, 156), but concentrations do not correlate with disease severity, prognosis, pulmonary function test results, or other biomarkers of PAP (9, 108). While GM-CSF autoantibodies are ubiquitous in individuals without PAP, albeit at far lower concentrations (109), a serum GM-CSF autoantibody test can reliably identify autoimmune PAP and can accurately distinguish it from other PAP-causing diseases, other lung diseases, and people without lung disease (Figure 4A). This diagnostic approach has been in use for two decades, and its sensitivity and specificity for a diagnosis of autoimmune PAP are both reported to be 100% (155, 156). GM-CSF autoantibody diagnostic testing is provided by specific centers located in the United States, Europe, China, and Japan.
Figure 4.

Blood-based tests used to diagnose autoimmune pulmonary alveolar proteinosis (PAP). (A) Measurement of serum GM-CSF (granulocyte/macrophage colony–stimulating factor) autoantibody concentration by the serum GM-CSF autoantibody test. Shown are results for individuals with the indicated diseases and interpretive ranges for the test results. Each symbol represents the test result for one individual determined by ELISA using a patient-derived, affinity-purified, polyclonal GM-CSF autoantibody reference standard as previously reported (155). The reference ranges used for test result interpretation (normal, ⩽3.1 μg/ml; indeterminate, >3.1 to <10.2 μg/ml; and abnormal, ⩾10.2 μg/ml) were determined according to guidelines established by the Clinical and Laboratory Standards Institute (234), and updated on April 5, 2019, on the basis of results for 153 healthy individuals (median, 0.33 μg/ml; 90% confidence interval [CI], 0.3–0.4 μg/ml) and 339 patients with autoimmune PAP (median, 84 μg/ml; 90% CI, 10.2–499 μg/ml). Test results within the indeterminate range are typically confirmed by evaluation of GM-CSF signaling. (B) Measurement of GM-CSF signaling by the GM-CSF signaling index (GM-CSF-SI) test. Shown are results for individuals with autoimmune PAP and healthy people and the interpretive ranges for the test results. Each symbol represents the test result for one individual determined by incubating heparinized whole blood with recombinant human GM-CSF (10 ng/ml, 30 min) followed by flow cytometry to quantify phosphorylated STAT5. Results are expressed as GM-CSF-SI, calculated as the mean fluorescence intensity (MFI) in GM-CSF–stimulated cells minus the MFI of unstimulated cells divided by the MFI of unstimulated cells and multiplied by 100, as previously reported (75). The reference ranges used for test result interpretation (normal, ⩾216 units; indeterminate, >20 to <216 units; abnormal, ⩽20 units) were determined according to guidelines established by the Clinical and Laboratory Standards Institute (234) and updated on April 8, 2019, on the basis of results for 77 healthy individuals (median, 506 units; 90% CI, 483–564 units) and 80 patients with autoimmune PAP (median, 2.2 units; 90% CI, 0–4.2 units). STAT5 = signal transducer and activator of transcription 5.
GM-CSF Signaling Tests
Because autoantibodies cause PAP by blocking GM-CSF signaling, several measures of the GM-CSF–neutralizing capacity of blood have been developed into diagnostic tests. On such tests, the GM-CSF signaling index (GM-CSF-SI) test measures the ability of exogenously added GM-CSF to stimulate STAT5 phosphorylation in leukocytes in freshly obtained heparinized blood (77, 157, 158). This test also readily identifies patients with autoimmune PAP (Figure 4B). Because GM-CSF neutralization can be caused by high concentrations of autoantibodies of moderate GM-CSF–binding affinity or lower concentrations of autoantibodies of high GM-CSF–binding capacity, this test can be a useful complement to the GM-CSF autoantibody test by providing functional data related to the GM-CSF–neutralizing capacity of blood. Other such tests to measure the neutralization of GM-CSF signaling in whole blood include the CD11b stimulation index test (159) and the GM-CSF signaling EC50 test (effective [GM-CSF] concentration required to achieve a GM-CSF signaling response) (160).
Other Laboratory Tests
Routine laboratory tests are typically normal in autoimmune PAP except for serum lactate dehydrogenase, which is nonspecific but is elevated in proportion to disease severity as measured by A-aDO2 (3, 143). Serum concentrations of SPs (SP-A–SP-D), YKL-40 (chitinase-3-like protein 1), MCP-1, Cyfra21-1 (cytokeratin 19 fragment), and KL-6 (Krebs von den Lungren protein-6) are often elevated in autoimmune PAP, and the degree of increase correlates with disease severity (9, 108, 161–165), but they can also be increased in individuals with other lung diseases (166, 167), thereby limiting their diagnostic value. Nevertheless, these serum biomarkers can be useful to monitor disease activity over time; indeed, KL-6 has been shown to better predict disease progression and therapeutic need than serum lactate dehydrogenase or PaO2 (168).
Genetic Tests
Although an association with two histocompatibility alleles has been reported (16, 17), no genetic tests are available to diagnose autoimmune PAP.
Lung Biopsy
The cardinal features of lung histopathology in autoimmune PAP include alveoli filled with granular eosinophilic material and well-preserved septa, extracellular debris, enlarged, foamy-appearing alveolar macrophages, cholesterol crystals, and interstitial lymphoid hyperplasia (1–3, 32). Importantly, however, although microscopic examination of a transbronchial or surgical lung biopsy can demonstrate that PAP is present, lung histopathology cannot specifically identify autoimmune PAP or any other PAP-causing disease. Furthermore, because of the patchy involvement of the lung in autoimmune PAP, lung biopsies fail to identify the presence of PAP in a substantial number of cases, 29% in one report (169).
Diagnosis
The diagnosis of autoimmune PAP is challenged by its low prevalence, nonspecific symptoms and radiographic findings, minimal physical examination findings, and normal routine laboratory test results. It is usually misdiagnosed as pneumonia until the failure of several courses of “appropriate” therapy (antibiotics) prompts diagnostic reconsideration, referral, and disease-specific testing (2, 4, 7, 9, 170). Results from the U.S. National PAP Registry indicate that an accurate diagnosis of PAP is delayed by a median of 1.5 years (interquartile range, 0.5–13.5 years) after onset of symptoms (171). Thus, heightened awareness is important for a timely and accurate diagnosis.
PAP should be suspected in an afebrile patient with progressive dyspnea of insidious onset with or without cough, chest congestion, scant production of whitish sputum, and diffuse bilateral infiltrates on chest radiography. HRCT of the chest can confirm the characteristic radiographic findings and aid in determining disease severity. The “cloudy” gross appearance and “dirty” microscopic appearance of the BAL fluid can help confirm the presence of PAP. Although a transbronchial or surgical lung biopsy can confirm that PAP is present, a biopsy cannot identify the PAP-causing disease, fails to confirm the presence of PAP in a substantial proportion of cases, and is associated with significant morbidity. Therefore, a lung biopsy is not necessary in most cases and is recommended only for more difficult cases after other tests fail to result in diagnosis. These findings are useful criteria for the diagnosis of autoimmune PAP (Table 1).
Table 1.
Criteria for Diagnosis of Autoimmune Pulmonary Alveolar Proteinosis*
| Essential criterion |
| Abnormal serum GM-CSF autoantibody test result† |
| Supporting criteria |
| Chest HRCT scan showing diffuse ground-glass opacification and superimposed septal thickening (“crazy-paving sign”)‡ |
| BAL cytopathology showing extensive, mostly extracellular, amorphous PAS-positive cell fragments/debris, ghost cells, and/or large foamy (PAS-positive, oil red O–positive) macrophages§ |
| Lung biopsy histopathology showing alveoli filled with eosinophilic (PAS-positive) granular sediment, enlarged foamy-appearing alveolar macrophages, and/or cholesterol crystals (clefts)ǁ |
Definition of abbreviations: GM-CSF = granulocyte/macrophage colony–stimulating factor; HRCT = high-resolution computed tomography; PAS = periodic acid–Schiff.
Diagnosis requires the presence of the essential criterion and any one of the supporting criteria.
Usually determined quantitatively by ELISA. The (laboratory-specific) cutoff value for an abnormal test result depends on the nature of the GM-CSF autoantibody reference standard and the assay protocol (see text for details).
Ground-glass opacification may occur without superimposed septal thickening in mild disease and usually but not always involves multiple lobes, with or without subpleural sparing.
BAL fluid usually appears opalescent and milky white (or brown in smokers) and contains a waxy sediment, which appears quickly on standing at room temperature or in the cold.
A lung biopsy is often unnecessary and should be performed only if clinically indicated (see text for details).
In patients with suspected or confirmed PAP, a serum GM-CSF autoantibody test (Figure 4A) should be performed because it is highly sensitive and specific for a diagnosis of autoimmune PAP, which accounts for 90% of all cases of PAP (2, 9, 171). Although the serum GM-CSF autoantibody concentration is markedly increased in most patients with autoimmune PAP (median, 84 μg/ml; 90% confidence interval, 10.2–499 μg/ml; n = 339) compared with healthy individuals (median, 0.33 μg/ml; 90% confidence interval, 0.3–0.4 μg/ml; n = 153) (B. Carey and B. Trapnell, unpublished results from the Cincinnati PAP Clinical Research Diagnostic Testing Program), for test results at the low end of the abnormal range, evaluation of the GM-CSF–neutralizing capacity of whole blood (by performing a GM-CSF-SI test) can be a useful follow-up confirmatory test (Figure 4B). These two measures of the disease-causing GM-CSF autoantibodies (concentration and neutralizing capacity) constitute a diagnostically useful complementary test pair because autoimmune PAP can be caused either by higher concentrations of lower-affinity autoantibodies or by lower concentrations of higher-affinity autoantibodies.
Autoimmune PAP can be differentiated from hereditary PAP caused by GM-CSF receptor mutations because in the latter, GM-CSF autoantibody test results are normal (negative) and GM-CSF signaling index test results are abnormal; further tests include measurement of serum GM-CSF (which is undetectable in autoimmune PAP and markedly increased in hereditary PAP). Follow-up testing for hereditary PAP includes evaluation for the presence of CD116 and CD131 on blood leukocytes by flow cytometry (77) and/or nucleotide sequencing to detect biallelic mutations in CSF2RA or CSF2RB (77, 158, 172, 173). Differentiation from secondary PAP is usually possible by the identification of an underlying clinical condition known to cause secondary PAP (137, 174–183), normal GM-CSF autoantibody test results, and normal GM-CSF signaling test results (Figure 5). Differentiation from congenital PAP is typically determined by genetic testing to identify mutations in in SFTPB, SFTBC, ABCA3, and NKX2.1 (40, 41, 184–186). The differential diagnosis of PAP syndrome has been reviewed (4, 7).
Figure 5.

Algorithm for diagnosis of pulmonary alveolar proteinosis (PAP). PAP is suspected on the basis of history and radiographic findings and can be confirmed by the gross and microscopic appearance of BAL fluid. A serum GM-CSF (granulocyte/macrophage colony–stimulating factor) autoantibody test is performed and is highly sensitive and specific for diagnosis of autoimmune PAP. Secondary PAP is diagnosed in individuals with a negative test result who have a condition known to cause PAP. Those with negative test results and no known PAP-causing disease should undergo blood GM-CSF signaling testing and serum GM-CSF testing to identify hereditary PAP caused by CSF2RA or CSF2RB gene mutations, followed by appropriate confirmatory genetic testing. Patients with normal concentrations of serum GM-CSF (e.g., low/undetectable) and normal GM-CSF signaling test results (e.g., detectable) should undergo genetic testing used to identify surfactant production disorders (also known as congenital PAP), including mutational analysis of SFTPC, SFTPB, ABCA3, and NKX2.1 (*). A transbronchial or surgical lung biopsy may be needed if all test results are negative, and further genetic testing may be needed to identify other ultrarare PAP-causing diseases (i.e., methionine-transfer RNA mutations [235], lysinuric protein intolerance [236], or lymphoid cell deficiency–related PAP [237]) Adapted from Reference 4.
Therapy
Soon after the description of PAP as a disorder of phospholipid/protein accumulation, multiple therapies were tested empirically, including antibiotics, corticosteroids, physical dissolution using potassium iodide, streptokinase, trypsin, heparin, acetylcysteine, and saline “washing” (for a review, see Reference 187). With the serendipitous discovery of PAP in GM-CSF–deficient mice and subsequent identification of GM-CSF–neutralizing autoantibodies in patients with (then) idiopathic PAP, therapeutics focused to GM-CSF augmentation, plasmapheresis, and anti–B lymphocyte therapy. Here, we focus on lung lavage and GM-CSF augmentation.
Therapeutic Lung Lavage
Initially, this procedure involved administration of saline through a transtracheal catheter at 50–60 drops/min for 1–2 hours four times daily for 2–3 weeks to induce cough-mediated clearance, which improved DlCO, radiographic appearance, and lung histopathology but was distressing, time consuming, and poorly accepted (188, 189). Subsequently, incorporation of a double-lumen endotracheal tube to isolate each lung, permitting one lung to be washed with large volumes of saline while ventilating the other lung, resulted in a procedure, WLL, that could be completed in a single session and was feasible, effective, and safe (190, 191). Despite further refinements, proposed modifications, and global acceptance as the standard therapy for PAP for nearly six decades, no single optimized protocol has been developed and adopted universally, nor have the indications for its therapeutic use or methods to assess treatment responses been standardized. Accordingly, the WLL procedure itself is described briefly together with indications for its clinical use, contraindications, complications, efficacy, and durability of the treatment response; for further procedural details, the reader is referred to published reports (including a global practice survey) (187, 192–197).
Optimally, WLL is performed by an experienced team that includes nursing, anesthesiology, respiratory therapy, and interventional pulmonary medicine or surgery staff members. The preoperative assessment and preparation are similar to those used for bronchoscopy and BAL, including the management of anticoagulation (187). After induction of general anesthesia, a double-lumen endotracheal tube is placed and the adequacy of endotracheal lung isolation confirmed (187). Although various body positions have been reported, few data are available to recommend any one position for WLL other than supine; repositioning the tube is difficult during WLL, and care must be taken to prevent spillage into the contralateral lung. The nontreated lung is denitrogenated by ventilating with 100% oxygen. In adults, a total saline volume of 30–50 L should be anticipated. Saline is warmed to 37°C, instilled in incremental volumes of 500–1,000 ml, and drained under gravity using a closed system. Chest percussion is performed to loosen and emulsify the accumulated sediment, either using a wraparound vest or by manual percussion (194, 198). Interestingly, a modified lavage technique involving the use of controlled manual ventilation during the infusion–recovery cycle resulted in significant improvement in sediment removal compared with WLL without the use of controlled manual ventilation (199). Initial returns are typically milky or turbid and form a sediment layer on standing, the thickness of which corresponds to the efficacy of sediment removal. Each cycle of infusion and drainage takes 3–5 minutes (longer if asthma is present). The process is continued until the effluent becomes clear or the optical density (λ = 405 nm) of the effluent is reduced to <0.4 (199). Intraoperative monitoring should include continuous measurement of peripheral oxygen saturation and serial readings of arterial blood gas concentrations, vital signs, and infusion and effluent volumes, as well as observation of leakage into the contralateral lung or pleural space. At the end of the procedure, residual saline is drained and aspirated from the lung, ventilation with 100% oxygen is resumed, the double-lumen tube is replaced with a single-lumen tube, remaining fluid is aspirated with a flexible bronchoscope if necessary, and monitoring is continued in the recovery unit until extubation. In patients with advanced disease and respiratory failure, WLL can be performed using extracorporeal membrane oxygen (200), or segmental lung lavage can be performed by bronchoscopy (201).
Although the indications for WLL vary among centers, the primary indication is limitation of daily activities because of dyspnea (194, 198). Other indications include decline in lung function, declining oxygenation, a shunt fraction greater than 10–12%, radiographic disease progression, and a desire to reduce the requirement for supplemental oxygen therapy (192). WLL should not be performed in patients with active microbial lung infection, because of the risk of disseminated infection, sepsis, and shock. Most centers perform WLL in separate sessions for each lung, with the most severely affected lung treated first and the other lung treated several days to weeks later (194, 198).
Although no randomized controlled trials have documented efficacy and safety, WLL therapy for PAP is widely regarded as the current standard of care and is capable of alleviating symptoms and improving radiographic findings and gas exchange in patients with PAP (202, 203). In one comprehensive study, the increased 5-year survival was higher in patients with PAP undergoing WLL than untreated patients (94 ± 2% vs. 85 ± 5%; n = 146 and 85; P = 0.04) (3). Most (70%) patients underwent WLL within 5 years of diagnosis; the median number of procedures was two, and among 55 treated individuals, the median duration of benefit was 15 months; and only 20% of patients did not require repeat WLL at 3 years. Among 41 patients with PAP, WLL improved arterial PaO2 by 20.1 mm Hg; improvement in other pulmonary function parameters was less impressive. WLL was effective in more than 95% of patients; however, a small fraction did not respond despite aggressive lavage, presumably because of pulmonary fibrosis. Treatment-related improvement results from the physical removal of the accumulated surfactant and can happen within hours of completion of therapy. Although it is generally considered safe, the potential complications of WLL include hypoxemia, hydropneumothorax, pneumonia, acute respiratory distress syndrome, and sepsis.
GM-CSF Augmentation
Within a year of the discovery of PAP in GM-CSF–deficient mice, Seymour and colleagues reported (204) that subcutaneous administration of GM-CSF in a patient with (then) idiopathic PAP for 70 days was associated with improvement in clinical, radiographic, and physiological manifestations of PAP (Table 2). These findings were confirmed in case reports (205–208) and two open-label dose-escalation studies of subcutaneous GM-CSF (5–20 μg/kg once daily for 1.5–3 mo or 5–18 μg/kg once daily for 6–12 mo, respectively), which reported improvement in A-aDO2 in 6 of 14 (165) or 12 of 25 (208, 209) autoimmune PAP patients, respectively. The latter study also reported improvement in DlCO, radiographic appearance, and the distance covered in a 6-minute-walk test (Table 2). Subcutaneous GM-CSF administration (8 μg/kg/d for 12 wk) was also noted to improve BAL cytology in patients with autoimmune PAP (32). Although injection site irritation was noted in 85% of patients in one study (208), no significant treatment-associated adverse events were noted in any of these studies (Table 2).
Table 2.
Clinical Trials of Granulocyte/Macrophage Colony–Stimulating Factor Augmentation Therapy Reported in the Medical Literature
| Year | First Author | Trial Design | Subjects (n) | Drug | Route | Duration | a-aDo2 | DlCO | CT/X-Ray | 6MWT-D | TEAE | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1996 | Seymour | First in human, single site, open label | 1 | Molgramostim | s.c. | 70 d | Improved | NR | Improved | NR | None | 204 |
| 2001 | Seymour | Phase I/II, single site, open label | 14 | Molgramostim/sargramostim | s.c. | 6–12 wk | Improved | Improved | Improved | Improved | None | 165 |
| 2004 | Arai | First in human single site, open label | 1 | Molgramostim | Inhaled | 24 wk | Improved | Improved | Improved | NR | None | 211 |
| 2005 | Tazawa | Phase I/II, single site, open label | 3 | Molgramostim | Inhaled | 24 wk | Improved | NR | NR | NR | None | 210 |
| 2006 | Wylam | Retrospective, observational | 12 | Sargramostim | Inhaled | Up to 64 wk | Improved | Improved | Improved | NR | None | 213 |
| 2006 | Venkateshiah | Phase I/II, single site, open label | 25 | Sargramostim | s.c. | 24 wk | Improved | Improved | Improved | Improved | None | 208 |
| 2010 | Tazawa | Phase I/II, multisite, open label | 35 | Sargramostim | Inhaled | 24 wk | Improved | Improved | Improved | Improved | None | 215 |
| 2012 | Ohashi | Phase II, single site, open label | 6 | Sargramostim | Inhaled | 12 wk | Improved | NR | NR | NR | None | 222 |
| 2014 | Papiris | Retrospective, observational | 6 | Sargramostim | Inhaled | 14–65 mo | Improved | Improved | Improved | NR | None | 214 |
| 2016 | Campo | Phase II, single site, open label | 18 | Sargramostim* | Inhaled | 55 mo | Improved | Improved | Not significant | NR | None | 227 |
| 2019 | Tazawa | Phase III, multisite, randomized, double blind, placebo controlled | 64 | Sargramostim | Inhaled | 24 wk | Improved | Improved | Improved | Not significant | None | 224 |
| 2020 | Tian | Phase II, multisite, randomized, open label | 36 | Molgramostim | Inhaled | 24 wk | No change | Improved | No change | No change | None | 223 |
| 2020 | Trapnell | Phase II, multinational, randomized, double blind, placebo controlled | 138 | Molgramostim | Inhaled | 24 wk | Improved | Improved | Improved | Not significant | None | 225 |
Definition of abbreviations: 6MWT-D = 6-minute-walk test distance (distance covered during test); a-aDo2 = alveolar–arterial difference in oxygen concentration; CT = computed tomography (of the chest); NR = not reported; s.c. = subcutaneous; TEAE = treatment-emergent adverse events.
Whole-lung lavage versus whole-lung lavage plus inhaled granulocyte/macrophage colony–stimulating factor.
The initial evaluations of inhaled GM-CSF in autoimmune PAP reported improvement in a-aDo2 and BAL cytology (210–212). One retrospective study of escalating doses (250–500 μg) of GM-CSF inhaled twice daily every other week for 64 weeks reported improvement in a-aDo2 and DlCO in 11 of 12 patients (213), while another evaluating a single dose (250 μg) of GM-CSF inhaled once daily for 4 days of every 8-day period for 64 weeks reported improvement in a-aDo2, DlCO, symptom scores, and radiographic findings in 6 of 6 patients with autoimmune PAP (214). A larger prospective, open-label study of inhaled GM-CSF (250 μg/d on Days 1–8 of every 14-d period for 12 wk, followed by 125 μg/d on Days 1–4 of every 14-d period for 12 wk) demonstrated improvement in a-aDo2 in 24 of 35 patients with autoimmune PAP and that it was safe and well-tolerated (215) (Table 2); a follow-up study showed that 23 of 35 patients had durable treatment responses (216). Additional case reports confirmed these results (217–222). One randomized, placebo-controlled, open-label study of inhaled GM-CSF (150 μg twice daily on alternating weeks for 24 wk) (or placebo) in 36 patients with mild to moderate autoimmune PAP in China reported improvement in DlCO and St. George’s Respiratory Questionnaire score but not a-aDo2, radiographic appearance, or requirement for WLL (223).
The first randomized, placebo-controlled, double-blind study of inhaled GM-CSF, the PAGE trial (Pulmonary Alveolar Proteinosis GM-CSF Inhalation Efficacy Trial in Japan), was conducted in 64 patients with autoimmune PAP with mild to moderate disease (defined as PaO2 < 75 mm Hg and >50 mm Hg) in Japan (224). Patients received blinded intervention (treatment group: GM-CSF 125 μg [on alternating weeks], inhaled twice daily; control group: placebo, inhaled twice daily) for 24 weeks (224). Results showed a significant reductions in a-aDo2 in the treatment group compared with placebo (−4.50 ± 9.03 vs. 0.17 ± 10.50 mm Hg, respectively; P = 0.02), CT lung densitometry scores (indicating a reduction in pulmonary surfactant accumulation), and biomarkers of PAP but no significant improvement in clinical or patient-reported outcomes (Table 2). Inhaled GM-CSF was well tolerated and was not associated with an increase in serious adverse events (Table 2).
A second (global) randomized, placebo-controlled, double-blind study in 138 patients with autoimmune PAP, the IMPALA (Inhaled Molgramostim in Autoimmune Pulmonary Alveolar Proteinosis Patients) trial, compared three interventions: continuous GM-CSF, intermittent GM-CSF, and placebo (continuous arm: 300 μg GM-CSF inhaled once daily; intermittent arm: 300 μg GM-CSF inhaled once daily every other week [alternating with placebo to maintain blinding]; placebo arm: placebo inhaled once daily); all interventions were administered for a 24-week (blinded treatment) period, which was followed by an open-label treatment extension period in which patients received intermittent GM-CSF for 48 or 72 weeks (225). Results showed significant improvements in a-aDo2, DlCO, chest CT ground-glass opacification score, St. George’s Respiratory Questionnaire total score, and PAP biomarkers in the continuous GM-CSF group compared with placebo (Table 2). Inhaled GM-CSF was well tolerated and not associated with an increase in serious adverse events (Table 2). An important outcome was the demonstration that continuous daily administration of GM-CSF resulted in greater clinical benefit than intermittent daily GM-CSF (225). A third global, randomized, placebo-controlled, double-blind (IMPALA-2) trial is currently under way to evaluate the safety and efficacy of long-term daily administration of GM-CSF therapy for autoimmune PAP.
Several case reports found that prior administration of WLL improved the response to inhaled GM-CSF in patients with autoimmune PAP (218, 226). In a randomized, open-label study in 18 patients with autoimmune PAP in Italy, administration of inhaled GM-CSF after WLL resulted in a marked reduction in the requirement for subsequent WLL therapy (227).
In summary, GM-CSF is a promising (currently off-label) pharmacotherapeutic approach for autoimmune PAP, which is supported by numerous small studies and two completed and one ongoing large, randomized, placebo-controlled, double-blind trials. Efficacy is greater when GM-CSF is administered by inhalation rather than subcutaneously and by daily administration rather than daily on alternating weeks. A treatment response occurs in one-half to two-thirds of patients when administered at 250–300 μg/d. Anecdotal observations have indicated the existence of a dose-dependent treatment response, but the relationship has yet to be defined. Importantly, no studies have identified drug-related safety concerns. Additional studies are needed to determine the optimal starting dose and treatment time and whether an induction–maintenance dosing strategy will be useful.
Other Treatment Approaches
Of the numerous other treatment approaches tested in autoimmune PAP, several deserve brief mention. Although no prospective trials evaluating corticosteroids in autoimmune PAP have been reported, this approach is often considered or used. Recently, a retrospective study in 31 patients with autoimmune PAP found that corticosteroid therapy worsened the lung disease and increased the risk of infection in patients with autoimmune PAP (228).
Theoretically, plasmapheresis to remove the GM-CSF–neutralizing autoantibodies is a sound approach that has been tried (229). However, the procedure must be repeated frequently (daily for 2 wk) to reduce the very high autoantibody concentrations present (B. Trapnell, unpublished results). Thus, this approach appears impractical and unviable in our opinion.
Anti–B lymphocyte therapy with rituximab is also theoretically attractive. Several case reports, a small phase II study, and a more recent retrospective study of rituximab therapy for autoimmune PAP have been reported (230–232). However, the results are not particularly strong or convincing, and no specific treatment approach has been identified as effective. Thus, the role for rituximab in autoimmune PAP should be reserved for patients who are refractory to other therapy.
Rosuvastatin administration was associated with significant clinical improvement in several patients with autoimmune PAP (52), but the available results are insufficient to recommend this approach at the present time.
In patients with autoimmune PAP who develop pulmonary fibrosis, WLL becomes ineffective and lung transplantation is required; however, the autoimmune PAP lung disease returns and must be treated (233).
Conclusions
In the two decades since the comprehensive review of PAP syndrome by Seymour and Presneill (3), our understanding has advanced considerably. Idiopathic PAP has been redefined as an autoimmune disease mediated by polyclonal GM-CSF autoantibodies that block GM-CSF signaling, reduce GM-CSF–dependent cholesterol clearance, and result in foamy, cholesterol-stuffed, poorly differentiated macrophages unable to clear surfactant or perform other functions. Research advances also identified GM-CSF as a regulatory molecule critical for surfactant homeostasis, alveolar structural integrity, lung function, and lung host defense, altogether suggesting that GM-CSF is an essential pulmonary hormone.
Autoimmune PAP accounts for 90% of all cases of PAP and has a prevalence of 7–10 per million in the general population. Nonspecific clinical presentation, radiographic appearance, and normal routine laboratory findings often result in misdiagnosis of autoimmune PAP as pneumonia and long delays in an accurate diagnosis. The observation of symptoms unexpectedly milder than expected based on extensive radiographic abnormalities present is of diagnostic utility and should raise the suspicion of PAP; heightened awareness is important for an accurate and timely diagnosis. A now commercially available diagnostic test, the serum GM-CSF autoantibody test, has revolutionized the diagnosis of autoimmune PAP and rendered lung biopsies unnecessary in most cases. Lung biopsies should be reserved for more complex cases of PAP syndrome.
The clinical course of autoimmune PAP varies among patients with unremitting/slowly progressive disease in most patients, spontaneous improvement in a small percentage (5–7%), and rapid progression and/or pulmonary fibrosis, respiratory failure, and death in others. Although WLL remains the first-line therapy for autoimmune PAP and is effective in most patients, it is invasive, inefficient, dependent on operator experience, and unavailable at many medical institutions, and it does not prevent ongoing surfactant accumulation. Numerous case reports, small studies, and two completed randomized, placebo-controlled trials support the safety, tolerability, and efficacy of inhaled GM-CSF as a promising pharmacotherapy for autoimmune PAP.
Despite enormous progress in our understanding, questions remain regarding etiology, pathogenesis, and therapy for autoimmune PAP. What causes the breakdown of immune tolerance leading to high concentrations of GM-CSF autoantibodies in autoimmune PAP? How does loss of GM-CSF signaling impair myeloid cell differentiation and functions? Regarding inhaled GM-CSF therapy, what are the dose–response relationship, optimal starting dose, and optimal treatment time? What is its mechanism of action, and why does it not increase the GM-CSF autoantibody concentration? Will prior WLL treatment improve efficacy or accelerate the treatment response? Validated outcome measures are needed to accelerate pharmacotherapeutic development, and clinical practice guidelines are needed to improve the diagnosis and treatment of patients with autoimmune PAP.
Acknowledgments
Acknowledgment
The authors express sincere thanks to the PAP patient community for their collaboration in studies that have led to the advances described herein.
Footnotes
Supported by NHLBI grant R01 HL 085453 (B.C.T.) and the National Center for Advancing Translational Science in collaboration with the NHLBI (grant U54 HL127672) (B.C.T.).
Originally Published in Press as DOI: 10.1164/rccm.202112-2742SO on February 28, 2002
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med . 1958;258:1123–1142. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 2. Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med . 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 3. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med . 2002;166:215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 4. Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers . 2019;5:16. doi: 10.1038/s41572-019-0066-3. [DOI] [PubMed] [Google Scholar]
- 5. Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M, et al. Pulmonary alveolar proteinosis. Eur Respir Rev . 2011;20:98–107. doi: 10.1183/09059180.00001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar A, Abdelmalak B, Inoue Y, Culver DA. Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach. Lancet Respir Med . 2018;6:554–565. doi: 10.1016/S2213-2600(18)30043-2. [DOI] [PubMed] [Google Scholar]
- 7.Trapnell BC, McCarthy C. In: Murry & Nadel’s textbook of respiratory medicine. 7th ed. Broaddus VC, Ernst JD, King TE, Lazarus SC, Sarmiento KF, Schnapp LM, et al., editors. Philadelphia, PA: Elsevier; 2021. Pulmonary alveolar proteinosis syndrome; pp. 1363–1377. [Google Scholar]
- 8. Nogee LM. Genetic basis of children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol . 2010;23:15–24. doi: 10.1089/ped.2009.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, et al. Japanese Center of the Rare Lung Diseases Consortium Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med . 2008;177:752–762. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. Prevalence and healthcare burden of pulmonary alveolar proteinosis. Orphanet J Rare Dis . 2018;13:129. doi: 10.1186/s13023-018-0846-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitamura N, Ohkouchi S, Tazawa R, Ishii H, Takada T, Sakagami T, et al. Incidence of autoimmune pulmonary alveolar proteinosis estimated using Poisson distribution. ERJ Open Res . 2019;5:00190-2018. doi: 10.1183/23120541.00190-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campo I, Mariani F, Rodi G, Paracchini E, Tsana E, Piloni D, et al. Assessment and management of pulmonary alveolar proteinosis in a reference center. Orphanet J Rare Dis . 2013;8:40. doi: 10.1186/1750-1172-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonella F, Bauer PC, Griese M, Ohshimo S, Guzman J, Costabel U. Pulmonary alveolar proteinosis: new insights from a single-center cohort of 70 patients. Respir Med . 2011;105:1908–1916. doi: 10.1016/j.rmed.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 14. Xu Z, Jing J, Wang H, Xu F, Wang J. Pulmonary alveolar proteinosis in China: a systematic review of 241 cases. Respirology . 2009;14:761–766. doi: 10.1111/j.1440-1843.2009.01539.x. [DOI] [PubMed] [Google Scholar]
- 15. Cummings KJ, Nakano M, Omae K, Takeuchi K, Chonan T, Xiao YL, et al. Indium lung disease. Chest . 2012;141:1512–1521. doi: 10.1378/chest.11-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anderson K, Carey B, Martin A, Roark C, Chalk C, Nowell-Bostic M, et al. Pulmonary alveolar proteinosis: an autoimmune disease lacking an HLA association. PLoS One . 2019;14:e0213179. doi: 10.1371/journal.pone.0213179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakaue S, Yamaguchi E, Inoue Y, Takahashi M, Hirata J, Suzuki K, et al. Genetic determinants of risk in autoimmune pulmonary alveolar proteinosis. Nat Commun . 2021;12:1032. doi: 10.1038/s41467-021-21011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pattle RE. Properties, function and origin of the alveolar lining layer. Nature . 1955;175:1125–1126. doi: 10.1038/1751125b0. [DOI] [PubMed] [Google Scholar]
- 19. Golde DW, Territo M, Finley TN, Cline MJ. Defective lung macrophages in pulmonary alveolar proteinosis. Ann Intern Med . 1976;85:304–309. doi: 10.7326/0003-4819-85-3-304. [DOI] [PubMed] [Google Scholar]
- 20. Stratton JA, Sieger L, Wasserman K. The immunoinhibitory activities of the lung lavage materials and sera from patients with pulmonary alveolar proteinosis (PAP) J Clin Lab Immunol . 1981;5:81–86. [PubMed] [Google Scholar]
- 21. Müller-Quernheim J, Schopf RE, Benes P, Schulz V, Ferlinz R. A macrophage-suppressing 40-kD protein in a case of pulmonary alveolar proteinosis. Klin Wochenschr . 1987;65:893–897. doi: 10.1007/BF01745499. [DOI] [PubMed] [Google Scholar]
- 22. Bury T, Corhay JL, Saint-Remy P, Radermecker M. Alveolar proteinosis: restoration of the function of the alveolar macrophages after therapeutic lavage [in French] Rev Mal Respir . 1989;6:373–375. [PubMed] [Google Scholar]
- 23. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A . 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science . 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 25. Tanaka N, Watanabe J, Kitamura T, Yamada Y, Kanegasaki S, Nakata K. Lungs of patients with idiopathic pulmonary alveolar proteinosis express a factor which neutralizes granulocyte-macrophage colony stimulating factor. FEBS Lett . 1999;442:246–250. doi: 10.1016/s0014-5793(98)01668-8. [DOI] [PubMed] [Google Scholar]
- 26. Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med . 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med . 2007;356:567–579. doi: 10.1056/NEJMoa062505. [DOI] [PubMed] [Google Scholar]
- 28. Sakagami T, Uchida K, Suzuki T, Carey BC, Wood RE, Wert SE, et al. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. N Engl J Med . 2009;361:2679–2681. doi: 10.1056/NEJMc0904077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakagami T, Beck D, Uchida K, Suzuki T, Carey BC, Nakata K, et al. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Crit Care Med . 2010;182:49–61. doi: 10.1164/rccm.201001-0008OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bendtzen K, Svenson M, Hansen MB. GM-CSF autoantibodies in pulmonary alveolar proteinosis. N Engl J Med . 2007;356:2001. doi: 10.1056/NEJMc070650. [DOI] [PubMed] [Google Scholar]
- 31. Caparros D, Steenhouwer F, Marquette CH, Carpentier F, Foulet A. Pulmonary alveolar proteinosis: a sequential analysis of the alveolar cell population after complete pulmonary lavage: apropos of a case [in French] Rev Mal Respir . 1994;11:63–66. [PubMed] [Google Scholar]
- 32. Schoch OD, Schanz U, Koller M, Nakata K, Seymour JF, Russi EW, et al. BAL findings in a patient with pulmonary alveolar proteinosis successfully treated with GM-CSF. Thorax . 2002;57:277–280. doi: 10.1136/thorax.57.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Costello JF, Moriarty DC, Branthwaite MA, Turner-Warwick M, Corrin B. Diagnosis and management of alveolar proteinosis: the rôle of electron microscopy. Thorax . 1975;30:121–132. doi: 10.1136/thx.30.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilmore LB, Talley FA, Hook GE. Classification and morphometric quantitation of insoluble materials from the lungs of patients with alveolar proteinosis. Am J Pathol . 1988;133:252–264. [PMC free article] [PubMed] [Google Scholar]
- 35. Perez-Gil J, Weaver TE. Pulmonary surfactant pathophysiology: current models and open questions. Physiology (Bethesda) . 2010;25:132–141. doi: 10.1152/physiol.00006.2010. [DOI] [PubMed] [Google Scholar]
- 36. Veldhuizen R, Nag K, Orgeig S, Possmayer F. The role of lipids in pulmonary surfactant. Biochim Biophys Acta . 1998;1408:90–108. doi: 10.1016/s0925-4439(98)00061-1. [DOI] [PubMed] [Google Scholar]
- 37. Goerke J. Pulmonary surfactant: functions and molecular composition. Biochim Biophys Acta . 1998;1408:79–89. doi: 10.1016/s0925-4439(98)00060-x. [DOI] [PubMed] [Google Scholar]
- 38. Pérez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim Biophys Acta . 2008;1778:1676–1695. doi: 10.1016/j.bbamem.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 39. Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med . 2010;61:105–119. doi: 10.1146/annurev.med.60.041807.123500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nogee LM, de Mello DE, Dehner LP, Colten HR. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med . 1993;328:406–410. doi: 10.1056/NEJM199302113280606. [DOI] [PubMed] [Google Scholar]
- 41. Nogee LM, Dunbar AE, III, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med . 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 42. Nogee LM. Alterations in SP-B and SP-C expression in neonatal lung disease. Annu Rev Physiol . 2004;66:601–623. doi: 10.1146/annurev.physiol.66.032102.134711. [DOI] [PubMed] [Google Scholar]
- 43. Crouch EC. Collectins and pulmonary host defense. Am J Respir Cell Mol Biol . 1998;19:177–201. doi: 10.1165/ajrcmb.19.2.140. [DOI] [PubMed] [Google Scholar]
- 44. Orgeig S, Daniels CB. The roles of cholesterol in pulmonary surfactant: insights from comparative and evolutionary studies. Comp Biochem Physiol A Mol Integr Physiol . 2001;129:75–89. doi: 10.1016/s1095-6433(01)00307-5. [DOI] [PubMed] [Google Scholar]
- 45. Bernardino de la Serna J, Perez-Gil J, Simonsen AC, Bagatolli LA. Cholesterol rules: direct observation of the coexistence of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J Biol Chem . 2004;279:40715–40722. doi: 10.1074/jbc.M404648200. [DOI] [PubMed] [Google Scholar]
- 46. Daniels CB, Barr HA, Power JH, Nicholas TE. Body temperature alters the lipid composition of pulmonary surfactant in the lizard Ctenophorus nuchalis. Exp Lung Res . 1990;16:435–449. doi: 10.3109/01902149009068819. [DOI] [PubMed] [Google Scholar]
- 47. Lopatko OV, Orgeig S, Palmer D, Schürch S, Daniels CB. Alterations in pulmonary surfactant after rapid arousal from torpor in the marsupial Sminthopsis crassicaudata. J Appl Physiol (1985) . 1999;86:1959–1970. doi: 10.1152/jappl.1999.86.6.1959. [DOI] [PubMed] [Google Scholar]
- 48. Doyle IR, Jones ME, Barr HA, Orgeig S, Crockett AJ, McDonald CF, et al. Composition of human pulmonary surfactant varies with exercise and level of fitness. Am J Respir Crit Care Med . 1994;149:1619–1627. doi: 10.1164/ajrccm.149.6.8004321. [DOI] [PubMed] [Google Scholar]
- 49. Hawgood S, Poulain FR. The pulmonary collectins and surfactant metabolism. Annu Rev Physiol . 2001;63:495–519. doi: 10.1146/annurev.physiol.63.1.495. [DOI] [PubMed] [Google Scholar]
- 50. Wright JR. Pulmonary surfactant: a front line of lung host defense. J Clin Invest . 2003;111:1453–1455. doi: 10.1172/JCI18650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sallese A, Suzuki T, McCarthy C, Bridges J, Filuta A, Arumugam P, et al. Targeting cholesterol homeostasis in lung diseases. Sci Rep . 2017;7:10211. doi: 10.1038/s41598-017-10879-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCarthy C, Lee E, Bridges JP, Sallese A, Suzuki T, Woods JC, et al. Statin as a novel pharmacotherapy of pulmonary alveolar proteinosis. Nat Commun . 2018;9:3127. doi: 10.1038/s41467-018-05491-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ikegami M, Ueda T, Hull W, Whitsett JA, Mulligan RC, Dranoff G, et al. Surfactant metabolism in transgenic mice after granulocyte macrophage-colony stimulating factor ablation. Am J Physiol . 1996;270:L650–L658. doi: 10.1152/ajplung.1996.270.4.L650. [DOI] [PubMed] [Google Scholar]
- 54. Griese M. Pulmonary surfactant in health and human lung diseases: state of the art. Eur Respir J . 1999;13:1455–1476. doi: 10.1183/09031936.99.13614779. [DOI] [PubMed] [Google Scholar]
- 55. Yoshida M, Ikegami M, Reed JA, Chroneos ZC, Whitsett JA. GM-CSF regulates protein and lipid catabolism by alveolar macrophages. Am J Physiol Lung Cell Mol Physiol . 2001;280:L379–L386. doi: 10.1152/ajplung.2001.280.3.L379. [DOI] [PubMed] [Google Scholar]
- 56. Griese M, Bonella F, Costabel U, de Blic J, Tran NB, Liebisch G. Quantitative lipidomics in pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 2019;200:881–887. doi: 10.1164/rccm.201901-0086OC. [DOI] [PubMed] [Google Scholar]
- 57. Gough NM, Gough J, Metcalf D, Kelso A, Grail D, Nicola NA, et al. Molecular cloning of cDNA encoding a murine haematopoietic growth regulator, granulocyte-macrophage colony stimulating factor. Nature . 1984;309:763–767. doi: 10.1038/309763a0. [DOI] [PubMed] [Google Scholar]
- 58. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood . 1980;56:947–958. [PubMed] [Google Scholar]
- 59. Lieschke GJ, Burgess AW. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor (2) N Engl J Med . 1992;327:99–106. doi: 10.1056/NEJM199207093270207. [DOI] [PubMed] [Google Scholar]
- 60. Lieschke GJ, Burgess AW. Granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor (1) N Engl J Med . 1992;327:28–35. doi: 10.1056/NEJM199207023270106. [DOI] [PubMed] [Google Scholar]
- 61. Gschwend J, Sherman SPM, Ridder F, Feng X, Liang HE, Locksley RM, et al. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J Exp Med . 2021;218:e20210745. doi: 10.1084/jem.20210745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol . 2005;25:405–428. doi: 10.1615/critrevimmunol.v25.i5.50. [DOI] [PubMed] [Google Scholar]
- 63. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol . 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 64. Cantrell MA, Anderson D, Cerretti DP, Price V, McKereghan K, Tushinski RJ, et al. Cloning, sequence, and expression of a human granulocyte/macrophage colony-stimulating factor. Proc Natl Acad Sci U S A . 1985;82:6250–6254. doi: 10.1073/pnas.82.18.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Donahue RE, Wang EA, Stone DK, Kamen R, Wong GG, Sehgal PK, et al. Stimulation of haematopoiesis in primates by continuous infusion of recombinant human GM-CSF. Nature . 1986;321:872–875. doi: 10.1038/321872a0. [DOI] [PubMed] [Google Scholar]
- 66. Mayer P, Lam C, Obenaus H, Liehl E, Besemer J. Recombinant human GM-CSF induces leukocytosis and activates peripheral blood polymorphonuclear neutrophils in nonhuman primates. Blood . 1987;70:206–213. [PubMed] [Google Scholar]
- 67. Munn DH, Garnick MB, Cheung NK. Effects of parenteral recombinant human macrophage colony-stimulating factor on monocyte number, phenotype, and antitumor cytotoxicity in nonhuman primates. Blood . 1990;75:2042–2048. [PubMed] [Google Scholar]
- 68. Nakata K, Akagawa KS, Fukayama M, Hayashi Y, Kadokura M, Tokunaga T. Granulocyte-macrophage colony-stimulating factor promotes the proliferation of human alveolar macrophages in vitro. J Immunol . 1991;147:1266–1272. [PubMed] [Google Scholar]
- 69. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity . 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 70. Berclaz PY, Shibata Y, Whitsett JA, Trapnell BCGM-CSF. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma-mediated molecular connection between innate and adaptive immunity in the lung. Blood . 2002;100:4193–4200. doi: 10.1182/blood-2002-04-1102. [DOI] [PubMed] [Google Scholar]
- 71. Berclaz PY, Zsengellér Z, Shibata Y, Otake K, Strasbaugh S, Whitsett JA, et al. Endocytic internalization of adenovirus, nonspecific phagocytosis, and cytoskeletal organization are coordinately regulated in alveolar macrophages by GM-CSF and PU.1. J Immunol . 2002;169:6332–6342. doi: 10.4049/jimmunol.169.11.6332. [DOI] [PubMed] [Google Scholar]
- 72. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med . 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity . 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity . 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, et al. Pulmonary macrophage transplantation therapy. Nature . 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Metcalf D, Nicola NA, Mifsud S, Di Rago L. Receptor clearance obscures the magnitude of granulocyte-macrophage colony-stimulating factor responses in mice to endotoxin or local infections. Blood . 1999;93:1579–1585. [PubMed] [Google Scholar]
- 77. Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med . 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Carraway MS, Ghio AJ, Carter JD, Piantadosi CA. Detection of granulocyte-macrophage colony-stimulating factor in patients with pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 2000;161:1294–1299. doi: 10.1164/ajrccm.161.4.9906080. [DOI] [PubMed] [Google Scholar]
- 79. Mortensen BT, Schifter S, Pedersen LB, Jensen AN, Hovgaard D, Nissen NI. Development and application of a sensitive radioimmunoassay for human granulocyte-macrophage colony-stimulating factor able to measure normal concentrations in blood. Exp Hematol . 1993;21:1366–1370. [PubMed] [Google Scholar]
- 80. D’Andrea RJ, Gonda TJ. A model for assembly and activation of the GM-CSF, IL-3 and IL-5 receptors: insights from activated mutants of the common beta subunit. Exp Hematol . 2000;28:231–243. doi: 10.1016/s0301-472x(99)00159-9. [DOI] [PubMed] [Google Scholar]
- 81. Quelle FW, Sato N, Witthuhn BA, Inhorn RC, Eder M, Miyajima A, et al. JAK2 associates with the beta c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol Cell Biol . 1994;14:4335–4341. doi: 10.1128/mcb.14.7.4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsuguchi T, Zhao Y, Lilly MB, Kraft AS. The cytoplasmic domain of granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor alpha subunit is essential for both GM-CSF-mediated growth and differentiation. J Biol Chem . 1997;272:17450–17459. doi: 10.1074/jbc.272.28.17450. [DOI] [PubMed] [Google Scholar]
- 83. Duronio V, Clark-Lewis I, Federsppiel B, Wieler JS, Schrader JW. Tyrosine phosphorylation of receptor beta subunits and common substrates in response to interleukin-3 and granulocyte-macrophage colony-stimulating factor. J Biol Chem . 1992;267:21856–21863. [PubMed] [Google Scholar]
- 84. Sakamaki K, Miyajima I, Kitamura T, Miyajima A. Critical cytoplasmic domains of the common beta subunit of the human GM-CSF, IL-3 and IL-5 receptors for growth signal transduction and tyrosine phosphorylation. EMBO J . 1992;11:3541–3549. doi: 10.1002/j.1460-2075.1992.tb05437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell . 2008;134:496–507. doi: 10.1016/j.cell.2008.05.053. [DOI] [PubMed] [Google Scholar]
- 86. Hercus TR, Broughton SE, Ekert PG, Ramshaw HS, Perugini M, Grimbaldeston M, et al. The GM-CSF receptor family: mechanism of activation and implications for disease. Growth Factors . 2012;30:63–75. doi: 10.3109/08977194.2011.649919. [DOI] [PubMed] [Google Scholar]
- 87. Watanabe S, Itoh T, Arai K. Roles of JAK kinases in human GM-CSF receptor signal transduction. J Allergy Clin Immunol . 1996;98:S183–S191. doi: 10.1016/s0091-6749(96)70065-9. [DOI] [PubMed] [Google Scholar]
- 88. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol . 2002;71:511–519. [PubMed] [Google Scholar]
- 89. Guthridge MA, Powell JA, Barry EF, Stomski FC, McClure BJ, Ramshaw H, et al. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. EMBO J . 2006;25:479–489. doi: 10.1038/sj.emboj.7600948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gearing DP, King JA, Gough NM, Nicola NA. Expression cloning of a receptor for human granulocyte-macrophage colony-stimulating factor. EMBO J . 1989;8:3667–3676. doi: 10.1002/j.1460-2075.1989.tb08541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hayashida K, Kitamura T, Gorman DM, Arai K, Yokota T, Miyajima A. Molecular cloning of a second subunit of the receptor for human granulocyte-macrophage colony-stimulating factor (GM-CSF): reconstitution of a high-affinity GM-CSF receptor. Proc Natl Acad Sci U S A . 1990;87:9655–9659. doi: 10.1073/pnas.87.24.9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Goodall GJ, Bagley CJ, Vadas MA, Lopez AF. A model for the interaction of the GM-CSF, IL-3 and IL-5 receptors with their ligands. Growth Factors . 1993;8:87–97. doi: 10.3109/08977199309046929. [DOI] [PubMed] [Google Scholar]
- 93. Robb L, Drinkwater CC, Metcalf D, Li R, Köntgen F, Nicola NA, et al. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc Natl Acad Sci U S A . 1995;92:9565–9569. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shima K, Arumugam P, Sallese A, Horia U, Ma Y, Trapnell C, et al. A murine model of hereditary pulmonary alveolar proteinosis caused by homozygous Csf2ra gene disruption. Am J Physiol Lung Cell Mol Physiol . 2022;322:L438–L448. doi: 10.1152/ajplung.00175.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Seymour JF, Lieschke GJ, Grail D, Quilici C, Hodgson G, Dunn AR. Mice lacking both granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF have impaired reproductive capacity, perturbed neonatal granulopoiesis, lung disease, amyloidosis, and reduced long-term survival. Blood . 1997;90:3037–3049. [PubMed] [Google Scholar]
- 96. LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J Clin Invest . 1999;103:563–569. doi: 10.1172/JCI5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lieschke GJ, Stanley E, Grail D, Hodgson G, Sinickas V, Gall JA, et al. Mice lacking both macrophage- and granulocyte-macrophage colony-stimulating factor have macrophages and coexistent osteopetrosis and severe lung disease. Blood . 1994;84:27–35. [PubMed] [Google Scholar]
- 98. Paine R, III, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, et al. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol . 2000;164:2602–2609. doi: 10.4049/jimmunol.164.5.2602. [DOI] [PubMed] [Google Scholar]
- 99. Gonzalez-Juarrero M, Hattle JM, Izzo A, Junqueira-Kipnis AP, Shim TS, Trapnell BC, et al. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J Leukoc Biol . 2005;77:914–922. doi: 10.1189/jlb.1204723. [DOI] [PubMed] [Google Scholar]
- 100. Reed JA, Ikegami M, Cianciolo ER, Lu W, Cho PS, Hull W, et al. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol . 1999;276:L556–L563. doi: 10.1152/ajplung.1999.276.4.L556. [DOI] [PubMed] [Google Scholar]
- 101. Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest . 1996;97:649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zsengellér ZK, Reed JA, Bachurski CJ, LeVine AM, Forry-Schaudies S, Hirsch R, et al. Adenovirus-mediated granulocyte-macrophage colony-stimulating factor improves lung pathology of pulmonary alveolar proteinosis in granulocyte-macrophage colony-stimulating factor-deficient mice. Hum Gene Ther . 1998;9:2101–2109. doi: 10.1089/hum.1998.9.14-2101. [DOI] [PubMed] [Google Scholar]
- 103. Kleff V, Sorg UR, Bury C, Suzuki T, Rattmann I, Jerabek-Willemsen M, et al. Gene therapy of beta(c)-deficient pulmonary alveolar proteinosis (beta(c)-PAP): studies in a murine in vivo model. Mol Ther . 2008;16:757–764. doi: 10.1038/mt.2008.7. [DOI] [PubMed] [Google Scholar]
- 104. Nishinakamura R, Miyajima A, Mee PJ, Tybulewicz VL, Murray R. Hematopoiesis in mice lacking the entire granulocyte-macrophage colony-stimulating factor/interleukin-3/interleukin-5 functions. Blood . 1996;88:2458–2464. [PubMed] [Google Scholar]
- 105. Arumugam P, Suzuki T, Shima K, McCarthy C, Sallese A, Wessendarp M, et al. Long-term safety and efficacy of gene/pulmonary macrophage transplantation therapy of pulmonary alveolar proteinosis in Csf2ra−/− mice. Mol Ther . 2019;27:1597–1611. doi: 10.1016/j.ymthe.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Suzuki T, McCarthy C, Carey BC, Borchers M, Beck D, Wikenheiser-Brokamp KA, et al. Increased Pulmonary GM-CSF causes alveolar macrophage accumulation: mechanistic implications for desquamative interstitial pneumonitis. Am J Respir Cell Mol Biol . 2020;62:87–94. doi: 10.1165/rcmb.2018-0294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Uchida K, Nakata K, Trapnell BC, Terakawa T, Hamano E, Mikami A, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood . 2004;103:1089–1098. doi: 10.1182/blood-2003-05-1565. [DOI] [PubMed] [Google Scholar]
- 108. Seymour JF, Doyle IR, Nakata K, Presneill JJ, Schoch OD, Hamano E, et al. Relationship of anti-GM-CSF antibody concentration, surfactant protein A and B levels, and serum LDH to pulmonary parameters and response to GM-CSF therapy in patients with idiopathic alveolar proteinosis. Thorax . 2003;58:252–257. doi: 10.1136/thorax.58.3.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Uchida K, Nakata K, Suzuki T, Luisetti M, Watanabe M, Koch DE, et al. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood . 2009;113:2547–2556. doi: 10.1182/blood-2009-05-155689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Meager A, Wadhwa M, Bird C, Dilger P, Thorpe R, Newsom-Davis J, et al. Spontaneously occurring neutralizing antibodies against granulocyte-macrophage colony-stimulating factor in patients with autoimmune disease. Immunology . 1999;97:526–532. doi: 10.1046/j.1365-2567.1999.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Seymour JF, Begley CG, Dirksen U, Presneill JJ, Nicola NA, Moore PE, et al. Attenuated hematopoietic response to granulocyte-macrophage colony-stimulating factor in patients with acquired pulmonary alveolar proteinosis. Blood . 1998;92:2657–2667. [PubMed] [Google Scholar]
- 112. Wang Y, Thomson CA, Allan LL, Jackson LM, Olson M, Hercus TR, et al. Characterization of pathogenic human monoclonal autoantibodies against GM-CSF. Proc Natl Acad Sci U S A . 2013;110:7832–7837. doi: 10.1073/pnas.1216011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shibata Y, Berclaz P-Y, Trapnell BC. Arrest of alveolar macrophage differentiation, in vivo, in GM-CSF-deficient mice is associated with altered expression of transcription factors PU.1 and ICSBP [abstract] Am J Respir Crit Care Med . 2001;163:A790. [Google Scholar]
- 114. Bonfield TL, Raychaudhuri B, Malur A, Abraham S, Trapnell BC, Kavuru MS, et al. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. Am J Physiol Lung Cell Mol Physiol . 2003;285:L1132–L1136. doi: 10.1152/ajplung.00216.2003. [DOI] [PubMed] [Google Scholar]
- 115. Harris JO. Pulmonary alveolar proteinosis: abnormal in vitro function of alveolar macrophages. Chest . 1979;76:156–159. doi: 10.1378/chest.76.2.156. [DOI] [PubMed] [Google Scholar]
- 116. Nugent KM, Pesanti EL. Macrophage function in pulmonary alveolar proteinosis. Am Rev Respir Dis . 1983;127:780–781. doi: 10.1164/arrd.1983.127.6.780. [DOI] [PubMed] [Google Scholar]
- 117. de Villiers WJ, Smart EJ. Macrophage scavenger receptors and foam cell formation. J Leukoc Biol . 1999;66:740–746. doi: 10.1002/jlb.66.5.740. [DOI] [PubMed] [Google Scholar]
- 118. Maxfield FR, Wüstner D. Intracellular cholesterol transport. J Clin Invest . 2002;110:891–898. doi: 10.1172/JCI16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kennedy MA, Barrera GC, Nakamura K, Baldán A, Tarr P, Fishbein MC, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab . 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 120. Thomassen MJ, Barna BP, Malur AG, Bonfield TL, Farver CF, Malur A, et al. ABCG1 is deficient in alveolar macrophages of GM-CSF knockout mice and patients with pulmonary alveolar proteinosis. J Lipid Res . 2007;48:2762–2768. doi: 10.1194/jlr.P700022-JLR200. [DOI] [PubMed] [Google Scholar]
- 121. Igarashi M, Osuga J, Uozaki H, Sekiya M, Nagashima S, Takahashi M, et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ Res . 2010;107:1387–1395. doi: 10.1161/CIRCRESAHA.110.226613. [DOI] [PubMed] [Google Scholar]
- 122. Baldán A, Tarr P, Vales CS, Frank J, Shimotake TK, Hawgood S, et al. Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J Biol Chem . 2006;281:29401–29410. doi: 10.1074/jbc.M606597200. [DOI] [PubMed] [Google Scholar]
- 123. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol . 2014;15:1026–1037. doi: 10.1038/ni.3005. [DOI] [PubMed] [Google Scholar]
- 124. Malur A, Mccoy AJ, Arce S, Barna BP, Kavuru MS, Malur AG, et al. Deletion of PPAR gamma in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J Immunol . 2009;182:5816–5822. doi: 10.4049/jimmunol.0803504. [DOI] [PubMed] [Google Scholar]
- 125. Baker AD, Malur A, Barna BP, Ghosh S, Kavuru MS, Malur AG, et al. Targeted PPARgamma deficiency in alveolar macrophages disrupts surfactant catabolism. J Lipid Res . 2010;51:1325–1331. doi: 10.1194/jlr.M001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Baker AD, Malur A, Barna BP, Kavuru MS, Malur AG, Thomassen MJ. PPARgamma regulates the expression of cholesterol metabolism genes in alveolar macrophages. Biochem Biophys Res Commun . 2010;393:682–687. doi: 10.1016/j.bbrc.2010.02.056. [DOI] [PubMed] [Google Scholar]
- 127. Ikegami M, Jobe AH, Huffman Reed JA, Whitsett JA. Surfactant metabolic consequences of overexpression of GM-CSF in the epithelium of GM-CSF-deficient mice. Am J Physiol . 1997;273:L709–L714. doi: 10.1152/ajplung.1997.273.4.L709. [DOI] [PubMed] [Google Scholar]
- 128. Presneill JJ, Nakata K, Inoue Y, Seymour JF. Pulmonary alveolar proteinosis. Clin Chest Med . 2004;25:593–613. doi: 10.1016/j.ccm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 129. Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect . 2012;65:173–179. doi: 10.1016/j.jinf.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 130. Clague HW, Wallace AC, Morgan WK. Pulmonary interstitial fibrosis associated with alveolar proteinosis. Thorax . 1983;38:865–866. doi: 10.1136/thx.38.11.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Luisetti M, Bruno P, Kadija Z, Suzuki T, Raffa S, Torrisi MR, et al. Relationship among diffuse pulmonary fibrosis, alveolar proteinosis and anti-GM-CSF autoantibodies. Respir Care . 2011;56:1608–1610. doi: 10.4187/respcare.01054. [DOI] [PubMed] [Google Scholar]
- 132. Akira M, Inoue Y, Arai T, Sugimoto C, Tokura S, Nakata K, et al. Osaka Respiratory Diseases Symposia Group Pulmonary fibrosis on high-resolution CT of patients with pulmonary alveolar proteinosis. AJR Am J Roentgenol . 2016;207:544–551. doi: 10.2214/AJR.15.14982. [DOI] [PubMed] [Google Scholar]
- 133. Sha J, Langton D. Role of granulocyte-macrophage colony-stimulating factor in pulmonary fibrosis following pulmonary alveolar proteinosis. Respirol Case Rep . 2016;4:e00159. doi: 10.1002/rcr2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Agarwal PP, Seely JM, Perkins DG, Matzinger FR, Alikhan Q. Pulmonary alveolar proteinosis and end-stage pulmonary fibrosis: a rare association. J Thorac Imaging . 2005;20:242–244. doi: 10.1097/01.rti.0000160733.74997.2d. [DOI] [PubMed] [Google Scholar]
- 135. Moore BB, Coffey MJ, Christensen P, Sitterding S, Ngan R, Wilke CA, et al. GM-CSF regulates bleomycin-induced pulmonary fibrosis via a prostaglandin-dependent mechanism. J Immunol . 2000;165:4032–4039. doi: 10.4049/jimmunol.165.7.4032. [DOI] [PubMed] [Google Scholar]
- 136. Heppleston AG, Wright NA, Stewart JA. Experimental alveolar lipo-proteinosis following the inhalation of silica. J Pathol . 1970;101:293–307. doi: 10.1002/path.1711010402. [DOI] [PubMed] [Google Scholar]
- 137. Xipell JM, Ham KN, Price CG, Thomas DP. Acute silicoproteinosis. Thorax . 1977;32:104–111. doi: 10.1136/thx.32.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Dethloff LA, Gilmore LB, Brody AR, Hook GE. Induction of intra- and extra-cellular phospholipids in the lungs of rats exposed to silica. Biochem J . 1986;233:111–118. doi: 10.1042/bj2330111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. McCunney RJ, Godefroi R. Pulmonary alveolar proteinosis and cement dust: a case report. J Occup Med . 1989;31:233–237. doi: 10.1097/00043764-198903000-00008. [DOI] [PubMed] [Google Scholar]
- 140. deMello DE, Heyman S, Phelps DS, Hamvas A, Nogee L, Cole S, et al. Ultrastructure of lung in surfactant protein B deficiency. Am J Respir Cell Mol Biol . 1994;11:230–239. doi: 10.1165/ajrcmb.11.2.8049084. [DOI] [PubMed] [Google Scholar]
- 141. Hamvas A, Nogee LM, White FV, Schuler P, Hackett BP, Huddleston CB, et al. Progressive lung disease and surfactant dysfunction with a deletion in surfactant protein C gene. Am J Respir Cell Mol Biol . 2004;30:771–776. doi: 10.1165/rcmb.2003-0323OC. [DOI] [PubMed] [Google Scholar]
- 142. Hamvas A. Inherited surfactant protein-B deficiency and surfactant protein-C associated disease: clinical features and evaluation. Semin Perinatol . 2006;30:316–326. doi: 10.1053/j.semperi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 143. Martin RJ, Rogers RM, Myers NM. Pulmonary alveolar proteinosis: shunt fraction and lactic acid dehydrogenase concentration as aids to diagnosis. Am Rev Respir Dis . 1978;117:1059–1062. doi: 10.1164/arrd.1978.117.6.1059. [DOI] [PubMed] [Google Scholar]
- 144. Goldstein LS, Kavuru MS, Curtis-McCarthy P, Christie HA, Farver C, Stoller JK. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest . 1998;114:1357–1362. doi: 10.1378/chest.114.5.1357. [DOI] [PubMed] [Google Scholar]
- 145. Lee KN, Levin DL, Webb WR, Chen D, Storto ML, Golden JA. Pulmonary alveolar proteinosis: high-resolution CT, chest radiographic, and functional correlations. Chest . 1997;111:989–995. doi: 10.1378/chest.111.4.989. [DOI] [PubMed] [Google Scholar]
- 146. Wang BM, Stern EJ, Schmidt RA, Pierson DJ. Diagnosing pulmonary alveolar proteinosis: a review and an update. Chest . 1997;111:460–466. doi: 10.1378/chest.111.2.460. [DOI] [PubMed] [Google Scholar]
- 147. Johkoh T, Itoh H, Müller NL, Ichikado K, Nakamura H, Ikezoe J, et al. Crazy-paving appearance at thin-section CT: spectrum of disease and pathologic findings. Radiology . 1999;211:155–160. doi: 10.1148/radiology.211.1.r99ap10155. [DOI] [PubMed] [Google Scholar]
- 148. Ishii H, Trapnell BC, Tazawa R, Inoue Y, Akira M, Kogure Y, et al. Japanese Center of the Rare Lung Disease Consortium Comparative study of high-resolution CT findings between autoimmune and secondary pulmonary alveolar proteinosis. Chest . 2009;136:1348–1355. doi: 10.1378/chest.09-0097. [DOI] [PubMed] [Google Scholar]
- 149. Maygarden SJ, Iacocca MV, Funkhouser WK, Novotny DB. Pulmonary alveolar proteinosis: a spectrum of cytologic, histochemical, and ultrastructural findings in bronchoalveolar lavage fluid. Diagn Cytopathol . 2001;24:389–395. doi: 10.1002/dc.1086. [DOI] [PubMed] [Google Scholar]
- 150. Costabel U, Guzman J, Bonella F, Oshimo S. Bronchoalveolar lavage in other interstitial lung diseases. Semin Respir Crit Care Med . 2007;28:514–524. doi: 10.1055/s-2007-991525. [DOI] [PubMed] [Google Scholar]
- 151.Bonella F, Ohshimo S, Bauer P, Guzman J, Costabel U. In: Interventional pneumology. Strausz J, Bolliger CT, editors. Lausanne, Switzerland: European Respiratory Society; 2010. Bronchoalveolar lavage; pp. 59–72. [Google Scholar]
- 152. Milleron BJ, Costabel U, Teschler H, Ziesche R, Cadranel JL, Matthys H, et al. Bronchoalveolar lavage cell data in alveolar proteinosis. Am Rev Respir Dis . 1991;144:1330–1332. doi: 10.1164/ajrccm/144.6.1330. [DOI] [PubMed] [Google Scholar]
- 153. Bonfield TL, John N, Malur A, Barna BP, Culver DA, Kavuru MS, et al. Elevated monocyte chemotactic proteins 1, 2, and 3 in pulmonary alveolar proteinosis are associated with chemokine receptor suppression. Clin Immunol . 2005;114:79–85. doi: 10.1016/j.clim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 154. Trapnell BC, McCarthy C. The alveolar lipidome in pulmonary alveolar proteinosis: a new target for therapeutic development? Am J Respir Crit Care Med . 2019;200:800–802. doi: 10.1164/rccm.201905-1009ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Uchida K, Nakata K, Carey B, Chalk C, Suzuki T, Sakagami T, et al. Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis. J Immunol Methods . 2014;402:57–70. doi: 10.1016/j.jim.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kitamura T, Uchida K, Tanaka N, Tsuchiya T, Watanabe J, Yamada Y, et al. Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 2000;162:658–662. doi: 10.1164/ajrccm.162.2.9910032. [DOI] [PubMed] [Google Scholar]
- 157. Suzuki T, Maranda B, Sakagami T, Catellier P, Couture CY, Carey BC, et al. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J . 2011;37:201–204. doi: 10.1183/09031936.00090610. [DOI] [PubMed] [Google Scholar]
- 158. Suzuki T, Sakagami T, Young LR, Carey BC, Wood RE, Luisetti M, et al. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med . 2010;182:1292–1304. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kusakabe Y, Uchida K, Hiruma T, Suzuki Y, Totsu T, Suzuki T, et al. A standardized blood test for the routine clinical diagnosis of impaired GM-CSF signaling using flow cytometry. J Immunol Methods . 2014;413:1–11. doi: 10.1016/j.jim.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Krone K. Foley CL. Fishman MP. Vargus SO. Forbes LR. Al-Herz W. et al. Signal transducer and activator of transcription 5B (STAT5B) deficiency-associated lung disease. Am J Respir Crit Care Med . doi: 10.1164/rccm.202111-2527LE. [DOI] [PubMed] [Google Scholar]
- 161. Honda Y, Kuroki Y, Matsuura E, Nagae H, Takahashi H, Akino T, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med . 1995;152:1860–1866. doi: 10.1164/ajrccm.152.6.8520747. [DOI] [PubMed] [Google Scholar]
- 162. Takahashi T, Munakata M, Suzuki I, Kawakami Y. Serum and bronchoalveolar fluid KL-6 levels in patients with pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 1998;158:1294–1298. doi: 10.1164/ajrccm.158.4.9712003. [DOI] [PubMed] [Google Scholar]
- 163. Iyonaga K, Suga M, Yamamoto T, Ichiyasu H, Miyakawa H, Ando M. Elevated bronchoalveolar concentrations of MCP-1 in patients with pulmonary alveolar proteinosis. Eur Respir J . 1999;14:383–389. doi: 10.1034/j.1399-3003.1999.14b24.x. [DOI] [PubMed] [Google Scholar]
- 164. Kuroki Y, Tsutahara S, Shijubo N, Takahashi H, Shiratori M, Hattori A, et al. Elevated levels of lung surfactant protein A in sera from patients with idiopathic pulmonary fibrosis and pulmonary alveolar proteinosis. Am Rev Respir Dis . 1993;147:723–729. doi: 10.1164/ajrccm/147.3.723. [DOI] [PubMed] [Google Scholar]
- 165. Seymour JF, Presneill JJ, Schoch OD, Downie GH, Moore PE, Doyle IR, et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med . 2001;163:524–531. doi: 10.1164/ajrccm.163.2.2003146. [DOI] [PubMed] [Google Scholar]
- 166. Kuroki Y, Takahashi H, Chiba H, Akino T. Surfactant proteins A and D: disease markers. Biochim Biophys Acta . 1998;1408:334–345. doi: 10.1016/s0925-4439(98)00079-9. [DOI] [PubMed] [Google Scholar]
- 167. Brasch F, Birzele J, Ochs M, Guttentag SH, Schoch OD, Boehler A, et al. Surfactant proteins in pulmonary alveolar proteinosis in adults. Eur Respir J . 2004;24:426–435. doi: 10.1183/09031936.04.00076403. [DOI] [PubMed] [Google Scholar]
- 168. Bonella F, Ohshimo S, Miaotian C, Griese M, Guzman J, Costabel U. Serum KL-6 is a predictor of outcome in pulmonary alveolar proteinosis. Orphanet J Rare Dis . 2013;8:53. doi: 10.1186/1750-1172-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. McCarthy C, Carey B, Trapnell BC. Blood testing in the diagnosis of pulmonary alveolar proteinosis. Lancet Respir Med . 2018;6:e54. doi: 10.1016/S2213-2600(18)30372-2. [DOI] [PubMed] [Google Scholar]
- 170. McCarthy C, Carey BC, Nowell-Bostic M, Rieman-Klingler M, Chalk C, Toth A, et al. Differential diagnosis of pulmonary alveolar proteinosis: lung biopsy or blood test? [abstract] Am J Respir Crit Care Med . 2018;197:A1093. [Google Scholar]
- 171. McCarthy C, Carey B, Trapnell BC. Blood testing for differential diagnosis of pulmonary alveolar proteinosis syndrome. Chest . 2019;155:450–452. doi: 10.1016/j.chest.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hildebrandt J, Yalcin E, Bresser HG, Cinel G, Gappa M, Haghighi A, et al. Characterization of CSF2RA mutation related juvenile pulmonary alveolar proteinosis. Orphanet J Rare Dis . 2014;9:171. doi: 10.1186/s13023-014-0171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Suzuki T, Sakagami T, Carey BC, Chalk C, Wood RE, Black D, et al. Preclinical evaluation of pulmonary macrophages transplantation therapy of hereditary pulmonary alveolar proteinosis [abstract] Am J Respir Crit Care Med . 2011;183:A2505. [Google Scholar]
- 174. Ishii H, Seymour JF, Tazawa R, Inoue Y, Uchida N, Nishida A, et al. Secondary pulmonary alveolar proteinosis complicating myelodysplastic syndrome results in worsening of prognosis: a retrospective cohort study in Japan. BMC Pulm Med . 2014;14:37. doi: 10.1186/1471-2466-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ishii H, Tazawa R, Kaneko C, Saraya T, Inoue Y, Hamano E, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J . 2011;37:465–468. doi: 10.1183/09031936.00092910. [DOI] [PubMed] [Google Scholar]
- 176. Ladeb S, Fleury-Feith J, Escudier E, Tran Van Nhieu J, Bernaudin JF, Cordonnier C. Secondary alveolar proteinosis in cancer patients. Support Care Cancer . 1996;4:420–426. doi: 10.1007/BF01880639. [DOI] [PubMed] [Google Scholar]
- 177. Cordonnier C, Fleury-Feith J, Escudier E, Atassi K, Bernaudin JF. Secondary alveolar proteinosis is a reversible cause of respiratory failure in leukemic patients. Am J Respir Crit Care Med . 1994;149:788–794. doi: 10.1164/ajrccm.149.3.8118651. [DOI] [PubMed] [Google Scholar]
- 178. Hildebrand FL, Jr, Rosenow EC, III, Habermann TM, Tazelaar HD. Pulmonary complications of leukemia. Chest . 1990;98:1233–1239. doi: 10.1378/chest.98.5.1233. [DOI] [PubMed] [Google Scholar]
- 179. Schiller V, Aberle DR, Aberle AM. Pulmonary alveolar proteinosis: occurrence with metastatic melanoma to lung. Chest . 1989;95:466–467. doi: 10.1378/chest.95.2.466. [DOI] [PubMed] [Google Scholar]
- 180. Ruben FL, Talamo TS. Secondary pulmonary alveolar proteinosis occurring in two patients with acquired immune deficiency syndrome. Am J Med . 1986;80:1187–1190. doi: 10.1016/0002-9343(86)90683-2. [DOI] [PubMed] [Google Scholar]
- 181. Miller RR, Churg AM, Hutcheon M, Lom S. Pulmonary alveolar proteinosis and aluminum dust exposure. Am Rev Respir Dis . 1984;130:312–315. doi: 10.1164/arrd.1984.130.2.312. [DOI] [PubMed] [Google Scholar]
- 182. Webster JR, Jr, Battifora H, Furey C, Harrison RA, Shapiro B. Pulmonary alveolar proteinosis in two siblings with decreased immunoglobulin A. Am J Med . 1980;69:786–789. doi: 10.1016/0002-9343(80)90453-2. [DOI] [PubMed] [Google Scholar]
- 183. Carnovale R, Zornoza J, Goldman AM, Luna M. Pulmonary alveolar proteinosis: its association with hematologic malignancy and lymphoma. Radiology . 1977;122:303–306. doi: 10.1148/122.2.303. [DOI] [PubMed] [Google Scholar]
- 184. Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med . 2004;350:1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 185. Nogee LM. Genetics of pediatric interstitial lung disease. Curr Opin Pediatr . 2006;18:287–292. doi: 10.1097/01.mop.0000193310.22462.1f. [DOI] [PubMed] [Google Scholar]
- 186. Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Pathology Cooperative Group ChILD Research Co-operative. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med . 2007;176:1120–1128. doi: 10.1164/rccm.200703-393OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Awab A, Khan MS, Youness HA. Whole lung lavage-technical details, challenges and management of complications. J Thorac Dis . 2017;9:1697–1706. doi: 10.21037/jtd.2017.04.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Ramirez J, Schultz RB, Dutton RE. Pulmonary alveolar proteinosis: a new technique and rationale for treatment. Arch Intern Med . 1963;112:419–431. doi: 10.1001/archinte.1963.03860030173021. [DOI] [PubMed] [Google Scholar]
- 189. Ramirez J, Nyka W, McLaughlin J. Pulmonary alveolar proteinosis: diagnostic technics and observations. N Engl J Med . 1963;268:165–171. doi: 10.1056/NEJM196301242680401. [DOI] [PubMed] [Google Scholar]
- 190. Ben-Abraham R, Greenfeld A, Rozenman J, Ben-Dov I. Pulmonary alveolar proteinosis: step-by-step perioperative care of whole lung lavage procedure. Heart Lung . 2002;31:43–49. doi: 10.1067/mhl.2002.119831. [DOI] [PubMed] [Google Scholar]
- 191. Wasserman K, Blank N, Fletcher G. Lung lavage (alveolar washing) in alveolar proteinosis. Am J Med . 1968;44:611–617. doi: 10.1016/0002-9343(68)90062-4. [DOI] [PubMed] [Google Scholar]
- 192. Beccaria M, Luisetti M, Rodi G, Corsico A, Zoia MC, Colato S, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. Eur Respir J . 2004;23:526–531. doi: 10.1183/09031936.04.00102704. [DOI] [PubMed] [Google Scholar]
- 193. Moreira JP, Ferraz S, Freitas C, Morais A, Albuquerque RR, Fiuza C. Whole-lung lavage for severe pulmonary alveolar proteinosis assisted by veno-venous extracorporeal membrane oxygenation: a case report. Can J Respir Ther . 2018;55:9–12. doi: 10.29390/cjrt-2018-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Campo I, Luisetti M, Griese M, Trapnell BC, Bonella F, Grutters JC, et al. WLL International Study Group A global survey on whole lung lavage in pulmonary alveolar proteinosis. Chest . 2016;150:251–253. doi: 10.1016/j.chest.2016.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. McKenzie B, Wood RE, Bailey A. Airway management for unilateral lung lavage in children. Anesthesiology . 1989;70:550–553. doi: 10.1097/00000542-198903000-00030. [DOI] [PubMed] [Google Scholar]
- 196. Abdelmalak BB, Khanna AK, Culver DA, Popovich MJ. Therapeutic whole-lung lavage for pulmonary alveolar proteinosis: a procedural update. J Bronchology Interv Pulmonol . 2015;22:251–258. doi: 10.1097/LBR.0000000000000180. [DOI] [PubMed] [Google Scholar]
- 197. Wood RE. Pediatric bronchoscopy. Chest Surg Clin N Am . 1996;6:237–251. [PubMed] [Google Scholar]
- 198. Campo I, Luisetti M, Griese M, Trapnell BC, Bonella F, Grutters J, et al. WLL International Study Group Whole lung lavage therapy for pulmonary alveolar proteinosis: a global survey of current practices and procedures. Orphanet J Rare Dis . 2016;11:115. doi: 10.1186/s13023-016-0497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Bonella F, Bauer PC, Griese M, Wessendorf TE, Guzman J, Costabel U. Wash-out kinetics and efficacy of a modified lavage technique for alveolar proteinosis. Eur Respir J . 2012;40:1468–1474. doi: 10.1183/09031936.00017612. [DOI] [PubMed] [Google Scholar]
- 200. Cohen ES, Elpern E, Silver MR. Pulmonary alveolar proteinosis causing severe hypoxemic respiratory failure treated with sequential whole-lung lavage utilizing venovenous extracorporeal membrane oxygenation: a case report and review. Chest . 2001;120:1024–1026. doi: 10.1378/chest.120.3.1024. [DOI] [PubMed] [Google Scholar]
- 201. Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest . 2002;122:1480–1485. doi: 10.1378/chest.122.4.1480. [DOI] [PubMed] [Google Scholar]
- 202. Selecky PA, Wasserman K, Benfield JR, Lippmann M. The clinical and physiological effect of whole-lung lavage in pulmonary alveolar proteinosis: a ten-year experience. Ann Thorac Surg . 1977;24:451–461. doi: 10.1016/s0003-4975(10)63440-6. [DOI] [PubMed] [Google Scholar]
- 203. Rogers RM, Levin DC, Gray BA, Moseley LW., Jr Physiologic effects of bronchopulmonary lavage in alveolar proteinosis. Am Rev Respir Dis . 1978;118:255–264. doi: 10.1164/arrd.1978.118.2.255. [DOI] [PubMed] [Google Scholar]
- 204. Seymour JF, Dunn AR, Vincent JM, Presneill JJ, Pain MC. Efficacy of granulocyte-macrophage colony-stimulating factor in acquired alveolar proteinosis. N Engl J Med . 1996;335:1924–1925. doi: 10.1056/NEJM199612193352513. [DOI] [PubMed] [Google Scholar]
- 205. Kavuru MS, Sullivan EJ, Piccin R, Thomassen MJ, Stoller JK. Exogenous granulocyte-macrophage colony-stimulating factor administration for pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 2000;161:1143–1148. doi: 10.1164/ajrccm.161.4.9906044. [DOI] [PubMed] [Google Scholar]
- 206. Barraclough RM, Gillies AJ. Pulmonary alveolar proteinosis: a complete response to GM-CSF therapy. Thorax . 2001;56:664–665. doi: 10.1136/thorax.56.8.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. de Vega MG, Sánchez-Palencia A, Ramírez A, Cervera S, Aneiros J. GM-CSF therapy in pulmonary alveolar proteinosis. Thorax . 2002;57:837. doi: 10.1136/thorax.57.9.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Venkateshiah SB, Yan TD, Bonfield TL, Thomassen MJ, Meziane M, Czich C, et al. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest . 2006;130:227–237. doi: 10.1378/chest.130.1.227. [DOI] [PubMed] [Google Scholar]
- 209. Bonfield TL, Kavuru MS, Thomassen MJ. Anti-GM-CSF titer predicts response to GM-CSF therapy in pulmonary alveolar proteinosis. Clin Immunol . 2002;105:342–350. doi: 10.1006/clim.2002.5301. [DOI] [PubMed] [Google Scholar]
- 210. Tazawa R, Hamano E, Arai T, Ohta H, Ishimoto O, Uchida K, et al. Granulocyte-macrophage colony-stimulating factor and lung immunity in pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 2005;171:1142–1149. doi: 10.1164/rccm.200406-716OC. [DOI] [PubMed] [Google Scholar]
- 211. Arai T, Hamano E, Inoue Y, Ryushi T, Nukiwa T, Sakatani M, et al. Serum neutralizing capacity of GM-CSF reflects disease severity in a patient with pulmonary alveolar proteinosis successfully treated with inhaled GM-CSF. Respir Med . 2004;98:1227–1230. doi: 10.1016/j.rmed.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 212. Tazawa R, Nakata K, Inoue Y, Nukiwa T. Granulocyte-macrophage colony-stimulating factor inhalation therapy for patients with idiopathic pulmonary alveolar proteinosis: a pilot study; and long-term treatment with aerosolized granulocyte-macrophage colony-stimulating factor. A case report. Respirology . 2006;11:S61–S64. doi: 10.1111/j.1440-1843.2006.00811.x. [DOI] [PubMed] [Google Scholar]
- 213. Wylam ME, Ten R, Prakash UB, Nadrous HF, Clawson ML, Anderson PM. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J . 2006;27:585–593. doi: 10.1183/09031936.06.00058305. [DOI] [PubMed] [Google Scholar]
- 214. Papiris SA, Tsirigotis P, Kolilekas L, Papadaki G, Papaioannou AI, Triantafillidou C, et al. Long-term inhaled granulocyte macrophage-colony-stimulating factor in autoimmune pulmonary alveolar proteinosis: effectiveness, safety, and lowest effective dose. Clin Drug Investig . 2014;34:553–564. doi: 10.1007/s40261-014-0208-z. [DOI] [PubMed] [Google Scholar]
- 215. Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med . 2010;181:1345–1354. doi: 10.1164/rccm.200906-0978OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Tazawa R, Inoue Y, Arai T, Takada T, Kasahara Y, Hojo M, et al. Duration of benefit in patients with autoimmune pulmonary alveolar proteinosis after inhaled granulocyte-macrophage colony-stimulating factor therapy. Chest . 2014;145:729–737. doi: 10.1378/chest.13-0603. [DOI] [PubMed] [Google Scholar]
- 217. Satoh H, Tazawa R, Sakakibara T, Ohkouchi S, Ebina M, Miki M, et al. Bilateral peripheral infiltrates refractory to immunosuppressants were diagnosed as autoimmune pulmonary alveolar proteinosis and improved by inhalation of granulocyte/macrophage-colony stimulating factor. Intern Med . 2012;51:1737–1742. doi: 10.2169/internalmedicine.51.6093. [DOI] [PubMed] [Google Scholar]
- 218. Price A, Manson D, Cutz E, Dell S. Pulmonary alveolar proteinosis associated with anti-GM-CSF antibodies in a child: successful treatment with inhaled GM-CSF. Pediatr Pulmonol . 2006;41:367–370. doi: 10.1002/ppul.20347. [DOI] [PubMed] [Google Scholar]
- 219. Yamamoto H, Yamaguchi E, Agata H, Kandatsu N, Komatsu T, Kawai S, et al. A combination therapy of whole lung lavage and GM-CSF inhalation in pulmonary alveolar proteinosis. Pediatr Pulmonol . 2008;43:828–830. doi: 10.1002/ppul.20856. [DOI] [PubMed] [Google Scholar]
- 220. Rodríguez Portal JA, Rodríguez Becerra E, Sánchez Garrido A. Response to inhaled granulocyte-macrophage colony-stimulating factor in a patient with alveolar proteinosis [in Spanish] Arch Bronconeumol . 2009;45:150–152. doi: 10.1016/j.arbres.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 221. Robinson TE, Trapnell BC, Goris ML, Quittell LM, Cornfield DN. Quantitative analysis of longitudinal response to aerosolized granulocyte-macrophage colony-stimulating factor in two adolescents with autoimmune pulmonary alveolar proteinosis. Chest . 2009;135:842–848. doi: 10.1378/chest.08-1317. [DOI] [PubMed] [Google Scholar]
- 222. Ohashi K, Sato A, Takada T, Arai T, Nei T, Kasahara Y, et al. Direct evidence that GM-CSF inhalation improves lung clearance in pulmonary alveolar proteinosis. Respir Med . 2012;106:284–293. doi: 10.1016/j.rmed.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 223. Tian X, Yang Y, Chen L, Sui X, Xu W, Li X, et al. Inhaled granulocyte-macrophage colony stimulating factor for mild-to-moderate autoimmune pulmonary alveolar proteinosis–a six month phase II randomized study with 24 months of follow-up. Orphanet J Rare Dis . 2020;15:174. doi: 10.1186/s13023-020-01450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Tazawa R, Ueda T, Abe M, Tatsumi K, Eda R, Kondoh S, et al. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med . 2019;381:923–932. doi: 10.1056/NEJMoa1816216. [DOI] [PubMed] [Google Scholar]
- 225. Trapnell BC, Inoue Y, Bonella F, Morgan C, Jouneau S, Bendstrup E, et al. IMPALA Trial Investigators Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. N Engl J Med . 2020;383:1635–1644. doi: 10.1056/NEJMoa1913590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Ohkouchi S, Akasaka K, Ichiwata T, Hisata S, Iijima H, Takada T, et al. Sequential granulocyte-macrophage colony-stimulating factor inhalation after whole-lung lavage for pulmonary alveolar proteinosis: a report of five intractable cases. Ann Am Thorac Soc . 2017;14:1298–1304. doi: 10.1513/AnnalsATS.201611-892BC. [DOI] [PubMed] [Google Scholar]
- 227. Campo I, Mariani F, Paracchini E, Kadija Z, Zoretto M, Tinelli C, et al. Inhaled sargramostim and whole lung lavage (WLL) as therapy of autoimmune pulmonary alveolar proteinosis (aPAP) Eur Respir J . 2016;48:PA3870. [Google Scholar]
- 228. Akasaka K, Tanaka T, Kitamura N, Ohkouchi S, Tazawa R, Takada T, et al. Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: a retrospective cohort study. BMC Pulm Med . 2015;15:88. doi: 10.1186/s12890-015-0085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Kavuru MS, Bonfield TL, Thomassen MJ. Plasmapheresis, GM-CSF, and alveolar proteinosis. Am J Respir Crit Care Med . 2003;167:1036. doi: 10.1164/ajrccm.167.7.950. [DOI] [PubMed] [Google Scholar]
- 230. Kavuru MS, Malur A, Marshall I, Barna BP, Meziane M, Huizar I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J . 2011;38:1361–1367. doi: 10.1183/09031936.00197710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Soyez B, Borie R, Menard C, Cadranel J, Chavez L, Cottin V, et al. Rituximab for auto-immune alveolar proteinosis, a real life cohort study. Respir Res . 2018;19:74. doi: 10.1186/s12931-018-0780-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Borie R, Debray MP, Laine C, Aubier M, Crestani B. Rituximab therapy in autoimmune pulmonary alveolar proteinosis. Eur Respir J . 2009;33:1503–1506. doi: 10.1183/09031936.00160908. [DOI] [PubMed] [Google Scholar]
- 233. Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest . 1997;111:1457–1458. doi: 10.1378/chest.111.5.1457. [DOI] [PubMed] [Google Scholar]
- 234.Clinical and Laboratory Standards Institute. Defining, establishing, and verifying reference intervals in the clinical laboratory; approved guideline—third edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2010. [Google Scholar]
- 235. Lenz D, Stahl M, Seidl E, Schöndorf D, Brennenstuhl H, Gesenhues F, et al. Rescue of respiratory failure in pulmonary alveolar proteinosis due to pathogenic MARS1 variants. Pediatr Pulmonol . 2020;55:3057–3066. doi: 10.1002/ppul.25031. [DOI] [PubMed] [Google Scholar]
- 236. Torrents D, Mykkänen J, Pineda M, Feliubadaló L, Estévez R, de Cid R, et al. Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat Genet . 1999;21:293–296. doi: 10.1038/6809. [DOI] [PubMed] [Google Scholar]
- 237. Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med . 2011;208:227–234. doi: 10.1084/jem.20101459. [DOI] [PMC free article] [PubMed] [Google Scholar]