

Background and Aims:
Efficacy of chimeric antigen receptor (CAR) T cells for treating solid tumors, including HCC, remains a challenge. Nanobodies are emerging building blocks of CAR T cells due to their small size and high expression. Membrane proximal sites have been shown as attractive epitopes of CAR T cells. However, current CAR formats are not tailored toward nanobodies or targeting membrane distal epitopes.
Approach and Results:
Using hYP7 Fv (membrane proximal) and HN3 VH nanobody (membrane distal) as GPC3 targeting elements, we sought to determine how hinges and transmembrane portions of varying structures and sizes affect CAR T-cell function. We generated multiple permutations of CAR T cells containing CD8, CD28, IgG4, and Fc domains. We show that engineered HN3 CAR T cells can be improved by 2 independent, synergistic changes in the hinge and transmembrane domains. The T cells expressing the HN3 CAR which contains the hinge region of IgG4 and the CD28 transmembrane domain (HN3-IgG4H-CD28TM) exhibited high cytotoxic activity and caused complete HCC tumor eradication in immunodeficient mice. HN3-IgG4H-CD28TM CAR T cells were enriched for cytotoxic-memory CD8+ T cells and NFAT signals, and reduced β catenin levels in HCC cells.
Conclusion:
Our findings indicate that altering the hinge and transmembrane domains of a nanobody-based CAR targeting a distal GPC3 epitope, in contrast to a membrane proximal epitope, lead to robust T-cell signaling and induce swift and durable eradication of HCC tumors.
INTRODUCTION
HCC is a highly aggressive tumor with poor prognosis. HCC accounts for 90% of liver cancer cases and caused 30,230 deaths in 2021 in the US alone.1 Immunotherapy using chimeric antigen receptor (CAR)-engineered T lymphocytes has shown promising responses in hematopoietic malignancies.2 In solid tumors, however, a significant roadblock is the broad and nonspecific biomarker expression of tumor antigens within normal tissues resulting in off-target cytotoxicity during treatment.3–6 Glypican 3 (GPC3) is highly specific for HCC.7–9 Thus, targeting GPC3 uniquely allows therapeutic activity to be directed exclusively to HCC sites. In a recent preclinical orthotopic mouse model of GPC3 targeting CAR T cells, the humanized murine scFv (hYP7) significantly outperformed HN3 VH nanobody-based CAR T cells despite having comparably high affinities.10–12 This difference was thought to be due to targeting a membrane proximal epitope rather than a distal epitope, described in the CD22, GPC3, and mesothelin contexts.12–15
Two ways to improve existing CARs are inclusion of fully human sequences and optimization of the antigen binding domain. Therefore, we sought to redesign CAR T cells using the fully HN3 human VH, which targets a membrane distal epitope of GPC3. However, 2 major engineering issues hamper use of nanobodies, such as HN3, in conventional CAR formats. These include: (1) knowledge gaps in engineering nanobodies such as VHs within existing CAR formats which are more tailored to scF versus (2) unknowns about antibodies that target distal epitopes since the current dogma in the CAR T-cell field highlights the importance of membrane proximal epitopes.16,17
Despite these challenges, the HN3 VH has several features that make it an attractive candidate for CAR T-cell engineering including; the smaller size of the heavy chain variable domains (VH) and Wnt blocking capability.18–21 The reduced size of the antigen recognition domain is a key advantage for expression using gene therapy vectors since the size of transgenes affects the titer and overall expression of the CAR. Furthermore, the use of a smaller targeting element such as HN3 confers steric advantages over the scFv, enabling HN3 to reach small or hidden surfaces.19 In contrast to hYP7, which targets the membrane proximal C-lobe of GPC3, HN3 targets a hydrophobic cavity at the N-lobe containing the Wnt site.19 Since upregulation of Wnt signaling is a major driver of HCC pathogenesis, blockage of Wnt by leveraging the native binding region confers an inherent benefit and is of major clinical significance. Swift and direct Wnt blocking capability could serve to improve CAR T-cell efficacy and displace hYP7 as the best GPC3 targeting element.22–26 Considering all these features, we ventured to adapt HN3 to the CAR T context by using tailored engineering approaches to augment the binding capacity of HN3 and fully optimize efficacy. Our results indicate that tailoring the hinge and transmembrane domains improves HN3 potency in vitro and in vivo.
EXPERIMENTAL PROCEDURES
Cell models
Hep3B (HCC), HepG2 (hepatoblastoma), HEK-293T, and Jurkat cell lines were purchased from American Type Culture Collection. Hep3B and HepG2 were transduced with lentiviruses expressing luciferase.12 The luciferase-expressing Huh7 cell line was a generous gift from Dr. Andras Heczey at Baylor College of Medicine. The Hep3BKO GFP-luciferase cell line was established using a CRISPR/CAS9 strategy knockout as previously described (Figure S1, http://links.lww.com/HC9/A76).19 The Jurkat reporter, made in our laboratory, containing tdTomato gene under the control of the NFAT response element, was stably integrated into Jurkat cells. All cell lines were cultured using standard procedures as reported by our group.
Jurkat binding and Jurkat-NFAT reporter
Jurkat and Jurkat-NFAT reporter cells were transduced with CAR containing lentiviruses at multiplicity of infection (MOI) 5. After 7 days, the cell surface EGFRt was assessed using anti-human IgG-phycoerythrin (APC)-conjugated antibody. The cell surface binding the CAR on Jurkat cells to GPC3 protein tagged with hfc (ACRO Biosystems) was assessed using anti-human IgG-phycoerythrin (APC)-conjugated antibody (Jackson ImmunoResearch). Jurkat-tdTomato CAR T cells were cocultured with GFP expressing Hep3B cells at 1:1 ratio for the indicated intervals in and observed using a Zeiss confocal microscope. Cell count and size were assessed using existing ImageJ tools.
CAR T-cell production and cell killing studies
CAR T cells were produced as described previously.12 Briefly, HEK-293T cells were co-transfected with packaging plasmid psPAX2 (Addgene #12260) and enveloping plasmid pMD2.G (Addgene #12259) using Calfectin (SignaGen). Lentiviral particles were collected, concentrated, and the functional titer was assessed using the EGFTt marker. Peripheral blood mononuclear cells from healthy donors were stimulated for 24 hours using anti-CD3/anti-CD28 antibody-coated beads in the presence of IL-2 (Invitrogen). To track cell count and viability, cells were monitored over a period of 12 days.12 Cytolytic assays were performed using luciferase-expressing cell lines and T cells transduced with GPC3 CARs as described previously.12,27 Briefly, tumor cells were incubated with T cells at indicated effector-target ratios for 18 hours. Luminescence of the lysates was analyzed using the luciferase assay system (Promega) on a plate spectrophotometer (Victor; PerkinElmer).
Animal studies
All the procedures used in the animal studies were approved by the Institutional Animal Care and Use Committee at the NIH under the protocol (LMB-059). Mice were housed in softwood bedding and fed standard chow diets. All interventions were conducted during light cycles. Xenograft mouse models were established by i.p. injecting 3 × 106 Hep3B or 1 × 106 Huh7 GFP-luciferase cells in female NOD-SCID-Gamma (NSG) at 7 weeks of age. When the mean tumor bioluminescence reached 1 × 108, the mice were injected i.p. or i.v. with 5 × 106 untransduced human donor T cells, engineered HN3 CAR T cells with CD8H or IgG4H and CD8TM or CD28TM. The treated mice were imaged biweekly during weeks 1 to 4 and weekly after week 4 on a Xenogen IVIS-200 Spectrum camera. Mice were euthanized when they reached IACUC-approved morbidity endpoints including any sign of distress. For ex vivo analysis, T cells were isolated using a tumor dissociation kit (Miltenyi Biotec) as previously described.28 For blood analysis, blood was collected at the indicated time points and flow cytometry was conducted as previously described.26,28
Structural models
All models were constructed with AlphaFoldV2 and further analyzed in Chimera. Normal mode analysis was conducted as previously described.29 The IUPRED2 algorithm was used to measure disorder tendency.30,31 Normal mode analysis and volumetric calculations of the pdb structures enabled additional parameterization of structures.
Western blotting
Hep3B cells (0.5 × 106) and CAR T cells were cocultured at the indicated time intervals and cell lysates were harvested. The gel and membranes were run using standard protocols for wet transfer at room temperature (Genscript eBlot L1). The membranes were stained and probed overnight at 4°C using the antibodies listed in the Supplementary Data (Table S1, http://links.lww.com/HC9/A76).
RESULTS
IgG4H and CD28TM display a helical structure and minimize HN3 disorder tendency
To examine how each CAR T construct may fold, we made structural models of each permutation of the HN3, hYP7, hinge, and transmembrane domains by AlphafoldV2 (Figure 1A, B). Physical properties including volume and sphericity were measured based on these models.32 The HN3-IgG4H-CD28TM harbored the lowest surface area, volume, and sphericity, and these values were identical to those from HN3-IgG4H-CD8TM (Figure 1A, B). However, despite having the same physical properties, the HN3-IgG4H-CD28TM and hYP7-IgG4H-CD28TM, not HN3-IgG4H-CD8TM, displayed a structured transmembrane and hinge domain (Figure 1A). Prediction of key flexible and molecular vibrations by normal mode analysis, an algorithm to detect residues in motion by structural and experimental data, revealed that the transmembrane and hinge regions in the HN3-IgG4H-CD28TM and hYP7-IgG4H-CD28TM constructs more structured and rigid than CD8-containing constructs (Figure 1B).33 Furthermore, HN3 constructs containing the IgG4H-CD8TM, and CD8H-CD28TM did not contain the same helical folding patterns and had higher degrees of flexibility as indicated by the red regions than blue regions (Figure 1A). These modeling data point to the importance of physical and steric properties which may help to enhance HN3 binding to the GPC3 when paired with the appropriate stabilizing hinge and transmembrane domains.
FIGURE 1.
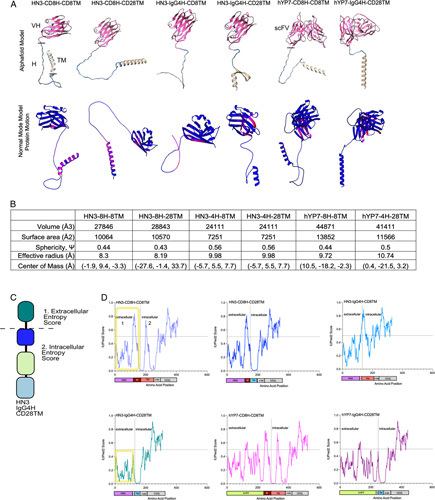
Structural modeling and entropy analysis reveals that the IgG4H and CD28TM display limited flexibility and low entropy. (A) Alphafold models and normal mode analysis of protein movement. The VH or scFV are coded in pink, the hinge (H) is in blue and the transmembrane (TM) is in tan. Residues with higher movement are listed in red, while restricted residues are in blue. (B) Physical characteristics of each CAR based on the models. Cell constructs are abbreviated as 8H-8TM (CD8H, CD8TM), 4H-28TM (IgG4H, CD28TM), 4H-8TM (IgG4H, CD8TM). (C) Diagram of extracellular and intracellular domains in the CAR T cell. The 4 main components include the targeting element, hinge, transmembrane, and t cell signaling portions. The intracellular and extracellular entropies are measured in 1 and 2. (D) Entropy scores using IUPRED2 algorithm. A higher score corresponds to higher entropy, while a lower score denotes lower entropy. CAR, chimeric antigen receptor.
To understand how another physical property may affect CAR-GPC3 binding, we also predicted intrinsically disordered regions, particularly in the extracellular region, by employing the IUPred2 algorithm.30 Using HN3 as the targeting element in all constructs, IUPred2 revealed high disorderness in the CD8H region (residues 110–140, Figure 1C, D). When CD8H was paired with CD28TM, the disorderness change was marginal (Figure 1D). Therefore, in terms of disorder tendency, CD8H-CD8TM and CD8H-CD28TM were similar. When IgG4H was paired with CD28TM, a dramatic change is seen as disorder tendency is minimized to low levels in the extracellular region where the interaction between the nanobody, hinge and transmembrane domain occurs (Figure 1D). This entropy minimization could affect HN3 binding capacity. Even when the IgG4H is paired with CD8TM, disorderness (residues 100–110) is increased, in contrast to the IgG4H-CD28TM. Altogether, these in silico analyses reveal that the orientation of the hinge-transmembrane domains, minimization of disorder, and flexibility in the targeting component are all critical features to consider in CAR T-cell design.
HN3 VH CAR Jurkat cells retained antigen specificity and binding capacity
To generate an array of CAR T cells, we developed T-cell lines (Jurkat) expressing CAR constructs containing the IgG4 hinge (IgG4H), CD8 hinge (CD8H), CD8 transmembrane (CD8TM), and CD28 transmembrane (CD28TM) domains (Figure 2A, B). To test the antigen specificity and binding capacity of the CAR component, we used the CAR Jurkat cells and assessed binding to GPC3 tagged with human Fc (hFc) (Figure 2C). We observed that all Jurkat-CAR constructs bound to GPC3-hFc efficiently (75.8%–98.6%) (Figure 2C). In this system, we observed only minimal enhancement of binding to GPC3-hfc by engineered constructs compared with the original CD8-containing constructs. All CAR T cells, including HN3-IgG4H-CD28TM, specifically bound GPC3-hFc.
FIGURE 2.
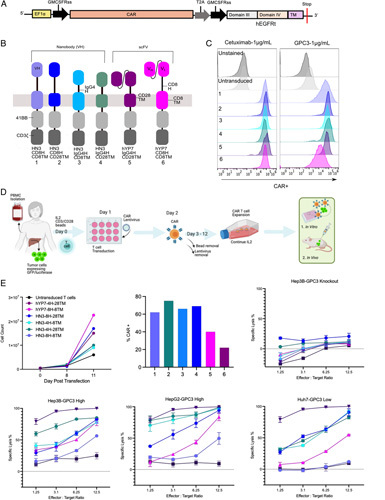
Engineered CAR T cells display increased cytoxicity in a panel of HCC line. (A) Map of CARs targeting GPC3 with varying hinge and transmembrane domains. (B) CAR constructs resulting in different permutations of CAR constructs. (C) Jurkat-CAR T cells binding to cetuximab, GPC3 at 1 μg/mL. (D) Overview of production of CAR T cells starting from donor PBMCs. (E) Cell count monitored over the course of 11 days. Transduction efficiency of all constructs used in the cytotoxicity experiment. Cell killing graphs showing cytotoxicity. CAR T cells were cultured with GPC3 expressing HCC cells lines (Hep3B, HepG2, and Huh7) and GPC3 knockout (KO) cells at different effector to target ratios. CAR, chimeric antigen receptor; PBMC, peripheral blood mononuclear cell.
HN3-IgG4H-CD28TM CAR T cells enhanced activity in high and low antigen density settings in vitro
CAR T cells were produced using human donor peripheral blood mononuclear cells and monitored over a period of 12 days to use in vitro or in vivo (Figure 2D). When we assessed the CAR surface expression, we found that the average expression of hYP7 constructs was 1.5- to 2-fold lower than that of HN3 constructs at the same MOI (Figure 2E). To examine CAR T-cell potency in high versus low antigen density contexts, we employed multiple HCC cell lines (Hep3B, HepG2, and Huh7).34 These cell lines were isolated from diverse patients with different levels of tumor aggressiveness and varying GPC3 expression level. We found that hYP7-IgG4H-CD28TM and HN3-IgG4H-CD28TM CAR T cells displayed the highest cell killing in all HCC lines, including in low antigen density Huh7 cells (Figure 2E).
Comparing the HN3 constructs containing different hinges and transmembrane domains, the HN3-CD8H-CD28TM CAR T cells demonstrated 40% improved killing from the original HN3-CD8H-CD8TM cells (Figure 2E). Our evaluation of HN3-CD8H-CD28TM versus HN3-IgG4H-CD28TM CAR T cells, revealed that inclusion of the IgG4 hinge domain (IgG4H) increases potency by 10% to 20% as well, especially at the lowest effector to target ratio. Due to this, we eliminated the HN3-CD8H-CD28TM from further in vivo investigation. All CAR T cells minimally reacted with the GPC3 knockout cell line, indicating low levels of nonspecific reactivity under these conditions. Taken together, this suggested that HN3 activity is maximized with the IgG4H and CD28TM together.
HN3-IgG4H-CD28TM CAR T cells eradicate GPC3 high HCC xenografts in mice
Having demonstrated both the specificity and cytotoxicity of engineered CAR T cells in vitro, we sought to test in vivo antitumor activity (Figure 3A). In line with the in vitro findings, HN3-IgG4H-CD28TM CAR T cells demonstrated high antitumor activity in vivo and large tumors were eradicated within 10 days (Figure 3B). In contrast, tumors in the HN3-IgG4H-CD8TM group responded only partially or continued to grow during the study (Figure 3B, C). Median survival of mice treated with untransduced T cells, HN3-CD8H-CD8TM, and hYP7-CD8H-CD8TM CAR T cells was 27, 29, and 30 days, respectively (Figure 3C). In contrast, all mice treated with HN3-IgG4H-CD28TM survived until the end of the study (day 35). The HN3-IgG4H-CD28TM group demonstrated swift reduction in tumor size starting at day 3 and ultimately remained tumor free for the full study period.
FIGURE 3.
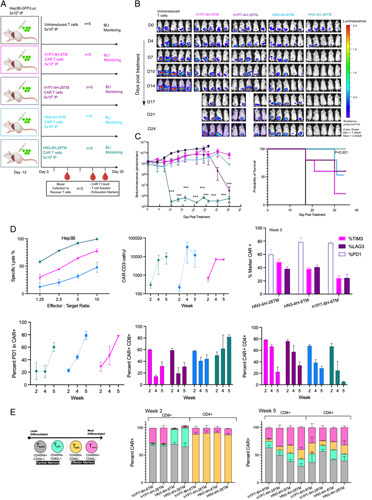
HN3-IgG4H-CD28TM CAR T cells eradicate tumors within 7 days in a Hep3B murine model. (A) Study timeline of tumor injection (i.p., 3 million), treatment of HCC tumors (i.p., 5 million) and blood collection in an NSG mouse model. Cell constructs are abbreviated using the previously described convention. (B) Bioluminescent imaging of tumors over the course of the study period. (C) Bioluminescent quantification. Values represent mean±SEM. million ***p < 0.001; ns, not significant. Survival curve for the full study period of 35 days. (D) Ex vivo T cells isolated from mouse spleen in Hep3B cells. Standardized CAR T-cell counts and CAR-positive percentage over 2 to 5 weeks. PD1, TIM3, and LAG3 panel at the end of study. PD1 indicating exhaustion in all groups over 2 to 5 weeks. CD4 and CD8 percentages over the 5-week period. (E) T-cell subsets consist of stem cell memory (Tscm), central memory (Tcm), effector memory expressing CD45RA (Temra), effector memory cells (Tem). Each group represents 3 animals on study (n = 3). CAR, chimeric antigen receptor; PD1, programmed cell death protein 1.
We next set out to compare the antitumor capacities of the HN3 VH construct to that of the more traditional scFv construct, hYP7. To address the issue of the scFv versus nanobody performance, we included the hYP7-IgG4H-CD28TM CAR T cells to compare with HN3-IgG4H-CD28TM. The hYP7-IgG4H-CD28TM construct was difficult to transduce in vitro with 25% to 30% transduction at most as well as lower expansion capability (Figure 2E). Despite these challenges, the construct performed well in cytotoxicity assays particularly in the Hep3B and HepG2 lines. However, in the xenograft model, the hYP7 construct induced delayed tumor regression only after day 30, while the lead engineered HN3 construct induced tumor regression within 3 days (Figure 3B, C). These data suggest that HN3-IgG4H-CD28TM is more potent than hYP7-IgG4H-CD28TM at the same dose level. Due to delayed antitumor response and expression challenges, we eliminated hYP7-IgG4H-CD28TM for further investigation in favor of HN3-IgG4H-CD28TM, which displayed the most potent activity in vivo and in vitro.
Having demonstrated tumor eradication while mice were on the study, we explored if the T cells retained the capacity to elicit cytotoxic activity ex vivo. We observed that T cells isolated from the spleens retrieved from mice on study reacted potently when cocultured with Hep3B cells. HN3-IgG4H-CD28TM CAR T cells were most potent in eliciting cytotoxic activity (Figure 3D).
CAR T cells targeting GPC3 display a large proliferative response
To monitor the number of CAR T cells and exhaustion markers, we analyzed blood retrieved from mice in the HCC tumor xenograft study (Figure 3A,D).35-37 Over the course of 28 days, CAR T-cell numbers increased by several logs from mice treated with the HN3-IgG4H-CD28TM, indicative of a large proliferative response in all groups (Figure 3D). The greatest enrichment of CAR T cells occurred between 2 and 4 weeks and plateaued at the 5-week time point. Furthermore, over the course of 2 to 5 weeks, average PD1 expression from 2 to 5 weeks and PD1 expression at the end of the study was lowest in the HN3-IgG4H-CD28TM group (Figure 3D).38 Importantly, PD1 expression remained lowin the HN3-IgG4H-CD28TM group, while CAR T cells rapidly expanded in mice from 2 to 4 weeks, only to increase at 5 weeks when there was no tumor (Figure 3D). At the end of study, exhaustion markers TIM3 and LAG3 were also lowest in the hYP7-CD8H-CD8TM group, although TIM3 and LAG5 were comparable across all 3 constructs tested. Altogether, these data demonstrated that in vivo CAR T-cell proliferation occurred within the first 4 weeks of treatment and CAR T cells continued to circulate and proliferated after tumor eradication. Limited exhaustion, measured by PD1, was seen until week 4, however exhaustion started to peak at week 5.
CAR T cells targeting GPC3 display enrichment of CD8+ and Temras
To understand the characteristics of the T-cell response driven by CAR T cells, we examined the proportions of CD4+ and CD8+ T cells as well as various subsets within these, including T Stem Cell Memory (Tscm), T Central Memory (Tcm), T Effector Memory (Tem), and T Effector Memory re-expressing CD45ra (Temra) (Figure 3E). The CAR T cells were recovered from animal blood at 3 time points during the study period (Figure 3A). At week 2, the hYP7 and HN3 groups contained similar numbers of CD8+ and CD4+ T cells (Figure 3D). In the hYP7 groups, the 2 major populations of CD8+ T cells were Tscm and Temra, while the major population of CD4+ T cells were Tem (Figure 3E). In contrast, at week 2, CD8+ T cells in the HN3 groups appeared to have predominantly Tscm and Tcm phenotypes, indicating that differentiation is slower in the HN3 group. The CD4+ T cells among the HN3 groups were the same, containing Tem much like the hYP7 group (Figure 3E). At week 5, most of the CAR T cells in the HN3-IgG4H-CD28TM group displayed the CD8+ phenotype and were observed to be engaged in effector functions (Figure 3E).
Of note, CAR T cells recovered from animal blood in the HN3-IgG4H-CD28TM group were overwhelmingly CD8+ Temra. (Figure 3E, Figure S3, http://links.lww.com/HC9/A76). Expansion of the CAR T cells over a 2- to 5-week period results in a clear transition to CD8+ Temra, which appear to be critical to the antitumor response. Central memory T cells also remained as a subset of the population indicating that both types of T cells are necessary to avert exhaustion and continue trafficking to the tumor site. This preponderance of CD8+ Temra was not seen in the other groups. In summary, this supports the idea that transition of CD8+ Temra cells are critical to inducing regression and durable responses.
IgG4H and CD28TM domains without Fc enhance HN3 potency in vitro and in vivo
To test whether the length of the hinge affects antigen binding, we incorporated Fc components and tested HN3 CAR T cells with short (IgG4H-CD28TM), intermediate (IgG4H-CD28TM-M, containing CH3) and long hinges (IgG4H-CD28TM-L, containing CH2CH3) (Figure 4A).16,17 Cell counts and transduction assays indicated that the long hinge construct was slower to expand and less effectively expressed at the same MOI (Figure 4B). All constructs minimally reacted with GPC3 knockout Hep3B cells, indicating an antigen-specific interaction (Figure 4B, E). The HN3-IgG4H-CD28TM performed outperformed intermediate and long hinge CAR T cells. Altogether, this confirmed that the HN3 nanobody can be engineered to induce potent killing in vitro using IgG4H and CD28TM together.
FIGURE 4.
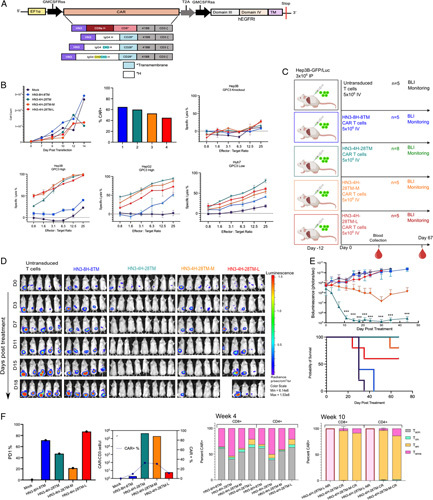
Fc containing CAR T cells do not improve efficacy of engineered CAR T cells in vitro or in vivo. (A) CAR T-cell construct map with Fc containing long (IgG4-CH2CH3H), intermediate (IgG4-CH3H), and short (IgG4H) hinge regions. (B) Cell count monitored over the course of 14 days. Transduction efficiency measured by CAR+ cells at day 8. CAR T cells were cultured with GPC3 expressing HCC cells lines (Hep3B, HepG2, and Huh7) and GPC3 knockout (KO) cells at different effector to target ratios to assess specific lysis. (C) Study diagram if injection (i.p., 3 million), treatment of HCC tumors (i.v., 5 million), and blood collection in an NSG mouse model Intermediate and long hinges containing Fc are indicated by M or L after the construct name. (D) Bioluminescence imaging results quantifying tumor size. (E) Bioluminescence quantification of tumor size. Values represent mean±SEM. million ***p < 0.001; ns, not significant. Survival curve over full course of study (67 days). (F) PD1 indicating exhaustion shows that in all constructs at week 4. Standardized CAR T-cell counts and CAR-positive percentage in all groups at week 4. All listed subsets of T cells were also quantified at 4 and 10 weeks by flow cytometry. CAR, chimeric antigen receptor; PD1, programmed cell death protein 1.
We next tested the function of Fc hinge-containing CAR T cells in the xenograft HCC tumor model (Figure 4C). Consistent with the in vitro findings, tumors in the HN3-IgG4H-CD28TM group regressed within 7 days (Figure 4D, E). All tumors in the HN3-IgG4H-CD28TM group were eradicated entirely and did not regrow after 67 days (Figure 4D, E). This lack of tumor can be observed in the abdominal cavity of a representative mouse at the end of study (Figure S2, http://links.lww.com/HC9/A76). Furthermore, when recovered spleens were compared, mice from the HN3-IgG4H-CD28TM group consistently had 2 to 3 times larger spleens and therefore, higher numbers of CAR+ cells within the spleens (Figure S2, http://links.lww.com/HC9/A76). Mice treated with CAR T cells containing Fc components showed partial responses (Figure 4D, E). Four of 5 mice in the HN3-IgG4-CH3H-CD28TM group had no tumors and 1 of 5 mice in the HN3-IgG4-CH2CH3H-CD28TM had no tumors. The HN3-CD8H-CD8TM group displayed no response to CAR T-cell treatment. When we considered a low antigen density model in vivo, we found striking tumor regression in 5 of 6 mice (Figure S4, http://links.lww.com/HC9/A76). All together, these data suggested that the HN3-IgG4H-CD28TM construct was the most potent in orchestrating a rapid and durable antitumor response.
We monitored the number of CAR T cells and exhaustion markers recovered from blood from the mice at 4 and 10 weeks. Consistent with the previous findings, standardized CAR T-cell counts were in the log range in mouse peripheral blood in the HN3-IgG4H-CD28TM and HN3-IgG4-CH3H-CD28TM groups (Figure 4F). Lower exhaustion by PD1 was observed in these same groups, at 48% and 20%, respectively (Figure 4E, F). Again, we sought to understand the characteristics of the T-cell response driven by CAR T cells by examining the proportions of CD4+ and CD8+ T cells as well as various T-cell subsets. By week 4, similar levels of CD8+ Temra were seen particularly in the group treated with HN3-IgG4H-CD28TM (Figure 4F). At the end of the study, we compared the T-cell phenotypes of complete responders to nonresponders. CD8+ Tem was the dominant phenotype observed in complete responders (Figure 4F). Based on the results of the in vitro and in vivo models, we concluded that extension of the hinge length using Fc was not necessary and possibly impeded the HN3-GPC3 interaction.
HN3-IgG4H-CD28TM block Wnt signaling and activate NFAT in HCC
Due to the potency observed in vivo, we examined the ability of the HN3 constructs to block tumor Wnt signaling when exposed to GPC3 expressing Hep3B cells (Figure 5A). We observed a reduction both active and total β catenin levels in the HN3-IgG4H-CD28TM when cocultured with Hep3B cells at 30 minutes and 3 hours. The engineered HN3 CAR T cells swiftly inhibited total β catenin and abolished active β catenin entirely at 3 hours (Figure 5A, B). Even at 30 minutes, engineered HN3 constructs had lower levels of total β catenin. These data confirmed that Wnt signaling is a direct target of the HN3-based CAR T cells and that within 3 hours HN3 directed CAR T cells directly inhibit β catenin activation.
FIGURE 5.
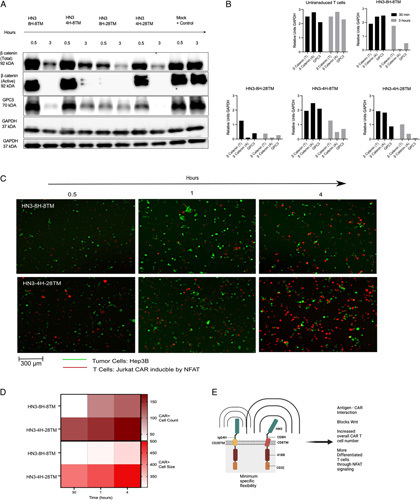
Cell signaling outcomes of Wnt and NFAT indicate swift β catenin blockage and NFAT upregulation. (A) In vitro coculture at 30 minutes and 3 hours indicating the level of total β catenin and active β catenin. (B) Quantification of western assays by GAPDH control. (C) Time course quantification of cocultured Hep3BGL and CAR Jurkat-NFAT cells. (D) Cell count and cell size which corresponds to intensity and pixels by ImageJ. (E) Proposed model of mechanism of HN3-4H-28TM CAR T cells in blocking Wnt signaling and inducing increased NFAT signaling. The brackets on each CAR construct indicate the sphere of motion. Each attribute listed on the right was measured or tested in each type of CAR construct. CAR, chimeric antigen receptor.
To understand how the CAR T cells act in vivo, we tested if NFAT played a role in the T-cell signaling.39–41 Over each time point, we observed gradually more NFAT signaling in the HN3-IgG4H-CD28TM CAR T cells compared with the original construct containing CD8H-CD8TM. Saturation of NFAT signaling reached a maximum at 4 hours when there were more CAR cells in the representative imaging window than Hep3B cells (Figure 5C). When the tdTomato signal was quantified by cell count and size, the differences between NFAT signaling in HN3-IgG4H-CD28TM versus HN3-CD8H-CD8TM were striking (Figure 5D). NFAT signaling in HN3-IgG4H-CD28TM treated Hep3B cells begins at a higher cell number in the 200 range overall and doubles within 4 hours with respect to cell number and intensity of the signal. These observations indicated that signaling via NFAT is more prevalent in T-cell activation in HN3-IgG4H-CD28TM than HN3-CD8H-CD8TM. Overall, we observed that NFAT is steadily upregulated in the HN3-IgG4H-CD28TM CAR T cells. Based on the full body of data, we propose that fine tuning the antigen-CAR interaction leads to these mechanistic changes in the cell thereby leading to potent and durable eradication of tumor (Figure 5E).
DISCUSSION
In this body of work, we engineered a VH guided by a structural rationale and developed a CAR T cell which more precisely fits in the CAR T format by leveraging a distal target epitope to achieve optimal efficacy in vitro and in vivo. Our evaluation of the fully human HN3 VH isolated from a phage display library, as the targeting element using precise engineering strategies has highlights the strong advantages of HN3 such as: (1) higher transduction, (2) efficacy in vitro and in vivo, and (3) Wnt blocking capability.
Unlike the hYP7 model, HN3 targets a completely different site on GPC3, distal to the cell membrane. Use of the distal site enables more efficient Wnt blocking capability, presumably leading to better CAR T-cell efficacy in contrast to hYP7. Here, we tested how selective engineering of extracellular and intracellular structural domains including the hinge and transmembrane portion are critical to induce efficient T-cell expansion and in vivo potency.42–44 More precisely, in the nanobody context, HN3 engineered CAR T cells perform best when combined with the short IgG4 hinge and CD28 transmembrane, inducing CD8+ Temra responses which lead to swift and durable tumor eradication.45–47
Structural modeling studies revealed that properties including entropy, flexibility, and movement can help characterize the type of interaction between the target antigen (GPC3) and the CAR T cell. These models and predictions of physical properties can facilitate analysis of in vitro and in vivo data. Both in the HN3 and hYP7 contexts, the angle and flexibility of the transmembrane domain relative to the targeting element affects how the CAR can orient with respect to the membrane. An alpha helix perpendicular to the cell membrane appears to improve CAR T activity in both contexts. As far as the structure of the hinge, a short, low entropy conformation appears to work best. Together, a restricted yet specific sphere of movement characterized by a short hinge and perpendicularly oriented transmembrane domain help stabilize both the HN3 and hYP7 CAR T cells. While we focused our analyses on structural models, the functional impact of CAR T-cell clustering due to IgG4 and CD28’s ability to form dimers could be another avenue of exploration. Based on the observed properties, the HN3 nanobody-based CAR is improved by the IgG4H-CD28TM, and the distal epitope seems to outperform the membrane proximal epitope.
In contrast to hYP7, HN3 has several desirable features. HN3 is appealing due to its ability to directly disrupt Wnt signaling through the native binding epitope and conserved VH only structure. Impressively, the engineered HN3-IgG4H-CD28TM CAR T cell is more potent than the previously published hYP7 CARs developed by our group and enables complete tumor eradication at the 5 million dose level within 7 to 10 days.12 To what extent the contribution of Wnt versus T-cell signaling is responsible for the potent effect is unclear without further investigation of competing ligands. Also, our data indicates HN3 constructs express more efficiently (between 60% and 80% more), which is at minimum 1.5-fold greater than the hYP7 group. Unlike hYP7, HN3 containing vectors can be more easily paired to develop bispecific or biparatopic modalities. We showed how HN3 engineered CAR T cells expanded in a log-fold fashion in mice, indicating that it is possible to boost expansion capacity. The HN3-IgG4-CD28TM CAR T cells also showed persistence and memory function as both Temra and naive T cells were present at the end of study. Overall, our data indicates that HN3 VH is an appealing targeting element when paired with the optimized hinge and transmembrane elements.
In our previous work, we showed that both the orthotopic and peritoneal models resemble the clinical environment.10–12 More specifically, both Hep3B and HepG2 tumors are associated with the mouse liver in both the previous and present study. Here, we opted for the peritoneal model since we tested numerous combinations of hinge and transmembrane components in cells and animal models by different routes of administration. Ideally, a genetically engineered mouse model with GPC3-positive HCC tumors would have been more clinically relevant to understand how mice with intact immune systems respond to treatment. Despite this, our work of testing all combinations of HN3-based CAR T cells serves as a basis for how engineering the extracellular components can boost CAR T-cell efficacy using a nanobody targeting a distal epitope using in an existing xenograft model. Surprisingly, these results challenge the existing dogma which focuses on scFv-based CARs and targeting proximal epitopes. In the HCC context, engineered HN3 VH-based CAR T cells display maximal efficacy and may mitigate challenges which plague solid tumors. As with many solid tumors, the therapeutic arsenal for HCC is still limited. HN3-IgG4H-CD28TM CAR T cells may harbor improved antitumor efficacy due to their unique capabilities.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Conception and design: A.K., N.L., and M.H. Development of methodology: A.K., D.L., N.L., Z.D., and M.H. Acquisition of data: A.K. Analysis and interpretation of data: A.K., D.L., N.L., L.R.R., and M.H. Writing: A.K. and M.H. Review and/or revision of manuscript: A.K. and M.H. Study supervision: M.H. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank the NCI CCR Animal Resource Program/NCI Biological Testing Branch for providing mice used in the present study, NCI CCR/Leidos Animal Facility assisting with training and animal care, and NCI CCR Flow Cytometry Core Facility for cell sorting. They also thank National Institutes of Health (NIH) Fellows Editorial Board for review.
CONFLICTS OF INTEREST
Nothing to report.
Footnotes
Funding information This work was supported by the Intramural Research Program of NIH, NCI (Z01 BC010891 and ZIA BC010891 to M.H.). This work was also supported by the NIH Mayo Hepatobiliary SPORE P50 CA210964 (L.R.R.). A.K. is a predoctoral fellow supported by the Mayo Clinic in Rochester, Minnesota (Clinical and Translational Science award UL1 TR000135), the Center for Cancer Research at the NCI, and the NIH Graduate Partnership Program.
Abbreviations: CAR, chimeric antigen receptor; GPC3, glypican 3; Tcm, central memory; Tscm, stem cell memory; Tem, effector memory; Temra, effector memory re-expressing CD45ra; VH, heavy chain variable domains.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Aarti Kolluri, Email: aarti.kolluri@nih.gov.
Dan Li, Email: dan.li2@nih.gov.
Nan Li, Email: nan.li@nih.gov.
Zhijian Duan, Email: zhijian.duan@nih.gov.
Lewis R. Roberts, Email: roberts.lewis@mayo.edu.
Mitchell Ho, Email: homi@mail.nih.gov.
REFERENCES
- 1.American Cancer Society. Cancer Facts and Figures 2021. 2021.
- 2.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (1991). 2013;36:133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu HC, Cheng W, Lai PL. Cloning and expression of a developmentally regulated transcript MXR7 in hepatocellular carcinoma: biological significance and temporospatial distribution. Cancer Res. 1997;57:5179–5184. [PubMed] [Google Scholar]
- 8.Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89–97. [DOI] [PubMed] [Google Scholar]
- 9.Ho M, Kim H. Glypican-3: a new target for cancer immunotherapy. Eur J Cancer. 2011;47:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phung Y, Gao W, Man YG, Nagata S, Ho M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. Mabs. 2012;4:592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YF, Ho M. Humanization of high-affinity antibodies targeting glypican-3 in hepatocellular carcinoma. Sci Rep. 2016;6:33878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Li N, Zhang YF, Fu H, Feng M, Schneider D, et al. Persistent Polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. 2020;158:2250–65.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassan R, Tomar S, Zhang J, Khanal M, Hong J, Venugopalan A, et al. Development of highly effective anti-mesothelin hYP218 chimeric antigen receptor T cells with increased tumor infiltration and persistence for treating solid tumors. Mol Cancer Ther. 2022;21:1195–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y-F, Phung Y, Gao W, Kawa S, Hassan R, Pastan I, et al. New high affinity monoclonal antibodies recognize non-overlapping epitopes on mesothelin for monitoring and treating mesothelioma. Sci Rep. 2015;5:9928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11:eaau7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23:757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng M, Gao W, Wang R, Chen W, Man YG, Figg WD, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci USA. 2013;110:E1083–E1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, Wei L, Liu X, Bai H, Ye Y, Li D, et al. A frizzled-like cysteine rich domain in glypican-3 mediates Wnt binding and regulates hepatocellular carcinoma tumor growth in mice. Hepatology. 2019;70:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao W, Kim H, Fau - Feng M, Feng M, Fau - Phung Y, Phung Y, et al. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology. 2014;60:576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao W, Tang Z, Zhang YF, Feng M, Qian M, Dimitrov DS, et al. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat Commun. 2015;6:6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95:8847–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763–73. [DOI] [PubMed] [Google Scholar]
- 24.Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43:515–24. [DOI] [PubMed] [Google Scholar]
- 25.Gao W, Ho M. The role of glypican-3 in regulating Wnt in hepatocellular carcinomas. Cancer Rep. 2011;1:14–19. [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–41.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, English H, Hong J, Liang T, Merlino G, Day C-P, et al. A novel PD-L1-targeted shark VNAR single-domain-based CAR-T cell strategy for treating breast cancer and liver cancer. Mol Ther Oncolytics. 2022;24:849–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov V, Lehnert U, Echols N, Milburn D, Engelman D, Gerstein M. Normal modes for predicting protein motions: a comprehensive database assessment and associated Web tool. Protein Sci. 2005;14:633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erdős G, Dosztányi Z. Analyzing protein disorder with IUPred2A. Curr Protoc Bioinformatics. 2020;70:e99. [DOI] [PubMed] [Google Scholar]
- 31.Mészáros B, Erdős G, Dosztányi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Voss NR, Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38:W555–W562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander JJ, Bey EM, Geddes EW, Lecatsas G. Establishment of a continuously growing cell line from primary carcinoma of the liver. S Afr Med J. 1976;50:2124–8. [PubMed] [Google Scholar]
- 34.Fu Y, Urban DJ, Nani RR, Zhang YF, Li N, Fu H, et al. Glypican-3-specific antibody drug conjugates targeting hepatocellular carcinoma. Hepatology. 2019;70:563–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen GM, Chen C, Das RK, Gao P, Chen C-H, Bandyopadhyay S, et al. Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors mediating long-term persistence of CAR T-cell therapy. Cancer Discov. 2021;11:2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma C, Cheung AF, Chodon T, Koya RC, Wu Z, Ng C, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discov. 2013;3:418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front Pharmacol. 2018;9:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson CL, Jurk D, Fullard N, Banks P, Page A, Luli S, et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat Commun. 2015;6:6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31:383–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Capurro M, Martin T, Shi W, Filmus J. Glypican-3 binds to Frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J Cell Sci. 2014;127:1565–75. [DOI] [PubMed] [Google Scholar]
- 42.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10:702–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu T, Li L, Huang C, Miao CG, Li J. MiR-520c-3p with therapeutic potential in hepatocellular carcinoma. Hepatol Res. 2014;44:825. [DOI] [PubMed] [Google Scholar]
- 46.Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduct Target Ther. 2021;6:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci USA. 2017;114:E6623. [DOI] [PMC free article] [PubMed] [Google Scholar]