

Abstract
Pediatric NAFLD has distinct and variable pathology, yet causation remains unclear. We have shown that maternal Western-style diet (mWSD) compared with maternal chow diet (CD) consumption in nonhuman primates produces hepatic injury and steatosis in fetal offspring. Here, we define the role of mWSD and postweaning Western-style diet (pwWSD) exposures on molecular mechanisms linked to NAFLD development in a cohort of 3-year-old juvenile nonhuman primates offspring exposed to maternal CD or mWSD followed by CD or Western-style diet after weaning. We used histologic, transcriptomic, and metabolomic analyses to identify hepatic pathways regulating NAFLD. Offspring exposed to mWSD showed increased hepatic periportal collagen deposition but unchanged hepatic triglyceride levels and body weight. mWSD was associated with a downregulation of gene expression pathways underlying HNF4α activity and protein, and downregulation of antioxidant signaling, mitochondrial biogenesis, and PPAR signaling pathways. In offspring exposed to both mWSD and pwWSD, liver RNA profiles showed upregulation of pathways promoting fibrosis and endoplasmic reticulum stress and increased BiP protein expression with pwWSD. pwWSD increased acylcarnitines and decreased anti-inflammatory fatty acids, which was more pronounced when coupled with mWSD exposure. Further, mWSD shifted liver metabolites towards decreased purine catabolism in favor of synthesis, suggesting a mitochondrial DNA repair response. Our findings demonstrate that 3-year-old offspring exposed to mWSD but weaned to a CD have periportal collagen deposition, with transcriptional and metabolic pathways underlying hepatic oxidative stress, compromised mitochondrial lipid sensing, and decreased antioxidant response. Exposure to pwWSD worsens these phenotypes, triggers endoplasmic reticulum stress, and increases fibrosis. Overall, mWSD exposure is associated with altered expression of candidate genes and metabolites related to NAFLD that persist in juvenile offspring preceding clinical presentation of NAFLD.

INTRODUCTION
NAFLD is one of the most prevalent noncommunicable liver diseases worldwide, affecting 10% of children in the US and up to 34% of obese youth.1,2 In rarer incidences, pediatric NAFLD can progress to NASH with accompanying inflammation and fibrosis, leading to end-stage liver disease and liver transplantation in early adulthood or sooner.3,4 However, the mechanisms driving the disease remain poorly understood. Pediatric NAFLD is present in 28% of obese children, yet it is also present at a rate of 8%–9.6% in nonobese children, suggesting that pediatric NAFLD onset can occur in the absence of obesity.5,6 Pediatric NAFLD in prepubertal children is associated with a higher risk for NASH progression.7,8 Identification of targets for halting pediatric NAFLD/NASH development is critical, as by the time pediatric NAFLD patients are admitted to the hospital, between 23% and 86% already have signs of NASH.8
The pathogenesis of pediatric NAFLD is multifactorial and distinct from the adult form of the disease.3,9 Pediatric NAFLD is more commonly associated with periportal steatosis and fibrosis rather than the typical pericentral or perisinusoidal fibrosis commonly seen in adult NAFLD.3,10 Recently, we demonstrated, in a nonhuman primate (NHP) model of maternal Western-style diet (mWSD) exposure, that fetal livers develop distinct periportal collagen deposition, steatosis, and stellate cell activation11 and have oxidative stress but no evidence of inflammation.12 Periportal collagen deposition was also present in 1-year-old (YO) offspring exposed to mWSD then switched to a chow diet (CD) postnatally.11 These offspring also have increased hepatic steatosis and increased hepatic macrophage activation in the absence of obesity, suggesting that mWSD exposure has long-lasting effects that impact hepatic metabolism and inflammation later in life.13 However, the molecular mechanisms for the pediatric form of the disease are unknown.
The developmental programming hypothesis states that adverse exposures during critical windows of development increase disease risk throughout the lifespan.9 Despite the existence of plausible mechanisms, the possibility that adverse perinatal exposures increase the risk of childhood NAFLD has received only limited attention.3,9 In mice exposed to both maternal and postweaning high-fat diet, NASH is accelerated.14 However, the molecular mechanisms underlying these effects are unclear,3,9 particularly in models that reflect the human condition. Our objective here was to examine the cellular, histologic, transcriptomic, and metabolomic pathways associated with development of NAFLD in 3YO juvenile NHP (equivalent to ∼9YO human) switched to a healthy CD at weaning. To discover similarities and differences in the mechanisms linking Western-style diet (WSD) exposure to NAFLD progression, we also compared the effects of postweaning Western-style diet (pwWSD) alone to continuous WSD exposure on histologic and molecular phenotypes in the liver of 3YO offspring.
MATERIALS AND METHODS
NHP model
All animal procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Oregon National Primate Research Center (ONPRC). The ONPRC abides by the Animal Welfare Act and Regulations enforced by the United States Department of Agriculture. Adult female Japanese macaques (Macaca fuscata) were fed a CD (14.7% calories from fat with 2.6% sucrose; Fiber Balanced 5000, Purina Mills) or WSD (36.3% calories from fat with 10% fructose and 8.8% sucrose; TAD diet 5L0P; Purina Mills) before and throughout pregnancy for 1–8 years, as reported previously.15 Dams were extensively phenotyped as described previously.12,15 Maternal phenotype data was measured in the early third trimester for each offspring’s corresponding pregnancy, including maternal body weight, fasting plasma insulin and glucose, and intravenous glucose tolerance test (ivGTT) results.12,15
Offspring were delivered naturally and kept with dams on their respective diets during lactation until weaning, at which point they were assigned to either a postweaning chow diet (pwCD) or pwWSD and maintained on that diet until 3YO. Within 2 months of necropsy, ivGTTs were performed. Offspring were killed, body and organ weights were collected, and blood was drawn from the abdominal aorta postpentobarbital and for analysis of blood chemistry and lipid profiles.15 Livers were removed, weighed, and pieces were flash frozen or fixed for histologic processing. Retroperitoneal white adipose tissue was also weighed.
Histopathologic analysis
Liver tissue samples were fixed in 10% zinc/formalin for 24 hours followed by storage in 70% ethanol. Samples were processed, paraffin-embedded, and 5-μm-thick sections prepared. Slides were stained with hematoxylin and eosin (H&E) or trichrome. Immunohistochemical detection of CD68-positive cells was performed11 with mouse CD68 (Agilent) and detection with an ImmPress HRP anti-mouse IGG peroxidase polymer detection kit (Vector Laboratories). Staining was visualized using an Olympus IX83 inverted microscope (Tokyo, Japan) and analyzed using ImageJ software. CD68-positive cells were counted, while blinded to the treatment group, within 2 sections per sample including 9–11 fields of view (FOV) surrounding one portal triad per section. Results are expressed as the average number of CD68-positive cells per FOV per animal.
SHG signal was acquired from unstained paraffin slides using a Zeiss 780 LSM laser-scanning confocal/multiphoton-excitation fluorescence microscope, as previously published.11 Image processing was performed using Zeiss ZEN 2012 software. Eight to 12 FOV containing 1 to 2 portal triads of similar size were analyzed for each liver sample using random blinded sampling. Images were analyzed and quantified with ImageJ software to obtain SHG signal intensity and area of SHG signal per FOV. The average intensity and area values for all measured FOV per animal are reported.
H&E-stained slides were scored by 2 independent, experienced pediatric hepatopathologists, blinded to the offspring groups, for features of NAFLD. Composite histologic activity was assessed using the validated NAFLD activity score (NAS) according to the NASH Clinical Research Network scoring system.16 NAS uses a semiquantitative scale to assess steatosis (0–3 score), lobular inflammation (0–3 score), and hepatocyte ballooning (0–2 score) to clinically grade liver histopathology.16
Liver tissue analysis
TBARS content was measured in liver tissue as previously described.17 Frozen liver tissue (∼50 mg) was homogenized in 500 μL ice-cold cell lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 1 mM EGTA, 1 mM EDTA) with phosphatase and protease inhibitors described previously13 using a bead mill homogenizer and ceramic beads. Capillary electrophoresis was performed on Jess Automated Simple Western System (ProteinSimple) according to manufacturer’s instructions with 0.5 mg/mL whole cell lysate and the following antibodies: HNF4α (1:10; Cell Signaling; 31135), BiP (1:20; Cell Signaling; 3183), and vinculin (1:1000; Cell Signaling; 13901). HNF4α and BiP were analyzed using RePlex Simple Western Assay to serially measure protein of interest and loading control (vinculin) within the same assay. Results were analyzed on Compass for Simple Western software.
Subset of offspring for multiomics analysis
For transcriptomic and metabolomic profiling experiments, we selected a subset of CD/CD and WSD/CD offspring based on the following criteria. In the CD/CD group, only offspring from dams with body fat measurements of < 26% were used. In the WSD/CD group, only offspring from dams with >30% body fat before pregnancy were used, with the exception of one animal that was born to a dam with 22% body fat. Given the smaller available sample numbers in the pwWSD cohorts, all offspring in the CD/WSD and WSD/WSD groups were included.
Bulk RNA sequencing and ingenuity pathway analysis (IPA)
RNA from the left liver lobe was isolated and quantitated on a Bioanalyzer system (Agilent). Total RNA (200–500 ng) and the Swift Rapid RNA Library kit (Integrated DNA Technologies) were used to prepare libraries. Libraries were sequenced as 2 × 150 bp reads on the NovaSeq. 6000 (Illumina) at the OUHSC Genomics Core. Derived sequences were analyzed with a custom computational pipeline consisting of the open-source GSNAP, Cufflinks, and R for sequence alignment and ascertainment of differential gene expression.18 Reads generated were mapped to the rhesus macaque genome (mmul10) by GSNAP,19 FPKM expression derived by Cufflinks,19 and differential expression analyzed with ANOVA in R. Differentially expressed genes (DEGs; p < 0.05 and Q < 0.1) were generated by comparing each group of WSD-exposed animals to CD/CD. Each of these lists, corresponding to each WSD group versus CD/CD, were then input into InteractiVenn software to determine the overlap of genes in each group (Venn diagrams) and to sort the DEGs into sets for downstream functional annotation (http://www.interactivenn.net/). Heatmaps were generated with Morpheus software (https://software.broadinstitute.org/morpheus). Data have been deposited into NCBI's GEO and are accessible through GEO series accession number GSE220102 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220102).
Lists of DEGs and the linear fold change compared with CD/CD were used as inputs in IPA (Qiagen) to identify predicted pathways and putative upstream regulators modified by mWSD and pwWSD exposure. Canonical pathways that met a threshold of −log(p value) of 5 or higher were declared significant. Positive Z-scores indicate predicted upregulation of the pathway, negative Z-scores indicate downregulation, Z-scores of 0 indicate neither upregulation nor downregulation, and pathways with no Z-score indicate that IPA was unable to calculate a Z-score. Predicted upstream regulators were declared significant with a p value of 1E-10 or lower and the list was filtered to include categories named in IPA as transcription regulator, ligand-dependent nuclear receptor, peptidase, group, kinase, growth factor, enzyme, transporter, and phosphatase.
Targeted metabolomic analysis
Frozen liver tissues were extracted at 15 mg/mL in cold 5:3:2 MeOH:MeCN:water with 30 minutes of vortexing at 4°C in the presence of glass beads as previously described.20 Homogenates were clarified by centrifugation (18,000g, 10 minutes, 4°C) and analyzed on a Thermo Vanquish UHPLC coupled to a Q Exactive MS using 5-minute C18 gradients in positive and negative ion modes (separate runs) as previously described.20 Metabolites were annotated and peaks integrated using MAVEN (Princeton University) in conjunction with the KEGG database. Zero readings (which were present in a maximum of 3 samples across all diet groups) were replaced with half of the minimum value detected for that metabolite across all diet groups. Metaboanalyst software (V5.0) was used for subsequent analysis. For comparisons of all 4 diet groups together, data were normalized by sum with range scaling, and CD/CD versus WSD/CD metabolite data were normalized by median, with auto-scaling. CD/WSD versus WSD/WSD were normalized by mean, with auto-scaling. Multivariate principal component analysis was performed with partial least squares discriminant analysis using metabolite profiles from all 4 diet groups or from CD/CD versus WSD/CD alone or CD/WSD versus WSD/WSD alone. The top 25 metabolites with the highest variable importance in projection (VIP) scores were loaded into clustered heatmaps (Pearson-Ward hierarchical clustering). Pathway enrichment analysis in the SMPDB database was performed using the top VIP metabolites in each dataset. Pathways were considered significant with p ≤ 0.05. In addition, for 2-group comparisons, and 4-group 4-way ANOVA comparisons, t tests were performed on normalized data to identify metabolites significantly different (p < 0.05) between groups.
Statistical analysis
Phenotypic data were analyzed by 2-way ANOVA with fixed effects of maternal diet, postweaning diet, sex, and interaction (SAS 9.4 or Prism V9; GraphPadA). Individual posttest comparisons (uncorrected Fisher least significant difference test) were made when the p value for maternal diet, postweaning diet, or interaction effect were p < 0.05. Posttest comparisons were considered significant if p < 0.05. NAS results were analyzed with a mixed-model ANOVA with fixed effects of maternal diet, postweaning diet, and sex, interaction effect (between maternal diet, postweaning diet, and sex), and random effect to account for repeated scores made by each pathologist. When significant, the effect of offspring sex is noted. When not significant, data for males and females were combined.
RESULTS
mWSD and pwWSD increase fasting insulin but not obesity in 3YO offspring
Body weight was not increased in offspring from either mWSD nor pwWSD groups, but pwWSD offspring had greater retroperitoneal adipose tissue and liver weights compared with pwCD groups (Table 1). Exposure to either mWSD or pwWSD increased fasting plasma insulin concentrations in 3YO offspring, while fasting glucose was lower in mWSD offspring weaned to CD (Table 1). In ivGTTs, glucose AUC was decreased and insulin AUC was greater in pwWSD offspring. No effect of mWSD was observed on glucose or insulin AUC. Offspring exposed to pwWSD had increased serum alanine transaminase concentrations, while aspartate transaminase concentrations were unaffected by mWSD or pwWSD (Table 1). Offspring from pwWSD groups had increased total serum cholesterol, LDL, and HDL levels (Table 1). Female offspring had higher circulating insulin levels, whereas male offspring had higher body weights, gamma-glutamyl transferase, and aspartate transaminase compared with female offspring (Table 1). Maternal phenotypes for the complete set of 3YO offspring, and maternal and offspring phenotypes for the subset of animals used for omics analyses are described in Tables 2–4, respectively.
TABLE 1.
Phenotype of 3-year-old offspring
| Maternal diet: | CD | CD | WSD | WSD | p values (ANOVA ) | |||
|---|---|---|---|---|---|---|---|---|
| Postweaning diet: | CD | WSD | CD | WSD | mWSD | pwWSD | INT | Sex |
| n | 26 | 6 | 27 | 9 | ||||
| Sex (male/female) | 10/16 | 4/2 | 17/10 | 4/5 | ||||
| Body weight (kg) | 6.13 ±0.18 | 5.63±0.32 | 6.42±0.26 | 5.82±0.28 | 0.58 | 0.10 | 0.44 | <0.05 |
| Male | 6.52±0.29 | 5.80±0.45 | 7.00±0.32 | 6.38±0.33 | ||||
| Female | 5.89±0.22 | 5.30±0.30 | 5.43±0.19 | 5.37±0.33 | ||||
| Average RWAT (g) | 0.45±0.05 | 2.38±1.04 | 0.85±0.13 | 2.04±0.44 | 0.51 | <0.0001 | 0.21 | 0.21 |
| Liver weight (g) | 135.1±4.1 | 116.9±7.8 | 142.0±4.8 | 122.9±6.9 | 0.57 | <0.05 | 0.69 | 0.06 |
| Glucose homeostasis | ||||||||
| Glucose (mg/dL)a | 57.4±1.8 | 60.5±5.3 | 51.3±1.7 | 54.8±2.4 | < 0.05 | 0.12 | 0.24 | 0.59 |
| Insulin (μU/mL)a | 4.4±0.6 | 21.8±7.4 | 6.4±1.2 | 10.0±1.9 | <0.005 | <0.0001 | <0.0001 | <0.05 |
| Male | 4.4±1.3 | 13.9±6.9 | 6.6±1.9 | 11.8±1.9 | ||||
| Female | 4.4±0.6 | 37.5±12.6 | 6.1±0.8 | 8.6±3.1 | ||||
| Glucose AUCb | 10008±201 | 8455±368 | 9020±337 | 8236±377 | 0.19 | <0.05 | 0.56 | 0.07 |
| Insulin AUCb | 1725±132 | 2192±329 | 1836±160 | 2264±244 | 0.94 | <0.05 | 0.07 | 0.67 |
| Blood chemistries | ||||||||
| ALT (IU/L)c | 44±2 | 52±5 | 44±1 | 65±4 | 0.10 | <0.0001 | 0.22 | 0.70 |
| AST (IU/L)c | 38±1 | 37±1 | 38±1 | 44±2 | 0.15 | 0.46 | 0.61 | 0.81 |
| GGT (IU/L)c | 90±2 | 87±3 | 102±4 | 97±5 | 0.07 | 0.53 | <0.1 | <0.05 |
| Male | 96±4 | 87±5 | 113±3 | 100±4 | ||||
| Female | 87±3 | 85±5 | 83±5 | 94±10 | ||||
| Alkaline phosphatase (IU/L)c | 411±14 | 457±36 | 463±22 | 470±48 | 0.26 | 0.30 | 0.24 | <0.0005 |
| Male | 434±26 | 493±43 | 521±22 | 539±97 | ||||
| Female | 396.6±17.6 | 387±38 | 363±25 | 415±33 | ||||
| Total cholesterol (mg/dL)c | 120±3 | 166±8 | 124±3 | 170±9 | 0.20 | <0.0001 | 0.09 | 0.57 |
| LDL (mg/dL)c | 63±2 | 80±8 | 67±3 | 82±10 | 0.29 | <0.05 | 0.22 | 0.71 |
| HDL (mg/dL)c | 55±1 | 87±5 | 55±1 | 89±4 | 0.25 | <0.0001 | <0.1 | 0.23 |
| TGs (mg/dL)a | 37±3 | 53±9 | 42±4 | 32±5 | 0.25 | 0.65 | 0.31 | 0.82 |
Data are represented as mean±SEM. Two-way ANOVA with fixed effect for maternal (mWSD) and postweaning (pwWSD) diet, interaction (INT) effect, and sex is shown. When sex effect for a variable has a p<0.05, mean±SEM for each sex is shown. p values in bold indicate significance.
Fasting plasma levels, obtained during necropsy.
Plasma levels, performed under fasting conditions within 2 months before necropsy.
Serum levels, obtained under fasting conditions during necropsy.
Abbreviations: ALT indicates alanine transaminase; AST, aspartate transaminase; CD, chow diet; GGT, gamma-glutamyl transferase; RWAT, retroperitoneal white adipose tissue, TG, triglyceride; WSD, Western-style diet.
TABLE 2.
Phenotype of the dams of the 3-year-old offspring
| Maternal diet: | CD | CD | WSD | WSD | p values (ANOVA) | ||
|---|---|---|---|---|---|---|---|
| Postweaning diet: | CD | WSD | CD | WSD | mWSD | pwWSD | INT |
| n | 26 | 6 | 27 | 9 | |||
| Age (years) | 10.1±0.7 | 9.6±0.4 | 8.1±0.4 | 6.7±0.6 | <0.05 | 0.22 | 0.54 |
| Years on diet | 0±0 | 0±0 | 2.9±0.3 | 2.5±0.7 | t test: p = 0.64 | ||
| Body weight (kg) | 9.4±0.3 | 9.4±0.5 | 10.4±0.4 | 9.1±0.8 | 0.56 | 0.24 | 0.27 |
| Body fat (%) | 22.3±1.7 | 20.0±3.8 | 27.9±1.9 | 16.1±2.1 | 0.77 | <0.05 | 0.10 |
| Body weight (kg)a | 10.2±0.3 | 10.0±0.5 | 10.4±0.3 | 9.7±0.7 | 0.91 | 0.39 | 0.69 |
| Glucose (mg/dL)a | 40.9±2.0 | 38.3±5.9 | 42.2±1.2 | 45.3±5.5 | 0.23 | 0.93 | 0.40 |
| Insulin (µU/mL)a | 33.9±8.4 | 14.6±1.5 | 31.7±4.8 | 33.7±8.3 | 0.44 | 0.43 | 0.34 |
| Insulin AUCa | 7415±771 | 4358±771 | 12415±1897 | 7316±1115 | 0.10 | 0.09 | 0.67 |
| Glucose AUCa | 6432±292 | 6934±167 | 6367±269 | 6137±419 | 0.36 | 0.77 | 0.44 |
Data are represented as mean±SEM. Two-way ANOVA with fixed effect for maternal and postweaning diet is shown. Student’s t test shown for years on WSD for WSD/CD versus WSD/WSD dams. p values in bold indicate significance.
Measurement taken during early third trimester of pregnancy.
Abbreviations: CD indicates chow diet; WSD, Western-style diet.
TABLE 4.
Phenotype of the subset of offspring used for RNA sequencing and metabolomics
| Maternal diet: | CD | CD | WSD | WSD | p values (ANOVA) | |||
|---|---|---|---|---|---|---|---|---|
| Postweaning diet: | CD | WSD | CD | WSD | mWSD | pwWSD | INT | Sex |
| n | 10 | 6 | 10 | 9 | ||||
| Sex (male/female) | 4/6 | 4/2 | 6/4 | 4/5 | ||||
| Body weight (kg) | 5.7±0.2 | 5.6±0.3 | 6.3±0.2 | 5.8±0.2 | 0.20 | 0.08 | 0.51 | <0.05 |
| Male | 5.9±0.1 | 5.8±0.4 | 6.6±0.2 | 6.3±0.3 | ||||
| Female | 5.6±0.4 | 5.3±0.3 | 5.7±0.1 | 5.3±0.3 | ||||
| Average RWAT (g) | 0.58±0.09 | 2.38±1.04 | 0.76±0.11 | 2.04±0.44 | 0.50 | <0.0005 | 0.48 | 0.20 |
| Liver weight (g) | 134.3±6.5 | 116.9±7.8 | 142.1±6.4 | 122.9±6.9 | 0.45 | <0.05 | 0.88 | 0.22 |
| Glucose homeostasis | ||||||||
| Glucose (mg/dL)a | 54.1±3.9 | 60.50±5.30 | 53.60±3.67 | 54.89±2.43 | 0.21 | 0.18 | 0.24 | 0.87 |
| Insulin (μU/mL)a | 3.52±0.88 | 21.82±7.40 | 7.26±2.30 | 10.05±1.92 | 0.05 | <0.0005 | <0.05 | 0.06 |
| Glucose AUCb | 10095±466 | 8455±368 | 9976±601 | 8236±377 | 0.75 | <0.005 | 0.76 | <0.05 |
| Male | 10320±842 | 8865±246 | 10771±625 | 8628±493 | ||||
| Female | 9915±582 | 7636±815 | 8783±972 | 7923±558 | ||||
| Insulin AUCb | 1577±213 | 2192±329 | 1618±169 | 2264±244 | 0.87 | <0.0005 | <0.05 | 0.23 |
| Blood chemistries | ||||||||
| ALT (IU/L)c | 42±3 | 52±5 | 46±2 | 65±4 | 0.10 | <0.005 | 0.81 | 0.79 |
| AST (IU/L)c | 38±2 | 37±1 | 40±2 | 44±2 | 0.13 | 0.73 | 0.77 | 0.86 |
| GGT (IU/L)c | 91±4 | 87±3 | 98±7 | 97±5 | 0.20 | 0.82 | <0.1 | <0.05 |
| Male | 92±7 | 87±5 | 113±4 | 100±4 | ||||
| Female | 90±6 | 85±5 | 76±7 | 94±10 | ||||
| Alkaline phosphatase (IU/L)c | 388±21 | 457±36 | 428±36 | 470±48 | 0.43 | 0.10 | 0.80 | <0.005 |
| Male | 427±33 | 493±43 | 496±25 | 539±97 | ||||
| Female | 362±23 | 387±38 | 326±51 | 415±33 | ||||
| Total cholesterol (mg/dL)c | 129±5 | 166±8 | 116±3 | 170±9 | 0.90 | <0.0001 | 0.14 | 0.63 |
| LDL (mg/dL)c | 66±5 | 80±8 | 60±3 | 82±10 | 0.93 | <0.05 | 0.39 | 0.52 |
| HDL (mg/dL)c | 57±1 | 87±5 | 52±1 | 89±4 | 0.88 | <0.0001 | 0.05 | 0.10 |
| TGs (mg/dL)c | 46±5 | 53±9 | 37±5 | 32±5 | 0.05 | 0.96 | 0.20 | 0.37 |
Data are represented as mean±SEM. Two-way ANOVA with fixed effect for maternal (mWSD) and postweaning (pwWSD) diet, interaction (INT) effect, and sex is shown. When sex effect for a variable has a p < 0.05, mean±SEM for each sex is shown. p values in bold indicate significance.
Fasting plasma levels, obtained during necropsy.
Plasma levels, performed under fasting conditions within 2 months before necropsy.
Serum levels, obtained under fasting conditions during necropsy.
Abbreviations: ALT indicates alanine transaminase; AST, aspartate transaminase; CD, chow diet; GGT, gamma-glutamyl transferase; RWAT, retroperitoneal white adipose tissue, TG, triglyceride; WSD, Western-style diet.
TABLE 3.
Phenotype of the dams of 3-year-old offspring used in RNA sequencing and metabolomics
| Maternal diet: | CD | CD | WSD | WSD | p values (ANOVA) | ||
|---|---|---|---|---|---|---|---|
| Postweaning diet: | CD | WSD | CD | WSD | mWSD | pwWSD | INT |
| n | 10 | 6 | 10 | 9 | |||
| Age (years) | 10.0±0.8 | 9.6±0.4 | 10.4±0.3 | 6.7±0.6 | 0.06 | <0.005 | <0.05 |
| Years on diet | 0±0 | 0±0 | 4.4±0.4 | 2.5±0.7 | t test: p = 0.06 | ||
| Body weight (kg) | 9.1±0.3 | 9.4±0.5 | 12.3±0.3 | 9.1±0.8 | <0.05 | <0.05 | <0.005 |
| Body fat (%) | 18.1±2.2 | 20.0±3.8 | 35.9±1.0 | 16.1±2.1 | <0.005 | <0.0005 | <0.0001 |
| Body weight (kg)a | 10.1±0.3 | 10.0±0.5 | 11.8±0.6 | 9.7±0.7 | 0.23 | 0.05 | 0.1 |
| Glucose (mg/dL)a | 40.3±3.2 | 38.3±5.9 | 38.7±1.4 | 45.3±5.5 | 0.53 | 0.59 | 0.32 |
| Insulin (µU/mL)a | 40.3±14.8 | 14.6±1.5 | 45.9±8.5 | 33.7±8.3 | 0.34 | 0.15 | 0.60 |
| Insulin AUCa | 8104±1213 | 4358±771 | 18855±3599 | 7316±1115 | <0.05 | <0.05 | 0.18 |
| Glucose AUCa | 6856±263 | 6934±167 | 6940±483 | 6137±419 | 0.46 | 0.46 | 0.37 |
Data are represented as mean±SEM. Two-way ANOVA with fixed effect for maternal (mWSD) and postweaning (pwWSD) diet is shown. The Student t test shown for years on WSD for WSD/CD versus WSD/WSD dams. p values in bold indicate significance.
Measurement taken during early third trimester of pregnancy.
Abbreviations: CD indicates chow diet; WSD, Western-style diet.
3YO mWSD offspring have increased periportal hepatic collagen deposition
We previously reported increased liver triglyceride content and periportal SHG signal, a highly sensitive measure of fibrillar collagen deposition,21 in 1YO offspring exposed to mWSD.11,13 Here, in 3YO offspring, H&E showed an absence of large lipid droplets in any of the groups (Figure 1A). Hepatic triglyceride content was not different in 3YO offspring exposed to either mWSD or pwWSD (Figure 1D). No change in CD68-positive macrophage count in mWSD offspring was observed but count increased by 28% with pwWSD (Figure 1B, E; p < 0.005). Both SHG signal intensity (Figure 1C, F) and area (Figure 1G) were greater in mWSD and pwWSD offspring compared with control offspring (p < 0.05). Hepatic TBARS content, a marker of oxidative stress, was increased in mWSD offspring with additive effects in pwWSD offspring (Figure 1H).
FIGURE 1.
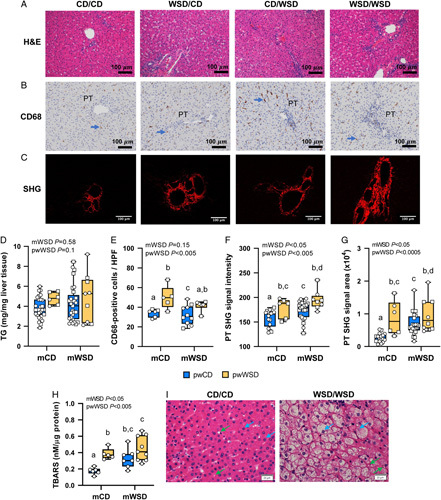
Liver phenotype in 3-year-old offspring. Representative images of hematoxylin and eosin (H&E)-stained livers (A), CD68-positive macrophage (brown) near portal triads (PTs); blue arrows indicate macrophage (B), and second harmonic generation (SHG) images of PTs with SHG signal in red (C). Scale bar: 100 μm. (D) Liver triglyceride (TG) content. (E) Mean number of CD68-positive macrophage per portal area in livers. SHG signal intensity (F) and area (G) in PT regions. (H) Thiobarbituric acid reactive substances (TBARS) content in livers. (I) Representative images from H&E-stained livers showing normal hepatocytes in CD/CD and hepatocyte ballooning in WSD/WSD. Blue arrows indicate hepatocytes and green, HSCs. Scale bar: 20 μm. (D–G) Symbols represent sample numbers per group, squares represent males, circles represent females. (D–H) Two-way ANOVA with effect for maternal (mWSD) and postweaning (pwWSD) diet is shown. Individual posttest comparisons are indicated when maternal, postweaning, or interaction effect are p < 0.05 and different letters represent differences between groups. n = 6–26 CD/CD, n = 10–27 WSD/CD, n = 5–6 CD/WSD, n = 6–9 WSD/WSD. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
To determine the clinical relevance of NAFLD severity in 3YO NHP offspring, liver sections were scored using NAS (Table 5). There were no increases in steatosis or lobular inflammation scores due to mWSD or pwWSD exposure. The hepatocyte ballooning score, a measure of hepatocyte apoptosis associated with NASH,22 was increased in association with mWSD and highest in WSD/WSD livers (INT p < 0.005; Figure 1I). The overall NAS was highest in the WSD/WSD group compared with the other 3 groups (INT p < 0.05). The fibrosis score was not different but, in general, males had overall higher scores than females irrespective of diet.
TABLE 5.
Three-year-old liver histopathology reads comprising NAS
| Maternal diet: | CD | CD | WSD | WSD | p values (ANOVA) | |||
|---|---|---|---|---|---|---|---|---|
| Postweaning diet: | CD | WSD | CD | WSD | mWSD | pwWSD | INT | Sex |
| n | 15–16 | 6 | 23–24 | 8 | ||||
| Sex (male/female) | ||||||||
| Steatosis | 0.35±0.09 | 0±0 | 0.19±0.06 | 0±0 | 0.23 | <0.0005 | 0.19 | 0.38 |
| Lobular inflammation | 0.65±0.12 | 0.67±0.19 | 0.79±0.1 | 0.75±0.19 | 0.41 | 0.93 | 0.69 | 0.10 |
| Hepatocyte ballooning | 0.32±0.09 | 0±0 | 0.24±0.07 | 0.75±0.23 | <0.05 | 0.53 | <0.005 | 0.72 |
| NAS | 1.32±0.16 | 0.67±0.19 | 1.21±0.13 | 1.5±0.24 | 0.05 | 0.32 | <0.05 | 0.56 |
| Fibrosis | 0.68±0.12 | 0.83±0.21 | 0.7±0.1 | 0.81±0.19 | 0.89 | 0.44 | 0.78 | <0.05 |
| Males | 0.91±0.22 | 0.87±0.29 | 0.79±0.12 | 0.87±0.29 | ||||
| Females | 0.52±0.11 | 0.75±0.25 | 0.55±0.14 | 0.75±0.25 | ||||
Data are represented as mean±SEM for both pathologists’ reads together. Two-way ANOVA with effect for maternal (mWSD) and postweaning (pwWSD) diet, interaction (INT), and sex is shown. When sex effect is significant, mean±SEM for the variable, broken down by males and females, is shown.
Abbreviations: CD indicates chow diet; NAS, NAFLD activity score; WSD, Western-style diet. p values in bold indicate significance.
Analysis comparing mWSD and pwWSD transcriptomics reveals distinct and common gene expression patterns
Given increased periportal collagen formation with mWSD exposures and hepatocyte ballooning score in WSD/CD offspring, we next sought to uncover potential molecular mechanisms that may be driving associations between WSD feeding and hepatic pathophysiology. There were distinct gene expression profiles in all 4 diet groups, as shown by clustering of samples within each group with principal component analysis (Figure 2A). To identify DEGs, gene expression in WSD/CD, WSD/WSD, and CD/WSD groups were compared with the CD/CD group. There were 861, 420, and 1269 DEGs n WSD/CD, WSD/WSD, and CD/WSD groups respectively compared with CD/CD (Figure 2B).
FIGURE 2.
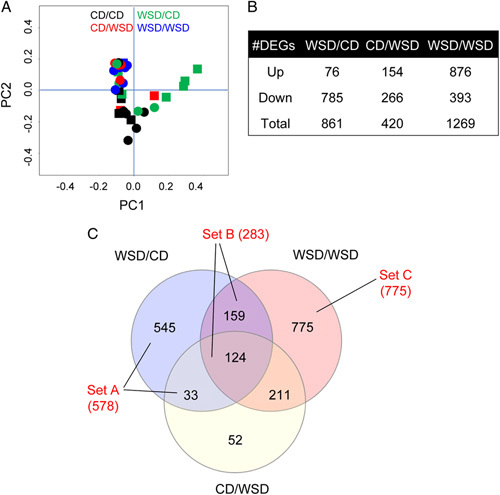
Liver transcriptomic analysis in 3-year-old offspring. (A) Two-dimensional principal component analysis plot showing samples in all diet groups. Black indicates CD/CD, green indicates WSD/CD, red indicates CD/WSD, and blue indicates WSD/WSD; squares indicate male and circles indicate female offspring. (B) The number of differentially expressed genes (DEGs) that are upregulated or downregulated and total DEGs in each group compared with CD/CD. (C) Venn diagram showing overlapping and distinct sets of DEGs. Blue circle indicates DEGs in WSD/CD versus CD/CD, red circle indicates DEGs in WSD/WSD versus CD/CD, yellow circle indicates DEGs in CD/WSD versus CD/CD. DEGs in set A are associated with maternal WSD (mWSD) in the absence of postweaning WSD (pwWSD). DEGs in set B are comprised of DEGs associated with mWSD regardless of postweaning diet. DEGs in set C are associated with mWSD only when combined with pwWSD. n = 10 CD/CD; n = 10 WSD/CD; n = 6 CD/WSD; n = 7 WSD/WSD. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
To compare the distinct and shared patterns of genes in mWSD- and pwWSD-exposed 3YO, we sorted these sets of DEGs (Figure 2C). First, there were 578 genes (545+33) with expression affected by mWSD exposure in the absence of pwWSD (set A). Second, there were 283 genes (159+124) with expression affected by mWSD in offspring regardless of their postweaning diet (set B). Third, there were 775 DEGs affected only in WSD/WSD offspring (set C). We used these DEG sets (A–C) in our subsequent functional annotation analysis.
mWSD regulates genes associated with mitochondrial function
In set A, there were 45 genes upregulated and 533 downregulated in WSD/CD compared with CD/CD (Figure 3A). The DEGs in set A, associated with mWSD exposure, were enriched for 20 predicted canonical pathways (Figure 3B). Pathways predicted to be downregulated included acute phase response, LXR/RXR activation, oxidative phosphorylation, fatty acid β-oxidation, complement system, and NRF2-mediated oxidative stress response. Other predicted pathways derived from set A DEGs included FXR/RXR signaling, mitochondrial dysfunction, glucocorticoid receptor signaling, and extrinsic prothrombin activation. We went on to identify a putative set of enriched upstream regulators corresponding to set A DEGs (Figure 3C). HNF4α and HNF1α, which can resolve steatosis and fibrosis in NAFLD,23,24 were predicted to be downregulated. Members of the PPAR family (PPARGC1α, PPARα, and PPARα), key factors for driving mitochondrial biogenesis and nutrient sensing, were predicted to be downregulated.25 TP53 (p53), insulin, insulin receptor (INSR), and NFE2L2 (NRF2) were also predicted to be downregulated. CLPP and LONP1, regulators driving the repair of misfolded or damaged mitochondrial proteins,26,27 were predicted to be upregulated.
FIGURE 3.
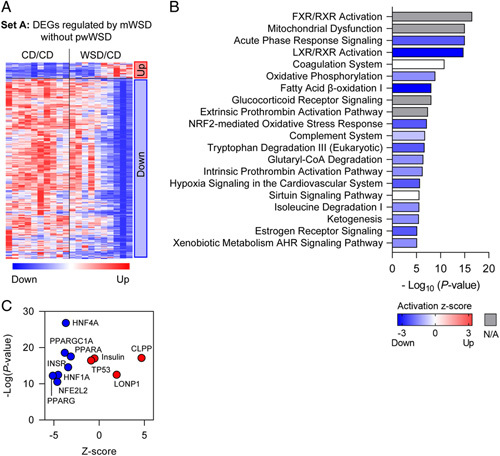
Differentially expressed genes (DEGs) associated with maternal WSD (mWSD) exposure. (A) Clustered heatmap showing upregulated and downregulated genes in set A. (B) Canonical pathways enriched in the DEGs are shown in order by p value. Bar shading indicates the predicted activation Z-score on a scale from highest (red) to lowest (blue), with white representing 0. For some pathways, ingenuity pathway analysis could not assign a Z-score (gray). (C) Predicted upstream regulators plotted with p value (x-axis) and predicted Z-score (y-axis). n = 10 CD/CD; n = 10 WSD/CD. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
mWSD decreases expression of antioxidant response genes, increases expression of mitochondrial dysfunction genes, and increases expression of endoplasmic reticulum (ER) stress response genes
The genes in set B were differentially expressed in both WSD/CD and WSD/WSD groups compared with CD/CD, yet, unexpectedly, the direction of differential expression was not the same for all genes. Within this set, we identified 2 subsets of DEGs (Figure 4A): one that had the same relative direction of change in both WSD/CD and WSD/WSD groups compared with CD/CD (197 DEGs) and another that had divergent expression between WSD/CD and WSD/WSD groups (85 DEGs). The genes that were similarly affected in both WSD/CD and WSD/WSD groups were enriched for pathways involved in oxidative phosphorylation, which was predicted to be downregulated, and mitochondrial dysfunction (Figure 4B). In the subset of DEGs with divergent expression in WSD/CD and WSD/WSD, there was no enrichment for canonical pathways, likely due to the small number of input DEGs. However, in this gene set, XBP1, the major transcriptional activator of the ER stress response,28 was predicted to be downregulated in WSD/CD and upregulated in WSD/WSD (Figure 4C).
FIGURE 4.
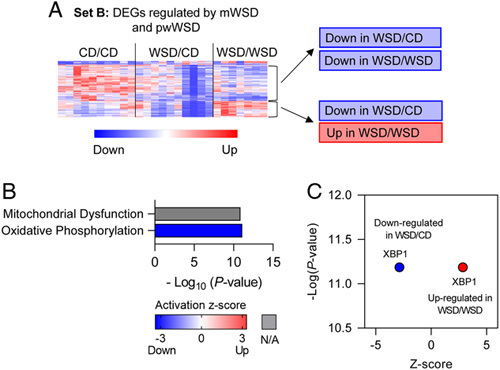
Differentially expressed genes (DEGs) associated with maternal WSD (mWSD) exposure in both postweaning CD (pwCD)and postweaning WSD (pwWSD) groups. (A) Clustered heatmap showing 2 sets of DEGs in set B. One with the same direction of fold change in WSD/CD and WSD/WSD and the other with opposite fold changes between WSD/CD and WSD/WSD. (B) Canonical pathways enriched in the DEGs are shown in order by p value. Bar shading indicates the predicted activation Z-score on a scale from highest (red) to lowest (blue), with white representing 0. For some pathways, ingenuity pathway analysis could not assign a Z-score (gray). (C) Predicted upstream regulator plotted with p value (x-axis) and predicted Z-score (y-axis). n = 10 CD/CD; n = 10 WSD/CD; n = 7 WSD/WSD. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
Continuous WSD exposure drives gene expression supporting the ER stress response and hepatic collagen deposition
The DEGs in set C represent genes differentially expressed only in WSD/WSD livers compared with CD/CD livers. In this set, there were 652 genes upregulated and 123 downregulated (Figure 5A). The canonical pathways enriched and predicted to be upregulated in this gene set included unfolded protein response, AMPK signaling, hepatic fibrosis signaling pathway, estrogen receptor signaling, and Rho family GTPase signaling. Other enriched pathways included glucocorticoid signaling, clathrin-mediated endocytosis, and CLEAR lysosomal signaling (Figure 5B). Upstream regulators enriched in DEGs from this set included ESR1, XBP1, HNF4A, PTEN, and TGFB1 and all were predicted to be upregulated (Figure 5C).
FIGURE 5.
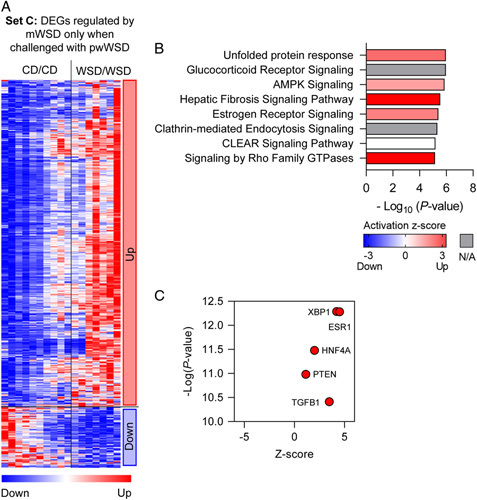
Differentially expressed genes (DEGs) associated with maternal WSD (mWSD) exposure when combined with postweaning WSD (pwWSD). (A) Clustered heatmap showing upregulated and downregulated genes in set C. (B) Canonical pathways enriched in the DEGs are shown in order by p value. Bar shading indicates the predicted activation Z-score on a scale from highest (red) to lowest (blue), with white representing 0. For some pathways, ingenuity pathway analysis could not assign a Z-score (gray). (C) Predicted upstream regulators plotted with p value (x-axis) and predicted Z-score (y-axis). n = 10 CD/CD; n = 7 WSD/WSD. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
mWSD decreases HNF4α protein expression and pwWSD increases BiP protein expression
HNF4α is a transcription factor commonly targeted as a NASH therapeutic and modulates pathways in the liver including antioxidant response and lipid metabolism, in part by stimulating mitochondrial functions such as β-oxidation.23,24,29 Protein expression of HNF4α was decreased only in WSD/CD livers (Figure 6A), consistent with the transcriptomics prediction (Figure 3C). However, WSD/WSD livers had HNF4α levels comparable to CD/CD, supporting that the predicted upregulation of HNF4α (Figure 5C) may overcome downregulation by mWSD. We also measured the protein expression of BiP (also known as GPR78), a key driver of the ER stress response.30 BiP expression was higher in offspring consuming the pwWSD (Figure 6B), supporting pwWSD effects on upregulation of ER stress pathways.
FIGURE 6.
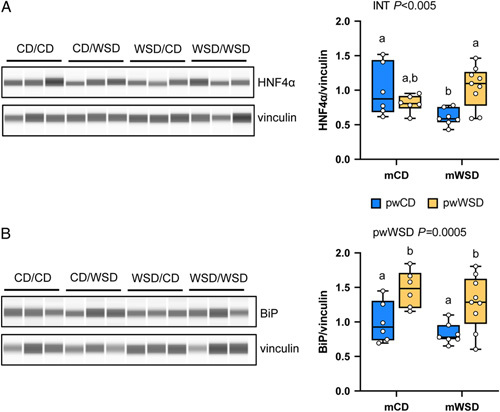
Protein expression of HNF4α and BiP. Representative images and analysis of protein capillary electrophoresis for HNF4α (A) and BiP (B) and loading control vinculin. Two-way ANOVA with effect for interaction effect (INT) or postweaning WSD (pwWSD) diet is shown. Individual posttest comparisons are indicated when maternal, postweaning, or interaction effect are p < 0.05 and different letters represent differences between groups. n = 6 CD/CD; n = 7 WSD/CD; n = 6 CD/WSD; n = 9 WSD/WSD. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
pwWSD alone drives changes in the liver metabolome supporting free fatty acid overload
To determine whether there were metabolite changes in 3YO offspring exposed to mWSD with or without pwWSD, we used a targeted metabolomic screen (181 metabolites were identified). partial least squares discriminant analysis of samples in all 4 groups showed distinct clustering by diet combination (Figure 7A). The 25 metabolites with the highest VIP scores driving separation of the 4 groups are shown in a clustered heatmap (Figure 7B). pwWSD had a greater effect than mWSD, as all 25 metabolites were different in pwWSD versus pwCD groups via one-way ANOVA. Within the top 25 metabolites between all 4 groups, 13 were increased in pwWSD groups and 12 were decreased. These 25 metabolites were enriched for 3 metabolic pathways: linoleic acid metabolism, phosphatidylcholine biosynthesis, and gluconeogenesis (Figure 7C). pwWSD livers had decreased unsaturated long-chain fatty acids including alpha-linoleic, eicosapentaenoic, docosapentaeonoic, docosahexaenoic, and octadecanoic acids, which are associated with anti-inflammatory actions.31 Further, short-chain and medium-chain acylcarnitines (methylmalonylcarnitine, hexanoylcarnitine, octenoylcarnitine, and proprionylcarnitine), which result from incomplete oxidation of free fatty acids,32 were increased in pwWSD livers (Figure 7B). In addition, there was decreased choline and increased S-adenosylmethionine (Figure 7B,C), suggesting epigenetic regulation.33,34 Glucose-6-phosphate, glucose, and lactate were increased, supporting increased glycolysis and/or increased gluconeogenesis (Figure 7B, C).
FIGURE 7.
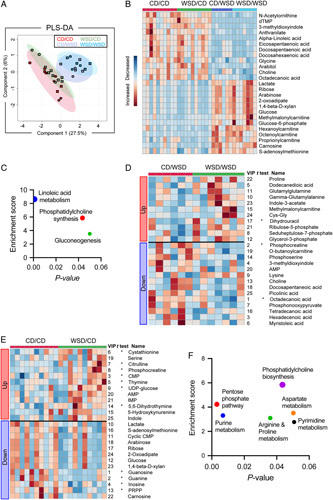
Metabolomics analysis of 3-year-old livers. (A) Two-dimensional partial least squares discriminant analysis plot showing separation of samples from CD/CD (red), WSD/CD (green), CD/WSD (light blue), and WSD/WSD (dark blue); squares indicate males and circles indicate females. (B) Heatmap showing hierarchical clustering of top 25 variable importance in projection (VIP) scoring metabolites between all 4 diet groups. (C) Metabolic pathways enriched in the top 25 VIP metabolites between all 4 groups. (D) Heatmap showing hierarchical clustering of the top 25 VIP metabolites between CD/WSD and WSD/WSD. (E) Heatmap showing hierarchical clustering of the top 25 VIP metabolites between CD/CD and WSD/CD. (F) Metabolic pathways enriched in the top 25 VIP metabolites from CD/CD versus WSD/CD. (B, D, E) Red indicates relative increase of each metabolite, and blue indicates relative decrease of each metabolite. (D and E) Asterisks indicate metabolites that are significantly (p < 0.05) different between the groups via unpaired t test. Numbers next to each metabolite indicate their relative VIP score rank. n = 10 CD/CD, n = 10 WSD/CD, n = 6 CD/WSD, n = 7 WSD/WSD. Abbreviations: CD indicates chow diet; PRPP, phosphoribosylpyrophosphate; WSD, Western-style diet.
mWSD combined with pwWSD has greater effects on pentose phosphate pathway activity and exacerbates effects on the metabolome
Next, to further evaluate the relationship between mWSD and pwWSD, we evaluated CD/WSD versus WSD/WSD liver metabolite profiles (Figure 7D). Ribulose-5-phosphate and seduheptulose-7-phosphate were increased in WSD/WSD livers, consistent with pentose phosphate pathway activation. In addition, methylmalonylcarnitine, a short-chain acylcarnitine, and glycerol-3-phosphate, a key substrate for triglyceride synthesis, were increased in WSD/WSD livers. Octadecanoic acid was further decreased compared with CD/WSD in WSD/WSD livers, as were other long-chain fatty acids including tetradecanoic acid, hexadecenoic acid, and myristoleic acid. Proline, a key intermediate for collagen synthesis, was also increased in WSD/WSD livers compared with CD/WSD. Together, these results indicate that the combination of mWSD with pwWSD has a greater impact on fatty acid metabolism and pentose phosphate pathway activity compared with animals that were fed a WSD beginning at weaning but not exposed to mWSD.
WSD/CD livers have increased purine synthesis
Last, we studied whether metabolite profiles were altered in livers from WSD/CD offspring versus CD/CD offspring. The top 25 VIP metabolites were enriched in purine synthesis, pentose phosphate pathway, arginine & proline metabolism, phosphatidylcholine biosynthesis, aspartate metabolism, and pyrimidine metabolism pathways (Figure 7E, F). Enrichment of these pathways was largely driven by increases in products of purine and pyrimidine synthesis (IMP, AMP, CMP), decreases in products of purine catabolism (guanine, guanosine, inosine), decreases in substrates of purine and pyrimidine synthesis (PRPP, ribose), and increases in products of pyrimidine breakdown (thymine, dihydrothymine) (Figure 7E). In addition, the methyl donor S-adenosylmethionine, shown to protect mitochondria from oxidative stress,35 and to regulate epigenetic pathways,33,34 was decreased in WSD/CD livers (Figure 7E).
DISCUSSION
Here, we demonstrate that mWSD exposure is associated with altered expression in many well-recognized signaling pathways and metabolites for NAFLD that promote fibrosis, oxidative stress, and mitochondrial dysfunction. Importantly, these effects persisted in 3YO juvenile offspring without advanced clinical histopathologic presentation of NAFLD. Histologically, we found increased hepatic portal collagen deposition, a major component of pediatric NAFLD that is more typical of pediatric versus adult forms of NAFLD.3 We used SHG imaging, a more sensitive method compared with standard histologic approaches,11,21 to identify this unique pattern of periportal collagen deposition in both mWSD and pwWSD 3YO offspring. Mechanistically, we identified robust changes in specific hepatic transcript and metabolite profiles in offspring exposed to mWSD, despite only slight increases in histologic-based NAS scoring, in the absence of hepatic steatosis or obesity. Importantly, our results demonstrate that these hepatic effects are present even when mWSD-exposed offspring are switched to a healthy diet at weaning, nearly 2.5 years earlier. Further, our results demonstrate that, unlike WSD initiated postnatally, continuous WSD exposure across the early lifespan had additive and potentially synergistic effects that worsened metabolite and gene signatures supporting fibrosis, and advanced histologic signs of NAFLD severity.
Increased portal collagen in mWSD-exposed livers implicates a perinatal origin for fibrosis patterning and longer term disease pathology. Previously, we showed that fetal and 1YO offspring exposed to mWSD have increased periportal collagen deposition.11 Fibrosis can not only progress, but can also regress spontaneously over time in NASH patients.36,37 Thus, the persistence of collagen deposition in 3YO mWSD-exposed offspring suggests that there is an impairment of pathways necessary to resolve fibrosis and/or an activation of pathways promoting fibrosis. With continuous WSD exposure, more profound transcriptional, metabolic, and histologic changes consistent with fibrosis were present, but there was no evidence of steatosis nor inflammation. This suggests that hepatocellular damage and fibrosis develops earlier in the pediatric NAFLD disease process than previously thought. Indeed, many well-known signaling pathways involved in pediatric NAFLD/NASH development are dysregulated early in the disease course, without histologic evidence of inflammation and fibrosis.38 Below, we discuss putative mechanisms driving collagen deposition supported by our transcriptomic and metabolomic profiling experiments.
Decreased HNF activity, mitochondrial dysfunction, and oxidative stress may be major mechanisms driving persistent collagen deposition following mWSD exposure. While WSD/CD livers did not have classical gene expression signatures of fibrosis, HNF4α and HNF1α pathways were identified as putative-enriched upstream regulators predicted to be downregulated. Indeed, protein expression of HNF4α was also decreased in WSD/CD livers. Since increased HNF activity can resolve fibrosis,24 likely through direct inactivation of HSCs, downregulation of HNF4α in WSD/CD may prevent resolution of collagen deposition. In WSD/CD livers, we also found enrichment of the mitochondrial dysfunction pathway and predicted downregulation of the oxidative phosphorylation pathway, as well as a predicted downregulation of NRF2, PPARα, and PPARGC1α, which together suggest impaired mitochondrial function, increased oxidative stress, and generation of reactive oxygen species.14,25,39 Increased hepatic TBARS content in WSD/CD livers supports the presence of oxidative stress. In addition to these effects on mitochondrial function, our data suggest an ongoing mitochondrial DNA repair response. Supporting this, WSD/CD livers had a metabolite profile consistent with increased purine synthesis (increased AMP, IMP, decreased PRPP) and decreased purine catabolism (decreased guanosine, guanine, inosine). Purine synthesis and availability is critical for DNA and RNA synthesis and repair.40 Further, mitochondrial dysfunction increases cellular purine pool size and modulates purine metabolism.40,41 Thus, increased purine synthesis in WSD/CD livers may be a consequence of mitochondrial dysfunction.41 WSD/CD livers also showed CLPP and LONP1 activation, which are pathways critical for mitochondrial repair and mitochondrial unfolded protein response.26 Reactive oxygen species are capable of driving HSC activation and fibrosis.42 We have previously shown oxidative stress, HSC activation, and portal collagen deposition in mWSD-exposed fetuses,11 depicted in Figure 8A, and we speculate that this persists at 3 years of age (Figure 8B). We speculate that reduced mitochondrial oxidative capacity and repair mechanisms through oxidative stress and mitochondrial dysfunction continue to support HSC activation or lack of collagen repair (Figure 8B).
FIGURE 8.
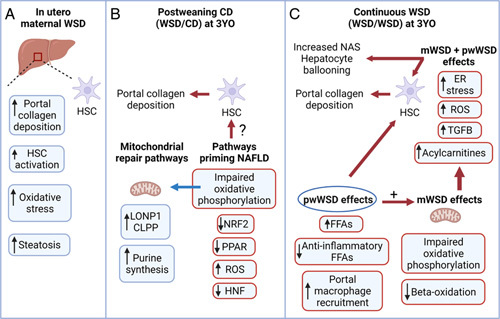
Long-term role for maternal WSD (mWSD) in the development of preclinical NAFLD phenotypes in juvenile nonhuman primates. (A) Exposure to mWSD drives oxidative stress, portal collagen deposition, HSC activation, and steatosis in fetal liver. (B) In mWSD 3-year-old (3YO) offspring weaned to CD (WSD/CD), livers show upregulated mitochondrial repair pathways including LONP1, CLPP, and purine synthesis. Pathways for NRF2, PPAR, and HNF are downregulated, which may activate HSCs and compromise regulatory pathways for portal collagen repair. (C) With continuous WSD exposure (offspring exposed to maternal and postweaning WSD, WSD/WSD), macrophages are recruited, free fatty acids (FFAs) are increased, and anti-inflammatory FFAs are decreased in the liver. Increased FFAs may activate HSCs and produce reactive oxygen species (ROS). Continuous WSD also increased acylcarnitines and upregulated pathways for TGFB activation, ROS production, and endoplasmic reticulum (ER) stress, which may contribute to increased portal collagen deposition, hepatocyte ballooning, and NAFLD activity scores (NAS). Created with BioRender.com. Abbreviations: CD indicates chow diet; WSD, Western-style diet.
WSD/WSD livers had more classical signs of fibrosis in the portal region, which may be due to distinct mechanisms versus WSD/CD or CD/WSD livers. We noted a significant increase in portal CD68 staining, along with increased plasma aspartate transaminase consistent with local macrophage infiltration and mild liver injury in pwWSD offspring. We speculate that bile acid metabolism also specifically contributes to portal fibrosis, a distinct characteristic of pediatric NAFLD.43 In WSD/WSD livers, there was predicted upregulation of the TGFβ1 pathway, a key driver of HSC activation and fibrosis,44 in addition to upregulation of the canonical hepatic fibrosis signaling pathway, comprised of collagen synthesis and HSC activation genes. Importantly, these gene signatures were specific to the WSD/WSD group and not the result of pwWSD alone. Further, we found that the activation state of the XBP1 pathway was predicted to be downregulated in WSD/CD, but upregulated in WSD/WSD. Protein expression of BiP, a target in the ER stress pathway, was increased by pwWSD. Thus, XBP1 activation and ER stress may be key features of mWSD exposure only when combined with pwWSD. This chronic ER stress may potentiate fibrosis and contribute to the hepatocyte ballooning observed in WSD/WSD livers (Figure 8C), as it can worsen oxidative stress and drive apoptosis.45,46 Proline was also increased in WSD/WSD livers, supporting collagen synthesis. In addition, excess dietary-free fatty acids from a pwWSD may directly stimulate HSCs to drive collagen deposition.47 When paired with mWSD-driven impairments in mitochondrial function, these excess free fatty acids may worsen oxidative stress through increased mitochondrial reactive oxygen species production and increased ER stress (Figure 8C). The decreased content of anti-inflammatory unsaturated long-chain fatty acids, which are inversely correlated with fibrosis,48 may set the stage for inflammation over time, while portal macrophage recruitment of CD688-positive cells in WSD/WSD livers may signify a role for a subset of macrophage in driving local collagen deposition through activation of portal HSCs, worsening fibrosis (Figure 8C). Collectively, in WSD/WSD livers, increased TBARS content, circulating alanine transaminase, NAS score, and hepatocyte ballooning suggest a potentially accelerated pathway for NAFLD/NASH.
Remarkably, despite evidence of reduced mitochondrial function and fuel overload in WSD/WSD offspring, hepatic steatosis was not detectable by hepatic triglyceride content or clinical steatosis scoring. This was surprising given our prior work demonstrating triglyceride accumulation in fetal12 and 1YO13 livers exposed to mWSD. However, over time and in combination with high physical activity and energy expenditure in these animals,49,50 the hepatic lipids may be dissipated to levels similar to controls by 3 years of age. Interestingly, the 3YO juvenile animals in this WSD/WSD cohort demonstrated higher physical activity and energy expenditure compared with CD/CD,49,50 suggesting that increased caloric load is matched with greater energy expenditure. However, pwWSD offspring did have increased retroperitoneal fat mass (visceral fat), which suggests that some excess fuels are being stored. Interestingly, despite the absence of hepatic steatosis, many pathways were primed towards lipid storage in WSD/CD livers (decreased β-oxidation, oxidative phosphorylation, HNF4α signaling, PPARGC1α, and PPARα) and WSD/WSD livers (decreased oxidative phosphorylation). Further, WSD/WSD livers had increased acylcarnitines and decreased anti-inflammatory fatty acids, supporting incomplete mitochondrial oxidation and processing of free fatty acids. In addition, WSD/WSD livers had increased glycerol-3-phosphate compared with CD/WSD, suggesting a possible increase in de novo lipogenesis. Thus, our data support that mWSD initiates gene expression changes that support priming of steatosis pathways.
Our results also support that maternal diet rather than maternal obesity contributes to the phenotypes we observed in the offspring liver. The chronic WSD used here has a variable effect on maternal obesity.13 However, we found no correlation between maternal percent body fat at time of conception with the degree of fibrosis in the offspring (data not shown). Further, we previously showed that switching obese dams to a healthy diet during pregnancy can prevent hepatic collagen deposition in fetal livers.11 Thus, this strongly suggests that maternal diet (a modifiable risk factor) can be targeted to prevent the developmental programming of pediatric NAFLD.
In summary, these findings demonstrate a long-term role for mWSD in the development of preclinical juvenile NAFLD phenotypes including portal collagen deposition, mitochondrial dysfunction, and oxidative stress, in the absence of obesity. The implications of local collagen deposition, oxidative stress, and mitochondrial dysfunction in juvenile offspring despite 2.5 years on a normal diet suggests that mWSD drives juvenile liver metabolic reprogramming. Further, the persistence of these phenotypes, even before pathologic evidence for inflammation and fibrosis, suggests increased susceptibility to more advanced NAFLD, and argue for an early intervention, beginning in utero, to prevent the development of childhood NAFLD.
ACKNOWLEDGMENTS
The authors thank the University of Colorado Anschutz Medical Campus Advanced Fluorescent Microscopy Core, Histology Core, and Metabolomics Core for assistance with SHG imaging, immunostaining of CD68, and metabolomics analysis, respectively. They also thank the University of Oklahoma Health Sciences Center Genomics Core for RNA sequencing analysis.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
Funding information Supported by the National Institute of Diabetes and Digestive and Kidney Diseases grants R24-DK090964 (to J.E.F., K.M.A., P.K.), R01-DK128416 (to J.E.F.), F30-DK122672 (to M.J.N.), R01-DK108910 (to S.R.W.), and the University of Colorado Nutrition Obesity Research Center P30-DK048520 (to J.E.F.). Microscopy experiments were performed in the Advanced Light Microscopy Core at University of Colorado Anschutz Medical Campus supported in part by Rocky Mountain Neurological Disorders Core grant P30-NS048154 and by Diabetes Research Center grant P30-DK116073.
Abbreviations: 3YO, 3-year-old; DEG, differentially expressed gene; ER, endoplasmic reticulum; FOV, fields of view; HSC, hepatic stellate cell; ivGTT, intravenous glucose tolerance tests; mWSD, maternal Western-style diet; NAFLD, nonalcoholic fatty liver disease; NAS, NAFLD activity score; NASH, nonalcoholic steatohepatitis; NHP, nonhuman primate; pwWSD, postweaning Western-style diet; SHG, second harmonic generation; VIP, variable importance in projection; WSD, Western-style diet
Jacob E. Friedman and Stephanie R. Wesolowski are co-senior authors.
Contributor Information
Michael J. Nash, Email: michael.nash@cuanschutz.edu.
Evgenia Dobrinskikh, Email: evgenia.dobrinskikh@cuanschutz.edu.
Rachel C. Janssen, Email: rachel-janssen@ouhsc.edu.
Mark A. Lovell, Email: Mark.Lovell@childrenscolorado.org.
Deborah A. Schady, Email: daschady@texaschildrens.org.
Claire Levek, Email: CLAIRE.LEVEK@CUANSCHUTZ.EDU.
Kenneth L. Jones, Email: ken-Jones@ouhsc.edu.
Angelo D’Alessandro, Email: Angelo.dalessandro@cuanschutz.edu.
Paul Kievit, Email: kievitp@ohsu.edu.
Kjersti M. Aagaard, Email: aagaardt@bcm.edu.
Carrie E. McCurdy, Email: cmccurd5@uoregon.edu.
Maureen Gannon, Email: maureen.gannon@vumc.org.
Jacob E. Friedman, Email: jed-friedman@ouhsc.edu.
Stephanie R. Wesolowski, Email: stephanie.wesolowski@cuanschutz.edu.
REFERENCES
- 1.Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: a systematic review and meta-analysis. PLoS ONE. 2015;10:e0140908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahota AK, Shapiro WL, Newton KP, Kim ST, Chung J, Schwimmer JB. Incidence of nonalcoholic fatty liver disease in children: 2009-2018. Pediatrics. 2020;146:e20200771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandala A, Janssen RC, Palle S, Short KR, Friedman JE. Pediatric non-alcoholic fatty liver disease: nutritional origins and potential molecular mechanisms. Nutrients. 2020;12:3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goyal NP, Schwimmer JB. The progression and natural history of pediatric nonalcoholic fatty liver disease. Clin Liver Dis. 2016;20:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–1393. [DOI] [PubMed] [Google Scholar]
- 6.Yu EL, Golshan S, Harlow KE, Angeles JE, Durelle J, Goyal NP, et al. Prevalence of nonalcoholic fatty liver disease in children with obesity. J Pediatr. 2019;207:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vajro P, Lenta S, Socha P, Dhawan A, McKiernan P, Baumann U, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr. 2012;54:700–713. [DOI] [PubMed] [Google Scholar]
- 8.Pardee PE, Lavine JE, Schwimmer JB. Diagnosis and treatment of pediatric nonalcoholic steatohepatitis and the implications for bariatric surgery. Semin Pediatr Surg. 2009;18:144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wesolowski SR, El Kasmi KC, Jonscher KR, Friedman JE. Developmental origins of NAFLD: a womb with a clue. Nat Rev Gastroenterol Hepatol. 2017;14:81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. 2005;42:641–649. [DOI] [PubMed] [Google Scholar]
- 11.Nash MJ, Dobrinskikh E, Newsom SA, Messaoudi I, Janssen RC, Aagaard KM, et al. Maternal Western diet exposure increases periportal fibrosis beginning in utero in nonhuman primate offspring. JCI Insight. 2021;6:e154093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009;119:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorn SR, Baquero KC, Newsom SA, El Kasmi KC, Bergman BC, Shulman GI, et al. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes. 2014;63:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. [DOI] [PubMed] [Google Scholar]
- 15.Elsakr JM, Zhao SK, Ricciardi V, Dean TA, Takahashi DL, Sullivan E, et al. Western-style diet consumption impairs maternal insulin sensitivity and glucose metabolism during pregnancy in a Japanese macaque model. Sci Rep. 2021;11:12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. [DOI] [PubMed] [Google Scholar]
- 17.Wesolowski SR, Mulligan CM, Janssen RC, Baker PR, II, Bergman BC, D’Alessandro A, et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Mol Metab. 2018;18:25–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baird NL, Bowlin JL, Cohrs RJ, Gilden D, Jones KL. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J Virol. 2014;88:5877–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemkov T, Reisz JA, Gehrke S, Hansen KC, D’Alessandro A. High-throughput metabolomics: isocratic and gradient mass spectrometry-based methods. Methods Mol Biol. 2019;1978:13–26. [DOI] [PubMed] [Google Scholar]
- 21.Liu F, Zhao JM, Rao HY, Yu WM, Zhang W, Theise ND, et al. Second harmonic generation reveals subtle fibrosis differences in adult and pediatric nonalcoholic fatty liver disease. Am J Clin Pathol. 2017;148:502–512. [DOI] [PubMed] [Google Scholar]
- 22.Lackner C. Hepatocellular ballooning in nonalcoholic steatohepatitis: the pathologist’s perspective. Expert Rev Gastroenterol Hepatol. 2011;5:223–231. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y, Zhu Y, Hu S, Xu Y, Stroup D, Pan X, et al. Hepatocyte nuclear factor 4α prevents the steatosis-to-NASH progression by regulating p53 and bile acid signaling (in mice). Hepatology. 2021;73:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang T, Poenisch M, Khanal R, Hu Q, Dai Z, Li R, et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J Hepatol. 2021;75:1420–1433. [DOI] [PubMed] [Google Scholar]
- 25.Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol. 2015;33:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibellini L, De Gaetano A, Mandrioli M, Van Tongeren E, Bortolotti CA, Cossarizza A, et al. The biology of Lonp1: more than a mitochondrial protease. Int Rev Cell Mol Biol. 2020;354:1–61. [DOI] [PubMed] [Google Scholar]
- 27.Lee YG, Kim HW, Nam Y, Shin KJ, Lee YJ, Park DH, et al. LONP1 and ClpP cooperatively regulate mitochondrial proteostasis for cancer cell survival. Oncogenesis. 2021;10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirota M, Kitagaki M, Itagaki H, Aiba S. Quantitative measurement of spliced XBP1 mRNA as an indicator of endoplasmic reticulum stress. J Toxicol Sci. 2006;31:149–156. [DOI] [PubMed] [Google Scholar]
- 29.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calder PC. Long-chain fatty acids and inflammation. Proc Nutr Soc. 2012;71:284–289. [DOI] [PubMed] [Google Scholar]
- 32.Bjørndal B, Alterås EK, Lindquist C, Svardal A, Skorve J, Berge RK. Associations between fatty acid oxidation, hepatic mitochondrial function, and plasma acylcarnitine levels in mice. Nutr Metab (Lond). 2018;15:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Friso S, Choi SW. Epigenetic mechanisms underlying the link between non-alcoholic fatty liver diseases and nutrition. Nutrients. 2014;6:3303–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mato JM, Martínez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol. 2013;12:183–189. [PMC free article] [PubMed] [Google Scholar]
- 35.King AL, Mantena SK, Andringa KK, Millender-Swain T, Dunham-Snary KJ, Oliva CR, et al. The methyl donor S-adenosylmethionine prevents liver hypoxia and dysregulation of mitochondrial bioenergetic function in a rat model of alcohol-induced fatty liver disease. Redox Biol. 2016;9:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw Open. 2019;2:e1912565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng CH, Xiao J, Lim WH, Chin YH, Yong JN, Tan DJH, et al. Placebo effect on progression and regression in NASH: Evidence from a meta-analysis. Hepatology. 2022;75:1647–1661. [DOI] [PubMed] [Google Scholar]
- 38.Yao K, Tarabra E, Sia D, Morotti R, Fawaz R, Valentino P, et al. Transcriptomic profiling of a multiethnic pediatric NAFLD cohort reveals genes and pathways associated with disease. Hepatol Commun. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fougerat A, Montagner A, Loiseau N, Guillou H, Wahli W. Peroxisome proliferator-activated receptors and their novel ligands as candidates for the treatment of non-alcoholic fatty liver disease. Cells. 2020;9:1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids. 2016;35:578–594. [DOI] [PubMed] [Google Scholar]
- 41.Nikkanen J, Forsström S, Euro L, Paetau I, Kohnz RA, Wang L, et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 2016;23:635–648. [DOI] [PubMed] [Google Scholar]
- 42.Gandhi CR. Oxidative stress and hepatic stellate cells: a paradoxical relationship. Trends Cell Mol Biol. 2012;7:1–10. [PMC free article] [PubMed] [Google Scholar]
- 43.Jahnel J, Zöhrer E, Alisi A, Ferrari F, Ceccarelli S, De Vito R, et al. Serum bile acid levels in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2015;61:85–90. [DOI] [PubMed] [Google Scholar]
- 44.Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-β in hepatic stellate cell activation and liver fibrogenesis—updated 2019. Cells. 2019;8:1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants (Basel). 2021;10:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Armandi A, Schattenberg JM. NAFLD and NASH: The metabolically diseased liver. Handb Exp Pharmacol. 2022;274:253–267. [DOI] [PubMed] [Google Scholar]
- 47.Marcher AB, Bendixen SM, Terkelsen MK, Hohmann SS, Hansen MH, Larsen BD, et al. Transcriptional regulation of hepatic stellate cell activation in NASH. Sci Rep. 2019;9:2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fridén M, Rosqvist F, Ahlström H, Niessen HG, Schultheis C, Hockings P, et al. Hepatic unsaturated fatty acids are linked to lower degree of fibrosis in non-alcoholic fatty liver disease. Front Med (Lausanne). 2021;8:814951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan EL, Grayson B, Takahashi D, Robertson N, Maier A, Bethea CL, et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J Neurosci. 2010;30:3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sullivan EL, Rivera HM, True CA, Franco JG, Baquero K, Dean TA, et al. Maternal and postnatal high-fat diet consumption programs energy balance and hypothalamic melanocortin signaling in nonhuman primate offspring. Am J Physiol Regul Integr Comp Physiol. 2017;313:R169–R179. [DOI] [PMC free article] [PubMed] [Google Scholar]