

Abstract
Mucopolysaccharidosis type IIIB (MPS IIIB) is an autosomal recessive lysosomal storage disease caused by mutations in the gene that encodes the protein N-acetyl-glucosaminidase (NAGLU). Defective NAGLU activity results in aberrant retention of heparan sulfate within lysosomes leading to progressive central nervous system (CNS) degeneration. Intravenous treatment options are limited by the need to overcome the blood–brain barrier and gain successful entry into the CNS. Additionally, we have demonstrated that AAV8 provides a broader transduction area in the MPS IIIB mouse brain compared with AAV5, 9 or rh10. A triple-capsid mutant (tcm) modification of AAV8 further enhanced GFP reporter expression and distribution. Using the MPS IIIB mouse model, we performed a study using either intracranial six site or intracisterna magna injection of AAVtcm8-codon-optimized (co)-NAGLU using untreated MPS IIIB mice as controls to assess disease correction. Disease correction was evaluated based on enzyme activity, heparan sulfate storage levels, CNS lysosomal signal intensity, coordination, activity level, hearing and survival. Both histologic and enzymatic assessments show that each injection method results in supranormal levels of NAGLU expression in the brain. In this study, we have shown correction of lifespan and auditory deficits, increased CNS NAGLU activity and reduced lysosomal storage levels of heparan sulfate following AAVtcm8-coNAGLU administration and partial correction of NAGLU activity in several peripheral organs in the murine model of MPS IIIB.
Introduction
Sanfilippo syndrome type B [mucopolysaccharidosis type IIIB (MPS IIIB)] is caused by a biallelic mutation on the NAGLU gene resulting in a deficiency in the enzyme N-acetyl-glucosaminidase (NAGLU). This causes a progressive accumulation of lysosomal glycosaminoglycans, specifically the linear polysaccharide, heparan sulfate. The accumulation of heparan sulfate in the lysosome leads to progressive central nervous system (CNS) degradation. In addition to a progressive decline of the CNS, clinical symptoms of Sanfilippo syndrome include a loss of motor function, hyperactivity, hearing deficiencies, hepatosplenomegaly, coarse facial features, ear nose and throat (ENT) abnormalities and cardiac abnormalities (1). The patient’s symptoms begin to manifest before age 5, although diagnosis can often be delayed and/or missed all together (2). Patients do not typically survive beyond the second decade of life as a result of the sequela of severe neurodegeneration caused by heparan sulfate accumulation (3,4). There is currently no known cure for Sanfilippo syndrome (5,6). There have been several recent therapeutic trials assessing interventions for lysosomal storage disorders, including enzyme replacement therapy and gene therapy (6,7). In MPS IIIB, clinical trials are underway or ongoing using a variety of approaches but the results to date have not resulted in marked improvement in disease course (NCT02618512, NCT04655911, NCT03784287, NCT02754076, NCT02324049, NCT03315182, NCT03300453) (6,8). Several recent trials including the Abeona Therapeutics AAV9-NAGLU IV trial have shown positive preliminary results (NCT04655911 AbeonaTherapeutics.com).
Gene therapy treatments done within mouse models have shown improvements in both severity and symptoms relating to MPS IIIB, although a complete correction of the disease has not yet been shown (7,9,10). The adeno-associated-virus (AAV) vector system has proven to be successful for transducing cells in the CNS because of its low immunogenicity, effective transduction efficiency and its specificity (11). Of the several AAV serotypes we have previously studied, AAV8 exhibited higher transduction of a green fluorescent protein (GFP) reporter into the brain than that of AAV5, 9, or rh10 when injected intracranially, via the intracranial six site (IC6) method used in this paper or in the bilateral thalamus, into 3- to 4-day old mice (12–14). Furthermore, site-specific amino acid modifications of the AAV8 capsid further enhanced brain transduction with GFP (15).
The most important aspects for creating a clinical therapy for MPS IIIB are quality and quantity of life. Our group has previously demonstrated success with intracranial AAV gene therapy resulting in improvements to motor function, auditory function and lifespan (16,17). Bone marrow transplants combined with AAV treatments have been found to be less effective than AAV alone for prolonging life (17), while addition of systemic lentiviral gene therapy confers moderate improvement (16). In this study, we demonstrate that parenchymal or cisternal delivery of a codon-optimized triple mutant AAV8 capsid (AAVtcm8-coNAGLU) corrects the MPS IIIB mouse levels of NAGLU activity and lysosomal storage to endogenous levels in the CNS, resulting in improvement in auditory function and lifespan.
Results
Treatments
MPS IIIB mice were injected with AAVtcm8-coNAGLU at 3–4 days old. Normal mice were +/− or +/+ for normal NAGLU sequence. A cohort of untreated normal and MPS IIIB mice were used as the comparator groups to determine the effect of therapy. Two routes of administration were evaluated: an intraparenchymal six-site injection (IC6, n = 24) and an intracisternal magna site (ICM, n = 24). Treated mice were injected with a total dose of 1.8e10 vg. All mice tolerated treatments well, and no adverse effects were observed post treatment.
Biochemical analysis of NAGLU activity
Cohorts of mice euthanized at 6 months of age as well as at lifespan endpoint euthanasia (predominantly ~ 640 days of age for normal and treated groups) were used for biochemical analysis. Using a tandem mass spectroscopy method of determining enzyme activity (18,19), NAGLU activity in the MPS IIIB NO TX mouse is approximately 3.5% that of the normal control animals in all brain regions (P < 0.0001). A similar level of background activity measurement is observed for the fluorometric assay in the liver and heart. The mass spectroscopy method is comparable to the fluorometric assay and allowed multiplex enzyme assay but was only used for the brains owing to cost constraints. As seen in Figure 2A and B, in the age matched 6-month-old animals, compared with the normal control group, NAGLU activity in the forebrain displayed a 190-fold increase (P < 0.0001) and a 5.8-fold increase (P < 0.05) in MPS IIIB IC6 and ICM mice, respectively. In the hindbrain, AAVtcm8-coNAGLU administration resulted in a 70-fold increase (P < 0.0.0001) with IC6 and 16.7-fold increase (P < 0.0001) with ICM in NAGLU activity in MPS IIIB mice. In the cerebellum, a 26- and 65-fold increase (P < 0.0001) in NAGLU activity was observed in the respective IC6 and ICM treatment groups compared with normal animals. Comparing treatment groups, IC6 and ICM activity differences were significantly elevated in the forebrain (P < 0.0001) and hindbrain (P < 0.05) but did not reach significance in the cerebellum.
Figure 2.

NAGLU activity assays measured in the forebrain, hindbrain and cerebellum by mass spectroscopy substrate cleavage based assay from IC6 or ICM treated MPS IIIB, MPS IIIB NO TX and normal control mice at 6 (A) and 21 (B) months of age; liver activity was measured by 4MU-based fluorescence assay at 6 and 21 months of age (C and D), and heart activity measured by 4MU-based fluorescence assay at 6 (E) and 21 (F) months of age. N = 4–6 at 6 months, 3 at 21 months for CNS and 3–8 for peripheral tissues, P-values are indicated by * (<0.05), ** (<0.01), ***(<0.0001), **** (<0.00001) or NS (not significant). All MPS IIIB NO TX mice reached endpoint before 13 months of age and are not included in the 21-month analysis. All CNS tissues were evaluated with the mass spectrometry method [A (13 males/8 females) and B (1 male/8 female)]. All peripheral tissues were evaluated using the 4MU-based assay [C (15 male/11 female), D (12 male/7 female), E (16 male/13 female) and F (12 male/15 female)].
At the ~ 640-day timepoint (Fig. 2B), NAGLU activity in forebrain, hindbrain and cerebellum, with the IC6 method were 193- (P < 0.0001), 133- (P < 0.001) and 8.8-fold (P = NS) above normal control. While 640-day NAGLU activity in forebrain, hindbrain and cerebellum with the ICM method were 7.8- (P = NS), 5.5- (P = NS) and 5.6-fold (P = NS) increased compared with normal control. The difference in activity between IC6 and ICM reaches P < 0.01 in forebrain and hindbrain but is NS in the cerebellum.
Peripheral organs including the heart and liver were assessed for NAGLU activity using the fluorometric enzymatic assay method. NAGLU activity assays run on liver supernatants showed that MPS IIIB IC6 and ICM mice had an improvement over MPS IIIB NO TX mice (Fig. 2C, P < 0.0001). Liver NAGLU also showed that normal mice were significantly different from MPS IIIB NO TX and from both treatment groups (P < 0.0001 vs. NO TX and P < 0.01 vs. IC6 and ICM).
Heart NAGLU activity showed that 6-month-old MPS IIIB NO TX mice were significantly lower than both normal and MPS IIIB ICM mice (Fig. 2E, P < 0.0001). Supraphysiological levels of NAGLU activity in the heart were observed for both treatment groups compared with 6-month-old normal animals (P < 0.01 for both) and were observed through 21 months of age in the ICM group (Fig. 2F, NS vs. IC6 and P < 0.05 for ICM).
Beta-glucuronidase activity
Brain and organ homogenates were assessed for effect of treatment on secondary elevation of beta-glucuronidase (GUSB). Several groups have observed compensatory expression of NAGLU and GUSB in which low or absent NAGLU results in elevated GUSB in the MPS IIIB mouse and vice versa in the MPS VII model, with return back toward normal as disease is corrected (16,17). Assessment of brain GUSB was performed using a mass spectrometry-based method as described later in multiplex with the NAGLU assays. At 6 months of age, the MPS IIIB NO TX animals had mean activity levels in all brain regions of 24.10 to 26.95 nmoles/mg/h, which was significantly elevated compared with all other groups (Fig. 3A, P < 0.0001) and normal animals had mean activity levels of 5.80–7.74 nmoles/mg/h which were not significantly different than either treatment group. IC6 and ICM means ranged from 6.29 to 8.33 and 7.44 to 8.42 nmoles/mg/h, respectively (Fig. 3A). At 21 months (Fig. 3B), there were no significant differences between the IC6 and ICM treatment groups and the normal animals with respect to brain GUSB activity.
Figure 3.

GUSB activity assays measured in the forebrain, hindbrain and cerebellum by mass spectroscopy substrate cleavage based assay from IC6 or ICM treated MPS IIIB, MPS IIIB NO TX and normal control mice at 6 (A) and 21 (B) months of age; liver activity by 4MU-based fluorescence assay at 6 and 21 months of age (C and D), and heart activity by 4MU-based fluorescence assay at 6 (E) and 21 (F) months of age. N = 4–6 at 6 months, 3 at 21 months for CNS and 3–8 for peripheral tissues, P-values are indicated by * (<0.05), ** (<0.01), ***(<0.0001), **** (<0.00001)) or NS (not significant). All MPS IIIB NO TX mice reached endpoint before 13 months of age and are not included in the 21-month analysis. All CNS tissues were evaluated with the mass spectrometry method [A (13 males/8 female) and B (1 male/8 females)]. All peripheral tissues were evaluated using the 4MU-based assay [C (15 male/11 female), D (12 male/7 female), E (16 male/13 female) and F (12 male/15 female)].
GUSB activity in liver homogenates, tested via the fluorescence assay, was not significantly different between groups at either 6 or 21 months of age (Fig. 3C and D).
Fluorometric-based determination of GUSB in the heart revealed that normal animals had significantly lower GUSB activity compared with MPS IIIB NO TX animals at 6 months (Fig. 3E, P < 0.001) and MPS IIIB IC6 treated animals at 21 months (Fig. 3F, P < 0.05).
Heparan sulfate and glycosaminoglycan levels
Forebrain, hindbrain and cerebellum region homogenates were assayed for heparan sulfate levels using both the endogenous biomarker and internal disaccharide mass spectroscopy methods described in the Methods section. These mass spectrometry methods are not affected by lipids in the homogenate and give a direct comparison of heparan sulfate levels. Complete correction of brain heparan sulfate levels in all treatment groups at 6 months (Fig. 4A, P < 0.001 to < 0.0001 for all groups vs. MPS IIIB NO TX group and NS for all other group comparisons) and correction at 21 months in all but one animal treated via the ICM method (Fig. 4B, P = NS for all comparisons) was observed. At 6 months, MPS IIIB NO TX animals display elevated heparan sulfate saccharides in the range of 16–150 pmoles/mg of protein whereas normal controls were 0–4.27 pmoles/mg of protein and IC6 treated MPS IIIB animals ranged from 0 to 2.8 pmoles/mg, and ICM treated MPS IIIB animals 0–3.3 pmoles/mg. At 21 months, all variability in the ICM animals was attributable to a single animal with elevated levels in the fore and hindbrain regions indicating inadequate anterior distribution in this animal. However, the remaining treated animals had levels in all brain regions below 2.9 pmoles/mg of protein. Of note, the animal with the higher anterior levels at 21 months performed normally on behavioral assessments and was asymptomatic at 21 months.
Figure 4.

Heparan sulfate saccharide level and DMMB measurement of total glycosaminoglycan assessments from IC6 or ICM treated MPS IIIB, MPS IIIB NO TX and normal control mice. Brain (forebrain, hindbrain, cerebellum) heparan sulfate internal and external disaccharide levels at 6 (A) and 21 (B) months of age; liver levels of DMMB measured glycosaminoglycan at 6 and 21 months of age (C and D), and heart levels of DMMB measured glycosaminoglycan at 6 (E) and 21 (F) months of age. N = 4–6 at 6 months, 3 at 21 months for CNS and 3–8 for peripheral tissues, P-values are indicated by * (<0.05), ** (<0.01), ***(<0.0001), **** (<0.00001)) or NS (not significant). All MPS IIIB NO TX mice reached endpoint before 13 months of age and are not included in the 21-month analysis. All CNS tissues were evaluated with the mass spectrometry method [A (13 males/8 female) and B (1 male/8 females)]. All peripheral tissues were evaluated using the DMMB-based assay [C (17 male/12 female), D (11 male/14 female), E (17 male/12 female) and F (11 male/14 female)].
Total glycosaminoglycan (GAG) levels in the liver were assessed using the dimethylmethylene blue (DMMB) method and revealed that MPS IIIB NO TX mice had significantly higher levels of total GAGs when compared with all other groups at 6 months of age (Fig. 4C, P < 0.05 vs. normal and P < 0.001 vs. IC6 and P < 0.01 vs. ICM). At 21 months, the normal and treated groups had similar total GAG content (Fig. 4D, P = NS).
Heart GAG content was significantly lower in normal and MPS IIIB IC6 than MPS IIIB NO TX mice at 6 months of age (Fig. 4E, P < 0.01 vs. normal and P < 0.05 vs. IC6). At both 6 and 21 months of age, heart tissue of normal animals did not have significantly different GAG content than either treatment group (Fig. 4E and F).
Immunohistochemical detection of lysosomal signal intensity
Lysosomal-associated protein 1 (LAMP1), a marker of the lysosomal membrane, was used to determine lysosomal distribution and signal intensity. LAMP-1 immunostaining revealed normalization of lysosomal signal intensity for MPS IIIB ICM and MPS IIIB IC6 mice when compared with MPS IIIB NO TX in the cerebellum and cerebral cortex (Fig. 5, P < 0.0001 vs. normal, IC6 and ICM). LAMP-1 staining showed a significant attenuation of lysosomal signal in both the cerebellum and cortex of ICM treated animals with no detectable differences from normal animals at 6 months of age. In addition, LAMP-1 staining in the cortex of IC6 treated animals showed a minimal difference in the cerebellum LAMP-1 area compared with normal animals (P < 0.05 vs. normal).
Figure 5.

Assessment of lysosomal distention in the caudal and rostral regions of the cerebral cortex, and the cerebellum. At 6 months post administration of AAVTCM8-coNAGLU, cortex and cerebellum were examined for LAMP1. (A) Detection of LAMP1 (green) was performed in separate representative specimens. (B) Quantification of ROI% positive area in the cortex (rostral), cortex (caudal) and cerebellum shows significant attenuation of lysosomal (LAMP1) signal in IC6 and ICM treated animals. P-values are indicated by * (<0.05), ** (<0.01), ***(<0.0001), **** (<0.00001) or NS (not significant).
Running wheel activity assessments
All mice tested (N = 8–10 per group; males only to eliminate variability related to estrus cycling) were housed inside ventilated, lighted chambers with a strictly maintained light schedule of 12 h on (7 a.m.–7 p.m.) and 12 h off (7 p.m.–7 a.m.). We previously identified an improvement in circadian rhythm, specifically in percentage of daytime (lights on) activity and time of activity onset (time after lights off) in MPS IIIB mice when compared with treated groups (16,17). Similar but not statistically significant differences in daytime activity proportion between MPS IIIB NO TX (~13%), normal (5–6%), MPS IIIB ICM (~9%) and MPS IIIB IC6 (6–9%) treatment groups were observed (Fig. 6). At 5 but not 4 months, time to activity onset (Fig. 6) was corrected to that of normal mice (19.38 h, or 22.8 min after lights out, P < 0.05 vs. MPS IIIB NO TX) for IC6 (19.39 h, 23.4 min, P < 0.01) and ICM mice (19.36 h, 21.5 min, P < 0.05) compared with MPS IIIB NO TX mice.
Figure 6.
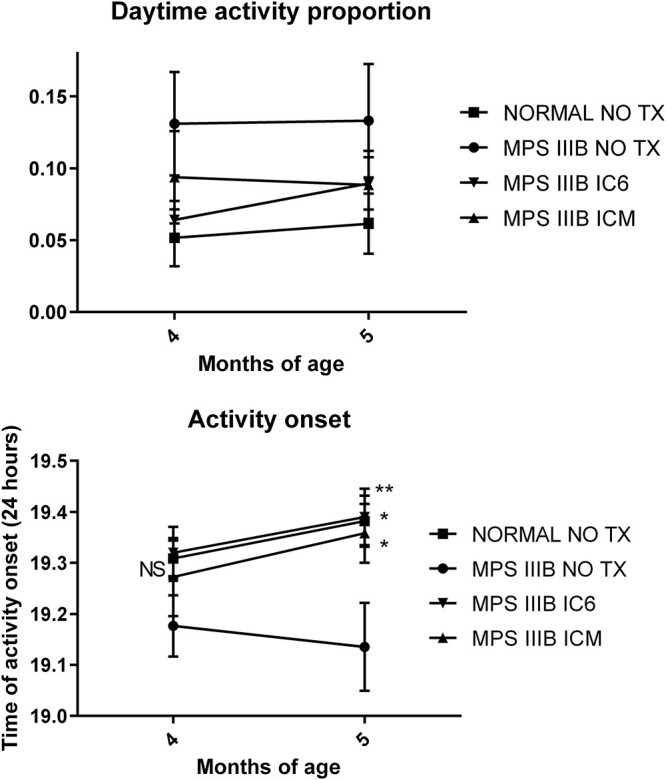
Proportion of running wheel activity during daytime from IC6 or ICM treated MPS IIIB, MPS IIIB NO TX and normal control mice at 4 to 5 months of age and time to activity onset (24 h) for mice of 4 to 5 months of age. N = 8 male mice per group. P-values are indicated by * (<0.05), ** (<0.01), ***(<0.0001), **** (<0.00001) or NS (not significant).
Motor function
Motor deficits in MPS IIIB mice typically appear at 8–10 months of age. We previously showed an improvement in motor deficits through a significantly delayed progression of motor function deficits in treated mice (16,17). In the current study, significant difference was not reached between groups. Animal services and veterinary care guidelines at UF maintain strict euthanasia protocols for animals which display declining body condition score. These requirements resulted in early euthanasia prior to effect on rotarod performance.
Auditory-evoked brainstem responses
Auditory-evoked brainstem responses (ABRs) were used to determine hearing sensitivity and deficits. ABR thresholds were measured at 8, 16 and 32 kHz with a tone burst stimulus. In concert with previous studies, we found a distinct deficit in MPS IIIB NO TX mice when compared with normal mice (16,17). Both routes of administration showed complete hearing normalization at 8 kHz (Fig. 7, ICM P < 0.0001 and IC6 P < 0.05 vs. MPS IIIB NO TX. Both NS vs. normal), while MPS IIIB ICM mice also display complete hearing correction at 16 Hz (P < 0.001 vs. MPS IIIB NO TX and NS vs. normal). In an overall group comparison, MPS IIIB ICM animals displayed superior auditory response compared with all other groups. No groups were statistically different from one another at 32 kHz, including the normal group reflecting the age-related high frequency hearing loss present in the background strain.
Figure 7.
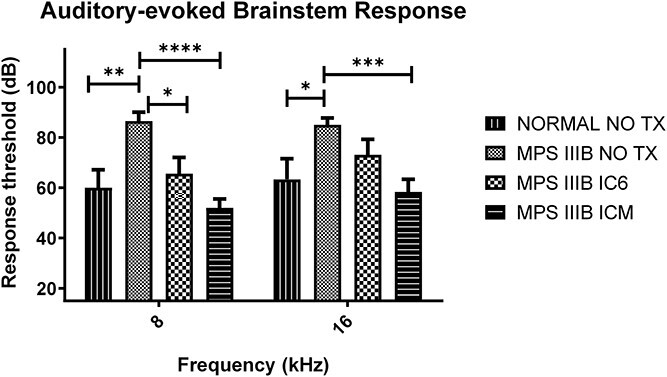
Auditory-evoked brainstem response thresholds from IC6 (N = 4 male/4 female) or ICM (N = 5 male/10 female) treated MPS IIIB, MPS IIIB NO TX (N = 6 male/7 female) and normal (N = 3 male/6 female) control mice at 8 to 9 months of age measured at 8 kHz and 16 kHz. P-values are indicated by * (<0.05), ** (<0.01), ***(<0.0001), **** (<0.00001) or NS (not significant).
Lifespan
We have previously shown that AAV treatment prolongs the life of MPS IIIB animals by > 100 days (16,17). In this study, AAVtcm8-coNAGLU treated mice show a complete correction of lifespan in comparison to MPS IIIB NO TX mice (Fig. 8). MPS IIIB IC6 mice median survival increased to 639 days (P < 0.0001 vs. NO TX) and MPS IIIB ICM mice median was not defined owing to the percent survival exceeding 50% at the end of the study (P < 0.0001 vs. NO TX). The median survival for normal mice was 644 days (P < 0.0001 vs. NO TX) and for MPS IIIB NO TX mice 320 days.
Figure 8.
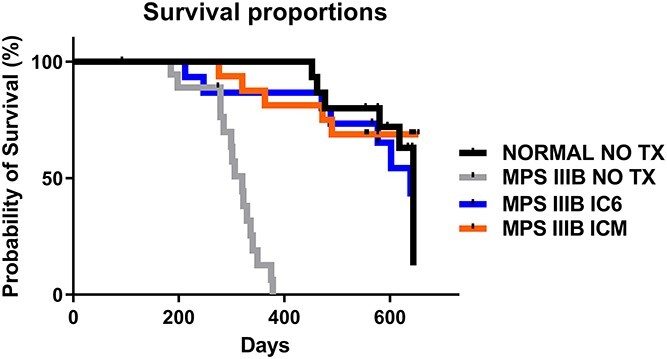
Kaplan–Meier survival curves from IC6 or ICM treated MPS IIIB, MPS IIIB NO TX and normal control mice. MPS IIIB no TX animals display a significant increase in mortality. AAVtcm8-coNAGLU administration by either IC6 or ICM results in normalization of median lifespan in MPS IIIB IC6 or ICM animals relative to normal.
Discussion
Despite Sanfilippo syndrome being a monogenic disease, patients face a variety of symptoms which primarily manifest in the CNS (3). Progressive neurodegeneration caused by heparan sulfate accumulation often causes death in MPS IIIB patients; however, patients face a number of other debilitating symptoms that contribute to an overall decline in health such as a loss of motor function, hearing deficiencies, hepatosplenomegaly, hyperactivity and sleep disruption.
The two most important goals in creating any clinical therapy are improvements in quality and quantity of life (20,21). Both pre-clinical and clinical studies of the mucopolysaccharidosis lysosomal storage diseases demonstrate the accumulation of GAGs at an early point in development that severely impacts neurologic function (21,22). For similar diseases such as MPS I, stem cell transplant has a demonstrable benefit for neurological manifestations in patients but only if initiated within the first 24 months of life (20). This is consistent with the general observation that earlier treatment is better for preventing irreversible neuronal damage (3). For this reason, this study aimed to treat mice at an earlier timepoint to achieve the best possible outcome for correction of the disease.
Previous studies (16,17) have shown that intraparenchymal AAV5-NAGLU brain injections significantly improve quality of life for MPS IIIB mice, but do not result in complete correction of the disease. Though above normal total brain NAGLU activity was attained, behavioral measures, lysosomal storage and the secondary lysosomal enzyme interaction seen for NAGLU and GUSB (16,17,23,24) were not normalized. This approach was subsequently evaluated in a clinical trial, which also demonstrated some improvement in disease course in the youngest patients treated despite lack of complete correction of heparan sulfate levels in the cerebrospinal fluid (CSF) (6). Intravenous AAV9-NAGLU (25) and Lentiviral-NAGLU transduced hematopoietic stem cell transplants (7) have also demonstrated improvements in the MPS IIIB mouse model, and clinical trials with these approaches have been started but are currently on hold. NAGLU enzyme replacement therapy given intravenously did not show clinical efficacy for neurological manifestations of the disease (8,26,27), but a CNS-directed NAGLU-IGFII fusion approach has shown promise pre-clinically (26,27) and is currently under evaluation in a clinical trial. In our study, the CNS was selected as the primary target for correction. Heparan sulfate levels and activity of NAGLU and GUSB served as markers to determine the degree of correction. Both GUSB and heparan sulfate levels represent measurable markers of disease that can be assessed by relatively non-invasive procedures through serum or CSF collection from patients. It is imperative to note that behavioral cognitive outcomes and measurements should not be ignored considering GUSB and heparan sulfate levels do not indicate a reversal of previous neuronal damage.
In the current study, we explored the effect of capsid modifications and route of administration on gene expression and enzyme distribution in the MPS IIIB mouse model. We had previously shown that IC6 was superior for widespread biodistribution in the brain (12,13,16,17,28) compared with other intraparenchymal and intravenous injection methods. Importantly, AAV8 had a particularly broad transduction across brain regions in the MPS IIIB model, compared with wild-type animals. Modification of the capsid to improve intracellular trafficking and reduce immune presentation further enhanced distribution across regions of the brain (15). The ICM route of administration is considerably less invasive for clinical applications compared with the IC6 route, which involves a six-site injection via skull burr holes with catheter placements into brain parenchyma as opposed to a single injection site performed percutaneously with image guidance into the cisterna magna.
Biochemical correction of disease involved assessment of NAGLU activity, compensatory changes in GUSB activity, LAMP1 staining and heparan sulfate or GAG levels in tissues at 6 months of age and near 21 months of age in subdivided brain regions to better assess regional distribution. As expected, NAGLU activity in the forebrain and hindbrain was higher with the IC6 method at both timepoints reflecting the direct administration of virus in these areas. However, normal to supranormal levels of activity were also present at both timepoints with the ICM route, and each route conferred similar activity in the cerebellum. The level of NAGLU activity with either route was sufficient to completely correct heparan sulfate levels, lysosomal distention (LAMP1 staining) and normalize GUSB activity in the brain, indicating achievement of desired treatment thresholds. All brain regions were assessed using mass spectrometry for heparan sulfate levels and enzymology. The DMMB method is prone to interference by the lipids present in the brain homogenates, but the mass spectroscopy method overcomes this limitation. Further, mass spectroscopy identifies heparan sulfate specifically rather than total GAG and could be multiplexed to do the NAGLU and GUSB enzyme activity in parallel. The mass spectroscopy method is substantially more resource intensive, so the more resource efficient 4MU-based enzyme activity assays and DMMB method was used preferentially in the other organs where it does not have lipid interference for GAG assessment. Correcting major CNS deficits was one of the chief goals of this study and as such we sought to obtain the most accurate data possible to assess and measure these outcomes in the brain.
Additionally, peripheral passage of vector was sufficient to transduce both the liver and heart to significant levels. Interestingly, though the liver had a mean of < 30% of normal activity in MPS IIIB mice receiving vector, this was adequate to reduce GUSB activity and completely correct liver GAG levels. These observations in the liver indicate a broad ability to cross correct progressive disease pathology with lower-than-normal NAGLU levels. In contrast, the heart had supranormal NAGLU activity but only intermediate correction of GUSB activity, while the heart GAG levels in the treated groups were not statistically different than the normal. This may indicate a less than complete cross-correction in the heart perhaps owing to the more fibrous and stratified layers of the heart relative to the liver that may limit diffusion of vector and cross correction of NAGLU between the layers.
We had previously described changes in the proportion of activity during the daytime and time to activity onset after the light period for the MPS IIIB animals. Affected patients display similar behaviors during phases of disease progression, including disruptions to sleep patterns for both children and caregivers; thus, effective treatments may serve as a dual benefit. Time to activity onset at 5 months of age was completely corrected by both treatment approaches. The proportion of daytime activity did not reach statistical significance in this study, but the trend toward improvement was similar in both treatment groups.
The affected children develop progressive difficulty communicating, and several studies have demonstrated not only cognitive impairments but also impaired hearing which can adversely affect communication. The MPS IIIB mouse is essentially deaf with no response to auditory stimulus up to the maximum decibel level of the ABR test. In this study, we were able to improve hearing levels in MPS IIIB mice with either injection route, though the ICM route appears to numerically correct beyond normal at both 8 and 16 kHz. This may reflect greater virus biodistribution to the ventral brain and thus the cranial nerves with a cisternal injection method.
The MPS IIIB mouse model has been well characterized and portrays many of the characteristics of the human disease, making it an excellent indicator for testing possible treatment methods (23). It is important to note that MPS IIIB untreated KO mice develop severe urinary retention that requires euthanasia; our treatment successfully rescues this trait. Motor function and balance skills are both affected by Sanfilippo syndrome, though the time to onset of these symptoms is often delayed until later years of life. This is reflected in the mouse model as symptoms do not manifest until around 10 to 12 months of age in the mice. Our group previously reported a significant delay in the progression of these symptoms. In the current study, statistical significance with rotarod testing was not achieved owing to more stringent euthanasia endpoint requirements of MPS IIIB NO TX mice at our facility resulting in early euthanasia of mice prior to the usual decline in rotarod performance.
Our most important clinically relevant outcome is extension in lifespan and overall health. This was achieved with either IC6 or ICM route of administration resulting in prolonged lifespan that was not statistically different from the normal mice out to at least 640 days of age. Our lifespan study was abbreviated at 640 days owing to the onset of mouse colony restrictions by the COVID pandemic at our institution. Although our study was abbreviated because of this restriction, we demonstrate that NAGLU levels remained at or above normal levels in both the 6- and 21-month cohorts.
We conclude that neonatal intracranial tcmAAV8-coNAGLU injections through IC6 and ICM administrations are relatively comparable for correction of neurological manifestations of MPS IIIB. Either route results in marked improvement in lifespan and is accompanied by improved or complete correction of hearing loss, and restoration of brain lysosomal heparan sulfate storage levels, and brain, heart and liver GUSB activity levels. Translation to clinical studies of either method to deliver tcm8-coNAGLU vector is promising, and additional studies should be conducted to determine the effectiveness of these administration routes in species more closely related to human brain volume and CSF circulatory flow. Both routes of administration of the modified vector normalized auditory measures and lifespan for MPS IIIB mice supporting a less invasive approach for future studies. Additional studies should also be conducted at later disease stages to better define the temporal window of treatment for MPS IIIB patients.
Materials and Methods
Study design
This study was designed to optimize and design an effective treatment for Sanfilippo syndrome using the murine model. The main goal of this study was to correct lifespan and CNS deficits in the MPSIIIB mouse to that of a normal mouse, through the use of an AAV8 triple capsid mutated virus, using a six-site intracranial or a cisternal injection. Behavioral tests were performed at various ages to determine relevant clinical outcomes and efficacy (Fig. 1). Longevity studies were performed to accurately determine if lifespan was altered by the treatments administered. At weaning mice were randomly allocated to the long-term study cohort and then to 6 month biochemistry cohort. Running wheel study mice were drawn from both allocations to allow adequate males. Rocking rotarod assessments and ABR testing were conducted only on mice within the long-term study. Owing to scheduling and nature of the assessments (anesthesia requirements for ABR testing), we were forced to stagger enrollment in specific behavior testing, as a result not all mice underwent all three behavior tests. Post ABR testing (if conducted), mice were housed until endpoints. Survival curves were done at this time.
Figure 1.

Study design. All behavior experiments and procedures conducted in the study have been arranged on a timeline scale to show an overview of the experiment. Created by BiRender.com.
Mice
The C57BL/6 NAGLU (B6.129S6-Naglutm1Efn/J) deficient strain (kind gift from Elizabeth Neufeld (29)) was used and maintained by strict sibling inbreeding. Tissues (toes) were collected from 1- to 4-day-old mice and subsequently genotyped following a NAGLU exon 6 and neomycin insertion cassette PCR protocol (30). All animal activities were performed in accordance with University of Florida Institutional Animal Care and Use Committee (IACUC) guidelines and policies (201803042).
Vector information
Briefly, the cassette contains the CMV enhancer/CBA promoter, kozak sequence, human codon-optimized NAGLU and a bGH poly A tail. Triple capsid mutant AAV8 vector was produced at the University of Florida Powell Gene Therapy Center Vector Core Laboratory (Gainesville, FL, USA) (15). Vector titer was determined by quantitative dot blot and then diluted and aliquoted before being stored at −80°C until treatments were administered.
Treatments
At 1–4 days, all mice were genotyped for the NAGLU exon 6 and neomycin gene insertion before allocation at day 3–4 of age in one of the following four groups: normal no treatment, which includes non-diseased wild-type and heterozygote mice (normal, n = 24, 10 male); mutant no treatment (MPS IIIB NO TX, n = 26, 13 male); MPSIIIB treated with IC6 injections (MPS IIIB IC6, n = 24, 12 males); and MPS IIIB treated with ICM injections (MPS IIIB ICM, n = 24, 10 males). MPS IIIB mice were randomly allocated to treatment group.
Mice were cryoanesthetized prior to injections. P3–P4 aged mice were placed in an aluminum foil enclosure surrounded by ice for approximately 2–4 min until movements were limited. It is important to note that the mice were not allowed to be directly placed on the ice and they were carefully monitored to avoid adverse reactions. Full anesthesia is difficult to obtain in neonatal mice owing to their small size and resulting problems in maintaining drug effects. Cryoanesthesia provides both a mild analgesic effect and an effective anesthetic method for newborn mice. MPS IIIB IC6 mice pups were injected freehand bilaterally into three intracranial regions (six sites, 2 μl per site): frontal (from bregma; 1 mm posterior, 2 mm lateral, 1.5 mm depth), temporal (from bregma; 3 mm posterior, 3 mm lateral, 2.5 mm depth) and cerebellum (from lambda; 1 mm posterior, 1 mm lateral, 3 mm depth) using a 10 μl Hamilton syringe (Part/REF:#: 25 μl, Neuros Syringe, Model 1702 RN, 33 gauge, Point Style 4) (5,16,17,30). Accurate depth was achieved using the protective needle sleeve blind stop that comes equipped on the Hamilton neuro-syringes to help ensure accurate depth measurements. MPS IIIB ICM pups were injected with 4 μl of viral aliquot directly into the cisterna magna at a depth of 3 mm. Suture lines were able to be clearly visualized owing to the young age of the mouse and the aid of a red light (Respironics Wee Sight Transilluminator, Nashville, TN, USA). Prior to ICM injections, mice were positioned prone with skin pulled taught allowing for visualization of the base of the skull and determination of the cisterna magna site (31). The total dose of virus administered was 1.8E10 vg for either injection route. The rate of infusion was ~ 1–2 μl/1 min with an additional 30 s before the needle was removed. On average, ICM injections took 4–6 min. IC6 injections took an average of 12–16 min. Pups were immediately placed on a heating block to return them to normal temperature following injections. When pups appeared awake, alert and warm, they were placed back with the dam and subsequently monitored over the following days. All mice tolerated the procedure well and recovered.
Tissue harvest
Mice were euthanized at 6 months of age or at endpoint (predominantly ~ 640 days of age for normal and treated groups) for biochemical analysis. Mice were euthanized between 181 and 729 days from all groups with the majority of aged animals collected at 640 days of age owing to COVID mandated colony restrictions at our institution. All mice were euthanized by CO2 asphyxiation and cervical dislocation or thoracotomy and were performed in accordance with the IACUC protocols. The following tissues were harvested: eye, brain, heart, lung, spleen, liver, kidney and muscle. Harvested tissue was then divided into two halves: one was flash frozen at −80°C immediately after harvest and the other was fixed into a 4% paraformaldehyde phosphate-buffered saline (PBS) solution for 24 h then transferred to a 1x PBS with 30% sucrose solution for storage at 4°C. The brain was cut into two sagittal halves: one half immediately fixed, and the other was subsequently divided into three regions forebrain, hindbrain and cerebellum (Fig. 1). Fixed tissues were then processed and embedded into paraffin before being sectioned for histological analysis. Tissues were stored at −80°C and 4°C until they were mechanically homogenized or embedded, respectively. Mechanical homogenization was completed using a hand-held tissue homogenizer; after tissue was added to a homogenization buffer containing 10 mm (7.5 pH) Tris Base, 150 mm NaCl, 1 mm DDT, 0.2% Triton X-100 and a protease cocktail inhibitor.
Histology
Tissues were processed and embedded after being washed in a 1x PBS solution. After being washed tissues were embedded in paraffin before being sectioned and placed on slides. Brain tissue was embedded in a sagittal orientation and slides were cut at a thickness of 4 μm. Slides were stained for LAMP-1 (Lysosomal Associated Membrane Protein 1). All slides were deparaffinized and underwent an antigen retrieval step. Briefly, sides were placed into a steamer containing water for 30 min and incubated at room temperature for 30 min before being placed into 1x PBS for 5 min. LAMP-1 used de-ionized water during the antigen retrieval step. A protein block was used before the slides were stained with antibodies to allow for optimal antibody coverage of tissue sections. Primary antibodies for staining included for LAMP-1 staining, and 1D4B anti-LAMP1 (AB_528127, DSHB, University of Iowa). Following an overnight primary antibody incubation (4°C) at a 1:1 concentration, the slides were exposed to a secondary antibody incubation at a 1:1000 concentration, for this step LAMP-1 d Gt anti-Rb 488 antibody (Invitrogen, Waltham, MA, USA; Catalog # A-11034). All slides were counterstained with a DAPI solution (Vectashield, H-1200-10). All slides were imaged with a Keyence BZ-X810 fluorescent microscope (Keyence, Itasca, IL, USA). Brains were imaged at 20x. Cisterna Magna space margin, brainstem, cerebellum, caudal cerebral cortex, rostral cerebral cortex, midbrain, substantia nigra, lateral ventricle, hippocampus and thalamus/striatum images were obtained. The brain regions were identified using a brain atlas. Images were quantified using ImageJ software by selection of a region of interest (ROI) on a stained slide, which was then quantified by percent positive area within the ROI. Each image was analyzed using ImageJ (NIH) software; all relative image calculations were also completed through ImageJ. The examiners conducting the histology were blinded until after histological procedures and data processing were finished.
Endogenous biomarker method of heparan sulfate level assessment in brains Heparan sulfate was measured using the endogenous biomarker method (32)
A 10 μl aliquot was taken from each sample homogenate (1:10 dilution) to assay and was measured using a Pierce BCA Protein Assay Kit from Thermo Scientific (Reference: 23225, Waltham, MA, USA). After aliquoting brain homogenates, samples/controls were mixed in 100 μl of the 0.25 M 3-methyl-1-phenyl-2-pyrazoline-5-one (PMP, CAS 89–25-8) solution (pH 9.510) containing 0.4 M ammonia and 1μm internal standard (ΔUA-GalNAc4S CAS 136144-56-4). Samples and controls were mixed with a pipe tman prior to incubating at 70°C for 90 min in a Peltier Thermal Cycler (PTC-200, Alameda, CA, USA).
Samples and controls were allowed to cool for 10 min after incubation and then were acidified with 100 μl 2 M formic acid (aq). PMP was washed from the acidified samples and controls by adding 200 μl chloroform and shaking vigorously by hand for 1 min. Samples and controls were then centrifuged for 1 min at 13 000 g to separate the layers and break up emulsions. A disposable polypropylene pipetman tip (200 μl) was wetted with chloroform, and then used to remove the bottom organic layer, which was disposed of in waste. The separation/washing step described before was performed a total of 4x per each sample/control to remove the PMP.
Finally, the samples and controls were centrifuged for an additional 5 min at 13 000 g to pellet particulates. Avoiding the pellet, the sample was transferred to a 250 μl pulled-point glass HPLC vial insert for LC–MS/MS analysis.
Analytes were separated by a Waters Acquity UPLC (Milford, MA, USA) equipped with flow-through needle and an Agilent Pursuit 3 PFP (2.0 x 100 mm, 3 μm) column (Santa Clara, CA, USA) held at room temperature. Solvent A was 0.1% formic acid in water; Solvent B was 0.1% formic acid in acetonitrile. Analytes were detected using an AB Sciex 6500 triple quad mass spectrometer (Framingham, MA, USA) in negative Turbo Spray mode.
Key for Biomarker Names (names of traces/MRMs):
ΔUA-GalNAc4S = Internal Standard.
(HNAc-UA)2 (1S) = MPS-IIIB.
Internal disaccharide method of brain heparan sulfate level assessment Heparan sulfate was measured using the internal disaccharide method (32)
A volume of each sample/control homogenate equal to 20 μg protein was taken to assay. Each aliquot was added to a 96-well plate and volumes were adjusted to 10 μl with MilliQ purified water. A blank well was prepared using 10 μl of MilliQ water. To each well containing a sample/control aliquot was added 90 μl of 50 mm Tris buffer with 11 mm calcium chloride (pH 7.0), 3 μl of 100 mm dithiothreitol, 10 μl of 0.1 mm chondrosine internal standard and 20 μl of enzyme solution. Enzyme solution was composed of 50 mIU/μl each of chondroitinase ABC and the heparanases I, II and III in 1% bovine serum albumin (BSA). The plate was covered with a sealing film and was incubated, shaking at 250 rpm at 37°C for 16 h. Note: IU = international unit. 1 IU is equal to amount of enzyme needed to convert 1 μmole substrate to product per minute (equivalent 600 units (UN) as defined by Sigma Aldrich). After incubation, 400 μl cold methanol was added to each sample/control and was mixed with a multichannel pipetman by pipetting up and down 20x to precipitate proteins. The plate was spun at 3000rcf for 15 min to pellet precipitates, and the supernatant was completely transferred to a new 96-well plate compatible with LC–MS/MS injection. Samples/controls were brought to dryness with a nitrogen stream and were reconstituted in 100 μl of MilliQ water. The mass spectrometer standard (MS-STD, see the following text for explanation) was prepared by diluting 10 μl of the MS-STD master stock to 100 μl with MilliQ water.
Analytes were separated by a Waters Acquity UPLC equipped with flow-through needle and a Hypercarb porous graphitic column (2.1 x 50 mm, 5 μm) held at 60°C. Solvent A was 148 mm ammonia in water; Solvent B was 100% acetonitrile. Analytes were detected using a Waters Xevo TQ-S triple quad mass spectrometer (Milford, MA, USA) in negative electrospray ionization mode.
Key for biomarker names:
Chondrosine = Internal Standard.
D0A0 = ΔUA-GlcNAc (Heparan Sulfate Biomarker).
D0S0 = ΔUA-GlcNS (Heparan Sulfate Biomarker).
A mass spectrometer standard was prepared (MS-STD) to correct for run-to-run variations in response factor between the IS and each biomarker. The concentrations in MS-STD master stock are as follows: 0.1 mm Chondrosine, 0.02 mm D0A0, 0.02 mm D0S0. Final results are corrected using the marker’s molar ratio compared with IS and response ratios from the MS-STD QC injections.
Brain mass spectrometry-based method NAGLU activity assay
Aliquots of 10 μl were taken in duplicate for each 10x diluted homogenate to assay. NAGLU activity was measured using the previously published method (33). Briefly, to each aliquot in a 96-well plate was added 30 μl of duplex assay cocktail (50 mm sodium citrate buffer, pH 5.0, containing 1.6 mm acetyl-CoA, 0.5 mm NAGLU substrate, 10 μm NAGLU internal standard, 0.5 μm HGSNAT substrate, 2 μm HGSNAT internal standard and 0.1 mm NAG-hiazoline). The plate was sealed with a silicone matt and shaken for 16 h at 37°C in an orbital shaker (250 rpm) incubator.
After incubation, 400 μl ethyl acetate was added and the well contents were mixed by pipetting up and down 10x. The plate was centrifuged at 3000rcf for 5 min, and then 150 μl of the upper layer was transferred to a shallow-well 96-well plate suitable for LC–MS/MS analysis. Solvent was removed from each well of the shallow-well plate with a jet of nitrogen, and the residue was reconstituted in 150 μl of water:acetonitrile (7:3) containing 0.1% formic acid. The plate was covered in aluminum foil in preparation for LC–MS/MS analysis.
Analytes were injected by a Waters Acquity binary solvent UPLC system equipped with fixed loop needle (Milford, MA, USA). Analytes were separated with a CSH C18 (2.1 x 50 mm, 1.7 μm) column and a CSH C18 VanGuard precolumn held at 22°C (Waters). Solvent A was 0.1% formic acid in water; Solvent B was 0.1% formic acid in acetonitrile. Analytes were detected using a Waters Xevo TQ triple quad mass spectrometer in positive electrospray ionization mode.
Organ biochemistry assays
Enzymatic activity assays were conducted to measure NAGLU (34) and GUSB (35). As has been observed previously in the MPS IIIB murine model low or absent NAGLU results in elevation of GUSB, creating an inverse correlation (16,17). NAGLU assays were conducted with supernatant samples mixed with 0.2 mm 4-MU-N-acetylalpha-D-glucosaminide (Sigma Aldrich) in a 0.1 m Na acetate, 0.5 mg/ml BSA, (pH 4.3) solution, and allowed to incubate at 37°C for 4 h. A 0.2 m Na2CO3/0.32 M Glycine solution was used to stop all reactions. A 4-methylumbelliferone solution was used to make a standard curve. Secondary lysosomal enzymatic activity (GUSB) was determined using a previously described protocol using the 4-methylumbelliferone enzymatic assay method. GUSB samples were mixed with a 10 mm 4-methylumbelliferyl−/3-glucuronide substrate (Sigma), in a 0.1 M sodium acetate buffer (pH 4.8) and allowed to incubate for 4 h at 37°C. The reactions were stopped using a 0.2 M Na2CO3/0.32 M glycine solution (pH 10.5) and immediately read at an absorbance of 365 nm and 460 nm. A protein assay (BioRad) was used to normalize both NAGLU and GUSB data. Samples were measured against a BSA standard curve and after incubating at room temperature for 15 min were measured for absorbance at 750 nm. Protein samples were diluted at 1:10 with homogenization buffer; this was factored into the final calculations for protein concentration. A Clariostar Plus microplate reader (BMG Labtech, Cary, NC, USA) was used to measure all samples.
GAGs were measured using a DMMB (Sigma-Aldrich, catalog number: C9819) absorbance assay as per manufacturer’s instructions. Total GAG levels were used as a surrogate for heparan sulfate levels in our model since heparan sulfate is the predominate GAG stored in the organs. Briefly, 100 μL of DMMB solution was added to 100 μL of supernatant samples, and then immediately transferred to and measured in a Clariostar Plus microplate reader at 525 nm. A chondroitin-6-sulfate standard (Chondrex) was used to build a standard curve.
Circadian activity
Long-term running wheel recordings were taken from mice to determine circadian rhythm activity. Male mice (N = 8 per group) were individually housed in specialized running wheel cages with climate and light controlled chambers from ages 3 to 5 months. Mice were randomly enrolled based on age (3–4 months). Males were selected based on previous data and testing, and we chose to limit the potential influence that estrus cycle could have on sleep variation.
Chambers were illuminated by timer activated fluorescent bulbs on a light–dark schedule (lights on at 7:00 a.m., lights off at 7:00 p.m.). Running wheel measurements were taken in 1 min intervals (Clocklab, Actimetrics, Evanston, IL, USA).
Phase angle of entrainment (relationship between offset of lights and onset of activity), total daily activity, daytime activity proportion and duration of daily activity were analyzed at 4 and 5 months of age. Statistical analysis was performed by repeated measures ANOVA.
Motor function—rotarod
Motor function was determined following a previously used protocol (36). All mice were trained on a rocking rotarod at a speed of 10 rpm, switching direction every full rotation for a 180 s maximum, three times per session. All mice were tested once every 30 days beginning at 6 months of age and continuing up through 548 days of age or endpoint of the mouse.
Auditory-evoked brainstem responses
ABR thresholds were measured with tone bursts at 8, 16 and 32, 48 kHz using an ABR recording system (Tucker-Davis Technologies) in normal NO TX (N = 3 male/6 female), MPS IIIB NO TX (N = 6 male/7 female), MPS IIIB IC6 (N = 4 male/4 female) and MPS IIIB ICM (N = 5 male/10 female) mice at 8–9 months of age (N = 8–15). Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) by intraperitoneal injection. Subdermal needle electrodes were placed at the vertex (active), ipsilateral ear (reference) and contralateral ear (ground). At each frequency, the sound level was reduced in 5–10 dB increments from 90 to 10 dB sound pressure level (SPL). A hearing threshold was defined as the lowest level that produced a noticeable ABR.
Statistics
GraphPad prism was used for statistical analysis. Repeated measures ANOVA was used for circadian rhythm assessment analysis. Two-way ANOVA with Bonferroni’s multiple comparisons tests was used to analyze ABR data, brain mass spectroscopy-based NAGLU, GUSB and heparan sulfate assays. One-way ANOVA was used for all other comparisons. Log or square root transformations of data were used for groups with unequal variance. Chauvenet’s criterion for exclusion of outliers was used for evaluation of and exclusion of a single outlier in the normal LAMP-1 staining % area group.
Lifespan
Log-rank test was performed to compare survival of the groups (n = 16 normal, 18 MPS IIIB NO TX, 16 MPS IIIB IC6, 15 MPS IIIB ICM). Kaplan–Meier curves were generated to demonstrate the effects of treatments on lifespan.
Acknowledgements
The Heldermon laboratory would like to acknowledge Sanfilippo Children’s Foundation, Sanfilippo Fundacja and Sanfilippo Initiative, Cure Sanfilippo and Lacerta Therapeutics. We would also like to acknowledge the Karyn Esser laboratory group for their assistance with circadian activity studies.
Conflict of Interest statement. H.K., Z.H., M.G. are cofounders or consult for GelbChemLLC. C.D.H. is the cofounder and stockholder for Lacerta Therapeutics Inc. C.J.R., E.B. and D.J.F. work for and have their own shares in Lacerta Therapeutics Inc.
Contributor Information
Courtney J Rouse, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA; Lacerta Therapeutics, Alachua, FL, USA.
Kimberley Hawkins, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Nadia Kabbej, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Justin Dalugdug, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Aishwarya Kunta, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Mi-Jung Kim, Department of Aging and Geriatric Research, University of Florida, Gainesville, FL, USA.
Shinichi Someya, Department of Aging and Geriatric Research, University of Florida, Gainesville, FL, USA.
Zachary Herbst, Department of Chemistry, University of Washington, Seattle, WA, USA.
Michael Gelb, Department of Chemistry, University of Washington, Seattle, WA, USA.
Isabella Dinelli, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Elizabeth Butterworth, Lacerta Therapeutics, Alachua, FL, USA.
Darin J Falk, Lacerta Therapeutics, Alachua, FL, USA.
Erinn Rosenkrantz, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Hamza Elmohd, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Hamid Khaledi, Department of Chemistry, University of Washington, Seattle, WA, USA.
Samar Mowafy, Department of Chemistry, University of Washington, Seattle, WA, USA; Pharmaceutical Chemistry Department, Misr International University, Cairo, Egypt.
Frederick Ashby, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Coy D Heldermon, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, USA.
Funding
Sanfilippo Children’s Foundation, Sanfilippo Fundacja and Sanfilippo Initiative, Cure Sanfilippo and Lacerta Therapeutics. National Institutes of Health/National Institute of Neurological Disorders & Stroke (R01NS102624 to C.D.H.); National Institute of Health; National Institute of Neurologic Disorders and Stroke; National Institute on Diabetes, Digestive and Kidney Disease; National Institute on Deafness and Communication Disorders (R01 DK067859 to M.G. and R03 DC011840, R01 DC012552 and R01 DC014437 to S.S.).
References
- 1. Parini, R. and Deodato, F. (2020) Intravenous enzyme replacement therapy in mucopolysaccharidoses: clinical effectiveness and limitations. Int. J. Mol. Sci., 21, 2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Delaney, K.A., Rudser, K.R., Yund, B.D., Whitley, C.B., Haslett, P.A. and Shapiro, E.G. (2013) Methods of neurodevelopmental assessment in children with neurodegenerative disease: Sanfilippo syndrome. JIMD Rep., 13, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gilkes, J.A. and Heldermon, C.D. (2014) Mucopolysaccharidosis III (Sanfilippo syndrome)- disease presentation and experimental therapies. Pediatr. Endocrinol. Rev., 12(Suppl 1), 133–140. [PubMed] [Google Scholar]
- 4. Lavery, C., Hendriksz, C.J. and Jones, S.A. (2017) Mortality in patients with Sanfilippo syndrome. Orphanet J. Rare Dis., 12, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilkes, J.A., Bloom, M.D. and Heldermon, C.D. (2016) Preferred transduction with AAV8 and AAV9 via thalamic administration in the MPS IIIB model: a comparison of four rAAV serotypes. Mol. Genet. Metab. Rep., 6, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tardieu, M., Zerah, M., Gougeon, M.L., Ausseil, J., de Bournonville, S., Husson, B., Zafeiriou, D., Parenti, G., Bourget, P., Poirier, B. et al. (2017) Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol., 16, 712–720. [DOI] [PubMed] [Google Scholar]
- 7. Holley, R.J., Ellison, S.M., Fil, D., O'Leary, C., McDermott, J., Senthivel, N., Langford-Smith, A.W.W., Wilkinson, F.L., D'Souza, Z., Parker, H. et al. (2018) Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain, 141, 99–116. [DOI] [PubMed] [Google Scholar]
- 8. Whitley, C.B., Vijay, S., Yao, B., Pineda, M., Parker, G.J.M., Rojas-Caro, S., Zhang, X., Dai, Y., Cinar, A., Bubb, G. et al. (2019) Final results of the phase 1/2, open-label clinical study of intravenous recombinant human N-acetyl-alpha-d-glucosaminidase (SBC-103) in children with mucopolysaccharidosis IIIB. Mol. Genet. Metab., 126, 131–138. [DOI] [PubMed] [Google Scholar]
- 9. Fu, H., Meadows, A.S., Ware, T., Mohney, R.P. and McCarty, D.M. (2017) Near-complete correction of profound Metabolomic impairments corresponding to functional benefit in MPS IIIB mice after IV rAAV9-hNAGLU gene delivery. Mol. Ther., 25, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murrey, D.A., Naughton, B.J., Duncan, F.J., Meadows, A.S., Ware, T.A., Campbell, K.J., Bremer, W.G., Walker, C.M., Goodchild, L., Bolon, B. et al. (2014) Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: toxicology, biodistribution, and immunological assessments in primates. Hum. Gene Ther. Clin. Dev., 25, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Naso, M.F., Tomkowicz, B., Perry, W.L., 3rd and Strohl, W.R. (2017) Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs, 31, 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gilkes, J.A., Bloom, M.D. and Heldermon, C.D. (2015) Mucopolysaccharidosis IIIB confers enhanced neonatal intracranial transduction by AAV8 but not by 5, 9 or rh10. Gene Ther., 23(3), 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gilkes, J.A., Bloom, M.D. and Heldermon, C.D. (2014) AAV8 is preferential candidate for neonatal gene transfer in murine MPS IIIB model. Mol. Ther., 22, S227–S227. [Google Scholar]
- 14. Gilkes, J.A., Bloom, M.D., Kolarich, A.R. and Heldermon, C.D. (2013) Differences in AAV serotype dependent distribution and tropism exist within the CNS of wild type and mutant mice in the MPSIIIB mouse model. Mol. Ther., 21, S128–S128. [Google Scholar]
- 15. Gilkes, J.A., Judkins, B.L., Herrera, B.N., Mandel, R.J., Boye, S.L., Boye, S.E., Srivastava, A. and Heldermon, C.D. (2020) Site-specific modifications to AAV8 capsid yields enhanced brain transduction in the neonatal MPS IIIB mouse. Gene Ther., 28(7–8):447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heldermon, C.D., Qin, E.Y., Ohlemiller, K.K., Herzog, E.D., Brown, J.R., Vogler, C., Hou, W., Orrock, J.L., Crawford, B.E. and Sands, M.S. (2013) Disease correction by combined neonatal intracranial AAV and systemic lentiviral gene therapy in Sanfilippo syndrome type B mice. Gene Ther., 20, 913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heldermon, C.D., Ohlemiller, K.K., Herzog, E.D., Vogler, C., Qin, E., Wozniak, D.F., Tan, Y., Orrock, J.L. and Sands, M.S. (2010) Therapeutic efficacy of bone marrow transplant, intracranial AAV-mediated gene therapy, or both in the mouse model of MPS IIIB. Mol Ther, 18, 873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elliott, S., Buroker, N., Cournoyer, J.J., Potier, A.M., Trometer, J.D., Elbin, C., Schermer, M.J., Kantola, J., Boyce, A., Turecek, F. et al. (2016) Dataset and standard operating procedure for newborn screening of six lysosomal storage diseases: by tandem mass spectrometry. Data Brief, 8, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elliott, S., Buroker, N., Cournoyer, J.J., Potier, A.M., Trometer, J.D., Elbin, C., Schermer, M.J., Kantola, J., Boyce, A., Turecek, F. et al. (2016) Pilot study of newborn screening for six lysosomal storage diseases using tandem mass spectrometry. Mol. Genet. Metab., 118, 304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Lee, J.H., Morton, J., Adams, H.R., Clarke, L., Eisengart, J.B., Escolar, M.L., Giugliani, R., Harmatz, P., Hogan, M., Kearney, S. et al. (2020) Therapy development for the mucopolysaccharidoses: updated consensus recommendations for neuropsychological endpoints. Mol. Genet. Metab., 131, 181–196. [DOI] [PubMed] [Google Scholar]
- 21. Derrick-Roberts, A., Kaidonis, X., Jackson, M.R., Liaw, W.C., Ding, X., Ong, C., Ranieri, E., Sharp, P., Fletcher, J. and Byers, S. (2020) Comparative analysis of brain pathology in heparan sulphate storing mucopolysaccharidoses. Mol. Genet. Metab., 131, 197–205. [DOI] [PubMed] [Google Scholar]
- 22. De Risi, M., Tufano, M., Alvino, F.G., Ferraro, M.G., Torromino, G., Gigante, Y., Monfregola, J., Marrocco, E., Pulcrano, S., Tunisi, L. et al. (2021) Altered heparan sulfate metabolism during development triggers dopamine-dependent autistic-behaviours in models of lysosomal storage disorders. Nat. Commun., 12, 3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li, H.H., Yu, W.H., Rozengurt, N., Zhao, H.Z., Lyons, K.M., Anagnostaras, S., Fanselow, M.S., Suzuki, K., Vanier, M.T. and Neufeld, E.F. (1999) Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. U. S. A., 96, 14505–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wolfe, J.H., Sands, M.S., Barker, J.E., Gwynn, B., Rowe, L.B., Vogler, C.A. and Birkenmeier, E.H. (1992) Reversal of pathology in murine mucopolysaccharidosis type VII by somatic cell gene transfer. Nature, 360, 749–753. [DOI] [PubMed] [Google Scholar]
- 25. Fu, H., Dirosario, J., Killedar, S., Zaraspe, K. and McCarty, D.M. (2011) Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol Ther, 19, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kan, S.H., Troitskaya, L.A., Sinow, C.S., Haitz, K., Todd, A.K., Di Stefano, A., Le, S.Q., Dickson, P.I. and Tippin, B.L. (2014) Insulin-like growth factor II peptide fusion enables uptake and lysosomal delivery of alpha-N-acetylglucosaminidase to mucopolysaccharidosis type IIIB fibroblasts. Biochem J, 458, 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kan, S.H., Aoyagi-Scharber, M., Le, S.Q., Vincelette, J., Ohmi, K., Bullens, S., Wendt, D.J., Christianson, T.M., Tiger, P.M., Brown, J.R. et al. (2014) Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. U. S. A., 111, 14870–14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kolarich, A., Heldermon, C. and Gilkes, J. (2013) Comparing IC6 and thalamic AAV5 injections on MPS IIIB model. Mol. Genet. Metab., 108, S55–S55. [Google Scholar]
- 29. Li, H.H., Yu, W.-H., Rozengurt, N., Zhao, H.-Z., Lyons, K.M., Anagnostaras, S., Fanselow, M.S., Suzuki, K., Vanier, M.T. and Neufeld, E.F. (1999) Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding N−acetylglucosaminidase. Proc. Natl. Acad. Sci. USA, 96, 14505–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gilkes, J.A., Bloom, M.D. and Heldermon, C.D. (2016) Mucopolysaccharidosis IIIB confers enhanced neonatal intracranial transduction by AAV8 but not by 5, 9 or rh10. Gene Ther., 23, 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chakrabarty, P., Rosario, A., Cruz, P., Siemienski, Z., Ceballos-Diaz, C., Crosby, K., Jansen, K., Borchelt, D.R., Kim, J.Y., Jankowsky, J.L. et al. (2013) Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS One, 8, e67680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herbst, Z.M., Urdaneta, L., Klein, T., Fuller, M. and Gelb, M.H. (2020) Evaluation of multiple methods for quantification of glycosaminoglycan biomarkers in newborn dried blood spots from patients with severe and attenuated mucopolysaccharidosis-I. Int. J. Neonatal. Screen., 6, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khaledi, H. and Gelb, M.H. (2020) Tandem mass spectrometry enzyme assays for multiplex detection of 10-mucopolysaccharidoses in dried blood spots and fibroblasts. Anal. Chem., 92, 11721–11727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marsh, J. and Fensom, A.H. (1985) 4-Methylumbelliferyl alpha-N-acetylglucosaminidase activity for diagnosis of Sanfilippo B disease. Clin. Genet., 27, 258–262. [DOI] [PubMed] [Google Scholar]
- 35. Sands, M.S., Barker, J.E., Vogler, C., Levy, B., Gwynn, B., Galvin, N., Sly, W.S. and Birkenmeier, E. (1993) Treatment of murine mucopolysaccharidosis type VII by syngeneic bone marrow transplantation in neonates. Lab Invest., 68, 676–686. [PubMed] [Google Scholar]
- 36. Heldermon, C.D., Hennig, A.K., Ohlemiller, K.K., Ogilvie, J.M., Herzog, E.D., Breidenbach, A., Vogler, C., Wozniak, D.F. and Sands, M.S. (2007) Development of sensory, motor and behavioral deficits in the murine model of Sanfilippo syndrome type B. PLoS One, 2, e772. [DOI] [PMC free article] [PubMed] [Google Scholar]