

Abstract
Usher syndrome (USH) is the most common form of hereditary deaf-blindness in humans. USH is a complex genetic disorder, assigned to three clinical subtypes differing in onset, course and severity, with USH1 being the most severe. Rodent USH1 models do not reflect the ocular phenotype observed in human patients to date; hence, little is known about the pathophysiology of USH1 in the human eye. One of the USH1 genes, USH1C, exhibits extensive alternative splicing and encodes numerous harmonin protein isoforms that function as scaffolds for organizing the USH interactome. RNA-seq analysis of human retinae uncovered harmonin_a1 as the most abundant transcript of USH1C. Bulk RNA-seq analysis and immunoblotting showed abundant expression of harmonin in Müller glia cells (MGCs) and retinal neurons. Furthermore, harmonin was localized in the terminal endfeet and apical microvilli of MGCs, presynaptic region (pedicle) of cones and outer segments (OS) of rods as well as at adhesive junctions between MGCs and photoreceptor cells (PRCs) in the outer limiting membrane (OLM). Our data provide evidence for the interaction of harmonin with OLM molecules in PRCs and MGCs and rhodopsin in PRCs. Subcellular expression and colocalization of harmonin correlate with the clinical phenotype observed in USH1C patients. We also demonstrate that primary cilia defects in USH1C patient-derived fibroblasts could be reverted by the delivery of harmonin_a1 transcript isoform. Our studies thus provide novel insights into PRC cell biology, USH1C pathophysiology and development of gene therapy treatment(s).
Introduction
Usher syndrome (USH) is a clinically and genetically heterogeneous disease that is the most frequent cause of hereditary deaf-blindness in humans affecting one in 6000–10 000 individuals, because of regional differences (1,2). USH is considered a retinal ciliopathy because of defects in photoreceptor cilia as well as other primary cilia (3–5). Among the three clinical subtypes of USH (USH1, USH2 and USH3), USH1 is the most severe form, characterized by profound congenital hearing impairment or deafness, vestibular dysfunction and prepuberal onset of progressive retinal degeneration in form of autosomal-recessive retinitis pigmentosa (6).
As of now, six genes have been identified for USH1 namely MYO7A (USH1B), USH1C, CDH23 (USH1D), PCDH15 (USH1F), USH1G and CIB2 (USH1J) (6). These USH1 genes encode proteins of divergent protein families that collectively function in dynamic networks at distinct subcellular locations in the eye and the inner ear (7). The human USH1C gene (ENSG00000006611; OMIM 276904) is located on chromosome 11 (chr 11:17493895-17 544 416 on GRCh38.p10) and encodes the harmonin protein. The gene consists of 28 exons spanning over 50 kb of the genomic region (Fig. 1A), with 11 distinct transcripts (splice variants) annotated in humans, so far (Ensembl ENSG00000006611) (8).
Figure 1.
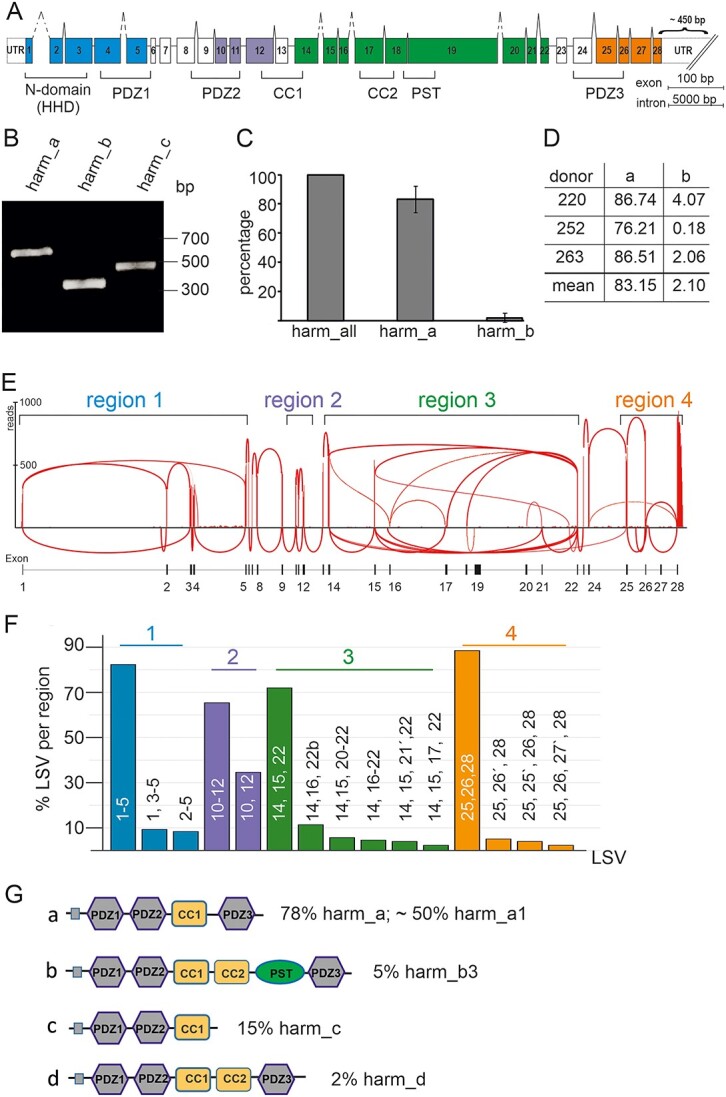
Expression of USH1C/harmonin a, b and c transcripts in the human retina. (A) Exon and intron structure of human USH1C. Exons are shown in boxes and denoted by the numbers 1–28. The different domains of the harmonin protein, namely N-terminal or HHD, PDZ (PSD-95, DLG, ZO-1) domains, PST (proline–serine–threonine rich) domain, CC (coiled-coil) domains, are marked by brackets below the gene structure. Colors indicate regions of LSV analyses. (B–D) RT-PCR analyses of USH1C/harmonin transcripts using isoform-specific primers in adult human retina. (B) RT-PCR analysis of USH1C/harmonin transcripts using isoform-specific primers in human retina. Transcripts of USH1C/harmonin isoforms a, b and c were detected. Equal amounts of amplified cDNA were loaded. (C) Quantitative RT-PCR of USH1C/harmonin isoforms. Primers detect either all USH1C/harmonin transcripts (a, b and c) or are specific for USH1C/harmonin_a or USH1C/harmonin_b transcripts, respectively. USH1C/harmonin_a isoforms are most prominent, whereas USH1C/harmonin_b isoforms are rarely expressed. (D) Percentages of transcripts in three different donors are shown. USH1C/harmonin_a-transcripts were most abundant (83.15%), USH1C/harmonin_b-transcripts were barely detected (2.1%). (E) Representative Sashimi plot of RNA-seq analysis of human retina. Splicing events were grouped into four regions (region 1–4). (F) Quantification of LSVs. LSVs of the same region are depicted by the same color. (G) Predicted domain structures of the USH1C/harmonin transcripts. Percentage of transcripts is based on the copy number of LSVs in regions 1–4. USH1C/harmonin class a is the most abundant class, with USH1C/harmonin_a1 being the most expressed isoform.
Ush1c transcripts in murine retinae exhibit extensive alternative splicing (9), with at least nine different isoforms of three different classes (a, b and c) based on their domain contents in the expressed harmonin protein (Fig. 1) (8,10) (www.ensembl.org). Characteristic sequences in all harmonin isoforms include 90–100 amino acid long PDZ domains, named for the three scaffold proteins originally recognized to contain these sequences: PSD-95, DLG and ZO-1 (11). PDZs contain a hydrophobic pocket that mainly associates with PDZ-binding motifs (PBM), present mostly at the C-terminus of target proteins. A majority of the interacting partners of harmonin bind to as many as three PDZ domains, making it a potent scaffold protein with a central position in the USH interactome (8,12). In addition to myosin VIIa (USH1B), cadherin 23 (USH1D) and SANS (USH1G), the USH2 proteins USH2A and VLGR1 (USH2C) bind to the PDZ1 domain in harmonin, whereas cadherin 23 (USH1D) and protocadherin 15 (USH1F) bind to the PDZ2 (8). SANS additionally interacts with PDZ3. Notably, all harmonin isoforms contain a CC (coiled-coil) domain and a conserved globular N-terminal domain, also named as the harmonin homology domain (HHD) (13), which increases the affinity of proteins binding to PDZ1, such as SANS, VLGR1 or USH2A (14–16). Harmonin_b, the longest isoform, is characterized by a second CC domain that is followed by a PST (proline–serine–threonine rich) domain that facilitates actin filament bundling (17). In addition, harmonin can interact with non-USH proteins in specific cellular compartments; e.g. the subunits of voltage-gated Ca2+-channels at ribbon synapses (18,19), and the GTPase regulator DOCK4 in stereocilia (20).
Alternative splicing of USH1C transcripts further augments functional properties of the harmonin scaffold protein, providing functional diversity to harmonin’s interaction profile in relevant cells. USH1C/harmonin isoforms are expressed in a wide range of tissues, yet studies on expression and subcellular localization of harmonin in rodent retinae have not been definitive (10,15,21–23). Based on the subcellular localization of harmonin isoforms in murine photoreceptor cells (PRCs), we and others have suggested differential scaffold functions of harmonin especially in the OS and at the synapses (8,15,21,24,25). A putative synaptic function has also been indicated in the murine retina (22), confirmed by zebrafish morpholino knockdowns (26) and recently by the USH1C pig model (27).
In the human retina, harmonin has been also been detected, along with other USH proteins, in the calyceal processes of cone and rod PRCs (23). These extensions of the PRC inner segment (IS) are suggested to stabilize the light-sensitive OS of the cell against mechanical forces (4,28). Interestingly, the PRCs of most vertebrates, except for rodents, reportedly possess calyceal processes. The absence of calyceal processes in mice is thought to be the reason why USH1 mouse models do not have a retinal phenotype observed in human USH1 patients (23,28).
Previous studies on the retina have indicated that USH1C/harmonin is mainly expressed in PRCs and to a lesser degree in secondary retinal neurons (RN) (8,21–23). However, harmonin was also shown to be expressed in Müller glia cells (MGCs) (26). More recent single-cell RNA-seq studies of human retinal cells even indicated the expression of USH1C almost exclusively in MGCs (29,30). MGCs are the radial glial cells of the retina extending from the outer limiting membrane (OLM) through all retinal layers to the extracellular inner limiting membrane (ILM) that separates the neural retina from the vitreous body of the eye. Functionally, MGCs not only maintain the structural integrity of the retina but are also essential for retinal homeostasis and physiology (31,32). However, nothing is known currently about the subcellular localization and function of harmonin in the MGCs.
Development of ocular gene augmentation therapies requires a detailed understanding of the expression profile of USH1C/ harmonin to delineate which isoform and into what retinal cell types the treatment has to be introduced (33). In this study, we investigated the expression of USH1C/harmonin at both the transcript and the protein level in the human retina, by RNA-seq and western blotting as well as correlative and in situ imaging techniques. We describe extensive splicing of USH1C and identify harmonin_a1 as the most prominent isoform in the human retina. We demonstrate USH1C/harmonin expression and subcellular localization in MGCs as well as in the retinal rod and cone PRCs. In addition, we provide evidence for novel interacting proteins of harmonin in distinct subcellular compartments. Finally, we identify the altered cilia length in fibroblasts from USH1C patients and demonstrate the reversal of this pathogenic phenotype by addition of harmonin_a1 to patient-derived cells.
Results
USH1C is frequently spliced in the human retina
To elaborate the transcriptomic profile of USH1C in the human retina, we first validated the expression of all three reported classes of USH1C/harmonin isoforms by reverse transcription polymerase chain reaction (RT-PCR) in five healthy human donor retinae (Fig. 1B–D, Supplementary Material, Tables S1–S3). RT-PCR demonstrated amplicons specific for all three isoform classes: a, b and c (Fig. 1B). Subsequent quantitative RT-PCRs (qRT-PCRs) of human donor retinas revealed the proportional presence of ~83% USH1C/harmonin_a and ~ 2% USH1C/harmonin_b splice variants of total USH1C/harmonin expression in human retina (Fig. 1C and D). The remaining isoform classes (c and d) are likely to account for ~15% of USH1C transcripts.
Next, we analyzed the local splicing events of USH1C in a human retina using RNA-seq. We generated an RNA-seq library of the human retina with ~90 million reads and 151 bp paired-end read-length and analyzed aligned RNA-seq data using Sashimi plots (34) (Fig. 1E). We observed extensive alternative splicing of USH1C in the human retina. For the analysis of the splicing events that are occurring on USH1C, we grouped the splicing events into four regions, namely exons 1–5 (region 1), 10–12 (region 2), 14–22 (region 3) and 25–28 (region 4), which were separated by canonically spliced exon–exon junctions (Fig. 1E). For the quantification of the observed local splice variants (LSV), we utilized 50 additional published RNA-seq samples (35). In order to increase the coverage for the detection and quantitation of rarely occurring LSVs, we consolidated the 50 samples into 10 fused samples with homogeneous numbers of reads. We then quantified each observed LSV in the merged samples. Only LSVs with a minimum of 20 raw counted reads in at least half of the merged samples were used for any further subsequent downstream analysis.
The LSV ex (1–5) is the most frequent LSV in region 1 of USH1C with a ~82% read coverage within region 1 (Fig. 1F). A novel LSV ex (1, 3–5), in which exon 2 is skipped (~9% read coverage), leads to a frameshift and a premature stop codon in exon 3. LSV ex (2–5) is characterized by an alternative start and had an abundance of ~ 8% among all LSVs in region 1.
In region 2 (exons 10–12), we observed two LSV that differ in the presence or absence of exon 11. Exon 11 was present in about two thirds of the reads. LSV ex (10, 12) does not shift the open reading frame and can be translated into a USH1C/harmonin isoform with a slightly shortened PDZ2 domain. Both LSVs are known and annotated transcripts for USH1C/harmonin_a1 (ENST00000318024) and a4 (ENST00000527020), respectively.
The alternative splicing in region 3 (exons 14–22) was very complex. At the protein level, this region encodes for the CC2 and PST domains, only present in USH1C/harmonin class_b isoforms. The most frequent LSV in region 3 was LSV ex (14, 15, 22) with a ~72% read coverage (Fig. 1F). This LSV lacks the CC2 and PST domains and therefore encodes for USH1C/harmonin class_a or _c. LSV ex (14, 16–22) was only covered with a very low number of reads in the RNA-seq samples. However, this LSV encodes for the CC2 and PST domains indicating a low expression of USH1C/harmonin isoform_b in the human retina. Thus, these RNA-seq data confirmed the qRT-PCR results (Fig. 1B–D). We further found two potentially new exons (16′ and 21′) located between exons 16 and 17, and 21 and 22, respectively, and a putative alternative 5′ splice site of exon 22.
Detailed analysis in region 4 revealed three LSVs; of these, LSV ex (25, 26, 28) is the most frequent splicing event with a read coverage of ~ 90%. Two novel exons (exons 25′ and 26′) that have not been previously described for USH1C were also observed (Fig. 1F). Exon 25′ was part of LSV ex (25,25′,26,28) and exon 26′ was part of LSV ex (25,26′,28), both with a read coverage of ~5%.
To translate our computational analyses, we combined the most abundant LSVs of regions 1, 2 and 4 with each LSV found in region 3 and subsequently used these in the online tool SMART (http://SMART.embl-heidelberg.de) to predict the domain structure (Fig. 1G). Most LSVs were assigned to the USH1C/harmonin classes a, b and c (8,10). In addition, we found a putative novel class for harmonin, which we named USH1C/harmonin_d that is similar to harmonin_a, but additionally carries a CC2 domain (Fig. 1G). Approximately 78% of all LSVs in region 3 were predicted to result in USH1C/harmonin_a isoforms, of which approximately two-thirds are USH1C/harmonin_a1 and the remaining harmonin_a4, depending on the splicing of exon 11 (region 2). This quantification indicated that ~50% of all harmonin expressed in the human retina is USH1C/harmonin_a1.
USH1C is highly expressed in both MGCs and RNs
To assign USH1C transcripts to specific cell types of the human retina, we performed bulk RNA-seq from sorted MGCs and RNs from human donor retinae (Supplementary Material, Tables S1 and S2). In bulk RNA-seq analyses, cross-contamination of sorted cell populations may occur because of biochromic magnetic-activated cell sorting (MACS) fractionation of MGCs and RNs. In the protocol applied <10% of cross-contamination was observed by immunofluorescence labeling and transcript analyses via quantitative PCR for cell-specific markers (see Materials and Methods and Supplementary Material, Fig. S1). As described above, we grouped the splicing events into four regions and set a cut-off of 5% for each LSV per region (Fig. 2A). In all four regions, UHS1C/harmonin transcripts were identified in RNs and MGCs (Supplementary Material, Table S2). In region 1, LSV ex (1–5) was the most frequent LSV, for both RNs and MGCs. Interestingly, the expression of LSV ex (1–5) was increased in MGCs compared with RNs. In addition to LSV ex (1, 3–5) and LSV ex (2–5), which were both expressed in the two cell types, we identified LSV ex (1, 5) in RNs only. In region 2, we observed the two LSVs (ex (10–12; ex (10, 12) having no differences in the number of LSV between RNs and MGCs. In region 3, our bulk RNA-seq revealed three LSVs, namely the most frequent transcript LSV ex (14, 15, 22), being slightly higher expressed in MGCs compared with RNs. LSV ex (14, 15, 20–22) and LSV ex (14, 15, 21, 22), encoding for the C-terminal part of the PST domain, were also detected. However, in contrast to the RNA-seq of the total retina, no LSV ex (18, 19) encoding the entire PST domain was detected, probably because of lower sequencing depth. In region 4, LSV ex (25, 26, 28) was the most abundant LSV and was expressed in both RNs and MGCs.
Figure 2.
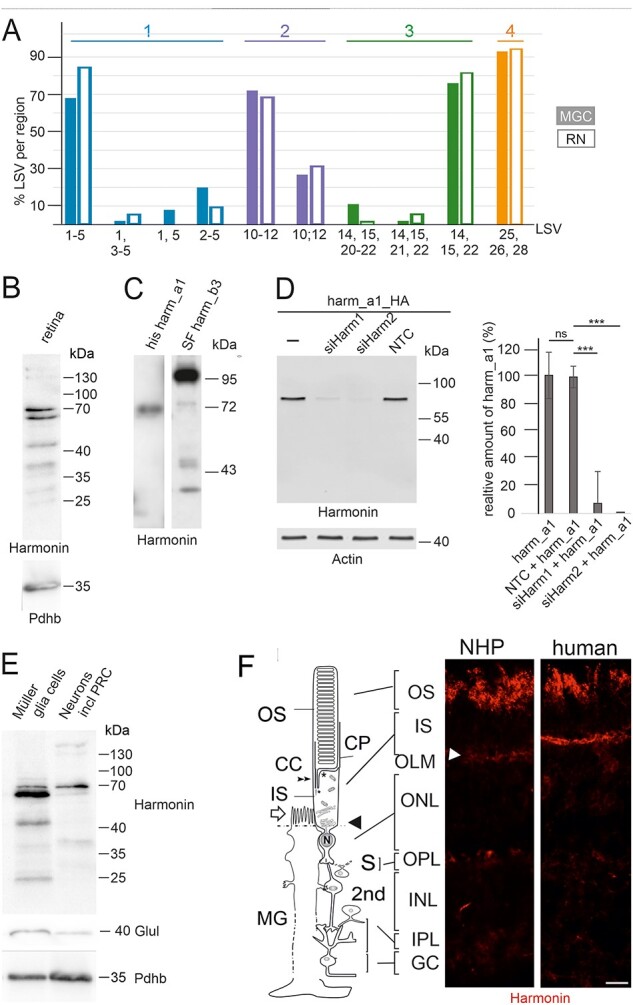
USH1C/harmonin expression in retinal cells. (A) Bulk RNA-seq of human RNs and MGCs. In RNs (filled bars) and MGCs (empty bars), USH1C/harmonin transcripts are detectable. (B) Western blot analysis of harmonin protein expression in human retina with affinity-purified polyclonal antibodies against harmonin (H3) showed two major bands at 64 and 70 kDa, which comigrate with harmonin_a1 in C. Lower bands may represent harmonin_c isoforms. (C) His-tagged harmonin a1 and SF-tagged harmonin b3 were transiently expressed in HEK293T cells. Pan antiharmonin H3 detected bands that co-migrate at the molecular weights of recombinant harmonin_a1 and harmonin_b3. Lower bands represent degraded products of harmonin_a and/or b. (D) Western blot analysis to validate the specificity of the harmonin antibody. A strong harmonin band is detected in HEK293T cells transfected with HA-tagged harmonin (Harm_a1_HA) or co-transfected with Harm_a1_HA and control siRNA (NTC). Harmonin is not detected in cells co-transfected with HA-tagged harmonin_a1 (Harm_a1_HA) and siHarm, indicating the specificity of the harmonin antibody. ***P-value < 0.001. (E) Antiharmonin (H3) western blot of MGCs and neurons including photoreceptors cells (RNs) isolated from human retina. Overall harmonin is higher expressed in MGCs, but a more prominent band is found at 70 kDa in RNs. Western blot verification with antiglutamate synthetase (Glul), a common marker for MGCs, indicated <10% contamination of the RN fraction with MGCs when quantified. Western blot for pyruvate dehydrogenase E1-β (Pdhb) served as the loading control, indicating that 1.5-fold more protein was loaded of the RN fraction when quantified. (F) Localization of harmonin in retina sections. Indirect immunofluorescence labeling of harmonin in a longitudinal section through the retina, the OS, the IS and the nuclei in the ONL and MGC of a NHP and human retina, respectively. In addition to the prominent labeling of the OLM (arrowhead), patchy harmonin staining was present in the layer of the photoreceptor OS. Faint staining was present in the IS, the OPL, the IPL and GCL. Scale bars: 10 μm.
In summary, our data demonstrate USH1C transcripts in both MGCs and RNs. The relative transcript levels of the different LSVs in RNs and MGCs appeared to be quite similar and comparable with the quantification of the whole retinal samples.
Harmonin protein expression and localization in human retinae
To assess harmonin protein expression in the human retina, we performed immunoblotting of protein lysates from human donor retinae using a harmonin antibody (H3), which was generated against a conserved N-terminal epitope (amino acids 1–89) of harmonin (21) to detect all five isoform groups of harmonin. Western blots demonstrated two major bands at a molecular mass close to 70 kDa, namely 70 and 64 kDa (Fig. 2B). The reference harmonin_a1 recombinantly expressed in HEK293T cells identified these bands as harmonin_a isoforms (Fig. 2C), consistent with our RNA-seq data (Fig. 1). These two bands can be either the two splice variants of USH1C/harmonin a1 and a4, which lacks exon 11 encoding for an calculated molecular weight of ~ 2.2 kDa or may represent posttranslational modifications of the same variant, such as phosphorylation, described in PhosphoSitePlus (https://www.phosphosite.org/proteinAction.action?id=5327601&showAllSites=true). Because (i) the intensities of both bands are almost equal, (ii) the RNAseq analysis revealed that USH1C/harmonin a1 transcripts are more abundant and (iii) the differences in molecular weight between the two bands are ~6 kDa, almost 3-fold compared with the calculated differences between the two splice variants USH1C/harmonin a1 and a4, we propose that both bands are posttranslational modifications of harmonin_a1. In addition, we observed much less prominent higher molecular mass isoforms (~130, ~150 kDa) and two prominent lower molecular mass proteins of ~ 45 and ~ 38 kDa. The higher bands may represent harmonin homodimers (8), whereas the lower bands either group c splice variants of harmonin (21,36) or protein degradation products, respectively. The absence of any harmonin band in the immunoblots of the harmonin-deficient tissue of a USH1C pig model (27) or siRNA-mediated knockdowns of USH1C/harmonin in HEK293T cells further validated the specificity of our H3 antibody (Fig. 2D).
Immunoblots of fractions enriched for different cell types revealed harmonin protein variants in fractions of both MGCs and RNs with an overall higher expression in MGCs (Fig. 2E). Nevertheless, differences in the band pattern in the western blots were striking: while the lower molecular weight band of harmonin_a ~64 kDa was much more prominent in the MGC, the higher molecular weight band at ~70 kDa was more intense in RNs. Accordingly, MGCs and RNs most probably express the same splice variant, but in RNs, it is likely to be additionally post translationally modified by phosphorylation. Additional low-molecular-weight bands specifically in MGCs probably represent harmonin_c and/or protein degradation products.
To localize harmonin protein in the human and non-human primate (NHP) retina, we performed immunostaining of harmonin in retinal cryosections (Fig. 2F). Fluorescence microscopy revealed intense harmonin immunofluorescence in the PRC OS layer and in the OLM of human and NHP retinae. In addition, we observed less intense immunostaining in the PRC IS, the outer plexiform layers (OPLs) and inner plexiform layers (IPLs) as well as the ganglion cell layer of human and NHP retinae.
Localization of harmonin at the OLM junction complexes
OLM is characterized by heterotypic cell–cell adhesions between photoreceptor and MGC membranes and consists of proteins from both adherens and tight junctions (37). Immunofluorescence double staining of harmonin with β-catenin, a component of adherens junctions (38), with the tight junction molecule JAM-B (39) and the actin-binding protein filamin A (40), respectively, showed partial co-localization with harmonin (Fig. 3A). GST-pull downs of the GST-tagged β-catenin, JAM-B and filamin A from HEK293T cell lysates demonstrated that the interaction of the C-terminal tails of all three OLM junction proteins with His-tagged harmonin_a1 (Fig. 3B–D). Reciprocal interaction pull-downs of GST-tagged PDZ domains revealed the binding of the C-terminal tail of β-catenin to the PDZ1 and PDZ3 domains of harmonin (Supplementary Material, Fig. S2A). Furthermore, yeast two-hybrid assays demonstrated the direct interactions of β-catenin, JAM-B and filamin A with harmonin (data not shown). β-catenin presumably bind to the PDZ1 and PDZ3 domains of harmonin via a C-terminal type-I PBM (DTDL) (41). In contrast, JAM-B contains a canonical type II PDZ targeting motif (FLV) at position 298–300 of its C-terminus, which is likely responsible for binding to harmonin (42).
Figure 3.
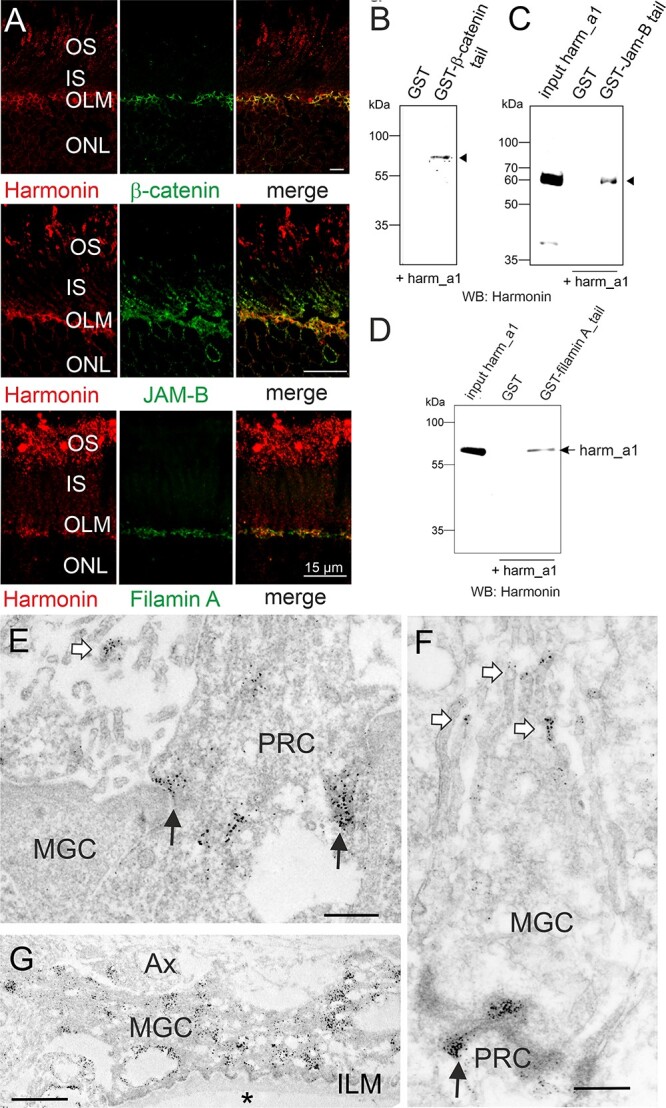
Subcellular harmonin localization at the outer limiting membrane of the human retina. (A) Indirect immunofluorescence double staining of harmonin and the adhesion junction molecule β-catenin, the tight junction molecule JAM-B, or the actin-binding protein filamin A. Merged images demonstrate an overlap of harmonin staining with β-catenin, JAM-B and filamin A staining at the outer limiting membrane (OLM). (B–D) GST-pulldown demonstrates the interaction of harmonin_a1 (harm_a1) with the C- terminal tails of β-catenin, JAM-B, and filamin A, respectively. (E–G) Immunoelectron microscopy analysis of harmonin labelling in a longitudinal section through the OLM of a human retina. Harmonin labelling concentrated in the electron dense adhesion junctions between Müller glia cells (MGCs) and photoreceptor cells (PRC) in the OLM (black arrows), microvilli of MGCs (white arrows) and MGC endfeet at the inner limiting membrane (ILM) which contacts the vitreous (asterisk). OS, outer segment; IS, inner segment; ONL, outer limiting membrane; AX, axon; Scale bars: Harmonin/β-catenin: 5 μm; Harmonin/JAM-B, Harmonin/Filamin_A: 15 μm; D, E: 0.5 μm; F: 1 μm.
Immunoelectron microscopy of ultrathin sections through the OLM of the human retina localized harmonin to both sides of the OLM junctions in the PRCs and MGCs (Fig. 3E and F). In addition, harmonin was labeled at the tips of microvilli (Fig. 3E and F) and abundantly in the endfeet of the MGCs at the ILM (Fig. 3G).
Differential expression of harmonin in rod and cone photoreceptor synapses
Next, we determined the localization of harmonin in photoreceptor synapses of the OPL. We extended our analyses to NHP retinas because synaptic regions turned out not to be well preserved in human donor retinas, possibly because of postmortem changes. Immunofluorescence double staining revealed a partial overlap of harmonin and RIBEYE, a marker of the synaptic ribbon (43) in photoreceptor synapses in both human and NHP sections (Fig. 4A). Immunoelectron microscopy of NHP retinae allowed us to confirm the localization of harmonin at the photoreceptor synapses and uncovered dense harmonin labeling in pedicles of cones, whereas the labeling in rod spherules was much weaker (Fig. 4B and C).
Figure 4.
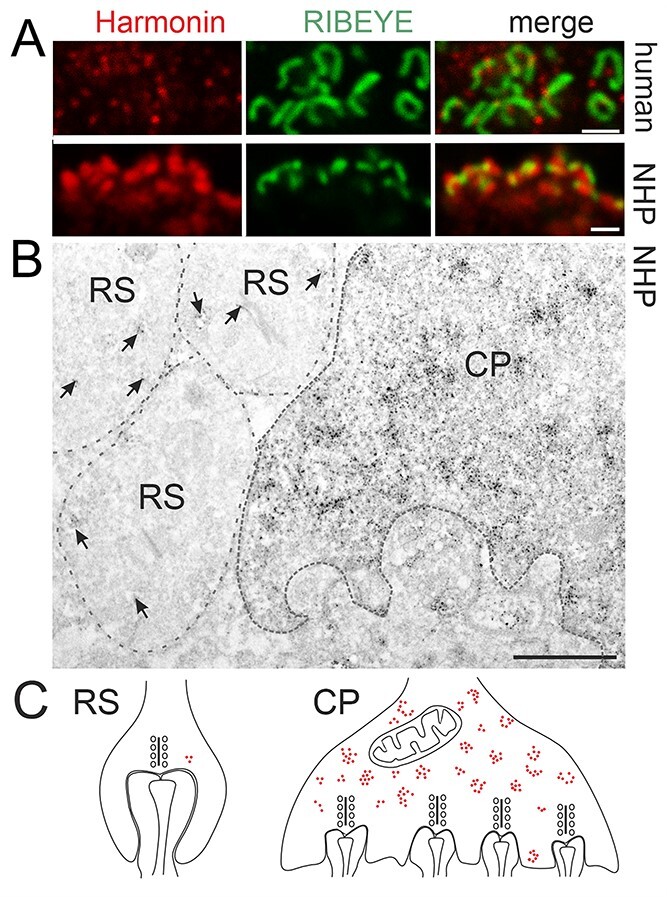
Harmonin localization at primate photoreceptor synapses. (A) Double immunofluorescence labeling of harmonin (red) and pre-synaptic protein RIBEYE (green) in human (upper panel) and NHP (lower panel) OPL synapses. Merged images revealed co-localization of harmonin and RIBEYE (yellow). (B) Immunoelectron microscopy analysis of harmonin in NHP photoreceptor synapses. Dense harmonin labeling was present in CP, but only weak harmonin labeling (arrows) was observed in rod spherule (RS). (C) Schematic representation of presynaptic harmonin labeling in RS and CP. Scale bars: A, B: 1 μm; E: 12 μm.
Differential expression of harmonin in the OS of the rod and cone PRCs
Immunofluorescence microscopy showed the presence of harmonin in the OS layer of the human retina (Figs 2F and5A). To discriminate between the rod OS (ROS) and cone OS (COS), we stained longitudinal cryosections through the human retina for harmonin and FITC-conjugated peanut agglutinin (PNA), a common marker for cones (44). Confocal analyses did not demonstrate a co-localization of harmonin and PNA (Fig. 5B). This was confirmed by maximum projections of confocal planes of fluorescence images and z-sections of maximum projections. Furthermore, the overlay of fluorescent intensity plots of harmonin and FITC-PNA staining revealed apparent dissimilarities in their peaks (Fig. 5C; low Pearson correlation coefficient values: Rr:0.18; and low Mander’s overlap coefficient values: M1:0.18; M2:0.19). In contrast, fluorescent intensity plots of harmonin and rod-specific arrestin-1 immunofluorescence staining showed apparent signal convergences (Fig. 6C; high Pearson correlation coefficient values: Rr:0.59; and Mander’s overlap coefficient M1:0.57; M2:0.42), suggesting harmonin expression in rod, but not in COS.
Figure 5.
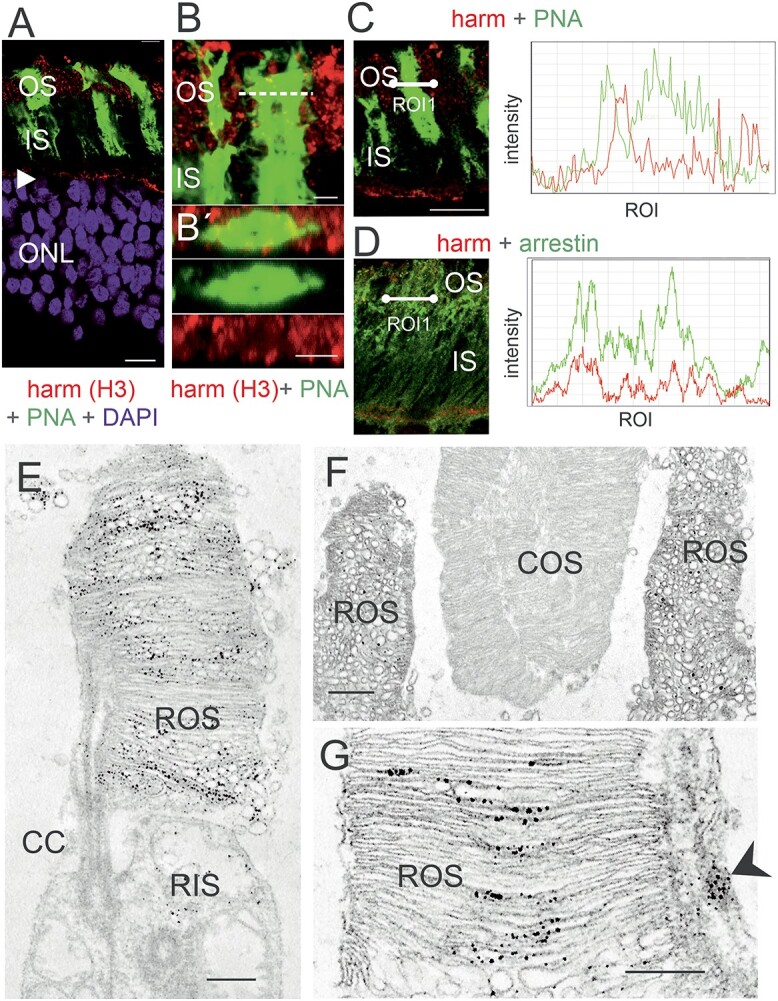
Harmonin in the OS of human PRCs. (A) Merged image of harmonin immunofluorescence (red) and fluorescent peanut agglutinin (PNA, green), a specific marker for the extracellular matrix sheath of cone photoreceptors in a longitudinal section through the photoreceptor layer, the OS, the IS and the nuclei in the ONL of a human retina. In addition to the prominent labeling of the OLM (arrowhead) patchy harmonin staining was present in the layer of the photoreceptor OS. (B, B´) Magnification of the OS region demonstrates no co-localization of harmonin and PNA. (B´) Confocal x,y scan image of harmonin and PNA labeling in the photoreceptor layer of a human retina. y, z scan at the dotted line in B at higher magnification. (C, D) Double labeling of harmonin (red) with PNA (green) (C) and rod-specific arrestin (green) (D) in the photoreceptor layer of human retina. Corresponding fluorescence intensity profiles of regions of interest (white lines) at the right panel demonstrated no co-localization of harmonin and PNA, but co-localization of harmonin and arrestin indicating the localization of harmonin in human rod OS but not cone OS. DAPI (blue): nuclear DNA. (E–G) Immunoelectron microscopy labeling of harmonin in a longitudinal section through human retinae. (E) In human rod PRCs, harmonin was labeled in ROS and was barely detected in rod inner segments. (F) Harmonin was labeled in ROS, but not in COS. (G) Harmonin is detected in ROS and calyceal processes (arrowhead). CC: connecting cilium. Scale bars: A: 10 μm, B: 2.5 μm, B’C, D: 5 μm, E, G: 0.5 μm, F: 1 μm
Figure 6.
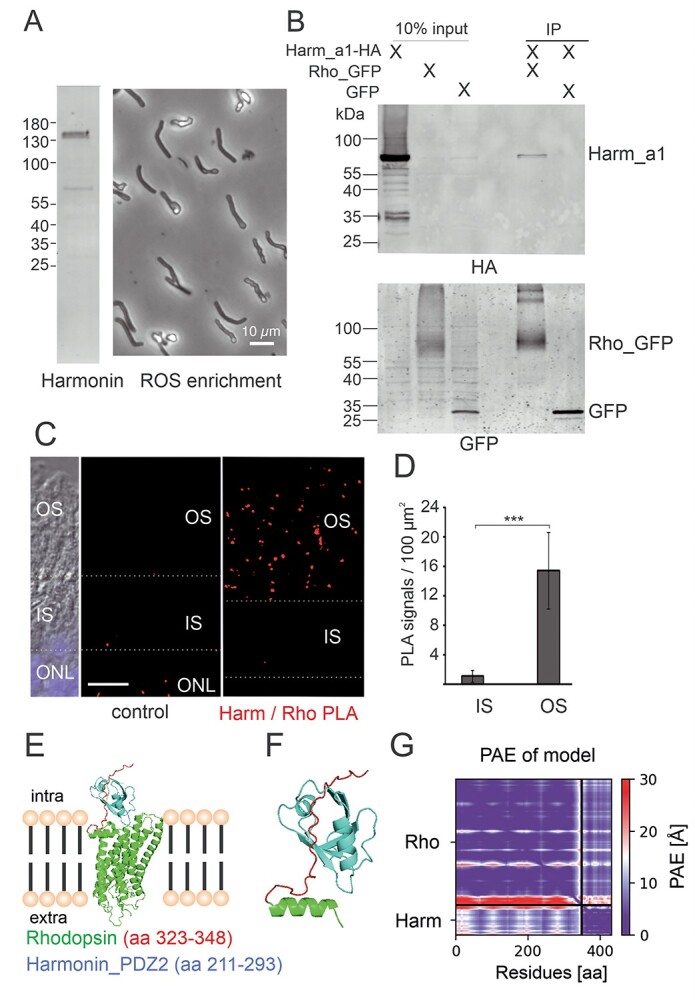
Harmonin is expressed in ROS and interacts with rhodopsin. (A) Harmonin is expressed in porcine photoreceptor OS. Left panel: western blot analysis of the isolated porcine ROS enriched fraction from density gradients revealed harmonin expression. Right panel: phase contrast picture of material used for the western blot. (B) GFP-Trap® demonstrates interaction between harmonin a1 (Harm_a1) and opsin-GFP (Rho_GFP) in transfected HEK293T cells. Harmonin_a1 was precipitated by immobilized opsin-GFP and not by GFP alone (replicates, n = 3). (C) PLA of harmonin (Harm) and rhodopsin in longitudinal cryosections through a human retina. PLA signals (red dots) indicate the interaction of harmonin (Harm) and rhodopsin in OS of human PRCs in situ. For a quantitative analysis, the IS/OS borders were defined based on differential interference contrast (DIC) microscopy image. Nuclear DAPI staining (blue) was used to define the ONL. ImageJ was adopted to define the different retina layers: OS and IS of PRCs, and ONL (white dashed lines). As the control, PLA was performed antiopsin antibodies and oligonucleotide-labeled antibodies, where almost no PLA signals were found. (D) Quantification of PLA signals in the OS and IS of PRCs. PLA signals were counted automatically in the different compartments and signals in controls were subtracted in three different samples. The number of PLA signals in OS were also significantly higher when compared with signals in IS. Scale bar: 10 μm. (E–G) Structure of the human rhodopsin-harmonin_a1_PDZ2 protein complex predicted by AlphaFold2. (E) Structure of full-length rhodopsin (Rho) (green) and harmonin (Harm)_PDZ2 (aa 211–293, blue). AlphaFold2 predicts binding of Harm_PDZ2 to intracellular C-terminus of Rho (aa323-348), enlarged in (F). (G) PAE for the modeled structure is very low for the predicted complex Harm_PDZ2-Rho (lower left and upper right rectangle), indicating high confidence of the protein complex. The heat map illustrates amino acid distances in Å (further explanation, see Supplementary Material, Figure S3).
Immunoelectron microscopy of human PRCs confirmed our confocal data: harmonin labeling was detected in the ROS but not of COS (Fig. 5E–G). In the ROS, harmonin immunostaining was observed at the base where OS discs are formed de novo and throughout the OS along the cytoplasmic side of the disc membranes (Fig. 5G). In addition, harmonin labeling was evident in the calyceal processes of PRCs, as previously described (23) (Fig. 5G, asterisk). Western blots of purified OS fractions of porcine retinae confirmed the expression of harmonin in PRC OS (Fig. 6A). Thus, our results show that harmonin is associated specifically with the disc membranes of ROS.
Harmonin interacts with rhodopsin
In the microvilli of the rhabdomes of Drosophila photoreceptors, functionally homologous to the OS discs of vertebrate PRCs, rhodopsin binds to the PDZ domain-containing scaffold protein INAD (45). To test whether the PDZ-protein harmonin also interacts with human rhodopsin, we performed in vitro co-immunoprecipitations assays with protein lysates from HEK293T cells expressing GFP-rhodopsin and HA-tagged harmonin or RFP-harmonin, respectively. Western blots of the recovered proteins revealed that harmonin_a1 co-immunoprecipitated with GFP-rhodopsin, but not in controls with GFP alone (Fig. 6B; Supplementary Material, Fig. S2B).
To examine a putative interaction of harmonin and rhodopsin in the human retina, we performed in situ proximity ligation assays (PLAs) applying anti-harmonin (H3) and monoclonal antibodies to rhodopsin in retinal cryosections (Fig. 6C). We observed positive PLA signals in the photoreceptor layer, predominately in the OS layer and a significant smaller numbers of signals in the IS (Fig. 6C and D). In contrast, almost no positive PLA signals were found in the negative controls (Fig. 6C).
In silico modeling of molecular interaction of harmonin and rhodopsin applying AlphaFold2/Multimer (46,47) predicted a complex of harmonin_PDZ2 and cytoplasmic C-terminus of rhodopsin with high confidence of the predicted alignment error (PAE) (Fig. 6E). In contrast, other domains of harmonin were not predicted to bind to rhodopsin (Supplementary Material, Fig. S3B–G). Moreover, modeling of the harmonin_PDZ2 of with the adrenergic receptor α-A1 did also not predict confident complex formation (Supplementary Material, Fig. S3H), further supporting the specificity of the harmonin_PDZ2-rhodopsin binding.
In summary, harmonin localization in ROS shown by complementary methods of light and electron microscopy, paired with the interaction of harmonin and rhodopsin in vitro, the close proximity of both proteins in the OS layer demonstrated by in situ PLAs as well as AlphaFold2 in silico predictions provide the first evidence for the interaction of rhodopsin and harmonin in ROS.
Retinal phenotype of USH1C patients
Two male siblings with confirmed mutations in USH1C (c.91C > T;p.(R31*), c.238dupC;p.(Arg80Profs*69)) were clinically examined at the age of 35 and 47 years, respectively (Fig. 7A–E; Supplementary Material, Fig. S4). Optical coherence tomography (OCT) analysis of the retina of a 35-year-old male patient revealed atrophy and thinning of the photoreceptor OS/IS and of the outer nuclear layer (ONL) up to the fovea. Outer retinal atrophy progressed to constriction of the visual field at around 5 degrees and a hyperfluorescent perifoveal ring on the fundus autofluorescence imaging (Fig. 7B). Additionally, epiretinal gliosis observed in OCT is a common complication of PRC degeneration, which also was a hallmark in the USH1C pig model (27). The posterior eye pole showed typical bone spicules in the mid periphery and periphery (Fig. 7A). Still, despite progressive peripheral retinal degeneration at 35 years, the central vision of the patient was relatively well preserved with visual acuity of 20/25 in both eyes and normal color perception (Fig. 7E). Electrophysiological tests showed much reduced amplitudes of the a and b waves in the full-field electro retinograms (ERGs) (recordings based on the international standards), or no measurable responses for both dark- and light-adapted protocols (data not shown).
Figure 7.
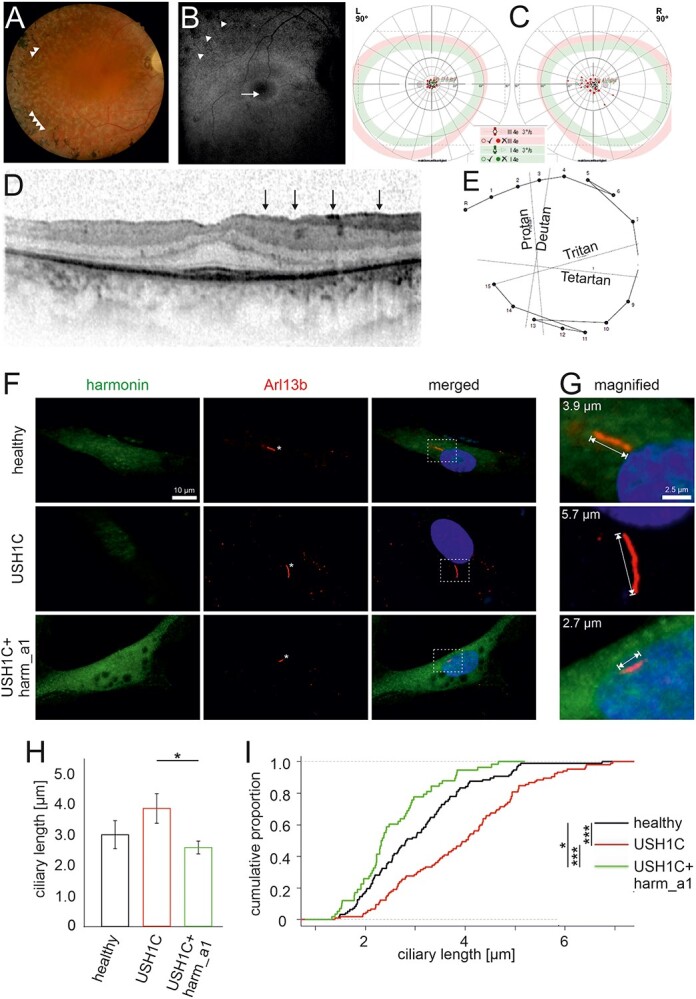
Retinal phenotype of an USH1C patient and ciliary phenotype of USH1C patient-derived cells. (A–E) Clinical findings of the retinal phenotype of a 35-year-old male with confirmed mutations in USH1C (c.91C > T;p.(R31*), c.238dupC;p.(Arg80Profs*69)). (A) Fundus photography showed bone spiculas in the mid-peripheral and peripheral retina (arrowheads), attenuated retinal vessels, waxy pallor optic disc and white spots of retinal pigment epithelium atrophy. (B) Fundus autofluorescence imaging displayed a hyperfluorescence ring around the fovea (arrow) and a disrupted hypofluorescence in the mid- and far periphery of the retina (arrowheads) corresponding to the outer retinal atrophy. (C) Kinetic visual field (90°): Concentric constriction for III4e and I4e markers with a central preserved area. (D) OCT showed epiretinal gliosis (marked as black vertical arrows), as well as gradual IS/OS loss up to the fovea. Only the foveal region displays a normal retinal structure with preserved photoreceptors and a central macula thickness of 269 μm. (E) Lanthony color test showed normal color perception. (F–I) Primary ciliary phenotype of USH1C patient-derived cells and rescue by harmonin_a1. (F, G) Immunofluorescence of harmonin (green) and ciliary marker Arl13b (red) in fibroblasts from a healthy donor (healthy), the clinically examined USH1CR80Pfs*69/R31*-patient (USH1C) and USH1C-fibroblasts transfected with harmonin_a1 (USH1C + harm_a1). (F) In healthy donor cells and harmonin_a1-transfected USH1CR80Pfs*69/R31* cells, harmonin is detectable. In untransfected USH1CR80Pfs*69/R31* cells, harmonin staining is barely visible. (G–I) Ciliary length measurements revealed longer cilia in USH1C patient-derived cells compared with control (healthy) and harmonin_a1-transfected USH1C cells. (H) Quantitative analysis of primary ciliary length reveals a significant decrease in the ciliary length in USH1C + harm_a1 fibroblasts towards the ciliary length of healthy donors. (I) Cumulative analysis of ciliary length and number of ciliated cells in fibroblasts of healthy donors (healthy), USH1CR80Pfs*69/R31* (USH1C) and USH1CR80Pfs*69/R31* fibroblasts transfected with harmonin a1 (USH1C + harm_a1) using R. Two-tailed Student’s t-test, *P ≤ 0.05, **P ≤ 0.01. Cells analyzed: healthy: n = 96; USH1C: n = 105; USH1C + harm_a1: 58, in three independent experiments. Scale bar: F: 10 μm, G: 2.5 μm.
Rescue of the ciliary phenotype in patient-derived fibroblasts
The photoreceptor OS resembles a highly modified primary sensory cilium that has been shown to be impaired in retinal ciliopathies such as USH (4,5,48). We analyzed primary cilia of dermal fibroblasts from one of the clinically characterized USH1CR80Pfs*69/R31* patients. After induction of ciliogenesis (3) primary cilia of USH1C patient fibroblasts were significantly longer compared with the primary cilia of healthy donor fibroblasts (Fig. 7F–I). Strikingly, this ciliary phenotype of USH1CR80Pfs*69/R31* patient fibroblasts was rescued by the re-expression of the harmonin_a1 (Fig. 7F and I). These data demonstrated that USH1C patient cells can serve potential cellular models for evaluating therapies for USH1C and that the harmonin_a1 isoform is a therapeutic active isoform, able to revert the USH1C phenotype to normal.
Discussion
Alternative splicing enhances the diversity of the transcriptome and proteome. Tissue- and cell-specific alternative splicing occurs frequently in mammals, and has previously been predicted for USH1C (36,49). In comparison with other tissues, the neural retina displays the highest level of alternative splicing events (50,51). Splicing programs are essential for retinal function and maintenance, and are regulated in an extremely complex manner by retina-enriched splicing factors (52–54). Here, we demonstrate extensive alternative splicing of USH1C in the human retina. We do not expect all variants observed to be translated into protein because of pre-translational processing of mRNAs (55). Nevertheless, we propose that several USH1C/harmonin isoforms are expressed, potentially with distinct patterns in the various human retinal cell types providing cell-specific functions ,as indicated in the cochlear and vestibular hair cells of the inner ear (49).
Harmonin is a scaffold protein that is modularly composed of well-defined domains and motifs, such as the N-terminal HHD, PDZs, CCs and a PBM (Fig. 1A,G) that all participate in protein–protein interactions (8,16). Identification of LSVs that include specific domains suggests that alternative splicing may affect binding of harmonin to target molecules in human retinal cells. For example, alternative splicing of exon 2 in region 1 is predicted to alter the globular N-domain, termed HHD, and influence the binding affinity of harmonin PDZ1 to target proteins, such as VLGR1/ADGRV1 (USH2C), USH2A and SANS (USH1G) (14,15). This hypothesis is supported by our recent results, indicating the reduced binding affinity of target proteins to harmonin lacking parts of the N-terminal HHD (Daniel Sturm and Kerstin Nagel-Wolfrum, unpublished). Furthermore, splicing out of exons 16–21 in region 3 would lead to the loss of CC2 and PST domains, which should impact the oligomerization of harmonin molecules and loss of actin filament bundling properties, respectively (17).
Differential binding of proteins to the numerous harmonin splice variants may imply differences in harmonin function in various retinal cell types. Accordingly, the regulation of alternative splicing of USH1C/harmonin in the retina is of importance. Previous studies have identified Musashi proteins (MSI1 and MSI2) as important regulators of alternative splicing in the retina (53,54,56). However, USH1C/harmonin has not been identified as a target for the splicing control machinery of Musashi proteins. Recently, we were able to show that the USHG1 protein SANS interacts with core components of the spliceosome and regulates splicing including constitutive and alternative splicing of USH1C/harmonin (57). In particular, the latter study shows that SANS causes the retention of exon 11, thereby leading to alternative expression of the human harmonin_a1 and _a4 transcripts. Further studies are needed to understand the interplay and interference of the two USH1 proteins, SANS and harmonin, in the retina at two different levels: in protein complexes (SANS-SAM/PBM binding to the N-terminal HHD/PDZ1 and PDZ3 of harmonin), possibly related to transport processes (14,16,58), and during splicing where SANS presumably modulate USH1C/ harmonin pre-mRNA (57).
The assignment of LSVs to USH1C/harmonin domains allowed us to quantify the expression of the harmonin classes in the human retina (Fig. 1G). This analysis demonstrates that harmonin_a is by far the most abundant class expressed in the human retina in concordance with our results by semi- and quantitative RT-PCR (Fig. 1B–C) and immunoblotting analyses (Fig. 2B–E) of donor retinae. Our data also indicate that the USH1C/harmonin_a1 transcript is more frequently expressed than USH1C/harmonin_a4 in the retina. Based on these findings, we concluded that harmonin_a1 is the most promising variant for gene replacement therapies. The rescue of the pathogenic phenotype in primary cilia of fibroblasts of USH1C patient after addition of harmonin_a1 further supports this conclusion (Fig. 7).
In contrast to the prevailing hypothesis of USH1C/harmonin expression in PRCs (21,23), our results demonstrate USH1C/harmonin expression in RNs (mainly PRCs) as well as MGCs of the human retina. Based on donor retina bulk RNA-seq, comparatively equal levels of USH1C RNA isoforms were evident in RNs and MGCs (Fig. 2A), which is also concordant with quantitative levels of transcripts. Notably, we did not observe substantial differences in LSV expression levels in RNs and MGCs. Our findings don’t support a recently published scRNA-seq study indicating USH1C expression almost exclusively restricted to MGCs in the human retina (29). The limited sequencing depth in scRNA-seq likely accounts for this discrepancy. In addition, scRNA-seq requires the dissociation of retinal cells prior to RNA-seq library preparation, which can have a huge impact on the gene expression profile (59). Although sorted MGCs and RNs contain to a certain degree (~10%) cross-contaminating cell types, our results from bulk RNA-seq profiling are validated by immunoblotting showing distinct band patterns of MGCs and RNs (with differently sized stronger bands in each fraction). Both findings are confirmed by our immunohistochemical studies using light and electron microscopy that demonstrate expression and subcellular localization of USH1C/harmonin in MGCs as well as PRCs.
Western blot analyses of the human retina and retinal cell fractions consistently demonstrate two major harmonin protein bands close to 70 kDa. As already outlined in the results, these are probably not different splice variants, but posttranslational modifications by phosphorylation of the same variant, namely USH1C/harmonin_a1. Which phosphorylation sites found in the harmonin molecule are phosphorylated in the human retina and which kinases are involved should be the subject of further investigation. Our data show that harmonin is overall higher expressed in MGCs than in RNs; however, the higher molecular weight band of ‘phosphorylated’ harmonin at 70 kDa is nevertheless much more conspicuous in the RNs fraction, which consists mainly of PRCs. It will be of great interest to find out in future studies what consequences the possible phosphorylation of harmonin has on its function in PRCs of the human retina.
The localization of harmonin in distinct subcellular compartments of human MGCs may provide insights into harmonin function. Immunoelectron microscopy revealed the localization of harmonin to the tips of the microvillar processes formed by their apical membrane of MGCs that project into the subretinal space surrounding the PRC IS (Fig. 3E and F), consistent with localization of harmonin in both the tip-link complex of stereocilia (which resemble modified microvilli) of auditory hair cells (60,61) and the very similar tip-link complex of the brush border microvilli of intestinal enterocytes (62). In contrast to the tip-link complex of stereocilia, in which harmonin interacts with other USH proteins, in the brush border microvilli tip-link, harmonin forms a stable ternary complex with ANKS4B and MYO7B for anchoring specific cadherins (62,63). A morphologic similarity between the microvilli of MGCs and the brush border microvilli is evident. Given that the absence of tip-link molecules in brush border microvilli leads to structural perturbations linked to ineffective epithelial transport (64), we suggest that harmonin may contribute to the structural arrangement of the microvilli populating the apical MGC membrane, being essential for their physiological function (31). Here, we provide first evidence for a tip-link complex in the microvilli of MGCs with a possible scaffolding function of harmonin. Additionally, we identified abundant harmonin expression in conical endfeet at the other pole of the MGCs adjacent to the ILM and vitreous humor (Fig. 3G). Harmonin is likely integrated into protein networks that mechanically stabilize the endfeet at the MGC base and can support anchoring of MGCs at the ILM.
Furthermore, we show a prominent submembranous localization of harmonin in MGCs and PRCs associated with the specialized heterotypic adhesion junctions of the OLM (Fig. 3). Partial co-localization of harmonin and β-catenin, a linker of cadherins to actin filaments, and the transmembrane protein JAM-B, together with the binding of the cytoplasmic tails of both proteins to harmonin strongly suggest that harmonin provides the molecular scaffold to anchor both proteins in the submembranous cytoplasm at the OLM junctions. Harmonin perhaps contributes to the linkage of the OLM junction complexes to the actin cytoskeleton. This is supported by the observed co-localization and binding of harmonin to the actin-binding protein filamin A, which has been described as an important component of the actin cytoskeleton in barrier-forming cell–cell adhesion complexes (40). Since harmonin is present in both adhesion and tight junction complexes, its deficiency may disrupt the structural integrity of the retina (adhesion component) and/or increase the permeability (tight junctional component) of the retinal barrier. In pathological conditions, the disruption of the retinal barrier function of the OLM would contribute to fluid accumulation in the macula, clinically manifesting as macular edema, which can be detected in many hereditary retinal degenerative diseases, including USH (65). In concordance, OCT of the two USH1C patients, reported here, not only show thinning of the PRC layer but also reveal macular alterations (Fig. 7A–E, Supplementary Material, Fig. S4). Further studies are required to elucidate whether macular lesions are caused by impaired leaky OLM adhesion junctions or by other processes, e.g. MGC gliosis or inflammation.
As in MGCs, we also observed the localization of harmonin in distinct subcellular compartments of cone and rod PRCs, namely in submembranous cytoplasm of the basal IS at the heterotypic OLM junctions, OS, calyceal processes and ribbon synapses. Previous studies indicate a multifaceted presynaptic role of harmonin in ribbon synapses, regulating L-type Ca2+ channels and neurotransmitter exocytosis in inner ear hair cells (18,19,66). Though harmonin is detected in the ribbon synapses of both PRC types, our immunoelectron microscopy data show that harmonin is more abundant in the synaptic pedicles of cones than in rod presynaptic terminals. This is consistent with the previously reported role of harmonin in synaptic maturation in cone pedicles of zebrafish (26) and with recent findings in our USH1C knock-in pig model indicating that the harmonin deficiency alters the width of cone synaptic pedicles (27). Notably, earlier studies have suggested selective modulation of pre-synaptic L-type calcium currents in cones to broaden the dynamic range of synaptic transfer by controlling transmitter release (67). Our data therefore support a putative role of harmonin contributing to this process.
The localization of harmonin in the calyceal processes of PRCs is consistent with previous reports (23). There is evidence that cadherin 23 (USH1D) and protocadherin 15 (USH1F) link the membrane of the calyceal processes to the plasma membrane of the OS via their long extracellular domains (28,68,69). In this scenario, harmonin is thought to anchor both cadherins in the cytoplasm of the calyceal processes mediated by binding of their C-terminal PBMs to harmonin’s PDZ2. More strikingly, we consistently found abundant harmonin expression in the OS of rod PRCs, whereas no harmonin is present in COS. There are obvious differences in disc morphogenesis and disc stacking between rod and cone PRCs. In contrast to cones, rod coin-roll-like stacked disc membranes become rimmed by the plasma membrane during disc neogenesis at the base of the OS (70,71). The localization of harmonin at the base of the OS and its ability to bind actin are consistent with its involvement in disc biogenesis, with the actin cytoskeleton playing a key role (72,73).
Harmonin lines up along the disc membranes of the entire rod outer segment (ROS) indicating a role in disc stacking. There is evidence for a network of cytoplasmic membrane–membrane tethers and/or spacers localized between the OS discs, but their molecular identity remains to be elusive (71). Interestingly, harmonin fulfills the criteria of such spacers: as a scaffold protein, harmonin facilitates protein networks, forms homomers, binds to membranes, directly to phospholipids and interacts with transmembrane proteins (8,74) such as rhodopsin, as shown herein. Our data on the interaction between harmonin and rhodopsin also suggest a role for harmonin in anchoring rhodopsin in disks by binding to its cytoplasmic C-terminus. Such roles of harmonin in disc morphogenesis and stacking are in line with our recent findings on the altered disc architecture of ROS in the USH1C pig model, revealing vertically oriented membrane discs, disk stacks interrupted by interstitial gaps and vesicle-like structures present at the OS base of rods in the absence of harmonin (27). Nevertheless, further studies are necessary to test and validate harmonin functions in the ROS.
The human retinal phenotype in the USH1C siblings included in this work confirms the retinal phenotype of retinitis pigmentosa with both morphological and impaired functions of both rod and cone PRC characteristics for USH1 patients (6). Our findings also agree with previous published findings on unrelated USH1C patients (22). With decreased functionality of the PRCs, we are not able to distinguish in which extent the reduction of the b-wave of the ERG—representing the signaling of the layer of the bipolar and MGCs—is caused by the lowered PRCs input or a dysfunction of MGCs or inner RNs. Thus, unfortunately, based on b-waves of the patients, we are not able to judge the actual functionality of the synaptic phenotype in humans.
In conclusion, we show that the scaffold protein harmonin is expressed in the human retina in form of numerous splice variants that localize in different subcellular compartments of PRCs and in MGCs. Defects in USH1C/harmonin would likely impair its various functions in distinct subcellular compartments of PRCs and MGCs. We hypothesize that these cellular dysfunctions have a cumulative effect, leading to progressive though slow retinal degeneration and vision impairment characteristic of USH1C patients. Understanding how these cellular defects interfere, potentially amplify and accumulate will provide new clues to cellular pathophysiology, elucidating potential targets for treatment and cure of USH1C patients. Although we cannot provide a fully elaborated therapeutic concept, we provide evidence that harmonin_a1 is the most promising splice variant for gene supplementation therapy and also identify both PRCs and MGCs as targets to treat vision loss in USH1C-patients.
Materials and Methods
Human subjects
Procedures adhered to the Declaration of Helsinki and were approved by the institutional review boards. Written informed consent was obtained from patients and probands.
All analyses of human retinae were performed using postmortem anonymized samples (Supplementary Material, Table S1). Donors had no known history of retinal disease and were either obtained from the Department of Ophthalmology, University Medical Center Mainz, Germany, the Department of Ophthalmology, Friedrich-Alexander-Universität, Erlangen-Nürnberg, Germany, the University of Utah, USA or from the National Disease Research Interchange (NDRI, Philadelphia, USA) and conformed to the tenets of the Declaration of Helsinki.
Clinical examination
Two sibling patients genetically diagnosed for USH1 with biallelic USH1C mutations (c.91C > T;p.(R31*), c.238dupC;p.(Arg80Profs*69) were examined clinically at the Center for Ophthalmology, University of Tübingen, including high-resolution retinal imaging and multimodal functional diagnostics. The clinical examination included a detailed medical history, best-corrected visual acuity testing, color perception evaluation using the Lanthony test, kinetic perimetry (Octopus 900; Haag-Streit International, Germany), slit-lamp examination, fundus examination in mydriasis with color fundus photography and 30 degrees fundus autofluorescence, as well as spectral domain OCT (Heidelberg Engineering GmbH, Germany).
Non-human primates
Eyes from three adult unaffected macaques (Macaque mulatta) were obtained from the German Primate Center (DPZ) where they were sacrificed as controls in other unrelated experiments.
Transcriptome analysis
RNA isolation from donor retinae was performed with ‘RNeasy Mini Kit’ (Qiagen, Hilden, Germany), according to company’s instruction. To increase the RNA yield, elution was performed twice. Libraries were generated with Illumina’s TruSeq Stranded mRNA Library Prep Kit and sequenced using Illumina HiSeq2500 sequencing platform as stranded, paired-end with a length of 151 nucleotides (mRNA datasets) (IMSB, Mainz, Germany). Using the ‘best practice’ pipeline from NEI commons using Trimmomatic (75), STAR (76) and Kallisto (77) (https://neicommons.nei.nih.gov/#/howDataAnalyzed), we aligned our sample to the Ensembl transcriptome version 89. In total, 81.4% of the 89.2 million reads derived from Donor 269-09 (Supplementary Material, Table S1) were aligned to the respective genome reference, out of which ~ 96% aligned uniquely. The mapped paired-end read length was 289.4 bp (of 302 bp) with a mismatch rate of 0.2%. Previously published RNA-seq samples (35) were downloaded as BAM-files from EBI’s ArrayExpress (E-MTAB-4377), converted into FASTQ files using SAMTOOLS (78). Sample converted files were processed using the same pipeline previously mentioned to investigate how many reads per sample were aligned to the genome. For LSV quantitation, samples were randomly assigned to 10 pools to create fused samples of ~165 million aligned fragments per pool. Individual sample files were processed using Kallisto with a FASTA file generated to the USH1C LSVs being investigated. Resulting estimated counts for the LSVs were summed according to the pooling strategy previously determined. For visualization, we used the Integrative Genomic Viewer (IGV; https://www.broadinstitute.org/igv/) (79,80). .
Reverse transcription, polymerase chain reactions, subcloning and identification of LSVs
The RNA isolation was performed using ‘TRIzol’ (Invitrogen, USA) according to the company instructions. The RT-PCR was performed with ‘Superscript II Reverse Transcriptase’ (Invitrogen), according to company instructions. The PCR was performed with ‘Taq DNA Polymerase with Standard Taq Buffer’ (NEB) according to company’s instructions. qPCR was performed on CFX96 real-time system (Bio-Rad) using the SYBRGreen iTAQ according to manufacturer’s instructions. Primers based on human USH1C sequences (http://www.ncbi.nlm.nih.gov/gene/10083) are listed in Supplementary Material, Table S3.
MACS of human MGCs and RNs
Retinal cell types were enriched using MACS as described previously (81,82). In brief, punches of 6 mm in the diameter of donor retinas were treated with a papain solution, washed and incubated with DNase I. Subsequently samples were triturated in extracellular solution. The resulting retinal cell suspensions were purified on antimouse/human ITGAM (alias CD11B) and antihuman CD31 microbeads (Miltenyi Biotec). Binding cells (microglia, vascular cells) were depleted from the retinal suspension using large cell (LS)-columns. For purification of MGCs, they were surface-labeled for CD29 (ITGB1) and CD29+ MGCs were separated using LS columns. Cells in the flow through the last sorting step, which were depleted of microglia, vascular cells and MGCs,were considered as the neuronal population. Sorted MGCs and RNs may suffer from cross-contaminating cell types, thus the purity of the fractions was determined by immunostaining and western blots with MGC markers such as antibodies against glutamine synthetase (Glul, MAB302, Merck Millipore) and drop sample analysis analyses demonstrating >90% purity of the fractions (Supplementary Material, Fig. S1). In previous studies, marker gene and protein expression profiles were analyzed in the fractions by RNAseq and mass spectrometry. While in MGC fractions established MGC markers such as glutamine synthetase (GLUL) and vimentin (VIM) were strongly enriched, neuronal markers including the vesicular glutamate transporter 1 (VGLUT1) or rhodopsin (RHO) were highest expressed in the neuronal cell populations (82).
Bulk RNA-sequencing on purified retinal cell populations
Total RNA was isolated from cell pellets after immunoseparation using the PureLink® RNA Micro Scale Kit (Thermo Fisher Scientific, Schwerte, Germany). RNA integrity validation and quantification was performed using the Agilent RNA 6000 Pico chip analysis according to the manufacturer’s instructions (Agilent Technologies). Enrichment of mRNA and library preparation (Nextera XT, Clontech), library quantification (KAPA Library Quantification Kit Illumina, Kapa Biosystems, Inc., USA) as well as sequencing on an Illumina platform (NextSeq 500 High Output Kit v2; 150 cycles) were performed at the service facility of the KFB Center of Excellence for Fluorescent Bioanalytics (Regensburg, Germany; www.kfb-regensburg.de). After de-multiplexing, at least 20 million reads per sample were detected. Quality control (QC) of the reads and quantification of transcript abundance was performed applying STAR (76), cutadapt was used to remove adapter sequences and several QC measures were queried with fastqc. Trimmed reads were aligned to the reference genome/transcriptome (mm10) applying STAR and transcript abundance was estimated with RSEM (83) expressed as Transcripts Per kilobase Million.
Western blot analyses
The immunoblots were performed as previously described (84). Briefly, one-fourth of a human retinae was placed in 125 μL HGNT-buffer, sonicated three times for 5 s and mixed with SDS-PAGE sample buffer (62.5 Mm Tris–HCL, pH 6.8; 10% glycerol, 2% SDS, 5% mercaptoethanol, 1 Mm EDTA and 0.025% bromphenol blue). As a molecular marker, PageRuler™ Prestained Protein Ladder ranging from 11 to 170 kDa was used (Fermentas). A total of 30 μg retina protein extracts were separated on 8% polyacrylamide gels and blotted onto PVDF transfer membranes (Millipore) followed by blocking with non-fat dried milk (Applichem). Cell pellets of enriched cell populations from retinal punches (6 Mm) were dissolved in reducing the Laemmli sample buffer, and denatured and sonicated. Samples were separated using a 12% SDS-PAGE. Detection was performed either by a chemiluminescence detection system (WesternSure PREMIUM Chemiluminescent Substrate, LI-COR) or using the Odyssey infra-red imaging system (LI-COR Biosciences).
Antibodies and fluorescent dyes
Primary antibodies used were as follows: anti-Arl13b (ab136648, Abcam), anti-JAM-B (SAB2501282, Sigma-Aldrich), anti-β-catenin (cs-7963, Santa Cruz), anti-filamin A (67133-1-ig, Proteintech) and anti-arrestin (SCT128, Santa Cruz Biotechnology), anti-his (27-4710-01, Amersham™), anti-actin (MA5-11869, Thermo Fisher Scientific), anti-GFP (gift from Clay Smith), anti-pyruvate dehydrogenase E1-β (Pdhb) (ab155996, Abcam), anti-glutamine synthetase (Glul) (MAB302, Merck Millipore) and anti-RFP (6G6, Chromotek). The antibody against harmonin (H3) was previously described (21). Monoclonal antibodies (mAbs) against bovine rod opsin B6-30a1, K16-155 and R2-15 were applied as previously described (85,86). Subcellular markers were RIBEYE (612 044, BD Bioscience), fluorescein-labeled lectin peanut agglutinin (FITC-PNA) and 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Secondary antibodies for immunofluorescence and western blot analysis were conjugated to Alexa 568 and Alexa 488 (Molecular Probes) or coupled to horseradish peroxidase (ECL Plus Western Blotting Detection System, GE Healthcare), respectively.
Immunofluorescence microscopy
After dissection human retinae and NHP retinae were cryofixed 11.5 to 31 h postmortem or directly, respectively, cryofixed in melting isopentane, cryosectioned and stained immunofluorescence as previously described (87,88). Immunofluorescence microscopy was performed with a Leica SP5 confocal laser scanning microscope (Leica microsystems) or a Leica DM6000B deconvolution microscope (Leica). Images were processed with Adobe Photoshop CS (Adobe Systems). Colocalization analysis were performed with the ImageJ (http://rsbweb.nih.gov/ij/) plugin JACoP (http://rsbweb.nih.gov/ij/plugins/track/jacop.html).
Immunoelectron microscopy
Human donor retina samples 199-09 and 121-10 were processed for pre-embedding immunolabeling as previously described (89). Ultrathin sections were analyzed with an FEI Tecnai 12 transmission electron microscope. Images were obtained with a charge-coupled device camera (SIS Megaview3; Surface Imaging Systems) acquired by AnalSIS (Soft Imaging System) and processed with Photoshop CS (Adobe Systems).
HEK293T cell culture
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal calf serum (FCS, Invitrogen) and 1% penicillin/streptomycin (Invitrogen) at 37°C and 5% CO2. Transfections of plasmids were performed with Lipofectamine® LTX and Plus Reagent (Invitrogen), siRNAs were transfected using Lipofectamin RNAiMAX, according to the manufacturer’s protocols, respectively.
Human primary fibroblast cultures
Dermal primary fibroblast lines were expanded from skin biopsies of human subjects (ethics volume: Landesärztekammer Rhineland-Palatinate to K.N.W.). Primary fibroblast lines were mycoplasma negative and cultured in DMEM, 10% FCS and 1% penicillin–streptomycin at 37°C and 5% CO2. For ciliogenesis, fibroblasts were starved in OPTIMEM reduced-serum medium (Invitrogen by Thermo Fisher Scientific) for 48 h and processed for immunocytochemistry as previously described (3). The length of Arl13b-positive primary ciliary axonemes were measured directly in images taken with a 63-x objective with a Leica DM6000B deconvolution microscope. For statistical analysis of ciliary length R was applied (https://www.rstudio.com/products/rstudio/download/).
siRNA knock-down
siRNAs against human USH1C/harmonin (siHarm1 and siHarm2) and non-targeting control siRNA (NTC) were purchased from IDT (TriFECTa® Kit, DsiRNA Duplex, IDT). For knock-downs, HEK293T cells were transfected with harmonin_a1-HA (pBI-CMV4-harm_a1-HA), and 20 nM non-targeted control. After 24 h, cells were lysed in Triton X-100 lysis buffer and protein concentrations were determined using Bicinchoninic acid (BCA) protein assay. Equal amounts of protein lysates were subjected to SDS-PAGE, followed by western blotting. Actin was used as a loading control. For quantification, the optical densities of harmonin_a1 (~ 80 kDa) bands were ascertained and normalized to the appropriate loading control. The percentage (%) of harmonin_a1 expression is shown in relation to harmonin a1-HA-transfected cells.
GST-pull downs
Constructs encoding harmonin domains were cloned in the pDEST17-vector (Gateway cloning system, Invitrogen, USA). The cDNAs encoding C-terminal tails of Mm JAM-B tail (amino acids 257-299), Mm β-catenin tail (amino acids 734-782) and filamin A tail (amino acids 2264-2648) were cloned in the vectors pDEST17 and pDEST15 and proteins were bacterially expressed. GST-pull down assays were performed as previously described (15).
GFP-Trap®
HEK293T cells were transfected with harmonin_a1-HA (pBI-CMV4-harm_a1-HA), GFP-Rhodopsin (pMT3-Rhodopsin-GFP) or GFP (pMT3-Rhodopsin-GFP), respectively, and lysed in Triton X-100 lysis buffer. Protein lysates of transfected cells were precleared using agarose beads. Equal amounts of precleared lysates were made in dilution buffer (10 Mm Tris/Cl pH 7.5, 150 Mm NaCl, 0.5 Mm EDTA). In total, 10% of the diluted amount was used as the input. GFP-fused polypeptides were immobilized at BSA blocked-Trap® agarose beads and used for co-precipitation assays according to the manufacturer’s protocol (ChromoTek). Precipitated protein complexes were eluted with an SDS-sample buffer at 65°C. Protein complexes were then subjected to SDS-PAGE and western blot.
Proximity ligation assay
For in situ PLA, the Duolink PLA probes anti-rabbitPLUS and anti-mouseMINUS, and the Detection Reagent Red was purchased from Sigma-Aldrich. PLAs were performed as previously described (90,91) according to the manufacturer’s protocol and adapted to our immunohistochemistry protocol for human retinae. As primary antibodies, anti-harmonin (H3) and a cocktail of three mAb opsin clones B6-30a1, K16-155 and R2-15) were applied. Negative control was were probed with anti-opsin antibodies and paired with the rabbit- and mouse-IgG-specific oligonucleotide-labeled antibodies. Mounted sections were analyzed on a Leica DM6000B microscope. For quantification, signals from controls were subtracted from signals of antibody combinations in four sections, each.
ROS enrichment
ROS were purified from five pig retinae using a method adapted from (92). Harvested pellets with the ROS were stored at −80°C before use in immunoblot analyses.
AlphaFold2 modeling
Modeling was done using the hetero-oligomer options of ColabFold (46,47). FASTA sequence of the respective protein was used from UniProt (UniProt 2020) and a colon between the respective sequences simulated complexes. The following sequences were used: Human USH1C/Harmonin_a1: Gene accession number: Q9Y6N9 harmonin_N-term aa 1-86, harmonin_PDZ1 aa 87-169; harmonin_PDZ2 aa 211-293, harmonin_CC-domain aa 310-377, harmonin_PDZ3 aa 452-537; rhodopsin: Gene accession number: P08100; Adrenergic receptor α-A1: Gene accession number: P35348. Alphafold2 notebook was used in the ColabFold version (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb#scrollTo=G4yBrceuFbf3, accessed last on 15 March 2022), default options were used but included a template mode. Structures were visualized, inspected and superimposed using PyMOL (Schrödinger, LLC), which was also used to make all figures.
Statistics
The statistical methods and the significance criteria for gene expression analysis and protein level analysis are listed in corresponding individual legends. Results are shown as the mean ± SEM of data from at least three separate experiments. Two-tailed Student’s t-test was used, significance was determined as: *P ≤ 0.05, **P ≤ 0.01.
Author contributions
K.N.W. and U.W. conceived, supervised the research project and wrote, reviewed and edited the manuscript. A.G., M.A.N. and A.S. supervised bioinformatics. B.F. generated RNA-seq libraries. B.F., T.M., M.B., M.S., T.A. and A.L. analyzed RNA-seq data. A.G. and L.K. performed and analyzed bulk RNA-seq. Data collection and analyses were performed by J.S. (ciliogenesis), M.B., T.G., K.N.W. (RT-PCR, PLAs, western blots, immunohistochemistry, TEM), K.A.W. (western blots), D.S., B.G. (Co-IP, pull-downs) and K.F. (ROS purification). J.F. performed Alphafold2 prediction analyses. K.S. and S.K. provided human skin biopsies and generated dermal primary fibroblasts. K.S. did clinical examination of USH1C patients. M.M.D., U.S.-S., I.K.K., L.A.O., M.A., J.M.V. and N.P. provided human donor eyes. Data were discussed with all coauthors.
Acknowledgements
We wish to thank Ulrike Maas, Elisabeth Sehn, and Gabriele B. Stern-Schneider for their excellent technical assistance. We thank Dr. Jan Reiners for contributing the initial pulldown experiments. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Conflict of Interest statement
None declared.
Funding
FAUN Foundation (Nuremberg) and USHER2020 Foundation to U.W., K.N.W.; Foundation Fighting Blindness (FFB PPA-0717-0719-RAD, TA-GT-0316-0694-JGU to U.W., K.N.W.); German Research Council/Deutsche Forschungsgemeinschaft in the framework of the SPP (SPP2127); Gene and Cell based therapies to counteract neuroretinal degeneration (project numbers: 399443882 to K.N.W., 399392938 to A.G., 39948788 to K.S., 398539671 to S.K., 399487434 to U.W.); European Union Seventh Framework Program (grant numbers 242013 TREATRUSH to U.W., 241955 SYSCILIA to U.W.); ProRetina Foundation Germany (Pro-Re/Seed/Kaplan-Grosche.8-2019 to A.G.) and NEI-IRP (ZIAEY000546 to A.S.).
Supplementary Material
This work is dedicated to H. Steffen Suchert (1945–2015).
Contributor Information
Kerstin Nagel-Wolfrum, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany; Institute of Developmental Biology and Neurobiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Benjamin R Fadl, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany; Neurobiology, Neurodegeneration and Repair Laboratory, National Eye Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Mirjana M Becker, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Kirsten A Wunderlich, Department of Physiological Genomics, BioMedical Center, Ludwig-Maximilian University Munich, 82152 Planegg-Martinsried, Germany.
Jessica Schäfer, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Daniel Sturm, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany; Institute of Developmental Biology and Neurobiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Jacques Fritze, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Burcu Gür, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Lew Kaplan, Department of Physiological Genomics, BioMedical Center, Ludwig-Maximilian University Munich, 82152 Planegg-Martinsried, Germany.
Tommaso Andreani, Computational Biology and Data Mining, Institute of Organismic & Molecular Evolution Biology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Tobias Goldmann, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Matthew Brooks, Neurobiology, Neurodegeneration and Repair Laboratory, National Eye Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Margaret R Starostik, Neurobiology, Neurodegeneration and Repair Laboratory, National Eye Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Anagha Lokhande, Neurobiology, Neurodegeneration and Repair Laboratory, National Eye Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Melissa Apel, Department of Ophthalmology, University Medical Centre Mainz, 55131 Mainz, Germany.
Karl R Fath, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany; Department of Biology, Queens College of CUNY, Kissena Blvd, Flushing, NY 11367, USA.
Katarina Stingl, University Eye Hospital, Centre for Ophthalmology, University of Tubingen, 72076 Tubingen, Germany.
Susanne Kohl, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tubingen, 72076 Tubingen, Germany.
Margaret M DeAngelis, Department of Ophthalmology and Ira G. Ross Eye Institute, Jacobs School of Medicine and Biomedical Sciences, University of Buffalo, NY 14209, USA.
Ursula Schlötzer-Schrehardt, Department of Ophthalmology, Friedrich-Alexander-Universität, Erlangen-Nürnberg, 91054 Erlangen, Germany.
Ivana K Kim, Retina Service, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, MA 02114, USA.
Leah A Owen, Department of Ophthalmology and Visual Sciences, University of Utah, Salt Lake City, UT 84132, USA.
Jan M Vetter, Department of Ophthalmology, University Medical Centre Mainz, 55131 Mainz, Germany.
Norbert Pfeiffer, Department of Ophthalmology, University Medical Centre Mainz, 55131 Mainz, Germany.
Miguel A Andrade-Navarro, Computational Biology and Data Mining, Institute of Organismic & Molecular Evolution Biology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
Antje Grosche, Department of Physiological Genomics, BioMedical Center, Ludwig-Maximilian University Munich, 82152 Planegg-Martinsried, Germany.
Anand Swaroop, Neurobiology, Neurodegeneration and Repair Laboratory, National Eye Institute, National Institutes of Health, Bethesda, MD 20892, USA.
Uwe Wolfrum, Institute of Molecular Physiology, Johannes Gutenberg University Mainz, 55128 Mainz, Germany.
References
- 1. Kimberling, W.J., Hildebrand, M.S., Shearer, A.E., Jensen, M.L., Halder, J.A., Trzupek, K., Cohn, E.S., Weleber, R.G., Stone, E.M. and Smith, R.J. (2010) Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet. Med., 12, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman, T.B., Schultz, J.M., Ahmed, Z.M., Tsilou, E.T. and Brewer, C.C. (2011) Usher syndrome: hearing loss with vision loss. Adv. Otorhinolaryngol., 70, 56–65. [DOI] [PubMed] [Google Scholar]
- 3. Samanta, A., Stingl, K., Kohl, S., Ries, J., Linnert, J. and Nagel-Wolfrum, K. (2019) Ataluren for the treatment of usher syndrome 2A caused by nonsense mutations. Int. J. Mol. Sci., 20, 6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. May-Simera, H., Nagel-Wolfrum, K. and Wolfrum, U. (2017) Cilia – the sensory antennae in the eye. Prog. Retin. Eye Res., 60, 144–180. [DOI] [PubMed] [Google Scholar]
- 5. Bujakowska, K.M., Liu, Q. and Pierce, E.A. (2017) Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb. Perspect. Biol., 9. 10.1101/cshperspect.a028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fuster-Garcia, C., Garcia-Bohorquez, B., Rodriguez-Munoz, A., Aller, E., Jaijo, T., Millan, J.M. and Garcia-Garcia, G. (2021) Usher syndrome: genetics of a human ciliopathy. Int. J. Mol. Sci., 22, 6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolfrum, U. (2011) Protein networks related to the Usher syndrome gain insights in the molecular basis of the disease. In Ahuja, S. (ed), Usher Syndrome: Pathogenesis, Diagnosis and Therapy. Nova Science Publishers Inc, USA, pp. 51–73. [Google Scholar]
- 8. Reiners, J., Nagel-Wolfrum, K., Jurgens, K., Marker, T. and Wolfrum, U. (2006) Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res., 83, 97–119. [DOI] [PubMed] [Google Scholar]
- 9. Zwaenepoel, I., Verpy, E., Blanchard, S., Meins, M., Apfelstedt-Sylla, E., Gal, A. and Petit, C. (2001) Identification of three novel mutations in the USH1C gene and detection of thirty-one polymorphisms used for haplotype analysis. Hum. Mutat., 17, 34–41. [DOI] [PubMed] [Google Scholar]
- 10. Verpy, E., Leibovici, M., Zwaenepoel, I., Liu, X.Z., Gal, A., Salem, N., Mansour, A., Blanchard, S., Kobayashi, I., Keats, B.J., Slim, R. and Petit, C. (2000) A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet., 26, 51–55. [DOI] [PubMed] [Google Scholar]
- 11. Kim, E. and Sheng, M. (2004) PDZ domain proteins of synapses. Nat. Rev. Neurosci., 5, 771–781. [DOI] [PubMed] [Google Scholar]
- 12. Kremer, H., van Wijk, E., Marker, T., Wolfrum, U. and Roepman, R. (2006) Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet., 15, R262–R270. [DOI] [PubMed] [Google Scholar]
- 13. Colcombet-Cazenave, B., Druart, K., Bonnet, C., Petit, C., Sperandio, O., Guglielmini, J. and Wolff, N. (2021) Phylogenetic analysis of harmonin homology domains. BMC Bioinform., 22, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adato, A., Michel, V., Kikkawa, Y., Reiners, J., Alagramam, K.N., Weil, D., Yonekawa, H., Wolfrum, U., El-Amraoui, A. and Petit, C. (2005) Interactions in the network of Usher syndrome type 1 proteins. Hum. Mol. Genet., 14, 347–356. [DOI] [PubMed] [Google Scholar]
- 15. Reiners, J., van Wijk, E., Marker, T., Zimmermann, U., Jurgens, K., te Brinke, H., Overlack, N., Roepman, R., Knipper, M., Kremer, H. et al. (2005) Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum. Mol. Genet., 14, 3933–3943. [DOI] [PubMed] [Google Scholar]
- 16. Yan, J., Pan, L., Chen, X., Wu, L. and Zhang, M. (2010) The structure of the harmonin/sans complex reveals an unexpected interaction mode of the two Usher syndrome proteins. Proc. Natl. Acad. Sci. USA, 107, 4040–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boeda, B., El-Amraoui, A., Bahloul, A., Goodyear, R., Daviet, L., Blanchard, S., Perfettini, I., Fath, K.R., Shorte, S., Reiners, J. et al. (2002) Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J., 21, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gregory, F.D., Bryan, K.E., Pangrsic, T., Calin-Jageman, I.E., Moser, T. and Lee, A. (2011) Harmonin inhibits presynaptic Cav1.3 Ca(2)(+) channels in mouse inner hair cells. Nat. Neurosci., 14, 1109–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gregory, F.D., Pangrsic, T., Calin-Jageman, I.E., Moser, T. and Lee, A. (2013) Harmonin enhances voltage-dependent facilitation of Cav1.3 channels and synchronous exocytosis in mouse inner hair cells. J. Physiol., 591, 3253–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan, D., Li, F., Hall, M.L., Sage, C., Hu, W.H., Giallourakis, C., Upadhyay, G., Ouyang, X.M., Du, L.L., Bethea, J.R. et al. (2006) An isoform of GTPase regulator DOCK4 localizes to the stereocilia in the inner ear and binds to harmonin (USH1C). J. Mol. Biol., 357, 755–764. [DOI] [PubMed] [Google Scholar]
- 21. Reiners, J., Reidel, B., El-Amraoui, A., Boeda, B., Huber, I., Petit, C. and Wolfrum, U. (2003) Differential distribution of harmonin isoforms and their possible role in Usher-1 protein complexes in mammalian photoreceptor cells. Invest. Ophthalmol. Vis. Sci., 44, 5006–5015. [DOI] [PubMed] [Google Scholar]
- 22. Williams, D.S., Aleman, T.S., Lillo, C., Lopes, V.S., Hughes, L.C., Stone, E.M. and Jacobson, S.G. (2009) Harmonin in the murine retina and the retinal phenotypes of Ush1c-mutant mice and human USH1C. Invest. Ophthalmol. Vis. Sci., 50, 3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sahly, I., Dufour, E., Schietroma, C., Michel, V., Bahloul, A., Perfettini, I., Pepermans, E., Estivalet, A., Carette, D., Aghaie, A. et al. (2012) Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol., 199, 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El-Amraoui, A. and Petit, C. (2005) Usher I syndrome: unravelling the mechanisms that underlie the cohesion of the growing hair bundle in inner ear sensory cells. J. Cell Sci., 118, 4593–4603. [DOI] [PubMed] [Google Scholar]
- 25. Millan, J.M., Aller, E., Jaijo, T., Blanco-Kelly, F., Gimenez-Pardo, A. and Ayuso, C. (2011) An update on the genetics of Usher syndrome. J. Ophthalmol., 2011, 417217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phillips, J.B., Blanco-Sanchez, B., Lentz, J.J., Tallafuss, A., Khanobdee, K., Sampath, S., Jacobs, Z.G., Han, P.F., Mishra, M., Titus, T.A. et al. (2011) Harmonin (Ush1c) is required in zebrafish Müller glial cells for photoreceptor synaptic development and function. Dis. Model. Mech., 4, 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grotz, S., Schafer, J., Wunderlich, K.A., Ellederova, Z., Auch, H., Bahr, A., Runa-Vochozkova, P., Fadl, J., Arnold, V., Ardan, T. et al. (2022) Early disruption of photoreceptor cell architecture and loss of vision in a humanized pig model of Usher syndromes. EMBO Mol. Med., 14, e14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schietroma, C., Parain, K., Estivalet, A., Aghaie, A., Boutet de Monvel, J., Picaud, S., Sahel, J.A., Perron, M., El-Amraoui, A. and Petit, C. (2017) Usher syndrome type 1-associated cadherins shape the photoreceptor outer segment. J. Cell Biol., 216, 1849–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cowan, C.S., Renner, M., De Gennaro, M., Gross-Scherf, B., Goldblum, D., Hou, Y., Munz, M., Rodrigues, T.M., Krol, J., Szikra, T. et al. (2020) Cell types of the human retina and its organoids at single-cell resolution. Cell, 182, 1623–1640.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu, L., Bolch, S.N., Santiago, C.P., Dyka, F.M., Akil, O., Lobanova, E.S., Wang, Y., Martemyanov, K.A., Hauswirth, W.W., Smith, W.C. et al. (2020) Clarin‐1 expression in adult mouse and human retina highlights a role of Müller glia in Usher syndrome. J. Pathol., 250, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bringmann, A., Pannicke, T., Grosche, J., Francke, M., Wiedemann, P., Skatchkov, S.N., Osborne, N.N. and Reichenbach, A. (2006) Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res., 25, 397–424. [DOI] [PubMed] [Google Scholar]
- 32. Gao, H., Luodan, L., Huang, X., Chen, X. and Xu, H. (2021) Muller glia-mediated retinal regeneration. Mol. Neurobiol., 58, 2342–2361. [DOI] [PubMed] [Google Scholar]
- 33. Nagel-Wolfrum, K., Moller, F., Penner, I. and Wolfrum, U. (2014) Translational read-through as an alternative approach for ocular gene therapy of retinal dystrophies caused by in-frame nonsense mutations. Vis. Neurosci., 31, 309–316. [DOI] [PubMed] [Google Scholar]
- 34. Katz, Y., Wang, E.T., Silterra, J., Schwartz, S., Wong, B., Thorvaldsdottir, H., Robinson, J.T., Mesirov, J.P., Airoldi, E.M. and Burge, C.B. (2015) Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics, 31, 2400–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pinelli, M., Carissimo, A., Cutillo, L., Lai, C.H., Mutarelli, M., Moretti, M.N., Singh, M.V., Karali, M., Carrella, D., Pizzo, M. et al. (2016) An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res., 44, 5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scanlan, M.J., Williamson, B., Jungbluth, A., Stockert, E., Arden, K.C., Viars, C.S., Gure, A.O., Gordan, J.D., Chen, Y.T. and Old, L.J. (1999) Isoforms of the human PDZ-73 protein exhibit differential tissue expression. Biochim. Biophys. Acta, 1445, 39–52. [DOI] [PubMed] [Google Scholar]
- 37. Omri, S., Omri, B., Savoldelli, M., Jonet, L., Thillaye-Goldenberg, B., Thuret, G., Gain, P., Jeanny, J.C., Crisanti, P. and Behar-Cohen, F. (2010) The outer limiting membrane (OLM) revisited: clinical implications. Clin. Ophthalmol., 4, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Golenhofen, N. and Drenckhahn, D. (2000) The catenin, p120ctn, is a common membrane-associated protein in various epithelial and non-epithelial cells and tissues. Histochem. Cell Biol., 114, 147–155. [DOI] [PubMed] [Google Scholar]
- 39. Daniele, L.L., Adams, R.H., Durante, D.E., Pugh, E.N., Jr. and Philp, N.J. (2007) Novel distribution of junctional adhesion molecule-C in the neural retina and retinal pigment epithelium. J. Comp. Neurol., 505, 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su, W., Mruk, D.D., Lie, P.P., Lui, W.Y. and Cheng, C.Y. (2012) Filamin a is a regulator of blood-testis barrier assembly during postnatal development in the rat testis. Endocrinology, 153, 5023–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DuChez, B.J., Hueschen, C.L., Zimmerman, S.P., Baumer, Y., Wincovitch, S. and Playford, M.P. (2019) Characterization of the interaction between β-catenin and sorting nexin 27: contribution of the type I PDZ-binding motif to Wnt signaling. Biosci. Rep., 39. 10.1042/BSR20191692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ebnet, K., Aurrand-Lions, M., Kuhn, A., Kiefer, F., Butz, S., Zander, K., Meyer Zu Brickwedde, M.K., Suzuki, A., Imhof, B.A. and Vestweber, D. (2003) The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: a possible role for JAMs in endothelial cell polarity. J. Cell Sci., 116, 3879–3891. [DOI] [PubMed] [Google Scholar]
- 43. tom Dieck, S. and Brandstatter, J.H. (2006) Ribbon synapses of the retina. Cell Tissue Res., 326, 339–346. [DOI] [PubMed] [Google Scholar]
- 44. Blanks, J.C. and Johnson, L.V. (1984) Specific binding of peanut lectin to a class of retinal photoreceptor cells. A species comparison. Invest. Ophthalmol. Vis. Sci., 25, 546–557. [PubMed] [Google Scholar]
- 45. Montell, C. (1999) Visual transduction in drosophila. Annu. Rev. Cell Dev. Biol., 15, 231–268. [DOI] [PubMed] [Google Scholar]
- 46. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Zidek, A., Potapenko, A. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., Žídek, A., Bates, R., Blackwell, S., Yim, J. et al. (2021) Protein complex prediction with AlphaFold-Multimer. bioRxiv. 10.1101/2021.10.04.463034. [DOI]
- 48. Chen, H.Y., Kelley, R.A., Li, T. and Swaroop, A. (2021) Primary cilia biogenesis and associated retinal ciliopathies. Semin. Cell Dev. Biol., 110, 70–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pan, B., Askew, C., Galvin, A., Heman-Ackah, S., Asai, Y., Indzhykulian, A.A., Jodelka, F.M., Hastings, M.L., Lentz, J.J., Vandenberghe, L.H., Holt, J.R. and Géléoc, G.S. (2017) Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol., 35, 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aisa-Marin, I., Garcia-Arroyo, R., Mirra, S. and Marfany, G. (2021) The Alter retina: alternative splicing of retinal genes in health and disease. Int. J. Mol. Sci., 22, 1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zelinger, L. and Swaroop, A. (2018) RNA biology in retinal development and disease. Trends Genet., 34, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murphy, D., Singh, R., Kolandaivelu, S., Ramamurthy, V. and Stoilov, P. (2015) Alternative splicing shapes the phenotype of a mutation in BBS8 to cause nonsyndromic retinitis pigmentosa. Mol. Cell. Biol., 35, 1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ling, J.P., Wilks, C., Charles, R., Leavey, P.J., Ghosh, D., Jiang, L., Santiago, C.P., Pang, B., Venkataraman, A., Clark, B.S. et al. (2020) ASCOT identifies key regulators of neuronal subtype-specific splicing. Nat. Commun., 11, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sundar, J., Matalkah, F., Jeong, B., Stoilov, P. and Ramamurthy, V. (2021) The Musashi proteins MSI1 and MSI2 are required for photoreceptor morphogenesis and vision in mice. J. Biol. Chem., 296, 100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vogel, C. and Marcotte, E.M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet., 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murphy, D., Cieply, B., Carstens, R., Ramamurthy, V. and Stoilov, P. (2016) The Musashi 1 controls the splicing of photoreceptor-specific exons in the vertebrate retina. PLoS Genet., 12, e1006256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yildirim, A., Mozaffari-Jovin, S., Wallisch, A.K., Schafer, J., Ludwig, S.E.J., Urlaub, H., Luhrmann, R. and Wolfrum, U. (2021) SANS (USH1G) regulates pre-mRNA splicing by mediating the intra-nuclear transfer of tri-snRNP complexes. Nucleic Acids Res., 49, 5845–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maerker, T., van Wijk, E., Overlack, N., Kersten, F.F., McGee, J., Goldmann, T., Sehn, E., Roepman, R., Walsh, E.J., Kremer, H. and Wolfrum, U. (2008) A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet., 17, 71–86. [DOI] [PubMed] [Google Scholar]
- 59. Fadl, B.R., Brodie, S.A., Malasky, M., Boland, J.F., Kelly, M.C., Kelley, M.W., Boger, E., Fariss, R., Swaroop, A. and Campello, L. (2020) An optimized protocol for retina single-cell RNA sequencing. Mol. Vis., 26, 705–717. [PMC free article] [PubMed] [Google Scholar]
- 60. Wu, L., Pan, L., Zhang, C. and Zhang, M. (2012) Large protein assemblies formed by multivalent interactions between cadherin23 and harmonin suggest a stable anchorage structure at the tip link of stereocilia. J. Biol. Chem., 287, 33460–33471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grillet, N., Xiong, W., Reynolds, A., Kazmierczak, P., Sato, T., Lillo, C., Dumont, R.A., Hintermann, E., Sczaniecka, A., Schwander, M. et al. (2009) Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron, 62, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li, J., He, Y., Lu, Q. and Zhang, M. (2016) Mechanistic basis of organization of the harmonin/USH1C-mediated brush border microvilli tip-link complex. Dev. Cell, 36, 179–189. [DOI] [PubMed] [Google Scholar]
- 63. Crawley, S.W., Shifrin, D.A., Jr., Grega-Larson, N.E., McConnell, R.E., Benesh, A.E., Mao, S., Zheng, Y., Zheng, Q.Y., Nam, K.T., Millis, B.A., Kachar, B. and Tyska, M.J. (2014) Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell, 157, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pinette, J.A., Mao, S., Millis, B.A., Krystofiak, E.S., Faust, J.J. and Tyska, M.J. (2019) Brush border protocadherin CDHR2 promotes the elongation and maximized packing of microvilli in vivo. Mol. Biol. Cell, 30, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Testa, F., Melillo, P., Bonnet, C., Marcelli, V., de Benedictis, A., Colucci, R., Gallo, B., Kurtenbach, A., Rossi, S., Marciano, E. et al. (2017) Clinical presentation and disease course of Usher syndrome because of mutations in Myo7a or Ush2a. Retina, 37, 1581–1590. [DOI] [PubMed] [Google Scholar]
- 66. Kersten, F.F., van Wijk, E., van Reeuwijk, J., van der Zwaag, B., Marker, T., Peters, T.A., Katsanis, N., Wolfrum, U., Keunen, J.E., Roepman, R. et al. (2010) Association of whirlin with Cav1.3 (alpha1D) channels in photoreceptors, defining a novel member of the usher protein network. Invest. Ophthalmol. Vis. Sci., 51, 2338–2346. [DOI] [PubMed] [Google Scholar]
- 67. Hosoi, N., Arai, I. and Tachibana, M. (2005) Group III metabotropic glutamate receptors and exocytosed protons inhibit L-type calcium currents in cones but not in rods. J. Neurosci., 25, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miles, A., Blair, C., Emili, A. and Tropepe, V. (2021) Usher syndrome type 1-associated gene, pcdh15b, is required for photoreceptor structural integrity in zebrafish. Dis. Model. Mech., 14, dmm048965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marshall, J.D., Paisey, R.B., Carey, C. and Macdermott, S. (1993) Alstrom Syndrome. In Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H. et al. (eds), GeneReviews®. University of Washington, Seattle, WA. [Google Scholar]
- 70. Burgoyne, T., Meschede, I.P., Burden, J.J., Bailly, M., Seabra, M.C. and Futter, C.E. (2015) Rod disc renewal occurs by evagination of the ciliary plasma membrane that makes cadherin-based contacts with the inner segment. Proc. Natl. Acad. Sci. USA, 112, 15922–15927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goldberg, A.F., Moritz, O.L. and Williams, D.S. (2016) Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res., 55, 52–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Corral-Serrano, J.C., Lamers, I.J.C., van Reeuwijk, J., Duijkers, L., Hoogendoorn, A.D.M., Yildirim, A., Argyrou, N., Ruigrok, R.A.A., Letteboer, S.J.F., Butcher, R. et al. (2020) PCARE and WASF3 regulate ciliary F-actin assembly that is required for the initiation of photoreceptor outer segment disk formation. Proc. Natl. Acad. Sci. USA, 117, 9922–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spencer, W.J., Lewis, T.R., Pearring, J.N. and Arshavsky, V.Y. (2020) Photoreceptor discs: built like ectosomes. Trends Cell Biol., 30, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bahloul, A., Michel, V., Hardelin, J.P., Nouaille, S., Hoos, S., Houdusse, A., England, P. and Petit, C. (2010) Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum. Mol. Genet., 19, 3557–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bolger, A.M., Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bray, N.L., Pimentel, H., Melsted, P. and Pachter, L. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 78. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Robinson, J.T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E.S., Getz, G. and Mesirov, J.P. (2011) Integrative genomics viewer. Nat. Biotechnol., 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Thorvaldsdottir, H., Robinson, J.T. and Mesirov, J.P. (2013) Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform., 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Grosche, A., Hauser, A., Lepper, M.F., Mayo, R., von Toerne, C., Merl-Pham, J. and Hauck, S.M. (2016) The proteome of native adult Müller glial cells from murine retina. Mol. Cell. Proteomics, 15, 462–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zauhar, R., Biber, J., Jabri, Y., Kim, M., Hu, J., Kaplan, L., Pfaller, A.M., Schäfer, N., Enzmann, V., Schlötzer-Schrehardt, U. et al. (2022) As in real estate, location matters: cellular expression of complement varies between macular and peripheral regions of the retina and supporting tissues. Front. Immunol., 13, 895519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li, B. and Dewey, C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform., 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schafer, N., Grosche, A., Schmitt, S.I., Braunger, B.M. and Pauly, D. (2017) Complement components showed a time-dependent local expression pattern in constant and acute white light-induced photoreceptor damage. Front. Mol. Neurosci., 10, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Adamus, G., Zam, Z.S., Arendt, A., Palczewski, K., McDowell, J.H. and Hargrave, P.A. (1991) Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vis. Res., 31, 17–31. [DOI] [PubMed] [Google Scholar]
- 86. Wolfrum, U. and Schmitt, A. (2000) Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil. Cytoskeleton, 46, 95–107. [DOI] [PubMed] [Google Scholar]
- 87. Wolfrum, U. (1991) Centrin-like and alpha-actinin-like immunoreactivity in the ciliary rootlets of insect sensilla. Cell Tissue Res., 266, 231–238. [Google Scholar]
- 88. Overlack, N., Kilic, D., Bauss, K., Marker, T., Kremer, H., van Wijk, E. and Wolfrum, U. (2011) Direct interaction of the Usher syndrome 1G protein SANS and myomegalin in the retina. Biochim. Biophys. Acta, 1813, 1883–1892. [DOI] [PubMed] [Google Scholar]
- 89. Sedmak, T., Sehn, E. and Wolfrum, U. (2009) Immunoelectron microscopy of vesicle transport to the primary cilium of photoreceptor cells. Methods Cell Biol., 94, 259–272. [DOI] [PubMed] [Google Scholar]
- 90. Bauss, K., Knapp, B., Jores, P., Roepman, R., Kremer, H., Wijk, E.V., Marker, T. and Wolfrum, U. (2014) Phosphorylation of the Usher syndrome 1G protein SANS controls Magi2-mediated endocytosis. Hum. Mol. Genet., 23, 3923–3942. [DOI] [PubMed] [Google Scholar]
- 91. Sorusch, N., Bauss, K., Plutniok, J., Samanta, A., Knapp, B., Nagel-Wolfrum, K. and Wolfrum, U. (2017) Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet., 26, 1157–1172. [DOI] [PubMed] [Google Scholar]
- 92. Papermaster, D.S. (1982) Preparation of retinal rod outer segments. Methods Enzymol., 81, 48–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.