

Abstract
Genomic imprinting results in gene expression bias caused by parental chromosome of origin and occurs in genes with important roles during human brain development. However, the cell-type and temporal specificity of imprinting during human neurogenesis is generally unknown. By detecting within-donor allelic biases in chromatin accessibility and gene expression that are unrelated to cross-donor genotype, we inferred imprinting in both primary human neural progenitor cells and their differentiated neuronal progeny from up to 85 donors. We identified 43/20 putatively imprinted regulatory elements (IREs) in neurons/progenitors, and 133/79 putatively imprinted genes in neurons/progenitors. Although 10 IREs and 42 genes were shared between neurons and progenitors, most putative imprinting was only detected within specific cell types. In addition to well-known imprinted genes and their promoters, we inferred novel putative IREs and imprinted genes. Consistent with both DNA methylation-based and H3K27me3-based regulation of imprinted expression, some putative IREs also overlapped with differentially methylated or histone-marked regions. Finally, we identified a progenitor-specific putatively imprinted gene overlapping with copy number variation that is associated with uniparental disomy-like phenotypes. Our results can therefore be useful in interpreting the function of variants identified in future parent-of-origin association studies.
Introduction
In contrast to most loci in the genome that have roughly equal expression from either parental chromosome, genomic imprinting leads to biased levels of gene expression or chromatin accessibility from either the maternal or paternal chromosome. Some genomic imprinting results in totally silenced expression of one parental allele and is more likely to be shared across multiple tissues (1). However, most imprinted genes exhibit some tissue and cell-type-specific imprinted expression (1–3). Some genes are imprinted specifically in humans (4) and many imprinted genes are also expressed in neural development or in the adult brain (5–7). Previous studies have identified imprinted genes in different human tissues or cells (5,8,9), but imprinted regulatory elements (IREs) have not been well defined during human neurogenesis nor have their cell-type specificity been assessed. Elucidation of cell-type-specific imprinting mechanisms during neurogenesis is critical for interpreting results of parent-of-origin association studies for neuropsychiatric disorders and subsequent therapeutic development (7,10–14).
The primary regulatory elements (REs) that control genomic imprinting, called imprinting control elements (ICEs), exhibit parental-specific DNA methylation. Some of these differentially methylated regions (DMRs) are inherited from the sperm or egg and are maintained postfertilization throughout development in all tissues (15–18). A separate class of DMRs acquires methylation after fertilization under the direction of germ-cell-specific DMRs and show tissue-specific methylation patterns (19,20). Cell-type-specific ‘reading’ of germline DMRs and other epigenetic alterations such as histone modifications (e.g. H3K27me3) without associated germ-cell-specific DMRs also contribute to regulation of imprinted expression in specific tissues/cell types (17,21–25). However, these tissue/cell-type-specific REs controlling imprinting are not well identified.
Genomic imprinting is most often studied in mice where reciprocal cross breeding is designed, parents are genotyped, and parent-of-origin-specific gene expression is measured in offspring using high-throughput sequencing data (26–28). Heterozygous genetic markers then allow the separation of maternal versus paternal expression (29–32). These studies have found that there are more genes with imprinted expression in the brain compared with non-brain tissues (33). In addition, genomic imprinting shows key functions in neurodevelopmental processes including neural progenitor expansion, migration, differentiation and cell polarization in mouse brains (7). However, in humans, it is difficult to obtain both cell-type-specific molecular phenotypes and genotype data from children, as well as genotype data from both parents on a large scale in order to demonstrate genomic imprinting. It is still possible to infer imprinting in humans where parental genotypes are unavailable. This can be achieved by detecting within-donor allelic imbalance unrelated to across-donor genotype in large datasets, where sequencing data for gene expression/chromatin accessibility and genotype data have been collected (Fig. 1A) (8,34). Here, we used the beta-binomial distribution to model allelic counts across a population of up to 85 genetically diverse donors, in order to estimate the dispersion of the allelic ratio (AR; reference allelic count to total count) in the population, for gene expression (RNA-seq) and chromatin accessibility (ATAC-seq). In the human genome, the reference allele can be either the paternal-inherited allele or the maternal-inherited allele. In datasets without the parental genotype information, we can calculate the AR of reads from the reference over the total count of reads, which will be a mix of high or low ARs across samples at imprinted sites. That is to say, if a single nucleotide polymorphism (SNP) is in an imprinted gene or RE, the AR will demonstrate a higher dispersion compared with SNPs in biallelically expressed genes or REs. Using this method, we identify putative IREs and genes in human neural progenitors (HNPs) and neurons.
Figure 1.

Identification of putatively imprinted genes and REs. (A) Schematic cartoon of experimental design and methods. Imprinted genes and REs can be detected using parental genotype data. In this study, we do not know parent-of-origin information, and we infer imprinted genes and REs using high dispersion at heterozygous SNPs. (B) Comparison of putatively imprinted genes and REs in neurons, progenitors and known imprinted genes/ICEs (left). Overlap between putatively imprinted genes and imprinted promoters in neurons or progenitors (right). (C) Ideogram of putative neuron/progenitor imprinted genes and REs on the human genome. The names of known imprinted genes and regions are bolded.
Results
Inference of IREs and imprinted genes in progenitors and neurons
We utilized primary human neural progenitor cell (phNPC) chromatin accessibility and expression quantitative trait loci datasets described in our previous work (35,36) to infer imprinted genes and REs. phNPCs were cultured in vitro as progenitor cells and also differentiated for 8 weeks, virally labeled, and sorted to obtain a homogeneous population of neurons. We then performed ATAC-seq and RNA-seq to obtain chromatin accessibility profiles (35) (N_Progenitor = 76 and N_Neuron = 61) and gene expression profiles (36) (N_Progenitor = 85 and N_Neuron = 74) in both cell types (Fig. 1A). We identified heterozygous genetic variants using imputed genotype data for all donors in progenitors and neurons in order to distinguish the two chromosomes, although we are unable to classify maternal or paternal origin (Fig. 1A). Allele-specific chromatin accessibility and gene expression were then calculated using read counts (for the reference allele and the alternative allele) at each accessible/expressed heterozygous SNP site (Fig. 1A).
The silenced allele in the imprinted genes, inherited from one parent, will, by definition, lead to decreased expression as compared with the expressed allele. Similarly, the silenced allele in the imprinted REs will lead to decreased chromatin accessibility as compared with the accessible allele. Here, we do not know the parental origin of each allele at a heterozygous locus. We expect that at an imprinted site, the AR will be either high (when the reference allele is the expressed allele) or low (when the reference allele is the silent allele) leading to high dispersion in estimates of the AR across donors (Fig. 1A). We estimated the dispersion of allelically biased chromatin accessibility or gene expression to identify the SNPs with highly variable (either high or low) AR in the population. We modeled allelic counts of chromatin accessibility or gene expression using a beta-binomial distribution at heterozygous SNPs (37,38) and evaluated the significance of the over-dispersion of AR using a likelihood ratio test. SNPs in known randomly monoallelic expression (RMAE) genes and randomly monoallelic chromatin accessible (RMACA) regions were removed (Supplementary Material, Table S1) (39,40). Finally, we kept SNPs with a significant AR over-dispersion (FDR < 0.05 across all heterozygous SNPs tested within a given cell type) (41). A minimal donor count for both high and low AR (ndonor ≥ 2 for AR ≥ 0.8 and ndonor ≥ 2 for AR ≤ 0.2) was used to exclude cases of allele-specific expression or chromatin accessibility driven by genetic effects (42) (Supplementary Material, Fig. S1A). Owing to our study lacking parental genotype data, we cannot exclude the possibility that allelic bias in newly identified imprinted genes/REs is owing to previously unidentified RMAE or RMACA rather than parental inheritance. Thus, we refer to these allele specifically expressed genes/REs as putatively imprinted.
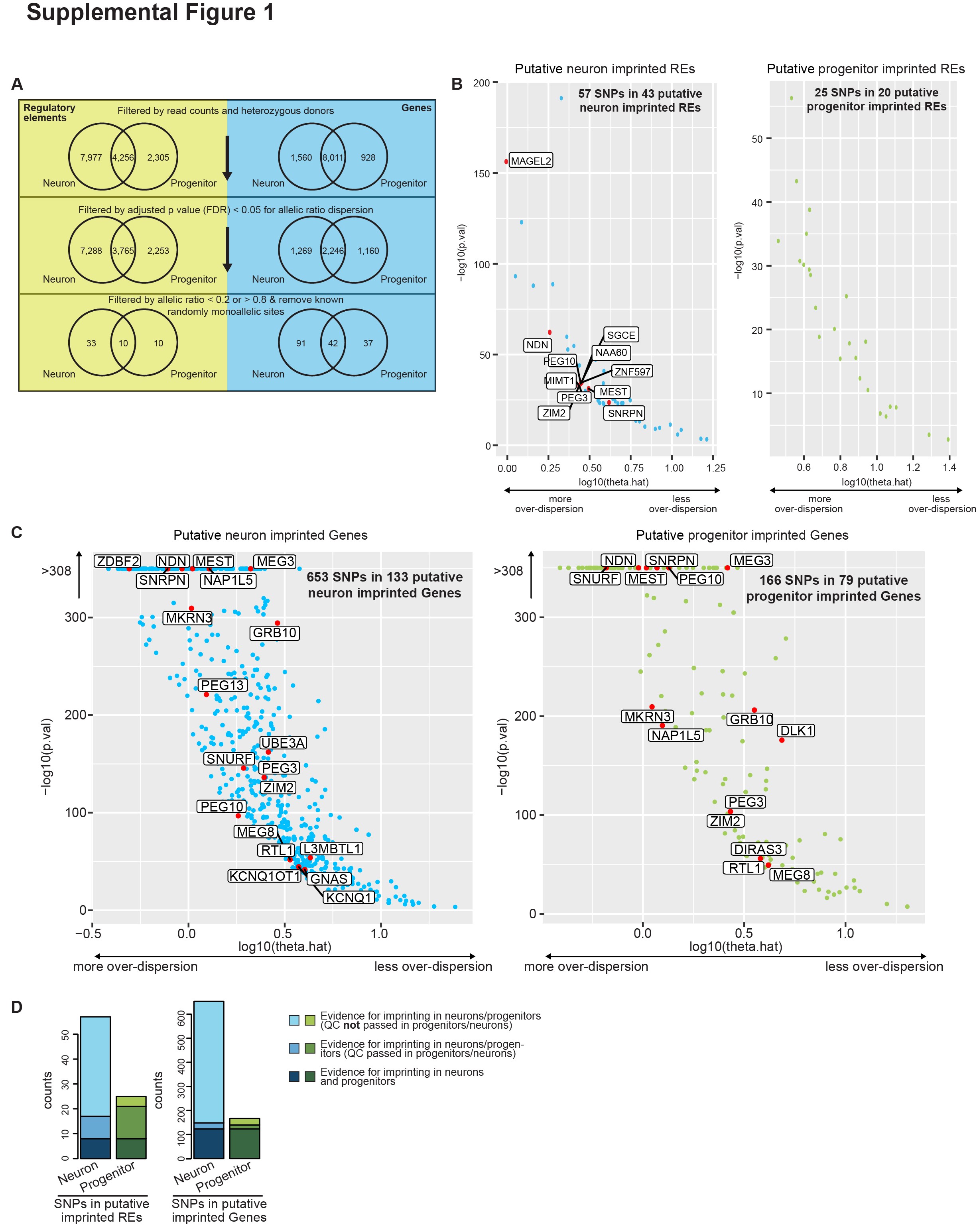
After these analyses and filtering steps, we identified 43 putative IREs containing 57 SNPs in neurons (nIREs) and 20 putative IREs containing 25 SNPs in progenitors (pIREs) (Fig. 1B and C; Supplementary Material, Fig. S1A, B and D; Supplementary Material, Table S2). We found three putative nIREs overlapped with known human ICEs for known imprinted genes PEG10, MEST and ZIM2/PEG3 (43), providing confidence in IRE calls. We also found 10 shared putative nIREs and pIREs overlapped with the promoters of 11 well-known imprinted genes (Supplementary Material, Fig. S1B; red labeled points) that are involved in neuronal development and differentiation: MAGEL2, NDN, SGCE, PEG10, NAA60, MIMT1, ZNF597, MEST, PEG3, ZIM2 and SNRPN (4,5,44–46). Surprisingly, we did not find any putative pIREs that overlapped with the promoters of well-known imprinted genes, so the pIREs identified here may represent a novel set of imprinted elements, such as the promoters of EIF2D, DDX11L2 and SPEG.
We identified 133 putative neuron imprinted genes (19 of which are previously known imprinted genes) containing 653 SNPs and 79 putative progenitor imprinted genes (14 of which are previously known imprinted genes) containing 166 SNPs (Fig. 1B and C; Supplementary Material, Fig. S1C; Supplementary Material, Table S3). Among these putatively imprinted genes, many genes have been previously reported as imprinted, such as UBE3A, NDN, PEG3, MEST, GRB10 and MEG3 (47–52) and some are known to have important functions in neurogenesis, such as DLK1 and ZDBF2 (53–56). We also found genes such as HM13, ZNF331, COPG2, DOC2B and PBX1 (Supplementary Material, Table S3) that have been previously described as imprinted in lymphoblastoid cell lines (57,58), but fit an imprinted expression pattern specifically in neurons in our study. However, we were not able to detect all of the known imprinted genes, such as IGF2, owing to lack of expression in these two cell types (Supplementary Material, Table S4) (59,60). To estimate the sensitivity of our method, we calculated the ratio of inferred imprinted genes to previously known imprinted genes that survived our quality control. We found 16 neuron putatively imprinted genes out of 40 known imprinted genes (40%) and 12 progenitor putatively imprinted genes out of 33 known imprinted genes (36%). In addition, we estimated whether our imprinting inferences may be affected by reads from the antisense strand. We found that the ratio of antisense strand reads to total transcribed reads is less than 10% for most of the SNPs in progenitors (252 out of 254 progenitor SNPs). In neurons, we found 148 out of 653 SNPs (23%) had antisense strand reads representing greater than 10% of total reads, but over 80% of these SNPs are from UBE3A where there is a known antisense RNA (61). The low amount of antisense transcripts at inferred imprinted locations indicates that this is unlikely to be a major source of bias in our method of imprinting inference.
We found 42 genes that show putative imprinting patterns in both progenitors and neurons (Fig. 1B, Supplementary Material, Table S3), including the well-known imprinted genes: GRB10, PEG10, MEST, MEG3, MEG8, NDN and SNHG14. Several of the putatively imprinted genes that are shared across cell types have not been previously identified as imprinted, such as PIANP and SNHG7. We identified 10 REs showing evidence of putative imprinting in both progenitors and neurons, at the promoters of EIF2D, DDX11L2, IL17RC, CRELD1, FAM86B3P, FAM86GP, FAM86FP, PIP4P1, EEF2KMT and ZNF826P. Many of these REs overlapped with promoters of genes that are not previously known as imprinted genes, such as CRELD1 and EIF2D, which could be new candidates for imprinting regulation. Although there was some overlap of putative imprinting between cell types (31.6%/53.2% putatively imprinted genes and 23.3%/50.0% putative IREs in neurons/progenitors), most putative imprinting was found only in one cell type. Often this was because the gene did not survive quality control (sufficient read count and number of heterozygous donors) in both cell types (Supplementary Material, Fig. S1D). Nevertheless, cell-type-specific imprinting patterns were still detectable when the same gene passed QC in both cell types. Overall, the detection of well-known imprinted genes and REs supports the statistical approach to identify imprinted candidates outlined here. Novel genes and REs may have been undetected in previous studies owing to the lack of a cell-type-specific or development system for studying these effects.
Dynamic imprinting between progenitors and neurons
Previous work has shown that dynamic changes of imprinting for some genes are critical for stereotypical neurogenesis (55). For example, Dlk1 is expressed from both alleles in adult neural stem cells, but is imprinted in surrounding tissue (55). We sought to determine whether the major cell types involved in human neurogenesis, progenitors and neurons also show dynamic cell-type-specific imprinting. This analysis requires that we focus on the genes and REs harboring heterozygous SNPs that passed our quality controls, sufficient read count and number of heterozygous donors, in both progenitor and neuron samples (see Materials and Methods). Several neurogenesis-associated genes, including DLK1, IGF2 and CDKN1C, did not pass one or more of these thresholds in at least one cell type, so we were not able to accurately call imprinting within at least one cell type and therefore were unable to determine if there is a dynamic change of imprinting during human neurogenesis (55,62,63).
Nevertheless, we found 52 genes that showed biallelic expression in progenitors but putatively imprinted expression in neurons (Supplementary Material, Table S5). Among these 52 genes, UBE3A and GNAS were identified as putatively imprinted genes in neurons but show biallelic expression in progenitors. UBE3A has been previously found to have neuron-specific imprinting, and GNAS has been previously identified to have tissue-specific imprinting (64,65). We also found eight REs showed a putative imprinting accessibility pattern in neurons but were biallelically accessible in progenitors (Supplementary Material, Table S5). These eight REs overlap with the promoters of 11 genes, which have not been previously reported as imprinted genes (47–52). Conversely, we identified 20 putatively imprinted genes in progenitors that showed biallelic expression in neurons (Supplementary Material, Table S5). These genes were not previously identified as imprinted (47–52). We did not find any pIREs with biallelic accessibility in neurons that were putatively imprinted in progenitors. Together, these results provide support that imprinting is a dynamic process during human cortical neurogenesis, and identified putative imprinted genes and REs that were previously unknown, perhaps owing to their dynamic nature.
We found 11 genes (MAGI2, PPP1R9A, GLIS3, INPP5F, OSBPL5, NTM, RB1, SMOC1, NAA60, DNMT1 and DGCR6L) that were previously known as imprinted genes but showed biallelic expression in both neurons and progenitors. We also found four previously known imprinted genes (PHLDA2, ANO1, ATP10A and GNAS) showed biallelic expression in progenitors and four previously known imprinted genes (DLX5, LRP1, BLCAP and GDAP1L1) showed biallelic expression in neurons. Many of these genes were shown to have functions related to brain development (66,67). This may indicate that relaxation of imprinting in these genes is important for stereotypical neurogenesis to occur.
Neuron/progenitor imprinting at known loci
A well-known example of an imprinted genomic cluster is the Prader–Willi/Angelman Syndrome (PWS/AS) region on human chromosome 15q11–q13 (68). Mutations in the PWS/AS region result in neurodevelopmental disorders, PWS and AS (69), in a parent-of-origin–dependent manner, demonstrating the important function of these imprinted genes during neural development. We identified putative neuron-specific IREs that overlap with the promoters of MAGEL2, NDN and SNRPN, the latter with multiple SNPs supporting the imprinting inference (Figs 1C and 2A and B). Of note, 72 SNPs in UBE3A passed our filtering criteria and showed evidence for neuron-specific imprinting of gene expression (Fig. 2A and C; Supplementary Material, Fig. S2A). This finding is in agreement with previous studies showing that UBE3A is expressed exclusively from the maternally inherited allele in neurons (70,71). In the PWS/AS region, we found more putative IREs and imprinted genes in neurons than in progenitors, which is also consistent with previous studies in iPSC-derived neurons (72,73).
Figure 2.

Putatively imprinted genes and REs at the AS/PWS locus. (A) Coverage plot of ATAC-seq in neurons and progenitors at the AS/PWS locus and SNPs in imprinted REs and genes. (B) Allelic ATAC-seq counts for selected heterozygous SNPs in putatively imprinted REs in (A) showing a characteristic imprinting pattern. (C) Allelic RNA-seq counts for selected SNPs in putatively imprinted genes in (A) also showing a characteristic imprinting pattern.
DNA methylation, transcription factor binding and histone modification at putative IREs
The methylation of CpG sites is an important epigenetic regulation of imprinting in mammals (74). To explore the relationship between methylation and putative IREs in neurons and progenitors, we first calculated the GC content of the putative IREs. We found the GC content is significantly higher within putative IREs as compared with biallelic REs (Fig. 3A). We found the putative IREs showed significantly higher overlap with human CpG islands than the biallelic REs in both neurons and progenitors (Fig. 3B). Enrichment of CpG sites at putative IREs supports a role for DNA methylation in genomic imprinting at these loci during neuronal differentiation.
Figure 3.

Epigenetic modifications and transcription factor binding at putatively imprinted REs. (A) GC content of putatively imprinted and biallelically accessible REs in neurons (left) and progenitors (right). (B) Overlap of putatively imprinted and biallelically accessible REs with human CpG islands in neurons (left) and progenitors (right). (C) Putative imprinted genes in progenitors and neurons overlapped with oocyte hyperDMR-related genes and oocyte hyperDMR-unrelated genes (25). (D) Putative nIREs and pIREs overlap with human morula H3K27me3 ChIP-seq peaks, which exhibit a strong bias toward the maternal allele (25). (E) Enriched TF motifs from JASPAR 2016 vertebrates core dataset within putatively imprinted REs as compared with biallelically accessible REs.
Another epigenetic marker involved in regulating genomic imprinting is H3K27me3 (25,75). A previous study found that maternal imprinting in morulae could be explained through either a methylation-dependent mechanism (17 oocyte hyper-DMR related genes) or a H3K27me3 mechanism (27 oocyte hyper-DMR unrelated genes) (25). As expected, the ICEs identified by methylation data from oocyte, morula and sperm (Supplementary Material, Fig. S2B) (76) showed higher overlap with the CpG islands than the H3K27me3 domains (Supplementary Material, Fig. S2F), because DNA methylation occurs at CpG islands. We found two oocyte hyper-DMR-related genes (PEG10 and SNRPN), and one oocyte hyper-DMR-unrelated gene (HLA-C) that overlapped the putative imprinted genes identified in both neurons and progenitors (Fig. 3C). In addition, we found that 8 out of 43 putative nIREs and 6 out of 20 putative pIREs overlapped with H3K27me3 domains in human morulae (Fig. 3D). These results indicate that the approach we used can identify both germline DNA methylation-dependent and germline DNA methylation-independent imprinted genes.
DNA-binding proteins (DBPs) binding to the IREs have important roles in maintaining imprinting by changing the methylation levels around their binding sites during early development (77,78). In addition, methylation at IREs may block some DBPs binding on the silence allele thereby decreasing expression of genes from the silence allele. However, it is unclear which DBPs are involved in either maintaining imprinting or the downstream consequences of imprinting during human neurogenesis, especially at cell-type resolution. To identify the DBPs that may bind to and regulate the putative IREs in neurons and progenitors, we performed an enrichment analysis of transcription factor (TF) motifs in the putative IREs using a binomial test (79). We used heterozygous SNPs to mark the putatively imprinted chromatin regions; however, we purposefully excluded SNPs, which have a consistent difference in chromatin accessibility across alleles (see Fig. 1A). Because of this, we did not aim to detect protein-binding motifs disrupted by heterozygous SNPs. Instead, we conducted a TF motif enrichment across the entirety of the putatively imprinted regions, not just at the heterozygous marking SNP, to determine if certain TFs are more likely to bind to imprinted regions (Fig. 3E). We retained only the TFs with significantly higher expression in the cell type tested for enrichment (Fig. 3E). Among TF motifs enriched in putative nIREs, we found ELK4, which is involved in upstream regulation of parent-of-origin-regulated genes in mice with sleep loss (80). We also found that CTCF TF motifs were enriched in putative pIREs. CTCF was shown to regulate the imprinted expression of KLD1 by binding to the ICE at KLD1-MEG3 locus in embryonic stem cells (81). Using these putative IREs, we are able to infer TFs implicated in imprinting gene regulation during human neurogenesis. We observed distinct TF motifs enriched in the inferred imprinting REs in the two cell types, which indicates different TFs binding to these chromatin regions and presumably having downstream functions on gene regulation during neurogenesis.
Putative IREs and imprinted genes indicate isoform-specific imprinting
Previous studies showed isoform-specific imprinted transcription for well-known genes, like PEG1 and MEST (82,83). However, the number of isoform-specific imprinted genes may still be underestimated, because isoform expression is highly cell-type-specific and imprinting has not been assessed in all cell types. Using putative IREs in neurons and progenitors, we predicted isoform-specific imprinted expression in each cell type. We found the putative nIREs overlapped with the promoter regions of 53 genes, among which the nIREs overlap specific isoforms at 40 of these genes. Previously reported genes show isoform-specific imprinting in our data, such as NAA60 and MEST (82,84). The putative pIREs overlapped with the promoter regions of 23 genes, 15 out of which are suggested to have putative isoform-specific imprinting.
In neurons, we found that the promoter region (chr21:39 385 651–39 386 540) of one isoform (ENST00000380713) of the gene GET1 (also known as WRB) showed a putative imprinting pattern at two SNPs (Fig. 4A and B). We did not find any SNP that passed QC in this RE in progenitors, therefore we are not able to test if this RE is a pIRE. This region in GET1 was previously reported as a new candidate–imprinted region according to DNA methylation analysis using peripheral blood samples (85,86), but to our knowledge, this is the first time this region was suggested as an IRE in neurons. GET1 is a receptor associated with protein transmembrane transport (87); but, its function in the human brain is unknown. This region overlaps with a DMR that is more methylated in oocyte and morula than sperm and maintains methylation in fetal and adolescent brains (Fig. 4C; Supplementary Material, Fig. S2B and C) (76,88). The DMR inherited from germ cells in the soma provides evidence that this putative IRE is an ICE that is maintained throughout development and potentially regulates maternal genomic imprinting of a specific GET1 isoform. No SNPs in the expression level data survived QC so we could not test whether GET1 isoform expression was imprinted (16,18).
Figure 4.
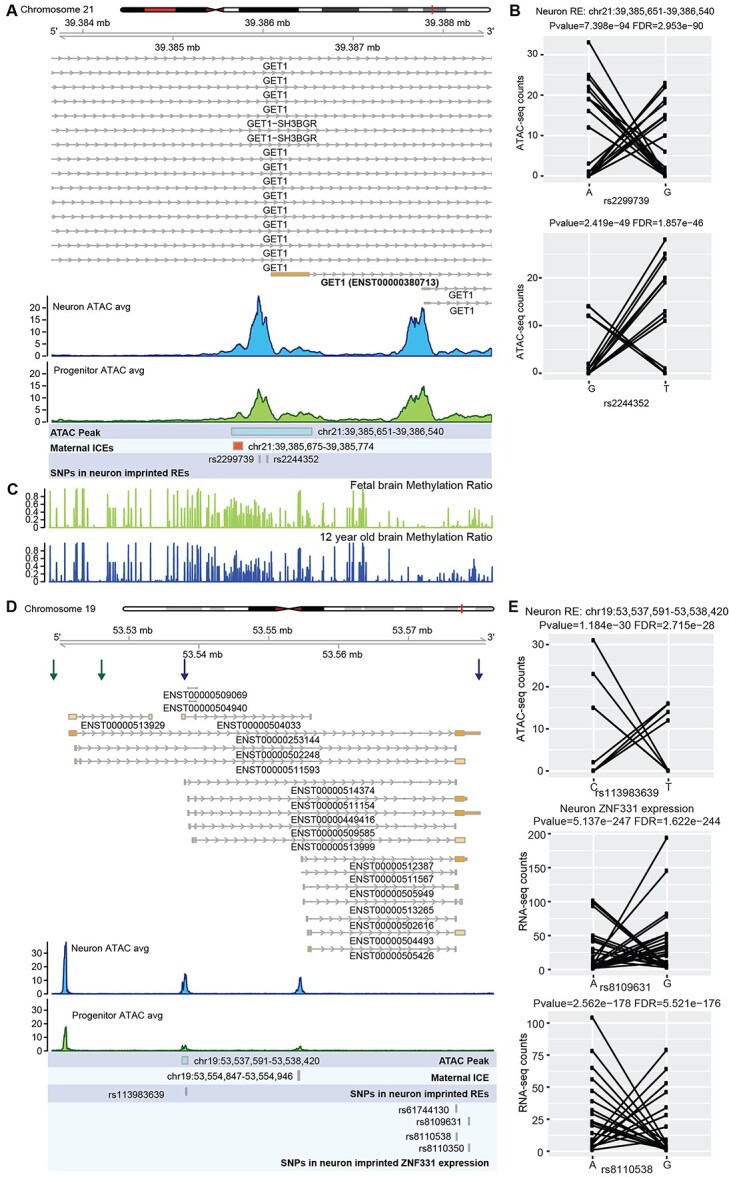
Putative imprinted REs and genes indicating isoform-specific imprinting in neurons. (A) Coverage plot of ATAC-seq in neurons and progenitors at GET1 locus. (B) Allelic ATAC-seq counts for the SNPs in the putatively imprinted RE in (A). (C) Methylation ratio in human fetal brain and 12-year-old brain (88) showing overlap with the ICE. (D) Coverage plot of ATAC-seq in neurons and progenitors at the ZNF331 locus. Boundaries of ZNF331 isoforms showed neuron-specific putatively imprinted expression patterns are indicated by blue arrows. Boundaries of ZNF331 imprinted isoforms in LCL are indicated by red arrows. (E) Allelic ATAC/RNA-seq counts for SNPs in the putatively imprinted RE and gene in (D).
We also found the gene ZNF331 had cell-type and isoform-specific imprinted expression patterns (Fig. 4D and E; Supplementary Material, Fig. S2D). We found a putative neuron-specific nIRE (chr19:53 537 591–53 538 420) overlapped with the promoter of a subset of isoforms of ZNF331 and these isoforms showed neuron-specific imprinted expression patterns (Fig. 4E, boundaries of the imprinted isoforms are indicated by blue arrows). We also found a germline DMR near the promoters of the isoforms that could serve as an ICE of this region (Supplementary Material, Fig. S2C and D) (76). The germline DMR is more methylated in oocyte and morula as compared with sperm (Supplementary Material, Fig. S2D), suggesting maternal imprinting. However, in progenitors, SNPs in this gene did not pass the threshold for significance and/or AR, implying biallelic expression. The SNP in the RE did not pass the threshold for read count, so we were not able to determine if there was a pIRE. Notably, isoform-specific imprinting of ZNF331 was previously reported in multiple cell types and tissues, including the brain and LCLs (boundaries of LCL imprinted isoforms are indicated by red arrows in Fig. 4D) (58,89–91). These results indicate cell-type-specific allelically biased isoform expression during neuronal development and differentiation.
Progenitor-specific DLK1 imprinting at Kagami Ogata/Temple syndrome paternal uniparental disomy locus
Uniparental disomy (UPD) results from homologous chromosomes, or parts of chromosomes, being inherited from only one parent (92). UPD and copy number variation mimicking UPD at imprinted sites results in abnormal expression. Copy number variation in the 14q32 imprinted gene cluster can lead to distinct maternal or paternal UPD phenotypes, named Temple syndrome and Kagami Ogata syndrome, respectively (93–96) (Fig. 5B: blue bars represent paternal deletion in the 14q32 imprinted gene cluster that cause Temple syndrome; red bars represent maternal deletion in the 14q32 imprinted gene cluster that cause Kagami Ogata syndrome). Individuals with Temple syndrome (UPD(14)mat) have characteristic features including pre- and postnatal growth retardation and developmental delay (97). Kagami Ogata syndrome (UPD(14)pat) results in prenatal overgrowth, developmental delay and facial abnormalities with full cheeks and protruding philtrum (98,99). Maternal deletions in the genomic region containing maternally expressed genes MEG3, MEG8 and RTL1 and sometimes containing paternally expressed gene DLK1 have been identified in individuals with Kagami Ogata syndrome (99). In general, individuals with maternal deletions in this region lack expression of the maternally expressed genes, but show overexpression of DLK1 in blood and placenta (98). In Temple syndrome, DLK1 expression is lost owing to the paternal deletion of this locus (100). We found evidence for putative imprinting of DLK1 gene expression in progenitors, but were unable to test for imprinting in neurons owing to insufficient read coverage (Fig. 5A, C and D; Supplementary Material, Fig. S2E). This imprinting pattern is distinct from adult neural stem cells where DLK1 is biallelically expressed, but is still consistent with the role of DLK1 in promoting neurogenesis in both mouse and human (55,56). In agreement with the function of DLK1, we found DLK1 showed a significantly higher expression level in progenitors than in neurons (log2FC = −3.67, FDR = 6.5e-244). Here, we suggest that DLK1 imprinting in progenitors contributes to the opposing phenotypes of overgrowth observed in Kagami Ogata syndrome, and growth retardation observed in Temple syndrome.
Figure 5.

Progenitor-specific DLK1 putative imprinting at Kagami Ogata syndrome paternal UPD locus. (A) Coverage plot of ATAC-seq in neurons and progenitors at Kagami Ogata syndrome paternal UPD locus. DLK1 showed putative progenitor-specific imprinted expression overlapping the copy number variation. (B) Copy number variation in the 14q32 imprinted gene cluster related to Temple syndrome and Kagami Ogata syndrome. (C) Allelic RNA-seq counts for the SNPs in MEG3 (rs10147988) and MEG8 (rs12879413) in neurons. (D) Allelic RNA-seq counts for the SNPs in MEG3 (rs10147988), MEG8 (rs12879413) and DLK1 in progenitors (rs1802710).
Discussion
Mutations at imprinted loci can lead to parent-of-origin–dependent inheritance for neurodevelopmental disorders. In order to better explain the mechanism underlying parent-of-origin disorders, imprinting must be detected within relevant cell types. By combining high-throughput sequencing data (RNA-seq and ATAC-seq) with genotype data, we identified putative cell-type-specific IREs and imprinted genes in two major cell types during human neuronal differentiation. We identified well-known IREs and genes in the PWS/AS region in neurons, providing confidence in our approach. We also identified putative cell-type-specific IREs and genes as new candidates for genomic imprinting. We found cell-type-specific REs may affect isoform-specific gene expression, as in neurons for GET1. Finally, we show that progenitor-specific putative imprinting of DLK1 overlaps with deletions causing Kagami Ogata syndrome, suggesting cortical neural progenitors contribute to neurobehavioral and growth changes observed in individuals with this syndrome.
Cell-type-specific imprinted gene expression and chromatin accessibility provide evidence that there are dynamic changes of genomic imprinting during human neuronal differentiation. In addition to putatively imprinted genes, we also identified putative IREs in neurons and progenitors, allowing us to explore the regulatory mechanisms of genomic imprinting. We found some genes only showed cell-type-specific putatively imprinted promoters but not imprinted expression, indicating that the IREs are established prior to imprinted expression. We identified the putative IREs that showed higher GC content and higher overlap with CpG islands and that some putative IREs overlap with H3K27me3. This suggests methylation of DNA or histone tails are likely regulatory mechanisms underlying the imprinted signal in chromatin accessibility. We also found different TF motifs enriched in the putative IREs in neurons and progenitors, providing new insights of the TFs that are involved in genomic regulation of imprinted loci during neurogenesis. However, we cannot determine if these TFs drive the allele-specific expression/chromatin accessibility or are impacted by the allele-specific chromatin.
Although previous studies showed loss of imprinting in human pluripotent stem cells cultured in vitro (101,102), another study on induced pluripotent stem cells suggested genomic imprinting is not erased at the AS/PWS locus (103). In this study, the well-known imprinted genes and promoters at AS/PWS locus were found in the neurons derived from primary neural progenitor cells (Fig. 2). The identification of these well-known imprinted genes indicated that the primary cell culture system used here was sufficient to study genomic imprinting at certain loci. However, it is unknown whether loss of imprinting at other sites occurred owing to cell culture conditions.
Here, we inferred putative genomic imprinting using ARs of RNA-seq and ATAC-seq reads. We separated genomic putative imprinting from allelic effects using a statistical test for dispersion and a threshold for the number of individuals with extreme AR. A difficulty of identifying genomic imprinting using next-generation sequencing data without genotype data from parents is to separate RMAE/RMACA from imprinted genes/REs. RMAE genes are mostly found in chromosome X owing to the X-chromosome inactivity, and there are less than 5% of genes showing RMAE on autosomes (2). In this study, we removed the previously known RMAE/RMACA (39,40) to increase the confidence of imprinting calls. However, we recognize that these datasets may miss human-specific or cell-type-specific random monoallelic sites in cell types relevant to this study and parental genotype data are necessary to completely disambiguate RMAE/RMACA from imprinting and so we refer to all novel imprinted genes/REs as putatively imprinted throughout the manuscript, and suggest that future experiments will be able to verify these sites using similar methods but with the addition of parental genotypes.
Additional experimental validation is also necessary to detect the molecular mechanisms of genomic imprinting during human neuronal differentiation. Genomic editing (deletion or modification) of IREs can be used to study their regulation of imprinting. Finally, we envision that combining the cell-type-specific genomic imprinting with well-powered parent-of-origin genome-wide association studies or parent-of-origin rare variant association studies will allow a better understanding of parent-of-origin effects on neurodevelopmental disorders.
Materials and Methods
Cell culture of primary human neural progenitor cells
We cultured and differentiated the phNPCs into neurons following the same methods in our previous work (35,104). Briefly, the cryopreserved HNPs with low number of passages (2.5 ± 1.8 s.d.) from 92 human fetal cortical tissue (14–21 gestation weeks) were thawed, captured and differentiated in ‘rounds’ of approximately 10. HNPs were cultured for 22 days with two splits on days 9 and 15, then on day 23 some of the HNPs were plated for ATAC-seq and RNA-seq library preparation of progenitor cells and the remainder of cells were differentiated into neurons (35,104). AAV2-hsyn1-eGFP virus for labeling neurons was added on day 64. Neurons were collected by FACS sorting on day 84 followed by immediate ATAC-seq and library preparation, and isolation of RNA for RNA-seq library preparation.
ATAC-seq and RNA-seq library preparation for human neural progenitors and neurons
ATAC-seq libraries were prepared immediately following cellular dissociation described in our previous methods (35,105) using 50 000 nuclei from each sample. All libraries were sequenced on an Illumina HiSeq2500 or MiSeq machine using 50 bp paired-end sequencing. RNA-seq libraries were prepared as previously described (36) and were sequenced on a NovaSeq S2 flow cell using 150 bp paired-end sequencing. In brief, RNA was isolated from progenitor cells and neuron cells using Qiagen miRNeasy Minelute kit and quantified by Qubit 2.0 fluorometer and Agilent Tapestation. The RNA-seq libraries were prepared using the Kapa Biosystems KAPA Stranded RNA-seq with Riboerase (HMR) kit, then the cDNA was fragmented to ~ 350 bp average insert size.
ATAC-seq, RNA-seq and genotype data pre-processing
Raw ATAC-seq and RNA-seq data were quality controlled and aligned to the human genome (GRCh38/hg38) using WASP to prevent mapping bias as previously described (35,36). Briefly, for ATAC-seq data, we removed the adapter sequence from fastq files, and then mapped sequence reads to the human genome using BWA MEM and WASP. Reads were removed if they were optical or PCR duplicates or mapped to mitochondrial genome, Y chromosome and ENCODE blacklist regions. Then, peaks were called for all samples using CSAW. For RNA-seq data, we removed the adapter sequence from fastq files, and then mapped sequence reads to the human genome using STAR/2.6.0a and WASP. We used Homo_sapiens.GRCh38.92 as the gene model for quantifying gene expression levels with the union exon.
Genotype data were preprocessed and imputed as previously described (35). In brief, Illumina HumanOmni2.5 or HumanOmni2.5Exome platform were used for genotyping for the 92 samples. Of note, 1 760 704 SNPs were saved for imputation after quality control and filtering to remove SNPs with Hardy–Weinberg equilibrium (<1e6), minor allele frequency (<0.01), individual missing genotype rate (>0.10) and variant missing genotype rate (>0.05). Imputation quality was assessed by retaining variants where imputation quality was high (R2 > 0.3 from Minimac4). After imputation to 1000 genomes reference panel, the high-quality imputed genotype data were filtered to remove SNPs using the same rules described before [Hardy–Weinberg equilibrium (<1e6), minor allele frequency (<0.01), individual missing genotype rate (>0.10) and variant missing genotype rate (>0.05)], resulting in ~13.6 million SNPs. Sex was determined using genotype data and ATAC-seq data, and we found 34% of donors are female and 66% are male.
Allele-specific read counts
We used GATK tools (106) to extract allele-specific read counts for every bi-allelic SNP (in accessible peaks or expression regions). We first filtered for SNPs within each donor that had sufficient read depth by retaining SNPs with total counts greater than or equal to 10 for neuron and progenitor samples, separately. Then to calculate allelic imbalance in chromatin accessibility and gene expression, we retained those SNPs with average read counts for all heterozygous donors greater than or equal to 15 for chromatin accessibility and 30 for gene expression. Finally, we retained only those SNPs that meet these previous thresholds for at least five heterozygous donors.
Before filtering, for ATAC-seq data, we observed an average of 2 471 263 (s.d. 266 777) heterozygous SNPs for neuron samples and 2 473 000 (s.d. 267 989) heterozygous SNPs for progenitor samples; for RNA-seq data, we observed 2 476 988 (s.d. 285 536) heterozygous SNPs for neuron samples and 2 474 622 (s.d. 271 657) heterozygous SNPs for progenitor samples. After filtering by read number, for ATAC-seq data, we retained on average of 26 337 SNPs (s.d. 2812) for 61 neuron samples and 26 367 SNPs (s.d. 2820) for 76 progenitor samples. These SNPs are located in 69 871 peaks, which is 77% of the total number of peaks (90 227). For RNA-seq data, we retained on average 382 874 SNPs (s.d. 44 384) for 74 neuron samples and 380 015 SNPs (s.d. 42 282) for 85 progenitor samples. These SNPs are located in 29 919 genes, which is 53% of the total genes in the gene model we used (56 852). Then we filtered the SNPs to retain those with sufficient number of heterozygous donors and average read counts for all heterozygous donors, for ATAC-seq data, we retained 19 960 SNPs in 12 233 peaks in neuron samples and 10 208 SNPs in 6561 peaks for progenitor samples. For RNA-seq data, we retained 80 811 SNPs in 9571 genes in neuron samples and 42 706 SNPs in 8939 genes for progenitor samples (Supplementary Material, Fig. S1A).
Estimation of over-dispersion and identification of putatively imprinted chromatin accessibility and gene expression
We identified over-dispersion using a likelihood ratio test based on the beta-binomial distribution for the allelic count (no. of reads from the reference allele given the no. of reads from both the reference allele and alternative allele) at each SNP. The allelic count can be modeled by a beta-binomial distribution, with the probability of expressing the parental-specific allele modeled by beta distribution (accounting for over-dispersion), and the number of reads observed modeled by a binomial distribution. The apeglm Bioconductor package was used to estimate parameters (107). The likelihood ratio test was used to determine significance for the over-dispersion parameter for the heterozygous SNPs. To identify putatively imprinted chromatin accessibility and gene expression, we used the following three conditions: (1) SNPs have at least two donors with an AR greater than or equal to 0.8; (2) SNPs have at least two donors with an AR less than or equal to 0.2 and (3) SNPs have significant over-dispersion of the AR (FDR < 0.05) (41).
SNPs in the previously defined RMAE gene body and promoter regions were removed (40). For RMACA from mouse cells (39), we converted the RMACA regions from mouse genome (mm9) to the human genome (hg38) using liftOver from UCSCtools via the R package (rtracklayer v1.44.0).
TFBS enrichment analysis
Potential transcription factor binding sites (TFBSs) were called in the human genome using TFBSTools from the JASPAR2016 core database for vertebrates as previously (35). This dataset includes both human and non-human vertebrates. We calculated the enrichment of TFBS in Neuron/Progenitor IREs using the binomial test (79). First, we found accessible regions (n) overlapping with all TFBSs for a given TF and calculated the fraction of base pairs of the motif compared with the overall base pairs of accessible peaks (p). Then, we counted the number of IREs (k) overlapping with TFBSs for this TF (k). The final step was to calculate P=Prbinom(x > = k|n,p) using the binomial test to determine the significance of the enrichment. We further filtered the enrichment results by differential expression from the same set of cells, and only kept the TFs with significantly higher cell-type-specific expression or accessibility in the tested cell type (36).
Identification of differential methylation regions among sperms, oocytes and morula
The genome-wide DNA methylation profiles for sperms (N = 4), oocytes (N = 2) and morula (N = 3) were downloaded via NCBI Gene Expression Omnibus (GEO) under accession number GSE49828. R package ‘MethylKit’ (v1.16.1) was used to analyze differential methylation regions for sperms versus oocytes and morula or oocytes versus sperm and morula. After removing the sites with <5 reads, methylation in 100-bp-tiles was called and the methylation level was estimated as previously described (108). A logistic regression model was used to identify the differential methylation tiles. The P-values were adjusted to q-values by the SLIM method (109). Tiles with q-value < 0.05 and percent methylation difference larger than 25% were assigned as differential methylation regions.
Peak calling for H3K27me3 CUT&RUN data in human morula
The H3K27me3 CUT&RUN data in human morula (N = 2) were downloaded via NCBI Gene Expression Omnibus (GEO) under accession number GSE123023. Peaks were called using SEACR (1.3) (110) and the peaks that occurred in both morulae were merged and kept as H3K27me3 domains. The H3K27me3 domains were converted between human genome builds (hg19 to hg38) using liftOver from UCSCtools via the R package (rtracklayer v1.44.0).
Data access
ATAC-seq/RNA-seq data and genotype data for neurons and progenitors are available via dbGAP (ph 001958 and phs2493). Human CpG island: https://genome.ucsc.edu/cgi-bin/hgTables?hgsid=578954849_wF1QP81SIHdfr8b0kmZUOcsZcHYr&clade=mammal&org=Human&db=hg38&hgta_group=regulation&hgta_track=knownGene&hgta_table=0&hgta_regionType=genome&position=chr9%3A133252000-133280861&hgta_outputType=primaryTable&hgta_outFileName=. Genome-wide DNA methylation profiles for sperms, oocytes and morula: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49828. Imprinted genes: https://geneimprint.com/site/genes-by-species. Genome-wide DNA methylation profiles for brain tissues: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE47966. H3K27me3 CUT&RUN profile in human morula: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123023.
Supplementary Material
{kind=link}
{kind=link}
Contributor Information
Dan Liang, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; UNC Neuroscience Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Nil Aygün, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; UNC Neuroscience Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Nana Matoba, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; UNC Neuroscience Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Folami Y Ideraabdullah, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Michael I Love, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Biostatistics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Jason L Stein, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; UNC Neuroscience Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Funding
National Institutes of Health (R00MH102357, U54EB020403, R01MH118349, R01MH120125); Brain Research Foundation (to J.L.S.).
Conflict of Interest statement. None declared.
Authors’ contributions
J.L.S. and M.I.L. conceived the study. J.L.S. directed and supervised the study. J.L.S. provided funding. N.A. performed pre-processing of allele-specific gene expression data. M.I.L. assisted with statistical identification of imprinting REs and genes. F.Y.I. assisted with interpretation of the results. D.L. performed pre-processing of ATAC-seq data and identification of imprinting REs and genes. J.L.S. and D.L. wrote the manuscript. All authors commented on and approved the final version of the manuscript.
References
- 1. Bonthuis, P.J., Huang, W.-C., Stacher Hörndli, C.N., Ferris, E., Cheng, T. and Gregg, C. (2015) Noncanonical genomic imprinting effects in offspring. Cell Rep., 12, 979–991. [DOI] [PubMed] [Google Scholar]
- 2. Kravitz, S.N. and Gregg, C. (2019) New subtypes of allele-specific epigenetic effects: implications for brain development, function and disease. Curr. Opin. Neurobiol., 59, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zink, F., Magnusdottir, D.N., Magnusson, O.T., Walker, N.J., Morris, T.J., Sigurdsson, A., Halldorsson, G.H., Gudjonsson, S.A., Melsted, P., Ingimundardottir, H. et al. (2018) Insights into imprinting from parent-of-origin phased methylomes and transcriptomes. Nat. Genet., 50, 1542–1552. [DOI] [PubMed] [Google Scholar]
- 4. Nakabayashi, K., Trujillo, A.M., Tayama, C., Camprubi, C., Yoshida, W., Lapunzina, P., Sanchez, A., Soejima, H., Aburatani, H., Nagae, G. et al. (2011) Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum. Mol. Genet., 20, 3188–3197. [DOI] [PubMed] [Google Scholar]
- 5. Babak, T., DeVeale, B., Tsang, E.K., Zhou, Y., Li, X., Smith, K.S., Kukurba, K.R., Zhang, R., Li, J.B., van der Kooy, D., Montgomery, S.B. and Fraser, H.B. (2015) Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet., 47, 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barlow, D.P. and Bartolomei, M.S. (2014) Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol., 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perez, J.D., Rubinstein, N.D. and Dulac, C. (2016) New perspectives on genomic imprinting, an essential and multifaceted mode of epigenetic control in the developing and adult brain. Annu. Rev. Neurosci., 39, 347–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baran, Y., Subramaniam, M., Biton, A., Tukiainen, T., Tsang, E.K., Rivas, M.A., Pirinen, M., Gutierrez-Arcelus, M., Smith, K.S., Kukurba, K.R. et al. (2015) The landscape of genomic imprinting across diverse adult human tissues. Genome Res., 25, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Santoni, F.A., Stamoulis, G., Garieri, M., Falconnet, E., Ribaux, P., Borel, C. and Antonarakis, S.E. (2017) Detection of imprinted genes by single-cell allele-specific gene expression. Am. J. Hum. Genet., 100, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ishida, M. and Moore, G.E. (2013) The role of imprinted genes in humans. Mol. Asp. Med., 34, 826–840. [DOI] [PubMed] [Google Scholar]
- 11. Nicholls, R.D. (2000) The impact of genomic imprinting for neurobehavioral and developmental disorders. J. Clin. Invest., 105, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mozaffari, S.V., DeCara, J.M., Shah, S.J., Sidore, C., Fiorillo, E., Cucca, F., Lang, R.M., Nicolae, D.L. and Ober, C. (2019) Parent-of-origin effects on quantitative phenotypes in a large Hutterite pedigree. Commun. Biol., 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brandler, W.M., Antaki, D., Gujral, M., Kleiber, M.L., Whitney, J., Maile, M.S., Hong, O., Chapman, T.R., Tan, S., Tandon, P. et al. (2018) Paternally inherited cis-regulatory structural variants are associated with autism. Science, 360, 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolter, J.M., Mao, H., Fragola, G., Simon, J.M., Krantz, J.L., Bazick, H.O., Oztemiz, B., Stein, J.L. and Zylka, M.J. (2020) Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature, 587, 281–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferguson-Smith, A.C. (2011) Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet., 12, 565–575. [DOI] [PubMed] [Google Scholar]
- 16. Hanna, C.W. and Kelsey, G. (2014) The specification of imprints in mammals. Heredity, 113, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnaud, P. (2010) Genomic imprinting in germ cells: imprints are under control. Reproduction, 140, 411–423. [DOI] [PubMed] [Google Scholar]
- 18. Plasschaert, R.N. and Bartolomei, M.S. (2014) Genomic imprinting in development, growth, behavior and stem cells. Development, 141, 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lopes, S., Lewis, A., Hajkova, P., Dean, W., Oswald, J., Forné, T., Murrell, A., Constância, M., Bartolomei, M., Walter, J. and Reik, W. (2003) Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum. Mol. Genet., 12, 295–305. [DOI] [PubMed] [Google Scholar]
- 20. Lucifero, D., Mertineit, C., Clarke, H.J., Bestor, T.H. and Trasler, J.M. (2002) Methylation dynamics of imprinted genes in mouse germ cells. Genomics, 79, 530–538. [DOI] [PubMed] [Google Scholar]
- 21. Prickett, A.R. and Oakey, R.J. (2012) A survey of tissue-specific genomic imprinting in mammals. Mol. Gen. Genomics, 287, 621–630. [DOI] [PubMed] [Google Scholar]
- 22. Andergassen, D., Dotter, C.P., Wenzel, D., Sigl, V., Bammer, P.C., Muckenhuber, M., Mayer, D., Kulinski, T.M., Theussl, H.-C., Penninger, J.M. et al. (2017) Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. elife, 6, e25125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang, X., Soloway, P.D. and Clark, A.G. (2011) A survey for novel imprinted genes in the mouse placenta by mRNA-seq. Genetics, 189, 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang, Q., Chow, J., Hong, J., Smith, A.F., Moreno, C., Seaby, P., Vrana, P., Miri, K., Tak, J., Chung, E.D. et al. (2011) Recent acquisition of imprinting at the rodent Sfmbt2 locus correlates with insertion of a large block of miRNAs. BMC Genomics, 12, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang, W., Chen, Z., Yin, Q., Zhang, D., Racowsky, C. and Zhang, Y. (2019) Maternal-biased H3K27me3 correlates with paternal-specific gene expression in the human morula. Genes Dev., 33, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang, X. and Clark, A.G. (2014) Using next-generation RNA sequencing to identify imprinted genes. Heredity, 113, 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oreper, D., Schoenrock, S.A., McMullan, R., Ervin, R., Farrington, J., Miller, D.R., de Villena, F.P.-M., Valdar, W. and Tarantino, L.M. (2018) Reciprocal F1 hybrids of two inbred mouse strains reveal parent-of-origin and perinatal diet effects on behavior and expression. G3, 8, 3447–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crowley, J.J., Zhabotynsky, V., Sun, W., Huang, S., Pakatci, I.K., Kim, Y., Wang, J.R., Morgan, A.P., Calaway, J.D., Aylor, D.L. et al. (2015) Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat. Genet., 47, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Babak, T., Deveale, B., Armour, C., Raymond, C., Cleary, M.A., van der Kooy, D., Johnson, J.M. and Lim, L.P. (2008) Global survey of genomic imprinting by transcriptome sequencing. Curr. Biol., 18, 1735–1741. [DOI] [PubMed] [Google Scholar]
- 30. Wang, X., Sun, Q., McGrath, S.D., Mardis, E.R., Soloway, P.D. and Clark, A.G. (2008) Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One, 3, e3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregg, C., Zhang, J., Weissbourd, B., Luo, S., Schroth, G.P., Haig, D. and Dulac, C. (2010) High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science, 329, 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laukoter, S., Pauler, F.M., Beattie, R., Amberg, N., Hansen, A.H., Streicher, C., Penz, T., Bock, C. and Hippenmeyer, S. (2020) Cell-type specificity of genomic imprinting in cerebral cortex. Neuron, 107, 1160–1179.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez, J.D., Rubinstein, N.D., Fernandez, D.E., Santoro, S.W., Needleman, L.A., Ho-Shing, O., Choi, J.J., Zirlinger, M., Chen, S.-K., Liu, J.S. and Dulac, C. (2015) Quantitative and functional interrogation of parent-of-origin allelic expression biases in the brain. elife, 4, e07860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reinius, B. and Sandberg, R. (2015) Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat. Rev. Genet., 16, 653–664. [DOI] [PubMed] [Google Scholar]
- 35. Liang, D., Elwell, A.L., Aygün, N., Krupa, O., Wolter, J.M., Kyere, F.A., Lafferty, M.J., Cheek, K.E., Courtney, K.P., Yusupova, M. et al. (2021) Cell-type-specific effects of genetic variation on chromatin accessibility during human neuronal differentiation. Nat. Neurosci., 24, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aygün, N., Elwell, A.L., Liang, D., Lafferty, M.J., Cheek, K.E., Courtney, K.P., Mory, J., Hadden-Ford, E., Krupa, O., de la Torre-Ubieta, L. et al. (2021) Brain-trait-associated variants impact cell-type-specific gene regulation during neurogenesis. Am. J. Hum. Genet., 108, 1647–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skelly, D.A., Johansson, M., Madeoy, J., Wakefield, J. and Akey, J.M. (2011) A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res., 21, 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castel, S.E., Levy-Moonshine, A., Mohammadi, P., Banks, E. and Lappalainen, T. (2015) Tools and best practices for data processing in allelic expression analysis. Genome Biol., 16, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu, J., Carter, A.C., Gendrel, A.-V., Attia, M., Loftus, J., Greenleaf, W.J., Tibshirani, R., Heard, E. and Chang, H.Y. (2017) Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat. Genet., 49, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gimelbrant, A., Hutchinson, J.N., Thompson, B.R. and Chess, A. (2007) Widespread monoallelic expression on human autosomes. Science, 318, 1136–1140. [DOI] [PubMed] [Google Scholar]
- 41. Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol., 57, 289–300. [Google Scholar]
- 42. Andergassen, D., Dotter, C.P., Kulinski, T.M., Guenzl, P.M., Bammer, P.C., Barlow, D.P., Pauler, F.M. and Hudson, Q.J. (2015) Allelome.PRO, a pipeline to define allele-specific genomic features from high-throughput sequencing data. Nucleic Acids Res., 43, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cowley, M., Skaar, D.A., Jima, D.D., Maguire, R.L., Hudson, K.M., Park, S.S., Sorrow, P. and Hoyo, C. (2018) Effects of cadmium exposure on DNA methylation at imprinting control regions and genome-wide in mothers and Newborn children. Environ. Health Perspect., 126, 037003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Watrin, F., Le Meur, E., Roeckel, N., Ripoche, M.-A., Dandolo, L. and Muscatelli, F. (2005) The Prader-Willi syndrome murine imprinting center is not involved in the spatio-temporal transcriptional regulation of the Necdin gene. BMC Genet., 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grütz, K., Seibler, P., Weissbach, A., Lohmann, K., Carlisle, F.A., Blake, D.J., Westenberger, A., Klein, C. and Grünewald, A. (2017) Faithful SGCE imprinting in iPSC-derived cortical neurons: an endogenous cellular model of myoclonus-dystonia. Sci. Rep., 7, 41156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ono, R., Nakamura, K., Inoue, K., Naruse, M., Usami, T., Wakisaka-Saito, N., Hino, T., Suzuki-Migishima, R., Ogonuki, N., Miki, H. et al. (2006) Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet., 38, 101–106. [DOI] [PubMed] [Google Scholar]
- 47. Dindot, S.V., Antalffy, B.A., Bhattacharjee, M.B. and Beaudet, A.L. (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet., 17, 111–118. [DOI] [PubMed] [Google Scholar]
- 48. Huntriss, J.D., Hemmings, K.E., Hinkins, M., Rutherford, A.J., Sturmey, R.G., Elder, K. and Picton, H.M. (2013) Variable imprinting of the MEST gene in human preimplantation embryos. Eur. J. Hum. Genet., 21, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blagitko, N., Mergenthaler, S., Schulz, U., Wollmann, H.A., Craigen, W., Eggermann, T., Ropers, H.H. and Kalscheuer, V.M. (2000) Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Hum. Mol. Genet., 9, 1587–1595. [DOI] [PubMed] [Google Scholar]
- 50. Zhang, X., Zhou, Y., Mehta, K.R., Danila, D.C., Scolavino, S., Johnson, S.R. and Klibanski, A. (2003) A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab., 88, 5119–5126. [DOI] [PubMed] [Google Scholar]
- 51. Jay, P., Rougeulle, C., Massacrier, A., Moncla, A., Mattei, M.G., Malzac, P., Roëckel, N., Taviaux, S., Lefranc, J.L., Cau, P. et al. (1997) The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet., 17, 357–361. [DOI] [PubMed] [Google Scholar]
- 52. Ho-Shing, O. and Dulac, C. (2019) Influences of genomic imprinting on brain function and behavior. Curr. Opin. Behav. Sci., 25, 66–76. [Google Scholar]
- 53. Duffié, R., Ajjan, S., Greenberg, M.V., Zamudio, N., Escamilla del Arenal, M., Iranzo, J., Okamoto, I., Barbaux, S., Fauque, P. and Bourc’his, D. (2014) The Gpr1/Zdbf2 locus provides new paradigms for transient and dynamic genomic imprinting in mammals. Genes Dev., 28, 463–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bouschet, T., Dubois, E., Reynès, C., Kota, S.K., Rialle, S., Maupetit-Méhouas, S., Pezet, M., Le Digarcher, A., Nidelet, S., Demolombe, V. et al. (2017) In vitro Corticogenesis from embryonic stem cells recapitulates the in vivo epigenetic control of imprinted gene expression. Cereb. Cortex, 27, 2418–2433. [DOI] [PubMed] [Google Scholar]
- 55. Ferrón, S.R., Charalambous, M., Radford, E., McEwen, K., Wildner, H., Hind, E., Morante-Redolat, J.M., Laborda, J., Guillemot, F., Bauer, S.R., Fariñas, I. and Ferguson-Smith, A.C. (2011) Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature, 475, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Surmacz, B., Noisa, P., Risner-Janiczek, J.R., Hui, K., Ungless, M., Cui, W. and Li, M. (2012) DLK1 promotes neurogenesis of human and mouse pluripotent stem cell-derived neural progenitors via modulating notch and BMP signalling. Stem Cell Rev. Rep., 8, 459–471. [DOI] [PubMed] [Google Scholar]
- 57. Pollard, K.S., Serre, D., Wang, X., Tao, H., Grundberg, E., Hudson, T.J., Clark, A.G. and Frazer, K. (2008) A genome-wide approach to identifying novel-imprinted genes. Hum. Genet., 122, 625–634. [DOI] [PubMed] [Google Scholar]
- 58. Jadhav, B., Monajemi, R., Gagalova, K.K., Ho, D., Draisma, H.H.M., van de Wiel, M.A., Franke, L., Heijmans, B.T., van Meurs, J., Jansen, R. et al. (2019) RNA-Seq in 296 phased trios provides a high-resolution map of genomic imprinting. BMC Biol., 17, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. GTEx Consortium (2020) The GTEx consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nowakowski, T.J., Bhaduri, A., Pollen, A.A., Alvarado, B., Mostajo-Radji, M.A., Di Lullo, E., Haeussler, M., Sandoval-Espinosa, C., Liu, S.J., Velmeshev, D. et al. (2017) Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science, 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rougeulle, C., Cardoso, C., Fontés, M., Colleaux, L. and Lalande, M. (1998) An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet., 19, 15–16. [DOI] [PubMed] [Google Scholar]
- 62. Laukoter, S., Beattie, R., Pauler, F.M., Amberg, N., Nakayama, K.I. and Hippenmeyer, S. (2020) Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat. Commun., 11, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ferrón, S.R., Radford, E.J., Domingo-Muelas, A., Kleine, I., Ramme, A., Gray, D., Sandovici, I., Constancia, M., Ward, A., Menheniott, T.R. and Ferguson-Smith, A.C. (2015) Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat. Commun., 6, 8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu, S., Yu, D., Lee, E., Eckhaus, M., Lee, R., Corria, Z., Accili, D., Westphal, H. and Weinstein, L.S. (1998) Variable and tissue-specific hormone resistance in heterotrimeric Gsprotein α-subunit (Gsα) knockout mice is due to tissue-specific imprinting of the Gsα gene. Proc. Natl Acad. Sci. U. S. A., 95, 8715–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamasaki, K., Joh, K., Ohta, T., Masuzaki, H., Ishimaru, T., Mukai, T., Niikawa, N., Ogawa, M., Wagstaff, J. and Kishino, T. (2003) Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet., 12, 837–847. [DOI] [PubMed] [Google Scholar]
- 66. Hong, G.-S., Lee, S.H., Lee, B., Choi, J.H., Oh, S.-J., Jang, Y., Hwang, E.M., Kim, H., Jung, J., Kim, I.-B. and Oh, U. (2019) ANO1/TMEM16A regulates process maturation in radial glial cells in the developing brain. Proc. Natl Acad. Sci. U. S. A., 116, 12494–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Perera, M., Merlo, G.R., Verardo, S., Paleari, L., Corte, G. and Levi, G. (2004) Defective neuronogenesis in the absence of Dlx5. Mol. Cell. Neurosci., 25, 153–161. [DOI] [PubMed] [Google Scholar]
- 68. Nicholls, R.D., Saitoh, S. and Horsthemke, B. (1998) Imprinting in Prader-Willi and Angelman syndromes. Trends Genet., 14, 194–200. [DOI] [PubMed] [Google Scholar]
- 69. Perk, J., Makedonski, K., Lande, L., Cedar, H., Razin, A. and Shemer, R. (2002) The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J., 21, 5807–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martins-Taylor, K., Hsiao, J.S., Chen, P.-F., Glatt-Deeley, H., De Smith, A.J., Blakemore, A.I.F., Lalande, M. and Chamberlain, S.J. (2014) Imprinted expression of UBE3A in non-neuronal cells from a Prader-Willi syndrome patient with an atypical deletion. Hum. Mol. Genet., 23, 2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hsiao, J.S., Germain, N.D., Wilderman, A., Stoddard, C., Wojenski, L.A., Villafano, G.J., Core, L., Cotney, J. and Chamberlain, S.J. (2019) A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl Acad. Sci. U. S. A., 116, 2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pólvora-Brandão, D., Joaquim, M., Godinho, I., Aprile, D., Álvaro, A.R., Onofre, I., Raposo, A.C., Pereira de Almeida, L., Duarte, S.T. and da Rocha, S.T. (2018) Loss of hierarchical imprinting regulation at the Prader-Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet., 27, 3999–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stanurova, J., Neureiter, A., Hiber, M., de Oliveira Kessler, H., Stolp, K., Goetzke, R., Klein, D., Bankfalvi, A., Klump, H. and Steenpass, L. (2016) Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci. Rep., 6, 30792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Paulsen, M. and Ferguson-Smith, A.C. (2001) DNA methylation in genomic imprinting, development, and disease. J. Pathol., 195, 97–110. [DOI] [PubMed] [Google Scholar]
- 75. Inoue, A., Jiang, L., Lu, F., Suzuki, T. and Zhang, Y. (2017) Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature, 547, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guo, H., Zhu, P., Yan, L., Li, R., Hu, B., Lian, Y., Yan, J., Ren, X., Lin, S., Li, J. et al. (2014) The DNA methylation landscape of human early embryos. Nature, 511, 606–610. [DOI] [PubMed] [Google Scholar]
- 77. Takahashi, N., Coluccio, A., Thorball, C.W., Planet, E., Shi, H., Offner, S., Turelli, P., Imbeault, M., Ferguson-Smith, A.C. and Trono, D. (2019) ZNF445 is a primary regulator of genomic imprinting. Genes Dev., 33, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sanli, I. and Feil, R. (2015) Chromatin mechanisms in the developmental control of imprinted gene expression. Int. J. Biochem. Cell Biol., 67, 139–147. [DOI] [PubMed] [Google Scholar]
- 79. McLean, C.Y., Bristor, D., Hiller, M., Clarke, S.L., Schaar, B.T., Lowe, C.B., Wenger, A.M. and Bejerano, G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tinarelli, F., Garcia-Garcia, C., Nicassio, F. and Tucci, V. (2014) Parent-of-origin genetic background affects the transcriptional levels of circadian and neuronal plasticity genes following sleep loss. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 369, 20120471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Llères, D., Moindrot, B., Pathak, R., Piras, V., Matelot, M., Pignard, B., Marchand, A., Poncelet, M., Perrin, A., Tellier, V., Feil, R. and Noordermeer, D. (2019) CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol., 20, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kosaki, K., Kosaki, R., Craigen, W.J. and Matsuo, N. (2000) Isoform-specific imprinting of the human PEG1/MEST gene. Am. J. Hum. Genet., 66, 309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stelzer, Y., Bar, S., Bartok, O., Afik, S., Ronen, D., Kadener, S. and Benvenisty, N. (2015) Differentiation of human parthenogenetic pluripotent stem cells reveals multiple tissue- and isoform-specific imprinted transcripts. Cell Rep., 11, 308–320. [DOI] [PubMed] [Google Scholar]
- 84. Morcos, L., Ge, B., Koka, V., Lam, K.C.L., Pokholok, D.K., Gunderson, K.L., Montpetit, A., Verlaan, D.J. and Pastinen, T. (2011) Genome-wide assessment of imprinted expression in human cells. Genome Biol., 12, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Docherty, L.E., Rezwan, F.I., Poole, R.L., Jagoe, H., Lake, H., Lockett, G.A., Arshad, H., Wilson, D.I., Holloway, J.W., Temple, I.K. and Mackay, D.J.G. (2014) Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J. Med. Genet., 51, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alves da Silva, A.F., Machado, F.B., Pavarino, É.C., Biselli-Périco, J.M., Zampieri, B.L., da Silva Francisco Junior, R., Mozer Rodrigues, P.T., Terra Machado, D., Santos-Rebouças, C.B., Gomes Fernandes, M. et al. (2016) Trisomy 21 alters DNA methylation in parent-of-origin-dependent and -independent manners. PLoS One, 11, e0154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vilardi, F., Lorenz, H. and Dobberstein, B. (2011) WRB is the receptor for TRC40/Asna1-mediated insertion of tail-anchored proteins into the ER membrane. J. Cell Sci., 124, 1301–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lister, R., Mukamel, E.A., Nery, J.R., Urich, M., Puddifoot, C.A., Johnson, N.D., Lucero, J., Huang, Y., Dwork, A.J., Schultz, M.D. et al. (2013) Global epigenomic reconfiguration during mammalian brain development. Science, 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Court, F., Tayama, C., Romanelli, V., Martin-Trujillo, A., Iglesias-Platas, I., Okamura, K., Sugahara, N., Simón, C., Moore, H., Harness, J.V. et al. (2014) Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res., 24, 554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Daelemans, C., Ritchie, M.E., Smits, G., Abu-Amero, S., Sudbery, I.M., Forrest, M.S., Campino, S., Clark, T.G., Stanier, P., Kwiatkowski, D. et al. (2010) High-throughput analysis of candidate imprinted genes and allele-specific gene expression in the human term placenta. BMC Genet., 11, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ben-David, E., Shohat, S. and Shifman, S. (2014) Allelic expression analysis in the brain suggests a role for heterogeneous insults affecting epigenetic processes in autism spectrum disorders. Hum. Mol. Genet., 23, 4111–4124. [DOI] [PubMed] [Google Scholar]
- 92. Robinson, W.P. (2000) Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays, 22, 452–459. [DOI] [PubMed] [Google Scholar]
- 93. Beygo, J., Elbracht, M., de Groot, K., Begemann, M., Kanber, D., Platzer, K., Gillessen-Kaesbach, G., Vierzig, A., Green, A., Heller, R., Buiting, K. and Eggermann, T. (2015) Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur. J. Hum. Genet., 23, 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Buiting, K., Kanber, D., Martín-Subero, J.I., Lieb, W., Terhal, P., Albrecht, B., Purmann, S., Gross, S., Lich, C., Siebert, R., Horsthemke, B. and Gillessen-Kaesbach, G. (2008) Clinical features of maternal uniparental disomy 14 in patients with an epimutation and a deletion of the imprinted DLK1/GTL2 gene cluster. Hum. Mutat., 29, 1141–1146. [DOI] [PubMed] [Google Scholar]
- 95. Chen, C.-P., Chern, S.-R., Lin, S.-P., Lin, C.-C., Li, Y.-C., Wang, T.-H., Lee, C.-C., Pan, C.-W., Hsieh, L.-J. and Wang, W. (2005) A paternally derived inverted duplication of distal 14q with a terminal 14q deletion. Am. J. Med. Genet. A, 139A, 146–150. [DOI] [PubMed] [Google Scholar]
- 96. Kagami, M., Sekita, Y., Nishimura, G., Irie, M., Kato, F., Okada, M., Yamamori, S., Kishimoto, H., Nakayama, M., Tanaka, Y. et al. (2008) Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat. Genet., 40, 237–242. [DOI] [PubMed] [Google Scholar]
- 97. Ioannides, Y., Lokulo-Sodipe, K., Mackay, D.J.G., Davies, J.H. and Temple, I.K. (2014) Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J. Med. Genet., 51, 495–501. [DOI] [PubMed] [Google Scholar]
- 98. Ogata, T. and Kagami, M. (2016) Kagami–Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet., 61, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rosenfeld, J.A., Fox, J.E., Descartes, M., Brewer, F., Stroud, T., Gorski, J.L., Upton, S.J., Moeschler, J.B., Monteleone, B., Neill, N.J. et al. (2015) Clinical features associated with copy number variations of the 14q32 imprinted gene cluster. Am. J. Med. Genet. A, 167A, 345–353. [DOI] [PubMed] [Google Scholar]
- 100. Prasasya, R., Grotheer, K.V., Siracusa, L.D. and Bartolomei, M.S. (2020) Temple syndrome and Kagami-Ogata syndrome: clinical presentations, genotypes, models and mechanisms. Hum. Mol. Genet., 29, R107–R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bar, S., Schachter, M., Eldar-Geva, T. and Benvenisty, N. (2017) Large-scale analysis of loss of imprinting in human pluripotent stem cells. Cell Rep., 19, 957–968. [DOI] [PubMed] [Google Scholar]
- 102. Frost, J., Monk, D., Moschidou, D., Guillot, P.V., Stanier, P., Minger, S.L., Fisk, N.M., Moore, H.D. and Moore, G.E. (2011) The effects of culture on genomic imprinting profiles in human embryonic and fetal mesenchymal stem cells. Epigenetics, 6, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chamberlain, S.J., Chen, P.-F., Ng, K.Y., Bourgois-Rocha, F., Lemtiri-Chlieh, F., Levine, E.S. and Lalande, M. (2010) Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl Acad. Sci. U. S. A., 107, 17668–17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Stein, J.L., de la Torre-Ubieta, L., Tian, Y., Parikshak, N.N., Hernández, I.A., Marchetto, M.C., Baker, D.K., Lu, D., Hinman, C.R., Lowe, J.K. et al. (2014) A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron, 83, 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Buenrostro, J.D., Wu, B., Chang, H.Y. and Greenleaf, W.J. (2015) ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol., 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M. and DePristo, M.A. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zitovsky, J.P. and Love, M.I. (2019) Fast effect size shrinkage software for beta-binomial models of allelic imbalance. F1000Research, 8, 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Akalin, A., Kormaksson, M., Li, S., Garrett-Bakelman, F.E., Figueroa, M.E., Melnick, A. and Mason, C.E. (2012) methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol., 13, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang, H.-Q., Tuominen, L.K. and Tsai, C.-J. (2011) SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics, 27, 225–231. [DOI] [PubMed] [Google Scholar]
- 110. Meers, M.P., Tenenbaum, D. and Henikoff, S. (2019) Peak calling by sparse enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin, 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.