

Abstract
The Warburg effect is the major metabolic hallmark of cancer. According to Warburg himself, the consequence of the Warburg effect is cell dedifferentiation. Therefore, reversing the Warburg effect might be an approach to restore cell differentiation in cancer. In this study, we used a mitochondrial uncoupler, niclosamide ethanolamine (NEN), to activate mitochondrial respiration, which induced neural differentiation in neuroblastoma cells. NEN treatment increased the nicotinamide adenine dinucleotide (NAD)+/NADH and pyruvate/lactate ratios and also the α-ketoglutarate (α-KG)/2-hydroxyglutarate (2-HG) ratio. Consequently, NEN treatment induced promoter CpG island demethylation and epigenetic landscape remodeling, activating the neural differentiation program. In addition, NEN treatment upregulated p53 but downregulated N-Myc and β-catenin signaling in neuroblastoma cells. Importantly, even under hypoxia, NEN treatment remained effective in inhibiting 2-HG generation, promoting DNA demethylation, and suppressing hypoxia-inducible factor signaling. Dietary NEN intervention reduced tumor growth rate, 2-HG levels, and expression of N-Myc and β-catenin in tumors in an orthotopic neuroblastoma mouse model. Integrative analysis indicated that NEN treatment upregulated favorable prognosis genes and downregulated unfavorable prognosis genes, which were defined using multiple neuroblastoma patient datasets. Altogether, these results suggest that mitochondrial uncoupling is an effective metabolic and epigenetic therapy for reversing the Warburg effect and inducing differentiation in neuroblastoma.
Introduction
One major metabolic cancer hallmark is the diversion of glucose flux from mitochondrial oxidation to fermentation and lactate production, known as the Warburg effect (1,2). Warburg reported that the consequence of this metabolic reprogramming is the conversion of normal differentiated cells to dedifferentiated (cancer) cells (3,4). Growing evidence has pointed out that metabolic reprogramming causes cell dedifferentiation through epigenetic remodeling (4–6).
The cancer epigenome generally displays global hypomethylation and CpG island hypermethylation compared to the epigenomes of corresponding healthy tissues (7). Particularly, CpG island hypermethylation in promoter regions leads to the silencing of tumor suppressors and cell differentiation markers and is associated with therapeutic resistance and poor prognosis in cancer patients (8,9). Unlike genetic mutations, epigenetic changes are reversible. Therefore, CpG island hypermethylation has become an attractive target for cancer therapy.
Recent reports have shown that mitochondria play a key role in epigenetic regulation and cell fate decisions by regulating the availability of certain metabolites (4–6). Ten-eleven translocation (TET) DNA demethylase uses α-ketoglutarate (α-KG) as a substrate to perform sequential oxidation of 5-methylcytosine, eventually removing the methylation (10). α-KG supplementation promotes pancreatic and colorectal cancer cell differentiation by reducing DNA methylation (11,12). However, in tumors, α-KG can be converted to the TET inhibitor 2-hydroxyglutarate (2-HG). While D-enantiomer (D-2-HG) is produced through gain-of-function point mutations in isocitrate dehydrogenases 1/2 (IDH1/2) (13,14), in hypoxic cancer cells such as neuroblastoma (NB) cells, lower nicotinamide adenine dinucleotide (NAD)+/NADH ratios favor the conversion of α-KG to L-enantiomer (L-2-HG) (15,16). Notably, lower NAD+/NADH ratios also favor the conversion of pyruvate to lactate, promoting the Warburg effect. Unlike D-2-HG, L-2-HG is produced via promiscuous substrate usage by lactate dehydrogenase (LDH), with additional contributions from malate dehydrogenase. While there are mutant IDH inhibitors to reduce D-2-HG levels, no effective therapeutic strategy inhibiting L-2-HG production is available. We hypothesized that CpG island hypermethylation could be reversed by increasing the intracellular NAD+/NADH and α-KG/2-HG ratios, correcting aberrant metabolic and epigenetic cancer cell phenotypes.
The electron transport chain (ETC) is the primary site for regenerating NAD+ from NADH in cells. Mitochondrial uncouplers facilitate proton influx across the mitochondrial inner membrane without generating adenosine triphosphate (ATP), leading to ETC activation. We hypothesized that ETC activation by mitochondrial uncouplers would increase the NAD+/NADH ratio, leading to an increased α-KG/2-HG ratio. In this study, we used a mitochondrial uncoupler, niclosamide ethanolamine (NEN), to reprogram the metabolism and epigenetic landscape of cancer cells. We show that NEN treatment increased intracellular NAD+/NADH, pyruvate/lactate, and α-KG/2-HG ratios. This metabolic reprogramming reduced DNA methylation in promoter regions and activated a neuronal differentiation program. Intriguingly, the differential methylation in gene bodies was highly associated with neuron differentiation-related genes. N-Myc, an oncogenic transcription factor often amplified in NB and associated with poorly differentiated histological features and worse patient survival, was downregulated by NEN treatment. Importantly, NEN treatment reduced 2-HG production, hypoxia-inducible factor (HIF) α subunit levels, and hypoxia-induced HIF target gene expression. Consistent with the in vitro data, NEN treatment reduced 2-HG levels and slowed tumor growth in an orthotopic NB mouse model. Altogether, these results uncover a new role of mitochondrial uncoupling as an intervention that reverses the Warburg effect and promotes NB differentiation.
Materials and Methods
Cell culture and reagents
MYCN-amplified NB cell lines SK-N-BE(2) (RRID:CVCL_0528), NB16 (RRID: CVCL_B323), and CHP134 (RRID:CVCL_1124) were obtained from Dr. John M. Maris’ laboratory (Children’s Hospital of Philadelphia) in 2016. A certificate of analysis is available from Dr. Maris’ group. Ovarian cancer cell line OVCAR3 was obtained from Dr. Erinn Rankin’s laboratory (Stanford University) in 2020. Lung cancer cell lines H29 and H82 were from Dr. Julien Sage’s laboratory (Stanford University) in 2020. Every two months, all cell lines were tested and found negative for mycoplasma (MycoAlert Mycoplasma Detection Kit; Lonza) (Last testing was performed on September, 2022). Cell lines within 5-15 passages were used for experiments. The authors did not authenticate these cell lines. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium (Caisson Labs; DFL15) supplemented with 1% penicillin-streptomycin (Gibco; 15140122), 10% fetal bovine serum (FBS; Sigma-Aldrich), and extra 1 mM glutamine (Gibco; 25030081). Dimethyl 2-oxoglutarate (DMKG; #349631) and 5-aza-2′-deoxycytidine (5-AZA; #A3656) were obtained from Sigma-Aldrich. NEN (#17118) was obtained from Cayman. All cell proliferation assays were performed with the indicated treatments and times in triplicate. Anti-N-Myc antibody (#sc-53993, RRID:AB_831602) and anti-p53 antibody (#sc-126, RRID:AB_628082) were obtained from Santa Cruz Biotechnology. Anti-β-tubulin III antibody (#5568S) and anti-histone H3 antibody (#3638S) were obtained from Cell Signaling Technology. Horseradish peroxidase-secondary antibody and goat anti-rabbit immunoglobulin G (H+L) cross-adsorbed secondary antibody Alexa Fluor 594 (#A-11012) were obtained from Invitrogen.
Protein extraction and immunoblotting
Cells were washed with ice-cold phosphate-buffered saline (PBS) buffer (Invitrogen; 20012050) and lysed with harvest buffer (10 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid [HEPES; pH 7.9], 50 mM sodium chloride [NaCl], 500 mM sucrose, 0.1 mM ethylenediaminetetraacetic acid [EDTA], and 0.5% Triton X-100) supplemented with 1% Halt inhibitors (Thermo Fisher Scientific; 78443) for 10 min. The cell lysate was centrifuged at 3000 rpm for 3 min at 4 °C, and the supernatant containing cytosolic proteins was transferred to a new tube. The pellet containing nuclear proteins was dissolved with nuclear lysis buffer (10 mM HEPES [pH 7.9], 0.5 M NaCl, 0.1 mM EDTA, 0.1 mM egtazic acid, and 0.1% NP40) supplemented with 1% Halt inhibitors (Thermo Fisher Scientific; 78443) for 5 min at 4°C. Then, the solution was sonicated at 4°C for six cycles (1 cycle = 30 s sonication [high intensity] and 30 s cooldown) using a Bioruptor Plus sonication (Diagenode; UCD-300). After sonication, the solution was centrifuged at 15,000 rpm for 10 min, and the supernatant was retained for nuclear extraction. Protein concentration was determined with the BCA assay (Thermo Fisher Scientific, 23227). Immunoblotting was performed as previously described (4). All the blots are representative of at least two independent experiments.
RNA isolation, reverse transcription, and real-time quantitative polymerase chain reaction (RT-qPCR)
The procedure was performed as previously described (17). Briefly, total RNA was isolated from 60 mm tissue culture plates according to the TRIzol Reagent (Invitrogen; 15596026) protocol. Next, 3 μg of total RNA was used in a reverse transcription reaction with the iScript cDNA Synthesis Kit (Bio-Rad). RT-qPCR amplification was performed using the Prism 7900 Sequence Detection System (Applied Biosystems) and TaqMan Gene Expression Assays (Applied Biosystems). Gene expression levels were normalized to the 18S ribosomal RNA. Data are presented as mean ± standard deviation (SD) across three RT-qPCR reactions, and representative values from three independent experiments are shown.
Next-generation RNA sequencing (RNA-seq) and bioinformatics analysis
The total RNA from treatment groups (control and NEN-treated; n = 3) was extracted using Trizol Reagent (Invitrogen; 15596026) according to the manufacturer’s instructions. The library was constructed and subjected to 150 bp paired-end sequencing on an Illumina sequencing platform at Novogene. RNA-seq analysis was performed using the kallisto and sleuth analytical pipelines. Briefly, a transcript index was generated based on Ensembl version 67 of build hg19 of the human reference genome sequence. Paired-end RNA-seq reads were pseudo-aligned using kallisto (v0.42.4) using this transcript index with 100 bootstraps (-b 100) to estimate the variance of estimated transcript abundances. Transcript-level estimates were aggregated to transcripts per million (TPM) estimates for each gene, with Ensembl gene names assigned using biomaRt. Differential gene expression analysis was performed using the sleuth R package across pairwise groups using Wald tests, with significant hits identified based on a sleuth q-value <0.05 and absolute log2 fold change > 0.693.
Gene set enrichment analysis (GSEA) was used to identify significantly enriched pathways by comparing the normalized data of the entire RNA seq TPM dataset between groups against the molecular signatures database (MSigDB v7.4). All annotated transcripts (∼21,427 total features) with expression values were uploaded to a locally-installed GSEA instance (v.4.1.0) and compared against the 50 Hallmark gene sets. Favorable or unfavorable prognosis gene sets were generated using a Kaplan–Meier assay (overall survival) for all genes independently and all 10 available NB patient databases from R2(18). Each gene that passed the Kaplan–Meier assay filter showed a significant correlation with prognosis (p < 0.05) in the relevant databases. Differentially expressed genes were functionally annotated by submitting them to the online Database for Annotation, Visualization and Integrated Discovery (DAVID) website (19). The DAVID analyses were performed using default parameters.
DNA methylation analysis
The SK-N-BE(2) cells were treated with dimethyl sulfoxide (DMSO) or 1 μM NEN for 24 h under normoxia and hypoxia (0.5%) before DNA extraction (n=3). DNA extraction was performed using the PureLink Genomic DNA Mini Kit (Gibco; K182001) according to the manufacturer’s protocol. DNA was quantified with a Nanodrop, and 3 μg of total DNA was used for bisulfite modification using the EZ DNA Methylation Kit (Zymo Research; D5001) according to the manufacturer’s protocol with the modification step recommended for array sample processing. Control PCR reactions were performed before array analysis to confirm successful DNA modification. Bisulfite-modified DNA (500 ng) was applied to the Infinium MethylationEPIC array (Illumina). According to the manufacturer’s protocol, after bisulfite DNA treatment, cytosines in the CpG sites genotype as C/T polymorphisms according to the manufacturer’s protocol. Raw array data was collected with ~866,000 probes for 12 samples across four groups: control and NEN-treated under normoxia or hypoxia. The fluorescence signals were measured from the BeadArrays using an Illumina BeadStation GX Scanner. The data analysis was performed in the R environment. A detection p-value for every probe in every sample, representing the signal quality, was generated using the minfi method (20). Poor probes with all samples’ p-values ≥0.01 were removed, leaving ~864,000 probes. Analyses used methylation level estimations based on M-Values (log2-based methylation over unmethylation signal). Differentially methylated probes were identified by first standardizing M-Values using a Student’s two-tailed t-test. Significant hits were identified based on a Student’s two-tailed t-test p < 0.05 and absolute M value difference > 0.5 between groups. Differentially methylated probes were annotated using the R CHAMP package.
TP53 mutation analysis
Mutational analysis for exons 5 to 9 of the TP53 gene was performed using direct sequencing. DNA extraction was performed using the PureLink Genomic DNA Mini Kit (Gibco; K182001) according to the manufacturer’s protocol. Each PCR was performed with an initial denaturation step of 3 min at 95°C, followed by 35 cycles of 95°C for 10 s, 58°C for 15 s, 72°C for 15 s, and a final extension step of 72°C for 5 min using the Phusion High-Fidelity PCR Kit (Thermo Fisher Scientific; #F553L). The PCR products were recovered after agarose gel electrophoresis. Both forward and reverse strands were sequenced to confirm TP53 mutations. The primers used are listed in Supplemental Table 4.
Neurite outgrowth assay
The quantification of neurite outgrowth was performed as previously described (4). Briefly, 1 × 104 SK-N-BE(2) and NB16 cells per well were inoculated into a 12 well-plate. After overnight incubation, cells were treated with the indicated conditions for 72 h. Then, images were taken by a Leica Florescent Microscope Dmi8 in phase contrast mode (20× magnification). Neurite lengths were traced and quantified using the ImageJ plugin NeuronJ (21). For each sample, total neurite length was measured and normalized to the number of cell bodies. Data are presented as mean ± SD across three biological replicates.
Immunofluorescence staining
SK-N-BE(2) and NB16 cells were seeded into 8-champer slides (Thermo Fisher Scientific; 154534) at 6 × 103 cells/well overnight and subjected to the indicated treatment for 72 h. The procedure was performed as previously described (4). Cells were fixed with 4% paraformaldehyde in PBS with 0.1% Tween-20 at room temperature (RT) for 30 min, followed by permeabilization with 0.1%Triton X-100 in PBS at RT for 10 min. Next, the cells were washed with PBS twice and blocked with 2.5% horse serum in PBS at RT for 1 h. Then, cells were subjected to immunofluorescence staining with primary antibody β3-Tubulin (CST; 5568) at 4°C overnight. Finally, after two washes with PBS, cells were incubated with Alexa Fluor 594-conjugated anti-rabbit secondary antibody (Life Technologies) at RT for 1 h, followed by staining with 4’,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, H-1800-2). Images were acquired with a Leica Dmi8 microscope. The data presented are representative of two independent experiments.
Metabolic analysis
The metabolomics and isotope tracing analyses were performed on an Agilent 1290 Infinity Liquid Chromatography (LC) System (Agilent) coupled to a Q-TOF 6545 mass spectrometer (MS; Agilent). Targeted analysis, isotopologues extraction, and natural isotope abundance correction were performed with Agilent Profinder B.10.00 (Agilent) as previously described (4). Data are presented as mean ± SD across three biological replicates.
Stable isotope tracing analysis
Glutamine isotope tracing under normoxia or hypoxia involved SK-N-BE(2) and NB16 cells being pretreated with DMSO or 1 μM NEN for 3 h. Next, the culture medium was changed to DMEM/F-12 without glutamine (Gibco; 21331020) or supplemented with 4 mM U-13C-glutamine (Cambridge Isotope Laboratories; CLM-1822-H) or 10% dialyzed FBS (Gibco; 26400044) under normoxia or hypoxia for 2 h. Data are presented as mean ± SD across three biological replicates.
Mouse orthotopic NB model
All mouse procedures were performed in accordance with Stanford University guidelines for the care and use of animals and were maintained and handled under protocols approved by the Institutional Animal Care and Use Committee. All procedures were performed with a 1:1 sex ratio of 7-week-old nude mice (Taconic). Procedures and ultrasound measurements (see below) were performed under general anesthesia using isoflurane inhalation. Orthotopic tumors within the adrenal gland were created as previously described (22). Briefly, a transverse incision was made on the left flank to locate the left adrenal gland, and 1 μL of PBS containing 105 SK-N-BE(2) or NB16 cells was injected into the adrenal gland using a 30G needle. Fascia and skin were closed in separate layers. Tumor formation was monitored by non-invasive ultrasound measurements, and diet intervention was provided once the tumor volume reached 5–10 mm3. A control diet (#D11112201) and a diet with 2000 ppm NEN (#D20101601) were obtained from Research Diets, Inc. The animals were euthanized when the tumor volume exceeded 1,500 mm3.
Monitoring tumor growth with high-frequency ultrasound
After securing the mouse in a prone position, a Vevo 2100 sonographic probe (Visual Sonics) was applied to the left flank to locate the left adrenal gland and the tumor. The monitoring of tumors by ultrasound was performed twice weekly. Serial cross-sectional images (0.076 mm between images) were taken. The tumor volume was measured using a 3D reconstruction tool (Vevo Software v1.6.0).
Histological and Immunohistochemical examination of mouse orthotopic tumors
Hematoxylin and eosin (H&E) and immunohistochemically stained sections were prepared from formalin-fixed and paraffin-embedded blocks of mouse orthotopic tumors. Immunostaining involved heating unstained sections for 30 min in Bond Epitope Retrieval Solution 2 (No. AR9640; Leica Biosystems Newcastle Ltd.) and incubating them with anti-human N-Myc rabbit monoclonal antibody (1:300 dilution; Cell Signaling Technology, #51705). Then, counter-staining with hematoxylin was performed.
Statistics
Cell proliferation and LC-MS experiments used three biological repeats for data analysis. Results are presented as mean ± SD. Mouse orthotopic NB experiment data are presented as mean ± standard error of the mean. Student’s t-tests and one- or two-way analysis of variance (ANOVA) tests were performed to assess the significance of differences between groups (two-tailed; unequal variance).
Data availability
The sequencing data (GSE209801) and methylation array data (GSE211558) were submitted to the NCBI GEO database.
Results
Mitochondrial uncoupling promotes NB cell differentiation and reduces MYCN expression
NEN is a salt form of the US Federal Drug Administration-approved mitochondrial uncoupler drug with an excellent safety profile (23,24). SK-N-BE(2) and NB16 cells were treated with NEN to investigate whether mitochondrial uncoupling could induce cell differentiation. Notably, NEN treatment induced neuron differentiation morphological changes in both SK-NE-BE(2) and NB16 cells based on neurite length measurements and immunofluorescence staining against the neuron differentiation marker β-tubulin III (Figure 1A, B). We confirmed that mitochondrial uncoupling could induce NB differentiation by treating NB cells with another mitochondrial uncoupler, BAM15 (25). Similarly, BAM15 treatment induced neuron differentiation morphological changes in both SK-NE-BE(2) and NB16 cells (Supplemental Figure 1A). In addition, NEN and BAM15 treatment induced cell proliferation arrest, which is usually associated with cell differentiation (Figure 1C; Supplemental Figure 1C). The half maximal inhibitory concentration (IC50) for NEN in inhibiting cell proliferation was ~0.31 μM in SK-N-BE(2) cells and ~0.70 μM in NB16 cells. The IC50 of BAM15 in inhibiting cell proliferation was ~2.76 μM in SK-N-BE(2) cells and ~4.53 μM in NB16 cells. We determined whether proliferation arrest was the cause of differentiation by treating cells with an antimetabolite, 5-fluorouracil, and a cyclin-dependent kinase 4/6 inhibitor, palbociclib. Both drugs arrested proliferation but did not cause neuron differentiation morphological changes (Supplemental Figure 2C, D), indicating that proliferation arrest was not the cause of cell differentiation.
Figure 1. NEN treatment promotes neuron differentiation and reduces MYCN expression.
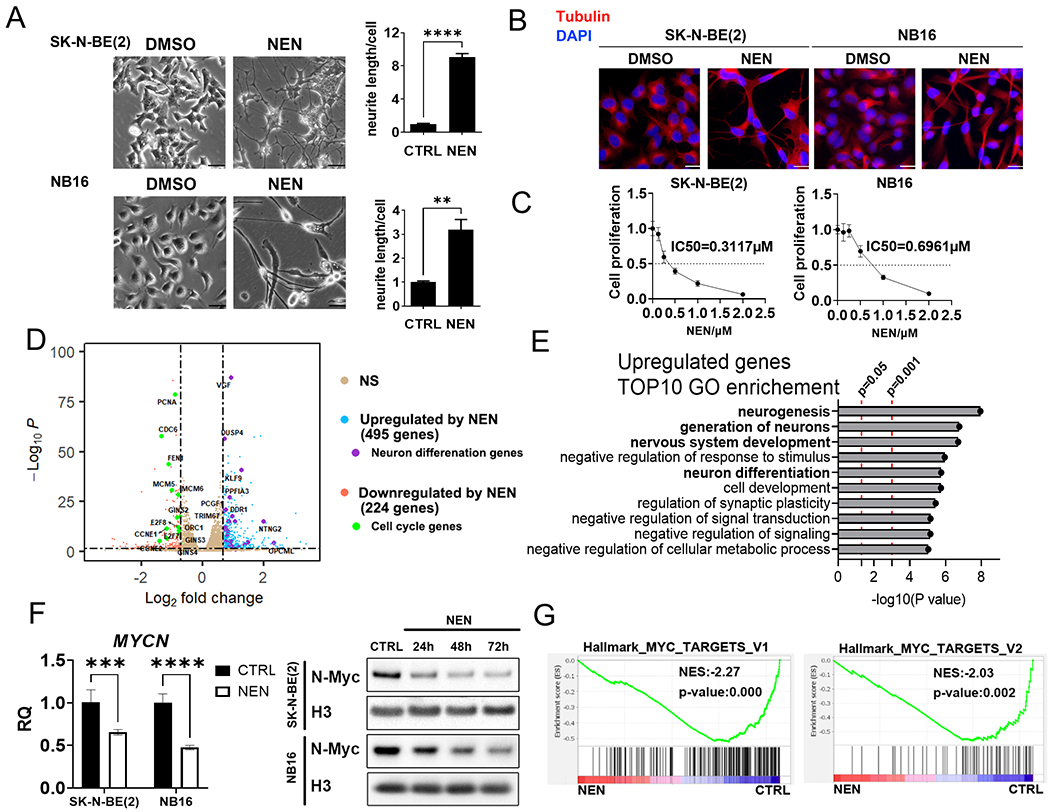
(A) Left: morphological features of NB cells treated with DMSO or 1 μM NEN for 96 h (scale bar: 50 μM). Right: Quantification of neurite outgrowth with NeuronJ. (B) Immunofluorescence staining of β-tubulin III (red) and DAPI (blue) in cells treated with DMSO or 1 μM NEN for 96 h (scale bar: 25μm). (C) Relative cell proliferation with NEN treatment for 3 d. (D) Volcano plot of gene expression distribution from the RNA-seq data (n=3) of SK-N-BE(2) cells treated with 1 μM NEN for 16 h. A horizontal dashed line denotes an adjusted p-value of 0.05. Vertical dashed lines denote an absolute log2 fold change of In(2). (E) The top 10 GO pathways enriched for NEN-upregulated genes in the DAVID analysis. (F) Left panel: MYCN mRNA levels in cells treated with 1 μM NEN for 24 h measured using RT-qPCR. Right panel: N-Myc protein levels in cells treated with NEN for the indicated time measured using immunoblots. (G) GSEA of N-Myc pathway genes from RNA-seq data in (D). Key: *, p < 0.05; **, p <0.01; ***, p < 0.001, ****, p < 0.0001 based on a Student’s two-tailed t-test. Please check the experimental replicates information in the Methods section.
Because cell differentiation requires transcriptome changes, we performed RNA-seq analysis in SK-N-BE(2) cells treated with DMSO or NEN for 16 h. We identified 495 significantly upregulated genes and 224 significantly downregulated genes with NEN treatment (Supplemental Table 1). The distribution of gene expression in the RNA-seq data is presented as a volcano plot (Figure 1D). The upregulated genes were enriched in multiple pathways, including neurogenesis, nervous system development, and neuron differentiation (Figure 1D and E; Supplemental Table 1), consistent with the neuron differentiation morphological changes. In contrast, the NEN-downregulated genes were enriched in pathways involved in DNA replication and cell cycle progression (Figure 1D; Supplemental Figure 2A; Supplemental Table 1), consistent with the inhibition of cell proliferation. In addition, GSEA of the RNA-seq gene sets showed that NEN treatment significantly reduced the expression of genes in two important cell division hallmarks, “E2F-TARGETS” and “G2M_CHECKPOINT” (Supplemental Figure 2B). MYCN, a key oncogenic transcription factor, is often amplified in NB, promoting pluripotency and unfavorable patient outcomes (26). Intriguingly, NEN treatment significantly reduced N-Myc mRNA and protein levels (Figure 1F). Consistent with this, GSEA results showed that NEN treatment significantly reduced the expression of genes in the hallmarks “MYC_TARGETS_V1” and “MYC_TARGETS_V2” (Figure 1G).
Mitochondrial uncoupling activates ETC and increases intracellular NAD+/NADH, pyruvate/lactate, and α-KG/2-HG ratios
Next, we examined whether mitochondrial uncoupling could induce cell differentiation through metabolic reprogramming. Mitochondrial uncoupling is a process that dissipates the proton gradient across the inner mitochondrial membrane, inhibiting ATP synthesis and activating the ETC to promote NADH oxidation (27,28). As expected, NEN treatment increased adenosine diphosphate (ADP)/ATP and adenosine monophosphate (AMP)/ATP ratios (Figure 2A; Supplemental Figure 3A), likely due to increased AMP and ADP levels caused by ATP synthesis inhibition (Supplemental Figure 4A, B). Importantly, NEN treatment increased the intracellular NAD+/NADH ratio in both SK-N-BE(2) and NB16 cells (Figure 2A; Supplemental Figure 3A). Also, in line with the observation that cell cycle arrest did not cause neuron differentiation morphological changes (Supplemental Figure 2C, D), 5-fluorouracil and palbociclib treatment did not alter the NAD+/NADH ratio (Supplemental Figure 2E). In addition, the pyruvate/lactate ratio, determined by the NAD+/NADH ratio (29), also increased with NEN treatment, indicating inhibition of the Warburg effect (Figure 2A; Supplemental Figure 3A).
Figure 2. NEN treatment increases the NAD+/NADH ratio to accelerate glutaminolysis.
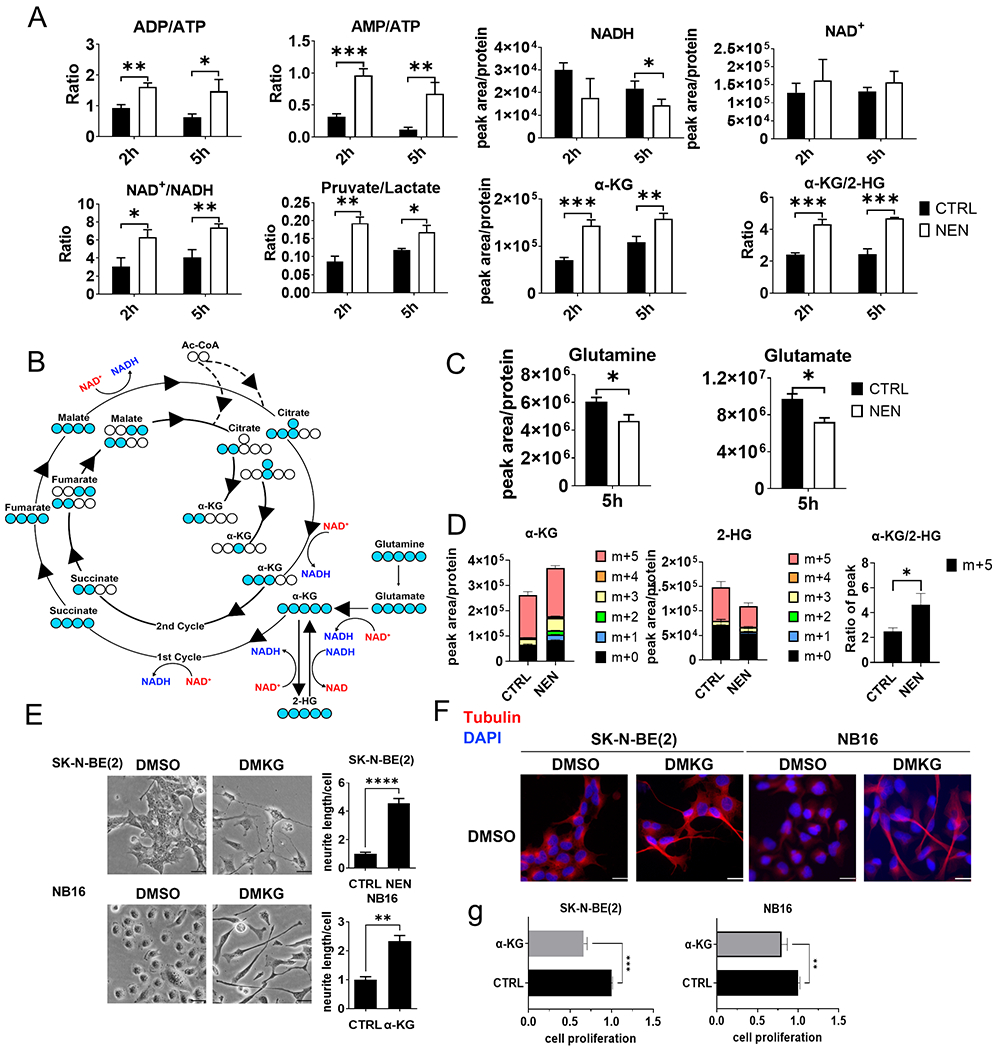
(A) Relative metabolite levels in SK-N-BE(2) cells treated with DMSO or 1 μM NEN measured using LC-MS. (B) 13C-labeling patterns of TCA cycle metabolites derived from U-13C-glutamine (Blue circle: 13C; hollow circle: 12C. (C) Relative L-glutamine and L-glutamate levels in SK-N-BE(2) cells treated with DMSO or 1 μM NEN for 5 h. (D) SK-N-BE(2) cells were pretreated with DMSO or 1 μM NEN for 3 h, then labeled with U-13C-glutamine for 2 h. (E) Left: morphological feature of cells treated with/without 3.5 mM DMKG for 96 h (scale bar: 50 μM). Right: Quantification of neurite outgrowth with NeuronJ. (F) Immunofluorescence staining for β-tubulin III (red) and DAPI (blue) in cells treated as in (E; scale bar: 25μm). (G) Cells treated with/without 3.5 mM DMKG for 3 d. All cell numbers were normalized to the control group. Key, *, p < 0.05, **, p < 0.01, ***, p < 0.001 based on a Student’s two-tailed t-test.
We also examined NEN’s metabolic reprogramming effect on other cancer cell lines, including an ovarian cancer cell line, OVCAR3, and two lung cancer cell lines, H29 and H82. All cell lines showed similar metabolic reprogramming effects, characterized by increased ADP/ATP and α-KG/2-HG ratios, indicating that NEN-induced metabolic reprogramming is not limited to NB cells (Supplemental Figure 6A, B and C). Similar to NEN, BAM15 treatment also increased NAD+/NADH and α-KG/2-HG ratios (Supplemental Figure 1B). Furthermore, NEN treatment did not appear to cause oxidative stress in our experimental setup since it did not reduce the glutathione/glutathione disulfide ratio in either SK-N-BE(2) or NB16 cells (Supplemental Figure 4A, B).
Because multiple key reactions in the tricarboxylic acid (TCA) cycle use NAD+ as an electron acceptor, a high NAD+/NADH ratio is the major driving force for the TCA cycle (Figure 2B). We next examined how mitochondrial uncoupling regulates TCA cycle metabolite levels. The levels of citrate, the first product of the TCA cycle, were unaffected by NEN treatment (Supplemental Figure 4A, B). Surprisingly, intracellular α-KG levels increased significantly in both cell lines after NEN treatment (Figure 2A; Supplemental Figure 3A). Succinate also accumulated under NEN treatment, while the α-KG/succinate ratio remained unchanged (Supplemental Figure 4A, B). No significant increases in fumarate or malate were observed under NEN treatment (Supplemental Figure 4A, B). Aspartate, derived from oxaloacetate, also accumulated under NEN treatment (Supplemental Figure 4A, B).
Because glutamate, derived from glutamine, is the major carbon source for generating α-KG in cultured cancer cells (30), we hypothesized that increased α-KG originated from glutamine. NEN treatment significantly reduced intracellular glutamine and glutamate levels, suggesting glutaminolysis acceleration via mitochondrial uncoupling (Figure 2C; Supplemental Figure 3B). We used the glutaminase inhibitor CB-839 to investigate whether glutaminolysis is necessary for NEN-induced differentiation. CB-839 treatment significantly reduced glutamate and downstream TCA cycle metabolite levels and the NAD+/NADH and α-KG/2-HG ratios (Supplemental Figure 5B), suggesting that glutaminolysis is essential for maintaining ETC activity in these cells. CB-839 treatment reduced neuron outgrowth length induced by NEN, indicating that glutaminolysis is necessary for NEN-induced NB cell differentiation (Supplemental Figure 5C).
We investigated how mitochondrial uncoupling alters glutamine flux into the TCA cycle using an isotope tracing assay with uniformly 13C-labeled glutamine ([U-13C]-glutamine; Figure 2B). Surprisingly, in SK-N-BE(2) cells, no significant increase in m+5 α-KG was observed. However, the labeling percentage of m+3 (2nd cycle) and m+1/m+2 (3rd cycle) α-KG significantly increased under NEN treatment (Figure 2D). In addition, the labeling fraction of m+2 (2nd cycle) and m+1 (3rd cycle) succinate and aspartate (from oxaloacetate) significantly increased under NEN treatment (Supplemental Figure 3C), confirming that NEN treatment accelerates TCA cycle flux in the oxidative direction. Importantly, the m+5 labeling abundance of 2-HG was significantly decreased by NEN treatment in SK-N-BE(2) cells (Figure 2D). The ratio of m+5 α-KG/m+5 2-HG increased under NEN treatment (Figure 2D), indicating that NEN treatment reduced the conversion of α-KG to 2-HG. No significant reduction of m+5 2-HG was observed in NB16 cells, possibly due to lower 2-HG generation in this cell line (Supplemental Figure 3D), while the m+5 α-KG/m+5 2-HG ratio still increased. The increased m+3/2/1 labeling in SK-N-BE(2) cells or m+5/3/1 labeling in NB16 cells reflected the α-KG increase associated with increased corresponding labeling in succinate and aspartate (Supplemental Figure 3C, D). Altogether, these data suggest that NEN treatment increased cellular α-KG through two potential mechanisms: accelerated glutaminolysis and blocked α-KG to 2-HG conversion.
We assessed whether increased intracellular α-KG levels were sufficient to induce NB cell differentiation by adding cell permeable α-KG (DMKG) to the cells and monitoring their morphological changes. Indeed, α-KG supplementation, but not two other dimethyl ethers (dimethyl carbonate and dimethyl glutarate), was able to induce a neuron differentiation morphological change in both SK-NE-BE(2) and NB16 cells, indicated by neurite length measurements and immunofluorescence staining for neuron differentiation marker β-tubulin III (Figure 2E, F; Supplemental Figure 5A), suggesting that increased intracellular α-KG levels can induce NB differentiation. Furthermore, the α-KG treatment also reduced cell proliferation in NB cells (Figure 2G).
Mitochondrial uncoupling remodels the epigenetic landscape in NB cells
Because NEN treatment increased α-KG, the TET substrate, and the α-KG/2-HG ratio, we hypothesized that NEN treatment alters the DNA methylation landscape to induce neuronal differentiation gene expression. We used an Illumina human Infinium MethylationEPIC array to study loci-specific DNA methylation changes. Genomic annotations of differentially methylated sites under NEN treatment were categorized according to their genomic location. NEN treatment generated more hypomethylated sites than hypermethylated sites in CpG islands (Figure 3A; Supplemental Table 2). In addition, NEN treatment generated more hypomethylated sites than hypermethylated sites in regions close to transcription start sites (TSS), including TSS1500, TSS200, 5’-untranslated region, and first exon (Figure 3B, Supplemental Table 2), where DNA hypermethylation is correlated with transcription suppression (7). To investigate the biological functions of this epigenetic reprogramming, we performed a gene ontology (GO) analysis of genes harboring differential methylation under NEN treatment. Under NEN treatment, genes containing hypomethylated CpG islands in the promoter region (TSS1500 and TSS200), multiple neuron differentiation, and cell death-related pathways were enriched. However, they were not within the top 10 enrichments (Figure 3C; Supplemental Table 2). In contrast, genes containing differentially methylated probes in the gene body CpG islands and with functions in pathways involved in nervous development, neuron projection morphogenesis, neuron differentiation, and axonogenesis were among the top 10 enrichments. These findings indicate that NEN treatment induced dramatic methylome reprogramming in the bodies of neuron differentiation-related genes (Figure 3D, Supplemental Table 2). We determined whether DNA demethylation was sufficient to induce cell differentiation in NB cells by treating cells with the DNA methyltransferase inhibitor 5-azacytidine (5-AZA). We found that 5-AZA treatment induced neuron differentiation morphological changes, suggesting the neuron differentiation genes could be silenced through DNA methylation, consistent with previous observations (31) (Figure 3E).
Figure 3. NEN treatment remodels the epigenetic landscape.
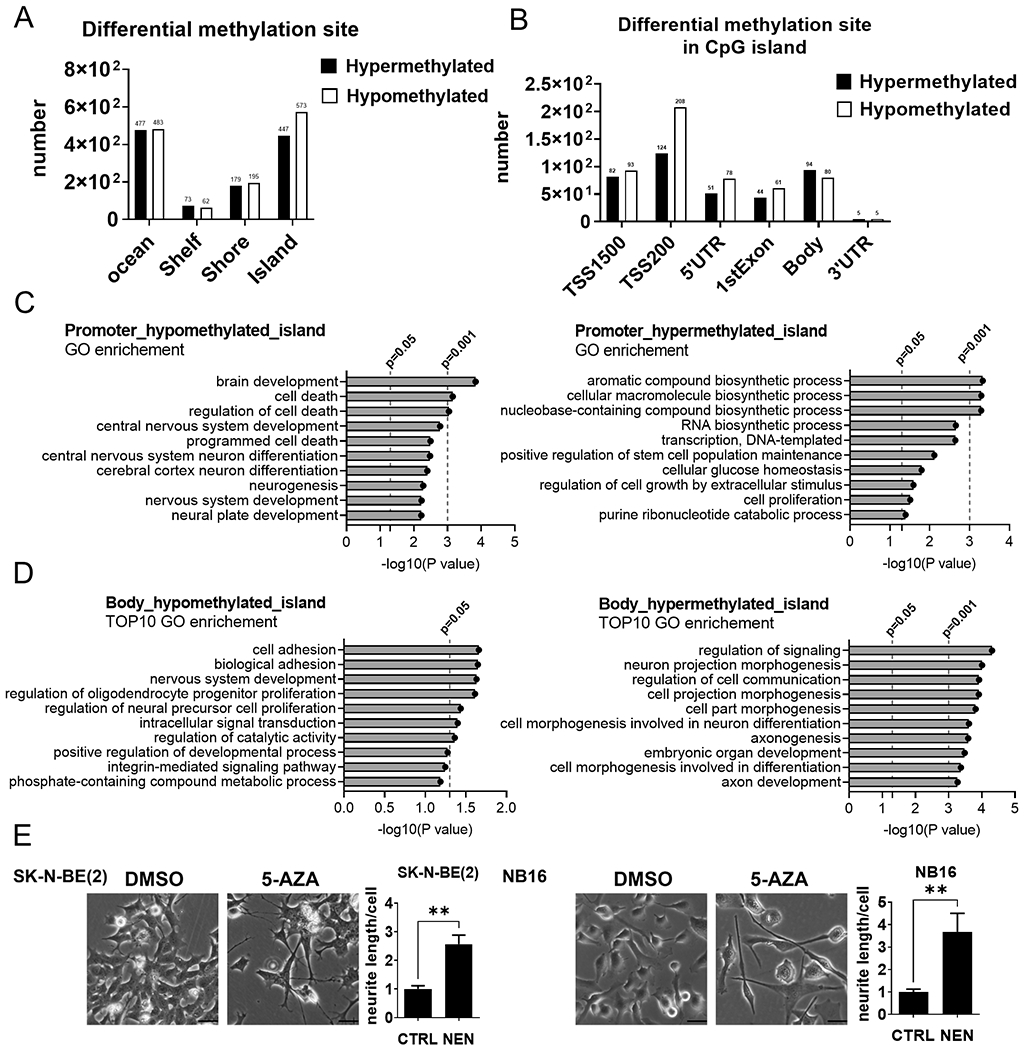
(A, B)The regional distribution of differentially methylated probes on the Illumina MethylationEPIC array in SK-N-BE(2) cells treated with DMSO or 1 μM NEN for 24 h (n=3). (C, D) GO pathway enrichment of differentially methylated probes in promoter or gene body regions. (E) Morphological feature of cells treated with DMSO or 2 μM 5-AZA for 96 h (scale bar: 50 μM). Neurite outgrowth was quantified using NeuronJ. Key: *, p < 0.05; **, p < 0.01 based on a Student’s two-tailed t-test.
It has been reported that α-KG treatment activates p53 and inhibits the Wnt/β-catenin pathway while promoting cell differentiation (11,32). Therefore, we hypothesized that NEN treatment also modulates these two pathways. Indeed, NEN treatment reduced the expression of canonical Wnt/β-catenin target genes and upregulated the expression of Wnt/β-catenin pathway inhibitors, including TP53 (33) and catenin beta interacting protein 1 (CTNNBIP1) (34), in our RNA-seq data (Supplemental Figure 7A). The downregulation of Wnt/β-catenin pathway genes transcription factor 7 (TCF7), baculoviral IAP repeat containing 5 (BIRC5), and cyclin D1 (CCND1) and upregulation of TP53 and CTNNBIP1 were validated using RT-qPCR (Supplemental Figure 7B, C). In addition, GSEA results showed that NEN treatment significantly increased the expression of genes in the hallmark “p53_pathway” (Supplemental Figure 7D). Consistent with gene expression changes, NEN treatment reduced β-catenin and increased p53 protein levels (Supplemental Figure 7E, F). Notably, 5-AZA also increased p53 protein levels, indicating that TP53 upregulation by NEN was due to DNA demethylation (Supplemental Figure 7E, F). However, 5-AZA treatment did not reduce β-catenin expression, suggesting that NEN treatment can regulate β-catenin expression at multiple levels (Supplemental Figure 7E, F).
We determined the TP53 mutation status by performing genomic sequencing of exons 5–9 in SK-N-BE(2), NB16, and another p53 wildtype (WT) cell line, CHP134. We found that SK-N-BE(2) and NB16 harbor TP53 mutations C135F and R248W, respectively (Supplemental Figure 7G), which could disrupt normal p53 function (35), suggesting that WT p53 is not necessary for NEN-induced cell differentiation. In addition, inhibition of cell proliferation and induction of neuron length outgrowth was also observed in the p53 WT NB cell line CHP134 under NEN treatment (Supplemental Figure 7H, I, J). Altogether, these data suggested that TP53 mutation status (at least for C135F and R248W) is not a key determinant in NEN-induced NB differentiation.
Mitochondrial uncoupling inhibits 2-HG generation, promotes DNA demethylation and NB differentiation, and downregulates HIF signaling under hypoxia
Clinically, tumor hypoxia is a significant obstacle to treatment because hypoxic tumor cells are more resistant to radiation (36) and chemotherapy (37). Under hypoxia, α-KG is reduced and converted to L-2-HG due to a reduced NAD+/NADH ratio (15,16), repressing α-KG-dependent dioxygenases (38), including DNA demethylase TET, and prolyl hydroxylases that hydroxylate the HIFα subunits. We found that NEN treatment could partially restore NAD+/NADH and pyruvate/lactate ratios under hypoxia (Figure 4A). Importantly, NEN treatment partially restored α-KG levels and significantly reduced 2-HG levels under hypoxia (Figure 4A). Surprisingly, NEN treatment induced dramatic global DNA demethylation changes under hypoxia, including methylation sites located in “ocean,” “shelf,” and “shore,” and especially in CpG islands (Figure 4B). NEN treatment consistently induced neuron differentiation morphological changes (Figure 4C). Importantly, 4 d of hypoxia pretreatment increased proliferation when cells were replated under normoxia conditions (without treatment). When cells were pretreated under hypoxia with NEN, this gain of proliferation advantage from hypoxia pretreatment was blocked (Figure 4D). Additionally, NEN treatment reduced the accumulation of HIFα subunits (Figure 4E) and the expression of HIF target genes such as pyruvate dehydrogenase kinase 1(PDK1), pyruvate dehydrogenase kinase 3 (PDK3), phosphoglycerate kinase 1 (PGK1), and lactate dehydrogenase A (LDHA) (Supplemental Figure 8A). In contrast, α-KG supplementation increased HIF-1α levels in SK-N-BE(2) cells (Figure 4E), possibly because supplemented α-KG was converted to 2-HG (15,39) (Supplemental Figure 8B, C). In NB16 cells, which have much lower 2-HG levels than SK-N-BE(2) cells (Supplemental Figure 8B, C), α-KG supplementation did not significantly upregulate HIF-1α (Figure 4E). Altogether, these data suggested that NEN treatment effectively promotes DNA demethylation and inhibits HIF signaling, even under hypoxic conditions.
Figure 4. NEN treatment inhibits 2-HG generation, promotes DNA demethylation, and inhibits HIF signaling under hypoxia.
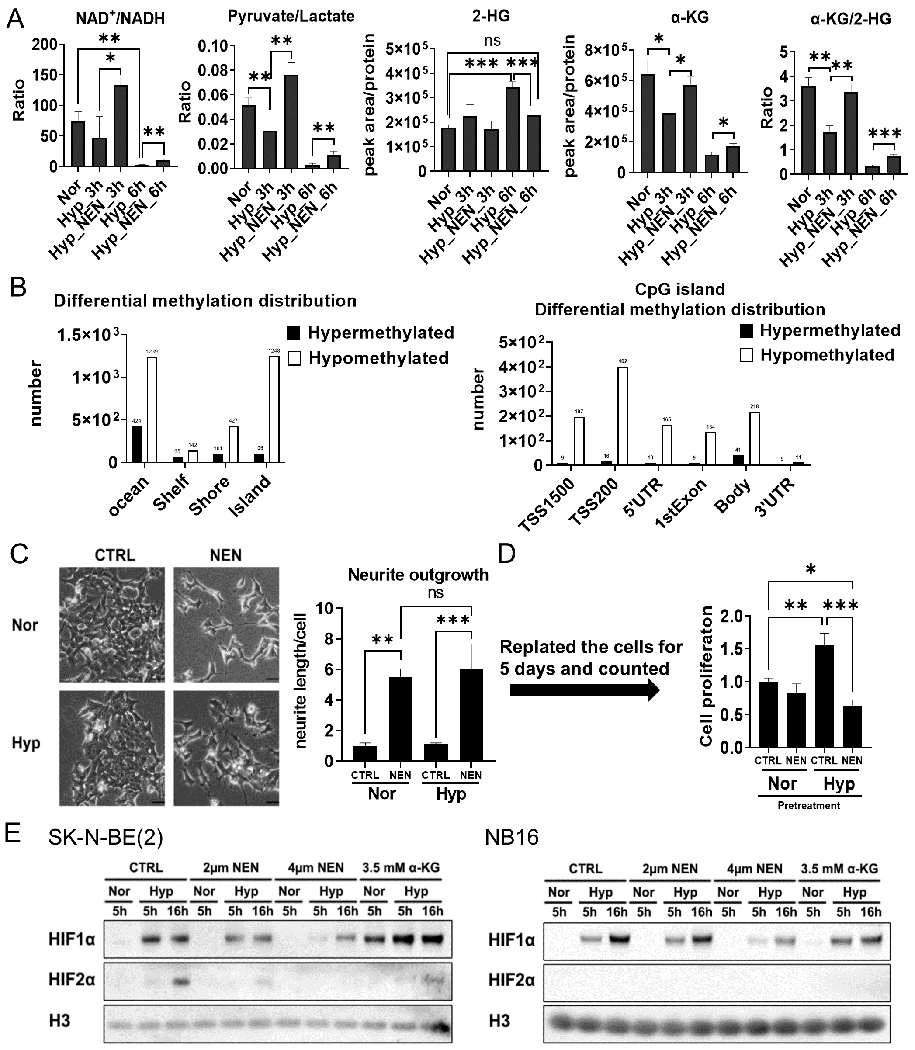
(A) LC-MS-based relative metabolite levels or ratios in SK-N-BE(2) cells treated with DMSO or 1 μM NEN under hypoxia (0.5% oxygen) for 3 h or 6 h. (B) The regional distribution of differentially methylated CpG sites on the Illumina MethylationEPIC array in SK-N-BE(2) cells treated with DMSO or 1 μM NEN under hypoxia for 24 h (n=3). (C) Left: Morphological features of SK-N-BE(2) cells treated with DMSO or 1 μM NEN under normoxia or hypoxia (0.5% oxygen) for 4 d (scale bar: 50 μM). Right: Quantification of neurite outgrowth with NeuronJ. (D) The cells in (C) were trypsinized, plated in 12-well plates without treatment, and counted after 5 d. (E) Protein levels of HIFs in SK-N-BE(2) and NB16 cells treated with DMSO, 2 μM NEN, 4 μM NEN, or 3.5 mM α-KG for the indicated time under normoxia or hypoxia (0.5% oxygen). Key: ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001 based on a one-way ANOVA test.
NEN supplementation inhibits orthotropic NB growth in vivo
Next, we examined whether NEN supplementation inhibits tumor growth in vivo using an orthotopic NB xenograft model (Figure 5A). Because intraperitoneal injection of NEN shows poor serum pharmacokinetics (Supplemental Figure 9A), we used a diet containing 2000 ppm NEN for treatment delivery (40). LC-MS analysis indicated that dietary NEN supplementation led to abundant niclosamide accumulation in mouse serum (~1 μM after 24 h and ~3 μM at the endpoint; Supplemental Figure 9B, C). A previous study showed that the liver and kidney are the two organs with the highest niclosamide accumulation (40). NEN accumulation in orthotopic NB xenograft tumors was comparable to levels found in the kidney but lower than in the liver (Supplemental Figure 9C). NEN supplementation significantly decreased SK-N-BE(2) and NB16 cell-derived tumor growth compared to the control group (Figure 5C, D; Supplemental Figure 9E). Importantly, NEN treatment significantly prolonged mouse survival (Figure 5C, D) without changing body weight or serum glucose level (Figure 5C, D; Supplemental Figure 9D). In addition, NEN treatment increased the NAD+/NADH ratio in the tumor (Figure 5B). Importantly, 2-HG levels were reduced, α-KG levels did not change, and together the α-KG/2-HG ratio was increased in the tumors in the NEN-treated group (Figure 5B; Supplemental Figure 9F). Furthermore, tumor cells from the NEN-treated group had much fewer enlarged prominent nucleoli (Figure 5E), an indicator of active ribosome biogenesis and worse prognosis in NB patients (26). Importantly, N-Myc and β-catenin protein levels were significantly reduced in NEN-treated xenografts (Figure 5E), consistent with our observations from in vitro assays (Figure 1F; Supplemental Figure 7E, F).
Figure 5. NEN supplementation reduces NB growth in vivo.
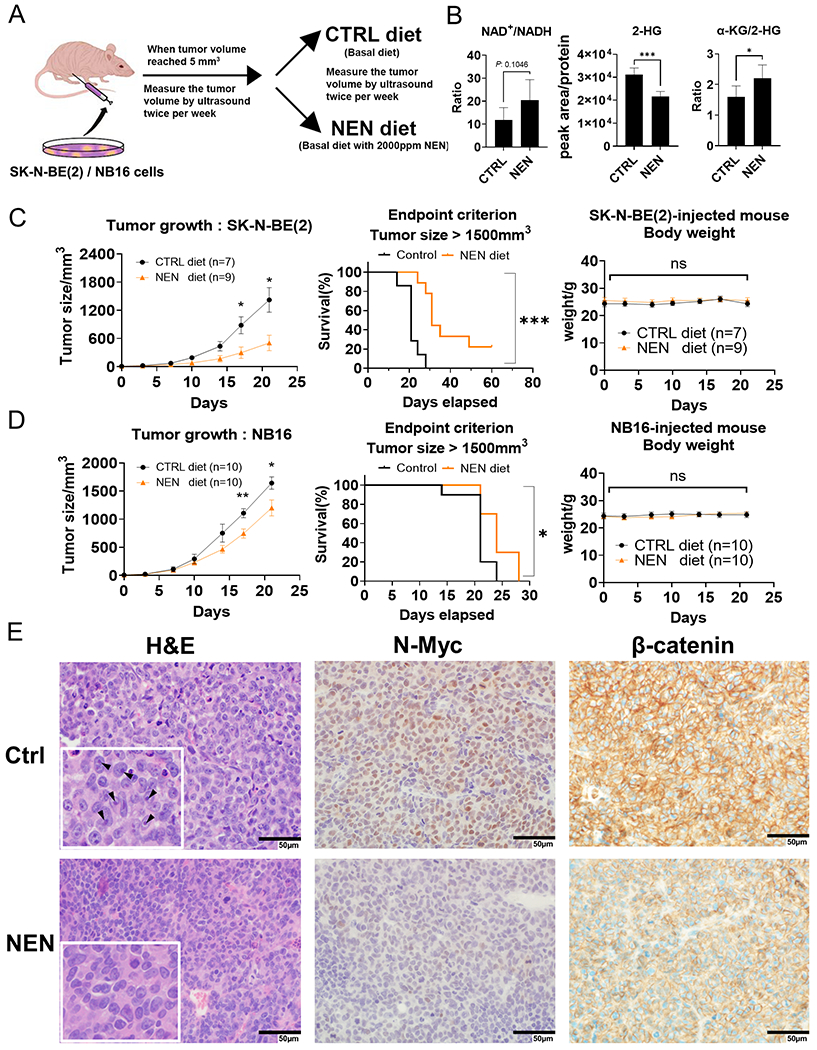
(A) Schematic of in vivo orthotopic NB xenograft experiment. (B) LC-MS-based relative metabolite levels in tumor samples (n=5). (C, D) The mice’s tumor growth, survival, and body weight curves. (E) H&E-stained tumors in control and NEN groups stained for N-Myc and β-catenin (scale bar: 50 μm); arrows indicate prominent enlarged nucleolar formations. Key: ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001 based on a Student’s two-tailed t-test.
NEN-induced gene expression changes indicate a favorable prognosis in NB patients
We further explored the clinical relevance of NEN-induced gene expression profile changes with an integration analysis using the gene expression profile from the NEN treatment experiment and 10 NB patient gene expression profiles from the R2 database (Supplemental Table 3A and B)(18). We generated gene sets indicating favorable or unfavorable prognosis from the 10 patient datasets in the R2 database (the Kaplan–Meier analysis of each gene had p < 0.05). Then, we constructed sets of overlapping genes from seven gene sets containing >1000 genes: an overlapped “favorable prognosis gene set” with 306 genes and an overlapped “unfavorable prognosis gene set” with 289 genes (Figure 6A and B; Supplemental Table 3C and D). These two gene sets were subjected to pathway enrichment analysis. The overlapped favorable prognosis gene set was enriched for multiple neuron differentiation terms, while the overlapped unfavorable prognosis gene set was enriched for cell-cycle related terms (Figure 6A and B; Supplemental Table 3D and E).
Figure 6. NEN treatment induces gene expression profile changes that indicate a favorable prognosis in NB patients.
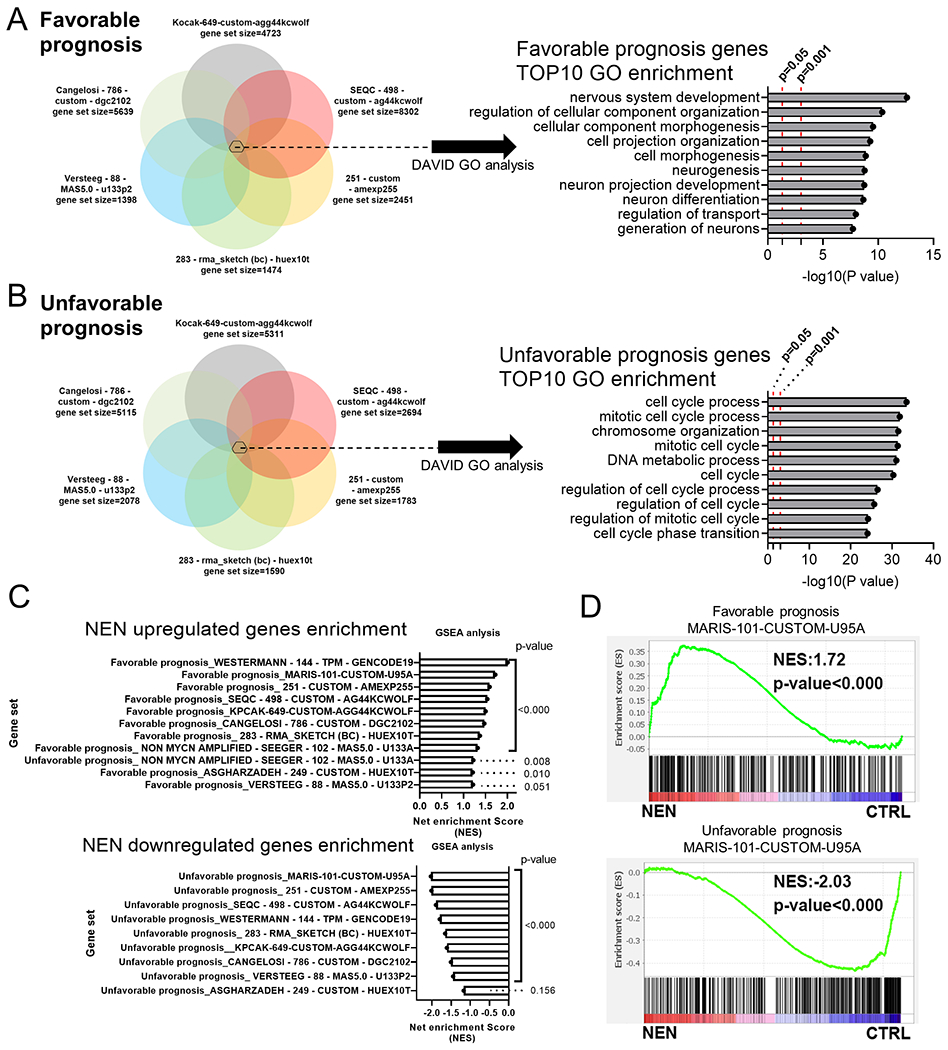
GO enrichment of (A) overlapping favorable prognosis genes (p < 0.05; >1000 genes) and (B) overlapping unfavorable prognosis genes (p < 0.05; >1000 genes) from seven available NB datasets submitted for DAVID analysis. (C) GSEA enrichment in RNA-seq gene expression data (n=3). The gene sets (favorable or unfavorable prognosis; p < 0.05) were created from 10 available NB datasets in the R2 database. (D) Representative GSEA analysis plot from (C).
Next, favorable prognosis and unfavorable prognosis gene signatures from each study were defined as pathways for GSEA analysis. Surprisingly, NEN-upregulated genes were more enriched in all the favorable prognosis gene pathways except one (Figure 6C and D; Supplemental Table 3F). In contrast, NEN-downregulated genes were more enriched in unfavorable prognosis gene pathways (Figure 6C and D; Supplemental Table 3G). The only exception was that NEN-upregulated genes were enriched in an unfavorable prognosis gene set derived from MYCN non-amplification NB. A potential explanation for this discrepancy is that our RNA-seq data was generated from SK-N-BE(2) cell line, which is an MYCN-amplified cell line (Figure 1F). We also compared our RNA-seq data with a previous study that identified niclosamide as an effective drug to target NB with a “high-risk gene signature” comprising 99 genes (41). Most of the 94 genes upregulated in the high-risk tumor signature were downregulated under NEN treatment. Among the five genes downregulated in the high-risk tumor signature, three were upregulated under NEN treatment (Supplemental Figure 10A, B). GO analysis of the “high-risk gene signature” found that multiple cell cycle processes and DNA replication pathways were enriched (Supplemental Figure 10C), consistent with the GO analysis of the NEN-downregulated genes (Supplemental Figure 2A). Altogether, these data suggest that NEN treatment reprograms the transcriptome to a gene expression profile associated with a favorable prognosis in patients with MYCN-amplified NB.
Discussion
Since Otto H. Warburg discovered that cancer cells have a high glucose consumption rate and produce large amounts of lactate in the 1920s (1,2), oncologists have been interested in how cancer cells alter metabolic pathways to gain advantages during cancer progression. In one of his milestone reviews, Warburg proposed that the cause of the Warburg effect was respiration injury and, consequently, cell dedifferentiation (3). At that time, he was aware that respiration inhibition led to metabolic reprogramming and insightfully proposed a connection between metabolic reprogramming and cell dedifferentiation. However, the underlying mechanism remained unclear due to the field’s limited understanding of the metabolic control of epigenetics.
Respiration inhibition is essentially ETC inhibition, which oxidizes cellular NADH to NAD+. ETC inhibition leads to a decrease in the NAD+/NADH ratio. A previous study examining NAD+/NADH ratios using the SoNar fluorescence sensor in cell lines showed that most cancer cell lines have lower NAD+/NADH ratios than primary cell lines (42). A low NAD+/NADH ratio not only drives lactate production from pyruvate to enhance the Warburg effect but also disrupts TCA cycle flux, promoting the chemical reduction of α-KG to form L-2-HG (15,16). Similar to D-2-HG produced by tumors carrying IDH1/2 mutations (13,14), L-2-HG also inhibits α-KG-dependent dioxygenases, including TET and Jumonji C domain-containing enzymes (38), leading to DNA and histone hypermethylation (15,43). This hypermethylation phenotype has been shown to block cell differentiation, promoting tumor progression (44,45). Here, we showed that treatment using mitochondrial uncouplers NEN and BAM15 effectively increased cellular α-KG levels in NB cells to promote differentiation. NEN treatment increased the NAD+/NADH ratio, accelerating glutaminolysis to upregulate α-KG and blocking the conversion of α-KG to 2-HG. Surprisingly, even under hypoxia, NEN treatment was still effective in restoring α-KG and reducing 2-HG levels, suggesting that NEN supplementation may be effective in vivo.
The cancer epigenome generally shows global DNA hypomethylation and promoter CpG island DNA hypermethylation. DNA hypermethylation in the promoter region leads to tumor suppressor and differentiation gene silencing. NEN treatment induced DNA hypomethylation in the promoter region and activated neuron differentiation-related gene expression. Intriguingly, NEN-induced differential methylation sites are enriched in the body of neuron differentiation genes, implying that gene body methylation status may also play a critical role in cell fate commitment. A previous study showed that DNA methylation in the gene body is critical in regulating its expression (46). Unlike pan-demethylating agents such as 5-AZA, NEN redistributed the DNA methylation landscape, decreasing methylation in the promoter while increasing methylation in the gene body, making it a more effective and precise epigenetic intervention for cancer treatment. Altogether, these data indicate that Warburg’s original hypothesis was correct: ETC activity is essential for cell differentiation. Further studies are needed to examine whether DNA demethylation by NEN treatment depends on restoration of TET activity, since there is no direct evidence provided in this study.
It has been reported that niclosamide treatment can induce TP53 expression, and p53-deficient cancer cells are more sensitive to niclosamide treatment (47). Unlike in adult cancers, where TP53 is often mutated, pediatric cancers such as NB exhibit a very low TP53 mutation rate (48). Epigenetic silencing through promoter methylation is one major mechanism of p53 inhibition in NB (48). Both NEN and 5-AZA treatment increased p53 protein levels in NB16 and SK-N-BE(2) cells, suggesting that p53 is upregulated in NB cells through DNA demethylation.
MYCN is the major oncogene amplified in NB. High MYCN expression is one key prognosis marker indicating poor survival in NB patients. MYCN is downregulated upon induction of differentiation in NB cells (49), suggesting a negative correlation between MYCN transcription and NB cell differentiation. Here, we showed that mitochondrial uncoupler treatment could diminish the “undruggable” MYCN expression, highlighting its therapeutic potential in high-grade NB patients. A previous study suggested that niclosamide can be repurposed to treat high-risk NB by inducing apoptosis and cell cycle arrest (41). Our data suggest that inducing differentiation is an alternative mechanism of niclosamide-induced tumor suppression.
In addition to α-KG upregulation, mitochondrial uncoupler treatment caused global metabolic reprogramming, including increasing NAD+/NADH and AMP/ATP ratios. These metabolic changes may further alter other signaling pathways dependent on NAD+ or ATP to remodel epigenetics and change cell fate. For example, it was well established that NEN treatment activates AMP-activated protein kinase (27), which phosphorylates and stabilizes TET (50). ETC inhibition and induction of the Warburg effect alter metabolism, causing global epigenetic reprogramming and cell dedifferentiation rather than affecting a single gene or pathway. Therefore, activating the ETC with mitochondrial uncouplers is expected to be an effective approach for reversing the Warburg effect and restoring the global metabolome and epigenome, redirecting tumor cells into a differentiated state.
Supplementary Material
Statement of Significance.
Targeting cancer metabolism using the mitochondrial uncoupler niclosamide ethanolamine leads to methylome reprogramming and differentiation in neuroblastoma, providing a therapeutic opportunity to reverse the Warburg effect and suppress tumor growth.
Acknowledgments
This work was supported by a Stanford Maternal and Child Health Research Institute Research Scholar Award (2020) and an American Cancer Society Research Scholar Grant (RSG-20-036-01) to J.Y., and NIH funding (R01NS094218) to B.C. We thank Dr. Yuqin Dai (Stanford ChEM-H Institute Metabolomics Knowledge Center) for support and use of facilities, Vanita S. Natu (Stanford Genomics) for providing Infinium MethylationEPIC analysis service, Drs. Maximilian Diehn, Beverly S. Mitchell, Laura Attardi, and Roeland Nusse (Stanford University) for valuable advice and suggestions, and John M. Snyder (Radiation Oncology) for support on statistical analysis. The graphical abstract was created with BioRender.com.
Footnotes
Competing interests
H.J., Y.L. and J.Y. submitted a patent application related to this manuscript.
References
- 1.Warburg O, Posener K, Negelein E. On the metabolism of carcinoma cells. Biochem Z 1924;152:309–44 [Google Scholar]
- 2.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol 1927;8:519–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science 1956;123:309–14 [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Gruber JJ, Litzenburger UM, Zhou Y, Miao YR, LaGory EL, et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis 2020;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab 2012;16:9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nature Reviews Cancer 2016;16:694–707 [DOI] [PubMed] [Google Scholar]
- 7.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012;13:484–92 [DOI] [PubMed] [Google Scholar]
- 8.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41 [DOI] [PubMed] [Google Scholar]
- 9.Henrich KO, Bender S, Saadati M, Dreidax D, Gartlgruber M, Shao C, et al. Integrative Genome-Scale Analysis Identifies Epigenetic Mechanisms of Transcriptional Deregulation in Unfavorable Neuroblastomas. Cancer Res 2016;76:5523–37 [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nature Reviews Genetics 2017;18:517. [DOI] [PubMed] [Google Scholar]
- 11.Morris JPt, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019;573:595–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran TQ, Hanse EA, Habowski AN, Li H, Ishak Gabra MB, Yang Y, et al. α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nature Cancer 2020;1:345–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting alpha-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010;17:225–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab 2015;22:304–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab 2015;22:291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li AM, Ducker GS, Li Y, Seoane JA, Xiao Y, Melemenidis S, et al. Metabolic Profiling Reveals a Dependency of Human Metastatic Breast Cancer on Mitochondrial Serine and One-Carbon Unit Metabolism. Mol Cancer Res 2020;18:599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koster J, Volckmann R, Zwijnenburg D, Molenaar P, Versteeg R. R2:Genomics Analysis and Visualization Platform AACR; 2019.
- 19.Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 2009;4:44. [DOI] [PubMed] [Google Scholar]
- 20.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30:1363–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry Part A: the journal of the International Society for Analytical Cytology 2004;58:167–76 [DOI] [PubMed] [Google Scholar]
- 22.Chiu B, Coburn J, Pilichowska M, Holcroft C, Seib FP, Charest A, et al. Surgery combined with controlled-release doxorubicin silk films as a treatment strategy in an orthotopic neuroblastoma mouse model. Br J Cancer 2014;111:708–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrews P, Thyssen J, Lorke D. The biology and toxicology of molluscicides, Bayluscide. Pharmacology & therapeutics 1982;19:245–95 [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Mook RA Jr., Premont RT, Wang J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018;41:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenwood BM, Weaver JL, Bajwa A, Poon IK, Byrne FL, Murrow BA, et al. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab 2014;3:114–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niemas-Teshiba R, Matsuno R, Wang LL, Tang XX, Chiu B, Zeki J, et al. MYC-family protein overexpression and prominent nucleolar formation represent prognostic indicators and potential therapeutic targets for aggressive high-MKI neuroblastomas: a report from the children’s oncology group. Oncotarget 2018;9:6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alasadi A, Chen M, Swapna GVT, Tao H, Guo J, Collantes J, et al. Effect of mitochondrial uncouplers niclosamide ethanolamine (NEN) and oxyclozanide on hepatic metastasis of colon cancer. Cell Death Dis 2018;9:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell 2021;81:691–707 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patgiri A, Skinner OS, Miyazaki Y, Schleifer G, Marutani E, Shah H, et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat Biotechnol 2020;38:309–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences 2007;104:19345–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westerlund I, Shi Y, Toskas K, Fell SM, Li S, Surova O, et al. Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2alpha to tumor suppression. Proc Natl Acad Sci U S A 2017;114:E6137–E46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran TQ, Hanse EA, Habowski AN, Li H, Gabra MBI, Yang Y, et al. alpha-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat Cancer 2020;1:345–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadot E, Geiger B, Oren M, Ben-Ze’ev A. Down-regulation of β-catenin by activated p53. Molecular and cellular biology 2001;21:6768–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daniels DL, Weis WI. ICAT inhibits β-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Molecular cell 2002;10:573–84 [DOI] [PubMed] [Google Scholar]
- 35.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014;25:304–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hockel M, Schlenger K, Mitze M, Schaffer U, Vaupel P. Hypoxia and Radiation Response in Human Tumors. Seminars in radiation oncology 1996;6:3–9 [DOI] [PubMed] [Google Scholar]
- 37.Teicher BA, Holden SA, al-Achi A, Herman TS. Classification of antineoplastic treatments by their differential toxicity toward putative oxygenated and hypoxic tumor subpopulations in vivo in the FSaIIC murine fibrosarcoma. Cancer research 1990;50:3339–44 [PubMed] [Google Scholar]
- 38.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol 2017;13:494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med 2014;20:1263–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang CT, Hsieh CH, Lee WC, Liu YL, Yang TS, Hsu WM, et al. Therapeutic Targeting of Non-oncogene Dependencies in High-risk Neuroblastoma. Clin Cancer Res 2019;25:4063–78 [DOI] [PubMed] [Google Scholar]
- 42.Zhao YZ, Wang AX, Zou YJ, Su N, Loscalzo J, Yang Y. In vivo monitoring of cellular energy metabolism using SoNar, a highly responsive sensor for NAD(+)/NADH redox state. Nat Protoc 2016;11:1345–59 [DOI] [PubMed] [Google Scholar]
- 43.Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 2014;4:1290–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014;26:577–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar R, Coronel L, Somalanka B, Raju A, Aning OA, An O, et al. Mitochondrial uncoupling reveals a novel therapeutic opportunity for p53-defective cancers. Nat Commun 2018;9:3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7 [DOI] [PubMed] [Google Scholar]
- 49.Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985;313:404–6 [DOI] [PubMed] [Google Scholar]
- 50.Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018;559:637–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data (GSE209801) and methylation array data (GSE211558) were submitted to the NCBI GEO database.