

Abstract
Objective:
Identify molecular mechanisms in brain tissue of Rasmussen encephalitis (RE) when compared to people with non-RE epilepsy (PWE) and control cases using whole exome sequencing (WES), RNAseq, and proteomics.
Methods:
Frozen brain tissue (ages 2–19 years) was obtained from control autopsy (n=14), surgical PWE (n=10), and surgical RE cases (n=27). We evaluated WES variants in RE associated with epilepsy, seizures, RE, and human leukocyte antigens (HLAs). Differential expression was evaluated by RNAseq (adjusted p<0.05) and label-free quantitative mass spectrometry (false discovery rate<5%) in the three groups.
Results:
WES revealed no common pathogenic variants in RE, although several rare and likely deleterious variants of unknown significance (VUS; ANGPTL7/MTOR, SCN1A, FCGR3B, MTOR) and more common HLA VUS in >25% RE cases (HLA-DRB1, HLA-DQA2) all with allele frequency <5% in the general population. RNAseq in RE vs. PWE (1516 altered transcripts) revealed significant activation of crosstalk between dendritic and natural killer cells (p=7.94×10−6, z=2.65), in RE vs. control (7466 transcripts) neuroinflammation signaling activation (p=6.31×10−13, z=5.07), and in PWE vs. control (945 transcripts) phagosome formation activation (p=2.00×10−13, z=5.61). Proteomics detected fewer altered targets.
Significance:
In RE, we identified activated immune signaling pathways and immune cell type annotation enrichment that suggest roles of the innate and adaptive immune responses, as well as HLA variants that may increase vulnerability to RE. Follow up studies could evaluate cell type density and subregional localization associated with top targets, clinical history (neuropathology, disease duration), and whether modulating crosstalk between dendritic and natural killer cells may limit disease progression.
Introduction
Rasmussen encephalitis (RE) is a unilateral encephalitis characterized by treatment-resistant epilepsy and progressive cognitive and motor decline.1 MRI reveals inflammation and neuropathology reveals reactive astrocytes, microglial nodules, T cell infiltration, and neuronal loss.1,2
Immune mechanisms are implicated in RE. Brain-infiltrating cells include clonally expanded CD8+ T cells, suggesting T cell cytotoxicity responding to an antigen, and adjacent neuron and astrocyte loss.1,3–7 Th1 activation was supported by targeted transcript analysis in RE brain compared to cortical dysplasia cases8 and T cell cytokine release.9 Less frequently, natural killer (NK) and myeloid cells infiltrate brain.3,5 Targeted genetic analyses link human leukocyte antigen (HLA) haplotypes with RE (HLA-DQA1, HLA-DQB1, HLA-C, HLA-DRB1),10 and some (HLA-DQA1, HLA-DQB1) are more frequent in RE brain compared to temporal lobe epilepsy (TLE) on whole exome sequencing (WES).11 Despite abundant evidence of altered signaling pathways in RE brain and associated genetic variants, mechanisms remain poorly understood, and it is uncertain which are a part or result of the underlying cause.
We evaluated molecular mechanisms in surgical brain tissue from RE compared to cases from surgical people with non-RE epilepsy (PWE) and autopsy controls by WES, RNAseq, and proteomics.
Materials and Methods
Specimens.
Frozen brain tissue (Table 1, Supplemental Table 1) was obtained after Institutional Review Board approval and in accordance with the Declaration of Helsinki from NYU Grossman School of Medicine, Amsterdam UMC, and UCLA. Consent was provided by parent/legal guardian(s). Autopsy controls (postmortem interval<5hours) were obtained from Amsterdam UMC (n=9) and NIH NeuroBioBank (n=5). We excluded cases with seizure history, neurological disease, and neuropathology. Surgical PWE was obtained from NYU Epilepsy Brain Bank (n=10), with diagnoses excluding RE and an encephalomalacia with herpes simplex virus case. Surgical RE (n=27), based on diagnostic guidelines,12,13 was obtained from UCLA. To match RE, control and PWE included 2–19 year olds. RE demographics were limited, with sex identified by RNAseq (n=10) with XIST and DDX3Y.14
Table 1.
Case History Summary
| Group | Cases | Age | Sex |
|---|---|---|---|
| Control | 14 | 13.5 ± 5.4 | 6 F / 8 M |
| PWE | 10 | 11.4 ± 1.9 | 1 F / 9 M |
| RE | 27 | 8.7 ± 3.9 | 21 F / 6 M |
Mean ± standard deviation is indicated. Known demographic information indicated for RE cases (age, n = 22).
WES.
DNA was isolated from RE brain (10mg) with DNeasy Purification of Total DNA (Qiagen) by handheld pestle-equipped device, exome libraries prepared by Illumina DNA prep with Enrichment (Cat.20025524), capture with Illumina Human Exome Panel (45 Mb, Cat.20020183), and sequencing at NYU Genome Technology Center (GTC) on Illumina NovaSeq6000 over S1 200 and SP200 cycle flow cells, 100bp paired-end. Average coverage: 143X (range 81–174X, n=27; mean coverage ≥119X for n=25). Annotations were by GATK Haplotype caller: depth≥10, alternate counts≥5. Variants were reviewed in Qiagen Clinical Insight (QCI) with phenotype filters: RE, epilepsy, seizures with “include related diseases above.” Variants were further filtered by Combined Annotation Dependent Depletion (CADD) score (deleterious (>20), likely deleterious (10–19), unlikely deleterious (<10))15 and maximum population allele frequency<0.05.
RNAseq.
RNA was isolated from brain (10mg) with miRNeasy Purification of Total RNA (Qiagen), using Qiazol with handheld pestle-equipped device, 22-gauge needle. RNA quality and concentration was determined by Agilent Bioanalyzer. Libraries were created by NYU GTC with Illumina Stranded Total RNA Prep with Ribo-Zero Plus (Cat.20040529), and sequenced on Illumina NovaSeq6000 with S2 100-cycle flow cell, 50bp paired-end.
Principal component analysis (PCA) and differential expression was evaluated with NYU Applied Bioinformatics Laboratory. RNAseq was analyzed by sns rna-star pipeline (https://igordot.github.io/sns/routes/rna-star.html). Adapters and low quality bases were trimmed using Trimmomatic (v0.36).16 Reads were mapped to reference genome (hg38) using STAR aligner (v2.7.3),17 alignments guided by Gene Transfer Format (GTF), and mean read insert sizes and standard deviations calculated using Picard tools (v.2.18.20). Genes-samples counts matrix was generated using featureCounts (v1.6.3),18 normalized by library size factors using DEseq2,19 and differential expression analysis performed in R environment (4.0.3). The Read Per Million (RPM) normalized BigWig files were generated using deepTools (v.3.1.0).20 Venn diagrams were generated with InteractiVenn.21
Gene biotypes were determined with ensembldb, AnnotationHub, Ens.Db.Hsapiens.v100. Brain cell type annotations were determined,22–24 with 1066 possible annotations with each gene having one annotation (except when annotated as both excitatory and inhibitory, gene generally annotated as “neuron”). In comparing previous brain dataset (n=1066), immune cell dataset (n=318),25 and original brain dataset,24 genes with duplicate annotations were removed and left 1365 possible annotations. We annotated 1285 genes (1002 brain, 283 immune). Cell type enrichment among differentially expressed transcripts was determined by Fisher’s exact test. Pathways associated with altered genes were identified by Ingenuity Pathway Analysis (IPA, Qiagen), with Core Analysis enrichment at p<0.05 and activated/inhibited |z-score|≥2. RNAseq and proteomics were evaluated by Pearson correlation.
Label-free Quantitative Mass Spectrometry (MS).
MS was performed with NYU Proteomics Laboratory. Brain (10mg) was solubilized in 100% TFA by “SPEED” prep.26 Samples were incubated in TFA (3min, 73°C), neutralized by 9-fold excess (v/v) 2M Tris, 10mM TCEP, 20mM chloroacetamide (30min, 90°C), centrifuged, and supernatants collected. Samples were diluted 6-fold with water containing sequencing-grade modified trypsin (mass 50:1, protein to trypsin). Overnight digestion (37°C) was stopped by TFA (final 2%) and 1ug peptide/sample loaded on Evosep C18 Tips. Peptides were eluted onto analytical LC column coupled to mass-spectrometer and separated on C18 analytical column (15cm × 150um ID, packed with ReproSil-Pur C19 1,9A beads, Evosep V1106) over 88min gradient using extended Evosep One method (15SPD) on Evosep One LC system. Peptides were directly eluted into Orbitrap HF-X mass-spectrometer (ThermoFisher) in data-independent mode (DIA) with MS2 fragmentation across 22m/z windows every MS1 scan event. High resolution full MS spectra were acquired with 120,000 resolution, 3e6 AGC target, 60ms maximum ion injection time, and 350–1650m/z scan. Following each MS scan, data-independent HCD MS/MS scans were acquired at 30,000 resolution, 3e6 AGC target, stepped NCE of 22.5, 25 and 27.5. Data were analyzed using Spectronaut, searched in directDIA mode against SwissProt human Uniprot database. Database search was performed in integrated search engine Pulsar. Enzyme specificity: trypsin, 2 maximum missed cleavages. Oxidation of methionine was searched as variable modification; cysteine carbamidomethylation was searched as fixed modification. False discovery rate (FDR) for peptide, protein, and site identification was 1%. Protein quantification was performed on MS2 level using 3 most intense fragment ions/precursor. Data was log transformed and normalized using median intensity across all samples; common contaminants, particularly hemoglobin, were excluded to reduce contamination effect. Raw file MassIVE ID:MSV000089203.
Protein matrix (n=3,886) was filtered for proteins quantified in ≥48% cases in ≥1 group (RE, PWE, Control) to n=3,721. For PCA, missing values were imputed from normal distribution (0.3width, 1.8downshift, relative to measured protein intensity distribution). Two sample t-tests were performed in Perseus v.1.6.2.3, FDR<5% (permutation-based with 250 data randomizations). Cell type annotations determined as above. We annotated 444 proteins (402 brain, 42 immune). Pathways were identified with IPA as above.
Results
WES
RE brain WES (n=27) identified no common pathogenic or likely pathogenic variants. Seventeen nonsynonymous variants of unknown significance (VUS) were identified in seven genes from 11 cases with CADD>20 (n=6): ANGPTL7/MTOR, C5, SCN1A, FCGR3B; CADD 10–19 (n=6): FCGR2B, C4A, C5; and CADD<10 (n=5): FCGR2A, FCGR3B, C5. ANGPTL7/MTOR variants were present in the ANGPTL7 exon plus strand and in the MTOR intron minus strand. Among the VUS, all were heterozygous (except FCGR2B, rs2125684) and only one variant occurred in two cases (FCGR2A, rs382627; CADD<10). Of immune related genes (excluding MTOR, SCN1A), there were VUS in nine cases with CADD>10. Among variants with CADD>20, five variants occurred at maximum population allele frequency<0.05: ANGPTL7/MTOR, SCN1A, and FCGR3B (Supplemental Table 2).
We examined HLA variants, reportedly associated with RE.10,11 We found nonsynonymous VUS in 13 HLA genes. Of HLA VUS in >25% RE (n=7/27) and maximum population allele frequency<0.05, there were six variants in three genes: HLA-DQA2 (rs62623408, rs148720159), HLA-DQB2 (rs200716952), and HLA-DRB1 (rs2308775, rs17882455, rs36044702). All were CADD>20 except HLA-DQB2 (CADD 10–19), and all variants were heterozygous (except HLA-DRB1, rs17882455, n=1). All three HLA-DRB1 variants were present in six cases.
RNAseq Differential Expression
After RNAseq in control (n=14), PWE (n=10), and RE (n=25), PCA segregated RE from control in PCA1 (p=0.0040; Figure 1A–B). In PCA2, there was segregation of RE and PWE (p=0.0015) as well as PWE and control (p=0.023; Figure 1C). Differential expression identified altered transcripts in each pairwise comparison (Supplemental Tables 3–5) and overlap of some transcripts (Figure 1D). When comparing RE and PWE, there were 1516 altered (Figure 1E). For RE vs. control, there were 7466 altered (Figure 1F). For PWE vs. control, there were 945 altered (Figure 1G).
Figure 1. RNAseq PCA and differential expression analysis.
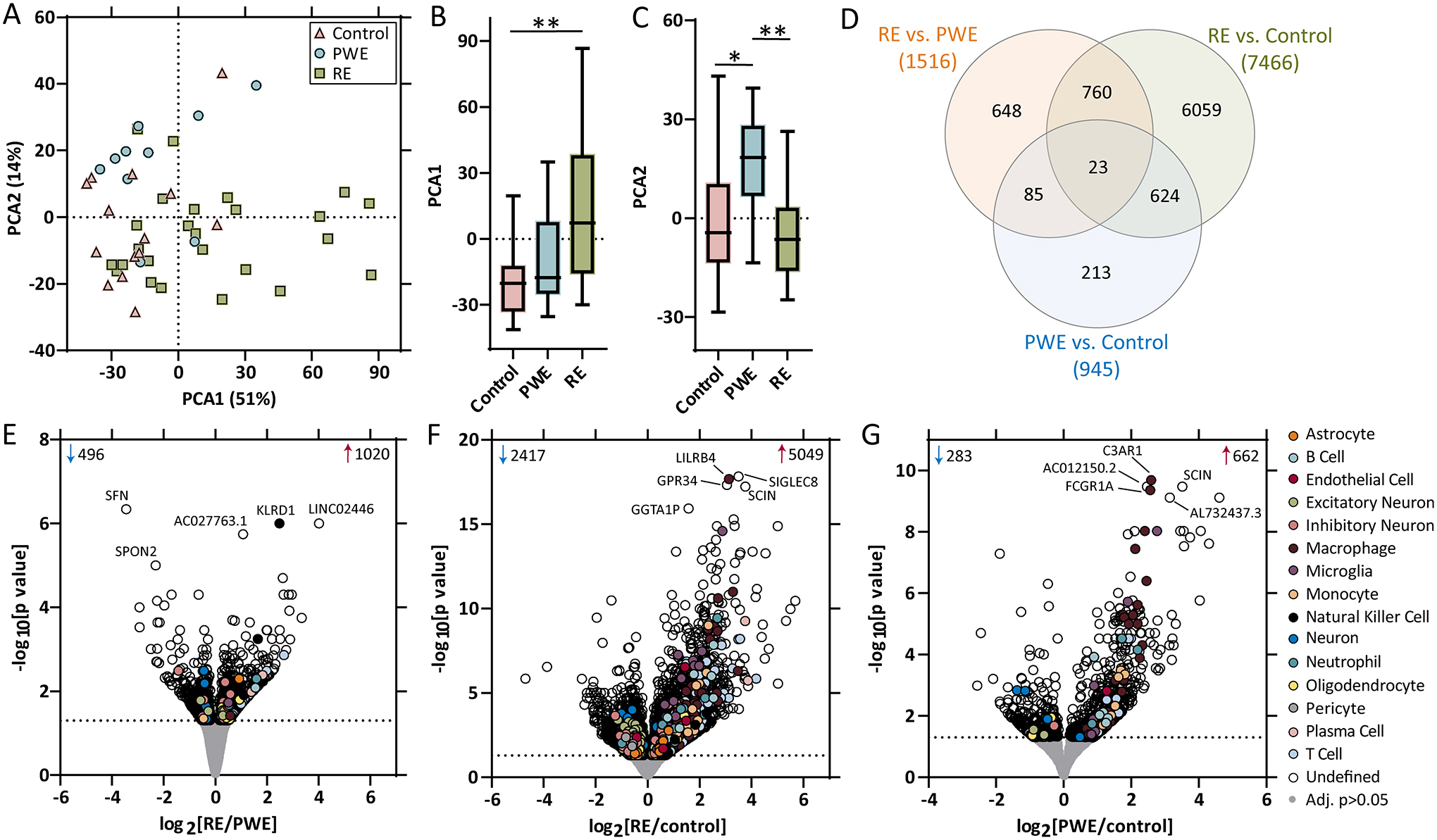
A-B) PCA of RNAseq (control: n = 14, PWE: n = 10, RE: n = 25) in brain tissue indicated segregation of the RE group from the control group in PCA1 (p = 0.0040). C) In PCA2, there was segregation of the control and PWE groups (p = 0.023), as well as the PWE and RE groups (p = 0.0015). D) Differential expression analysis for each pairwise comparison are indicated, as well as overlap in the number of significant transcripts, at an adjusted p value < 0.05 (dotted line), when comparing E) RE vs. PWE (1516 transcripts), F) RE vs. control (7466 transcripts), and G) PWE vs. control (945 transcripts). Annotations include the number of significantly increased (red arrow) and decreased (blue arrow) transcripts, top 5 altered transcripts are annotated by gene name, and brain and immune cell type annotations for each significant transcript are indicated.
RNAseq Cell Type Annotation
Most transcripts were ubiquitously expressed by multiple cell types or it was undetermined (Figure 1E–G, Supplemental Tables 3–5). In each pairwise comparison, there was enrichment for both brain and immune cell type annotations. Among the 1516 transcripts in RE vs. PWE, there was enrichment for immune cell type [T cell (p=3.46×10−6), NK cell (p=6.53×10−5), plasma cell (p=2.29×10−2)] and brain cell type annotations [neuron (p=2.00×10−3), excitatory neuron (p=4.85×10−2); Figure 2A]. For the 7466 transcripts in RE vs. control, there was enrichment for immune cell type [T cell (p<2.20×10−16), macrophage (p<2.20×10−16), monocyte (p=2.58×10−6), NK cell (p=5.00×10−4), B cell (p=2.51×10−2), neutrophil (p=3.91×10−2)] and brain cell type annotations [microglia (p=4.25×10−7), oligodendrocyte (p=1.00×10−4), excitatory neuron (p=6.90×10−3); Figure 2B]. For the 945 transcripts in PWE vs. control, there was enrichment for immune cell type [macrophage (p<2.20×10−16), monocyte (p=2.71×10−5), T cell (p=3.00×10−4), B cell (p=1.50×10−3)] and brain cell type annotations [microglia (p=4.27×10−6), neuron (p=1.20×10−2); Figure 2C].
Figure 2. Cell types and signaling pathways associated with differentially expressed transcripts by RNAseq.
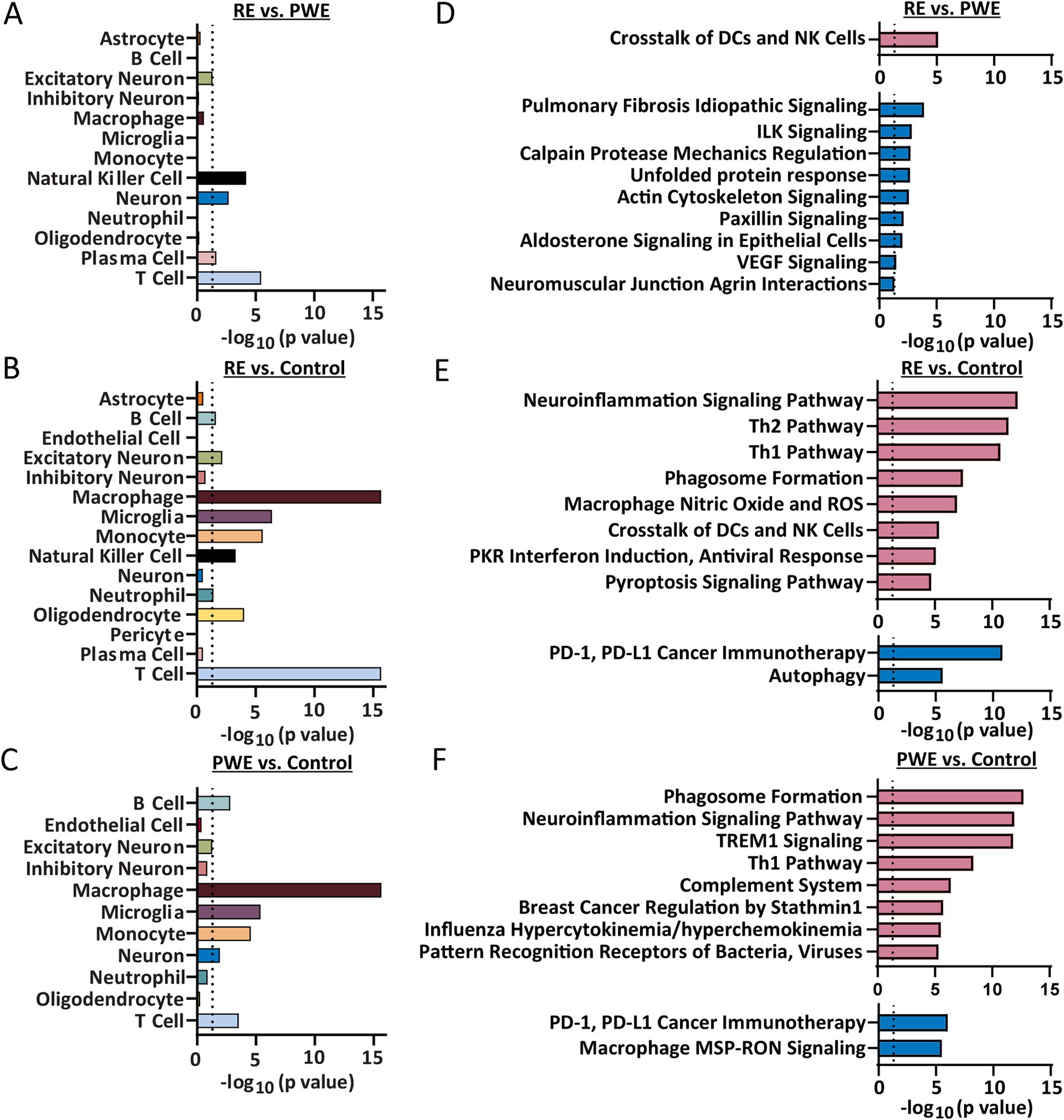
All cell type annotations associated with significantly altered transcripts are indicated for each pairwise comparison. A) Cell type annotation analysis of differentially expressed transcripts by Fisher’s exact test indicated enrichment (p < 0.05) for transcripts associated with T cell (p = 3.46 × 10−6), NK cell (p = 6.53 × 10−5), neuron (p = 2.00 × 10−3), plasma cell (p = 2.29 × 10−2), and excitatory neuron (p = 4.85 × 10−2) in RE vs. PWE. B) For RE vs. control, there was enrichment for transcripts with annotations for T cell (p < 2.20 × 10−16), macrophage (p < 2.20 × 10−16), microglia (p = 4.25 × 10−7), monocyte (p = 2.58 × 10−6), oligodendrocyte (p = 1.00 × 10−4), NK cell (p = 5.00 × 10−4), excitatory neuron (p = 6.90 × 10−3), B cell (p = 2.51 × 10−2), and neutrophil (p = 3.91 × 10−2). C) For PWE vs. control, there was enrichment for transcripts with annotations for macrophage (p < 2.20 × 10−16), microglia (p = 4.27 × 10−6), monocyte (p = 2.71 × 10−5), T cell (p = 3.00 × 10−4), B cell (p = 1.50 × 10−3), and neuron (p = 1.20 × 10−2). D) For RE vs. PWE, the 1516 altered transcripts were significantly associated with 1 activated pathway (red) and 9 inhibited pathways (blue), detailed further in Supplemental Table 6 (p value of overlap < 0.05, z score ≥|2|). The most significantly altered pathway was activation of crosstalk between DCs and NK cells (p value of overlap = 7.94 × 10−6, z = 2.65). E) For RE vs. control, the 7466 transcripts were associated with 23 activated and 18 inhibited pathways, detailed further in Supplemental Table 7. The top 10 significantly altered pathways are indicated, the most significant was activation of the neuroinflammation signaling pathway (p = 6.31 × 10−13, z = 5.07). F) For PWE vs. control, the 945 transcripts were associated with 27 activated and 3 inhibited pathways, detailed further in Supplemental Table 8. The top 10 significantly altered pathways are indicated, the most significant was activation of the phagosome formation pathway (p = 2.0 × 10−13, z = 5.61).
Among top enriched immune cell type annotations, T cell annotations were most significant in RE vs. PWE (15 increased transcripts) and RE vs. control (58 increased). By comparison in PWE vs. control, there were 8 increased and 1 decreased T cell transcripts. After T cell, NK cell transcripts were most significant in RE vs. PWE (5 increased). For RE vs. control, there were 8 increased NK cell transcripts. NK cell annotation enrichment was not present in PWE vs. control. Macrophage annotation enrichment was present in RE vs. control (55 increased) and PWE vs. control (32 increased), but not in RE vs. PWE.
The top two brain cell type annotations in each pairwise comparison included: for RE vs. PWE were neuron (7 decreased, 1 increased) and excitatory neuron (3 increased, 2 decreased), for RE vs. control were microglia (17 increased) and oligodendrocyte (39 increased, 2 decreased), and for PWE vs. control were microglia (7 increased) and neuron (78 decreased, 4 increased).
RNAseq Signaling Pathways
For RE vs. PWE, the 1516 transcripts were associated with ten signaling pathways (p<0.05, |z-score|≥2; 1 activated, 9 inhibited; Supplemental Table 6). Most significant was activation of crosstalk between dendritic cells (DCs) and NK cells (p=7.94×10−6, z=2.65; Figure 2D). For RE vs. control, the 7466 transcripts were associated with 41 pathways (23 activated, 18 inhibited; Supplemental Table 7). Among the top ten pathways, there were eight activated and two inhibited (Figure 2E). Most significant was neuroinflammation signaling activation (p=6.31×10−13, z=5.07). For PWE vs. control, the 945 transcripts were associated with 30 pathways (27 activated, 3 inhibited; Supplemental Table 8). Among the top ten pathways, there were eight activated and two inhibited (Figure 2F). Most significant was phagosome formation activation (p=2.00×10−13, z=5.61).
There was some overlap of altered pathways among pairwise comparisons. Only one pathway was common to all, crosstalk between DCs and NK cells. In RE vs. PWE, this included 14 transcripts (Figure 3). It was also activated in PWE vs. control to a lesser extent (p=3.24×10−5, z=3.32; Supplemental Table 8) with 11 differently altered transcripts. In RE vs. control, it was activated (37 transcripts; p=4.37×10−6, z=5.20; Supplemental Table 7), with transcripts overlapping other pairwise comparisons (15 unique transcripts). Among all pathways, 13 overlapped when comparing RE vs. control and PWE vs. control, and they were consistently activated or inhibited with some differently altered transcripts. For RE vs. PWE, there were eight pathways (paxillin signaling shared with RE vs. control) that had no overlap with other pairwise comparisons; all inhibited.
Figure 3. Altered transcripts in RE vs. PWE crosstalk between DCs and NK cells signaling pathway by RNAseq.
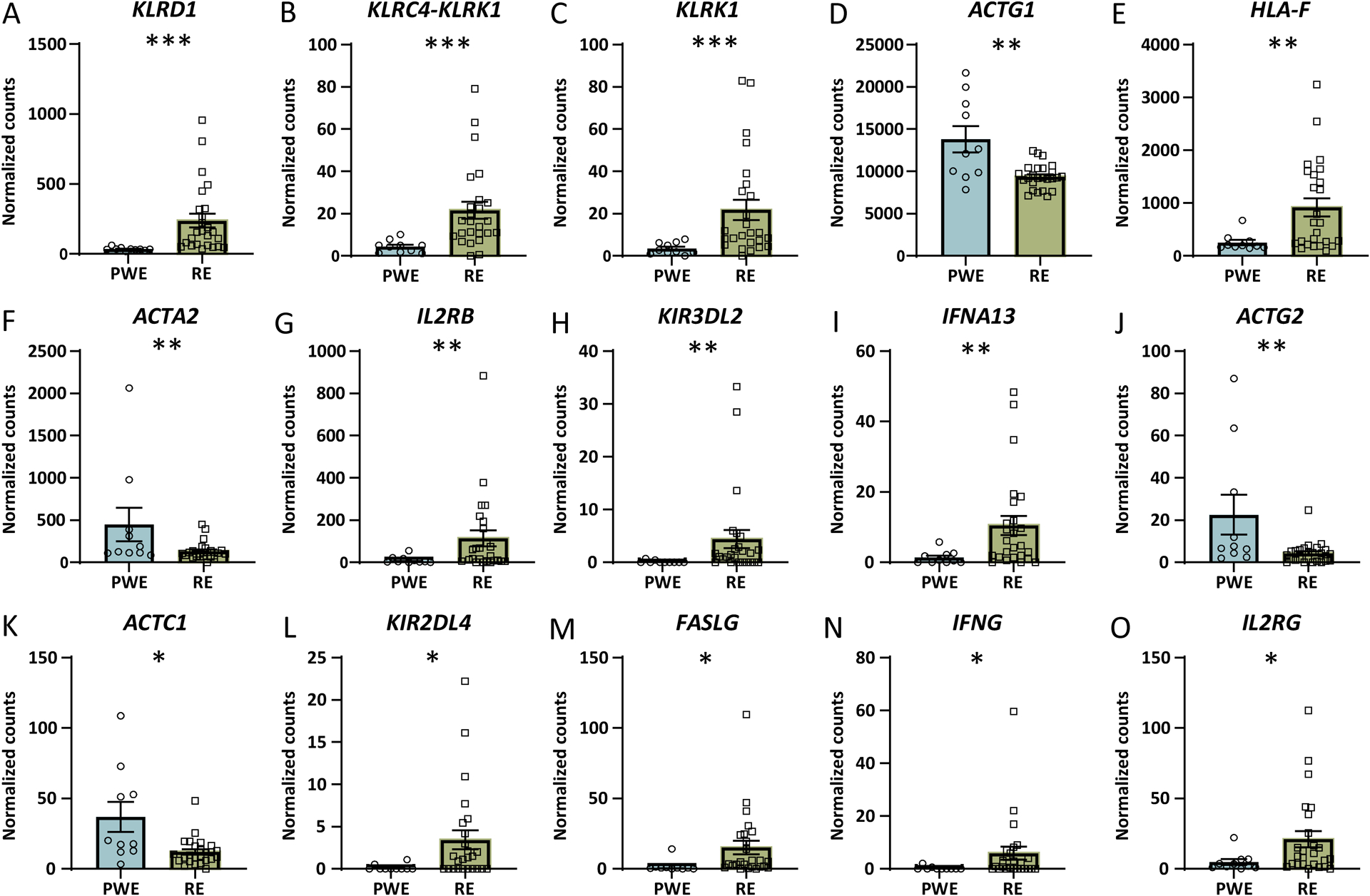
When comparing RE and PWE by RNAseq, the most significantly altered signaling pathway was activation of the crosstalk between DCs and NK cells signaling pathway (p value of overlap = 7.94 × 10−6, z = 2.65). A-O) The significantly altered transcripts associated with this pathway are depicted in order of decreasing significance (* adjusted p < 0.05, ** p < 0.01, *** p < 0.0001). Error bars indicate SEM.
T cells are involved in RE and were most enriched among significant transcripts in RE vs. PWE and control. However for RE vs. PWE, none of the 15 T cell transcripts were associated with altered pathways. In RE vs. control, 13/58 T cell transcripts were among 6/41 activated pathways (Th2 pathway, Th1 pathway, phagosome formation, macrophage NO and ROS production, crosstalk of DCs and NK cells, TREM1 signaling; Supplemental Table 7) and 2/41 inhibited pathways (paxillin signaling, cardiac hypertrophy signaling). Other pathways without T cell transcripts may modulate T cell activation;27 e.g., inhibition of the PD-1, PD-L1 cancer immunotherapy pathway in RE vs. control. For PWE vs. control, several T cell transcript related pathways were altered. There were 3/9 T cell transcripts represented among 8/30 activated pathways (phagosome formation, crosstalk of DCs and NK cells, NK cell signaling, DC maturation, leukocyte extravasation signaling, NFAT immune response regulation, hepatic fibrosis signaling, CD28 signaling in T helper cells; Supplemental Table 8).
Neuronal signaling was impacted in RE and PWE. Among the ten altered pathways in RE vs. PWE, several may impact neuronal signaling (e.g., altered cytoskeletal transcripts, ACTG1 in 7/10 pathways). Among the 41 pathways in RE vs. control, several may directly or indirectly impact neuronal signaling. The synaptogenesis pathway was inhibited (55 decreased transcripts, 24 increased, p=1.29×10−2, z=−3.08), with many overlapping ephrin signaling (decreased EPHB3, EFNB2, EFNA2, EFNB1, EPHA2, EPHA10, increased EPHA1; 61 altered transcripts, p=2.59×10−4, z=−1.18). Similarly, in PWE vs. control, among the 30 altered pathways, there were altered neuronal pathways that included G-protein coupled receptor signaling activation (p=7.94×10−5, z=3.12) and CREB signaling in neurons (p=2.24×10−4, z=2.92).
Proteomics Differential Expression
Label-free quantitative MS identified 3721 proteins from control (n=14), PWE (n=10), and RE (n=25). PCA indicated no segregation of groups in PCA1 or PCA2 (Figure 4A–C). Differential expression identified altered proteins in each pairwise comparison, as well as some overlap of proteins (Figure 4D). When comparing RE and PWE at FDR<5%, there were 3 altered proteins (2 up, 1 down; Figure 4E). For RE vs. control, there were 19 proteins (18 up, 1 down; Figure 4F). For PWE vs. control, there were 20 proteins (16 up, 4 down; Figure 4G).
Figure 4. Proteomics PCA and differential expression analysis.
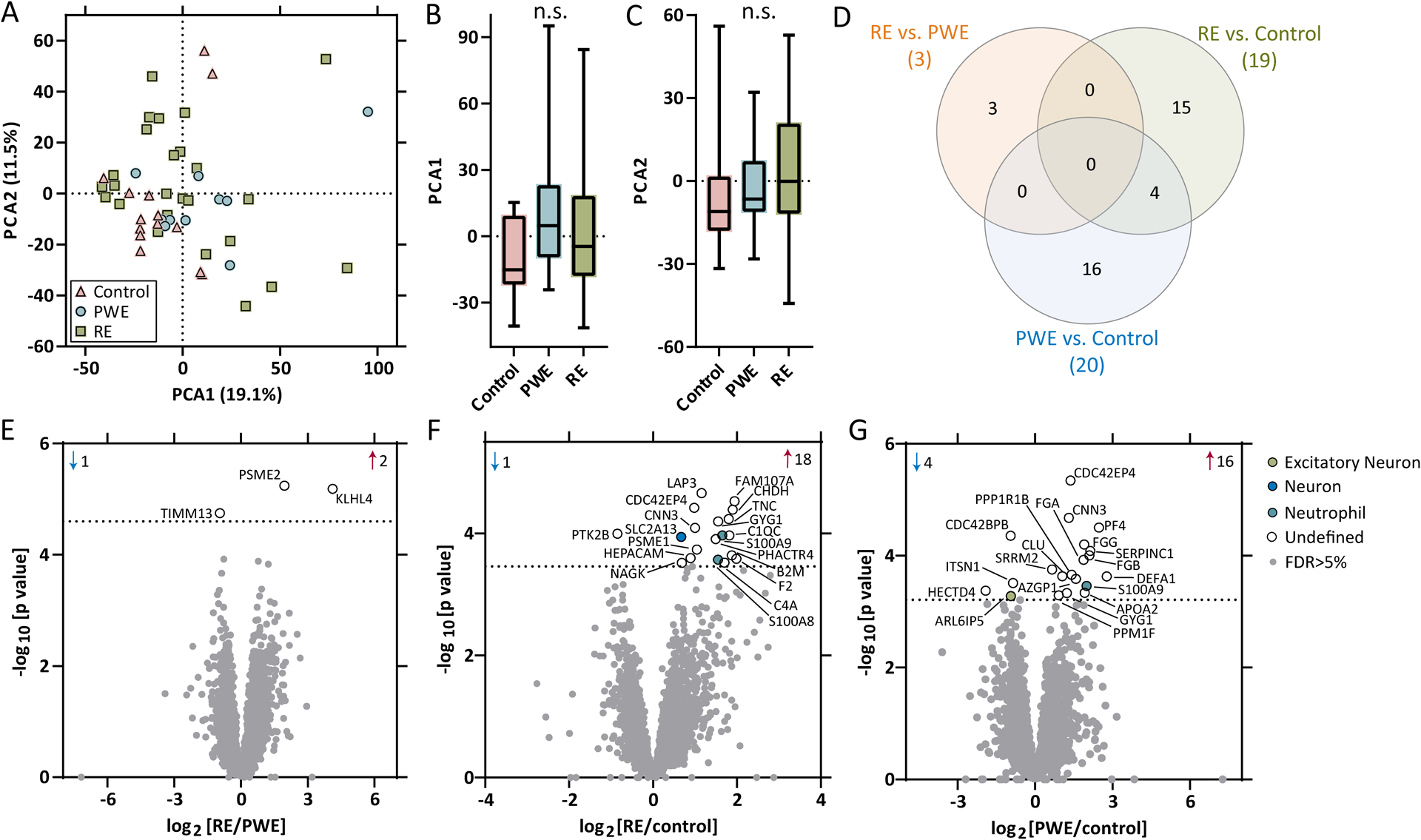
A-C) PCA of proteomics (control: n = 14, PWE: n = 10, RE: n = 25) in brain tissue indicated no segregation of groups. D) Differential expression analysis for each pairwise comparison are indicated, as well as overlap in the number of significant transcripts, at a 5% FDR (dotted line), when comparing E) RE vs. PWE (3 proteins), F) RE vs. control (19 proteins), and G) PWE vs. control (20 proteins). Annotations include the number of significantly increased (red arrow) and decreased (blue arrow) transcripts, all altered proteins are annotated by gene name, and brain and immune cell type annotations for each significant protein are indicated.
Most proteins were ubiquitously expressed by multiple cell types or it was undetermined (Figure 4E–G, Supplemental Table 9). There was no cell type enrichment among significant proteins. For each pairwise comparison, there were no pathways associated with altered proteins.
Correlation of RNAseq and Proteomics
Among altered transcripts in RE vs. PWE, 865/1516 were protein coding (393 lncRNA). For RE vs. control, 4582/7466 were protein coding (1869 lncRNA). For PWE vs. control, 620/945 were protein coding (227 lncRNA). However, many significant transcripts were not detected by proteomics, with 12.9% (196 proteins/1516 RNA) for RE vs. PWE, 13.3% (994 proteins/7466 RNA) for RE vs. control, and 9.8% (93 proteins/945 RNA) for PWE vs. control.
Of significant targets identified and detected in RNAseq or proteomics, correlation analyses determined similarities in expression. For RE vs. PWE, 49% (99/202) changed in the same direction but with no correlation between RNAseq and proteomics (p=0.15, R2=0.010; Supplemental Figure 1A). For RE vs. control, 70% (703/1001) changed in the same direction, and there was a positive correlation (p<0.0001, R2=0.22; Supplemental Figure 1B). For PWE vs. control, 69% (65/94) changed in the same direction, and there was a positive correlation (p=0.0028, R2=0.093; Supplemental Figure 1C).
Comparison of WES with RNAseq and Proteomics
Of the three genes with VUS in RE, one (FCGR3B) was among two of the activated RNAseq pathways in RE vs. control (Phagosome Formation, Fcγ Receptor-mediated Phagocytosis in Macrophages and Monocytes; Supplemental Table 2). The two HLA genes with VUS were among six altered pathways in RE vs. control (Neuroinflammation Signaling Pathway, Th2 Pathway, PD-1, PD-L1 cancer immunotherapy pathway, Th1 Pathway, Crosstalk between DC and NK Cells, MSP-RON Signaling In Macrophages Pathway). Of note, HLA transcript expression was altered in each pairwise comparison, with one increased (HLA-F) in RE vs. PWE, 23 increased in RE vs. control, and 11 increased in PWE vs. control (all overlapping with RE vs. control). HLA-DRB1 was increased 7.0-fold in RE vs. control (not in other pairwise comparisons), HLA-DQA2 increased 21.6-fold (also increased in PWE vs. control, 2.16-fold), and HLA-DQB2 increased 6.68-fold (not in other pairwise comparisons). No pathways identified by RNAseq for RE vs. PWE included the three genes with VUS, nor the two HLA genes.
By proteomics, of the three genes with VUS, MTOR was not differentially expressed (SCN1A, FCGR3B not detected). No HLA proteins were differentially expressed in any pairwise comparison (detected: HLA-A, HLA-B, HLA-C, HLA-DRA, HLA-DRB1).
Discussion
In RE brain, we performed the first RNAseq and proteomics analyses. We identified by RNAseq altered immune pathways, immune cell type annotation enrichment, and by WES HLA variants more common to RE. These findings demonstrate immune cell infiltration associated with innate and adaptive immune responses, as well as HLA variants that may increase vulnerability to RE. Most significantly there was activated crosstalk between DCs and NK cells in RE vs. PWE. Cell type annotation identified top enrichment in RE vs. PWE for T cell and NK cell, RE vs. control for T cell and macrophage, and in PWE vs. control for macrophage and microglia. Many significant transcripts identified by RNAseq were not detected by proteomics. There were few differentially expressed proteins and there was some correlation of expression levels among significant targets by RNAseq and proteomics.
In RE, we identified several VUS potentially related to a modified immune response or seizure threshold. We identified SCN1A (T297I), reported in epilepsy in combination with another variant28 and in a non-conserved amino acid.29 We also identified ANGPTL7/MTOR (Q175H, R140H). ANGPTL7 is involved in extracellular matrix organization, decreased in hepatocellular carcinoma with microvascular invasion.30 MTOR (N161S) was reported with other variants in cancer patients.31,32 The FCGR3B variant was present in an intron. FCGR3B variants and low copy number variation are associated with autoimmunity and infectious diseases.33 Additionally, HLA variants may be associated with RE,10,11 including HLA-DRB1 linked to autoimmune diabetes and multiple sclerosis. We identified two VUS (HLA-DRB1, HLA-DQA2) present in >25% RE, rare in the general population, more likely deleterious, and heterozygous except for one variant in one case. Previous RE brain WES evaluated non-silent variants in >80% of 15 RE cases and compared frequency to 10 TLE cases, associating deleteriousness to RE enriched variants, and reported variants that did not meet our filter thresholds.11 As with FCGR3B, follow up validation is needed in these highly variable regions associated with neuroinflammatory diseases like Alzheimer’s disease and multiple sclerosis,34 including correlation to allele expression levels that may influence disease susceptibility.35 Future studies may be of interest to identify low frequency mosaic variants with deeper sequencing, potentially in isolated brain-infiltrating lymphocytes that may provide better resolution and identification of more rare variants.
RNAseq identified immune cell transcripts in RE and PWE. CD8+ T cell cytotoxicity is implicated in RE;1,3–6 with an immune profile of three RE cases revealing mainly CD8+ T cells and fewer CD4+ T cells, NK cells, myeloid cells, and B cells.5 We similarly observed T cell, NK cell, macrophage, monocyte, plasma cell, B cell, and neutrophil transcript enrichment in RE compared to PWE and control. In PWE vs. control, macrophage, monocyte, T cell, and B cell transcripts were enriched. Epileptic foci may contain inflammation and infiltrating immune cells that are unrelated to infection or immune-mediated etiology, including monocytes after status epilepticus, macrophages and neutrophils during epileptogenesis, and perivascular T cells.36 In the immune profile of six pediatric focal cortical dysplasia (FCD) and tuberous sclerosis complex (TSC) cases (both identified in 3/10 of our PWE cases), the most abundant brain-infiltrating immune cells included myeloid and NK cells.5 Future studies may evaluate immune cell numbers (transcript numbers and expression levels do not necessarily correlate to cell number), histology (perivascular/infiltrating), and cell type distinction (macrophage/microglia/DCs).
Our RNAseq also identified altered brain cell type transcripts. With reported microglial nodules, neuronal and astrocyte loss, and white matter inflammation and damage,1,2,37 our observed cell type enrichment may reflect microgliosis, neuronal loss, or neuronal dysfunction. Oligodendrocyte enrichment may relate to neuronal loss, brain atrophy, compensatory response, or included adjacent white matter in specimens. Astrocyte transcripts were not enriched in RE vs. PWE or control, however some were differentially expressed in RE (most increased, without accounting for cell loss or proliferation). For PWE vs. control, microglial transcripts were increased and neuron transcripts decreased, which may reflect microgliosis, neuronal loss, and neuronal dysfunction. RNAseq in adult TLE cortex identified pathways associated with decreased neurons and synaptic signaling, with some immune related pathways activated.38 Another smaller RNAseq study in adult TLE also found increased astrocyte and microglia transcripts and decreased neuronal transcripts in hippocampus, with fewer differences in cortex.39 Our prior proteomic study identified decreased neuronal proteins in the hippocampus, and did not detect cortical immune pathway activation.22
RNAseq identified activated immune pathways in RE and PWE, most significantly crosstalk of DCs and NK cells in RE. DCs are professional antigen presenting cells that detect endogenous and exogenous molecules, link innate and adaptive immune responses, and promote NK and T cell cytotoxicity.40 NK cells are involved in innate and adaptive immunity to pathogens, autoimmunity, and tumor immunosurveillance.41,42 NK cell response is regulated by a receptor network, with activation resulting in lytic granule release; indirect lytic activity via pro-inflammatory cytokines that activates CD8+ T cells, antibody-dependent cell-mediated cytotoxicity via FcγRIIIa (FCGR3A) ligation that promotes DC maturation; activation via death-receptor ligation (Fas, TRAIL) resulting in apoptosis; and inhibition via killer cell immunoglobulin-like receptors (KIR) that recognize HLA to mediate autoimmunity and tumor immunosurveillance.41 In our dataset, NCR1 (NKp46), GZMB (granzyme B), FASLG, KLRD1, KIR3DL2, and KIR2DL4 were increased in RE vs. PWE and control (not PWE vs. control). FCGR3A was increased in RE and PWE vs. control (not RE vs. PWE). PRF1 (perforin) was increased in RE vs. control. Crosstalk of DCs and NK cells includes a subset of the aforementioned molecules and mechanisms, which can result in NK cell activation, DC maturation, and apoptosis.43,44 NK cells infiltrate RE brain, but DCs have not been investigated. However, occasional myeloid cells are reported.5 The DC and NK crosstalk pathway was also activated in PWE. DCs have been observed in FCD type II and an epilepsy animal model,45 and NK cells were detected in an immune profile of TSC and FCD.5 Overall we identified more significant DC and NK cell crosstalk activation in RE vs. PWE, although with some similarities to PWE, that suggests both active innate and adaptive immune responses in RE.
We identified altered neuronal pathways in RE and PWE. Synaptogenesis signaling was inhibited in RE vs. control, including general synaptic and ephrin transcripts. Ephrin signaling is related to both neuronal (axon guidance, dendrite spine morphogenesis) and immune cell types (infection, tumor, cell migration), as well as crosstalk among various cell types that express ephrin ligands and receptors resulting in bidirectional signaling.46,47 Neuronal damage caused by CD8+ T cell cytotoxicity and NK cells, found adjacent to neurons in RE,1 can result in permanent dysregulation of brain circuitry through phagocyte-mediated synaptic stripping and neuronal death.48,49
Our study had several limitations. Exome sequencing was performed at a depth that allowed for identification of variants in the RE cohort, but may not provide resolution to identify less frequent mosaic variants in bulk brain tissue. We compared autopsy and surgical sources and different cortical regions, potentially with inherent gene expression differences. Clinical variables may impact gene expression, i.e. medications, neuropathology, disease progression. The RE immune response may differ at various stages.8 Although we identified a 50–70% correlation of significantly detected transcripts and proteins, there are likely several factors that contributed to fewer differences observed by proteomics that include high abundance of hemoglobin that shifts detection of less abundant proteins below the detectable threshold, more disease/protein impact on a relatively small immune cell population that may be diluted in bulk brain tissue, and some heterogeneity of clinical variables (i.e. disease duration).
In summary, we identified activated immune pathways and immune cell type annotation enrichment in RE brain. Activated crosstalk of DCs and NK cells in RE suggests an active role of both innate and adaptive immune responses, in addition to inhibited synaptogenesis signaling that may contribute to neurological dysfunction. Further, we identified VUS that were associated with the altered pathways and thus may contribute to RE susceptibility. Future studies should evaluate how gene expression relates to clinical information as this may be associated with underlying etiology or the result of disease progression, and whether modulating crosstalk between DCs and NK cells can limit disease progression.
Supplementary Material
Key Points.
RNAseq in Rasmussen encephalitis (RE) brain tissue identified altered immune pathways and immune cell type transcript enrichment.
The most significantly altered pathway in RE vs. other epilepsies was activated crosstalk between dendritic and natural killer cells.
By whole exome sequencing, we identified HLA variants potentially deleterious and more common in RE.
These findings demonstrate innate and adaptive immune responses in brain, as well as HLA variants that may increase vulnerability to RE.
Acknowledgements
Funding was provided by Finding A Cure for Epilepsy and Seizures (FACES), Rasmussen Encephalitis Children’s Project, and the National Institutes of Health (NIH) P01AG060882 and P30AG066512. Gilad Evrony provided access to Qiagen Clinical Insight.
Conflicts of Interest/Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
The authors report no conflicts of interest. Daniel Friedman receives salary support for consulting and clinical trial related activities performed on behalf of The Epilepsy Study Consortium, a non-profit organization. Dr. Friedman receives no personal income for these activities. NYU receives a fixed amount from the Epilepsy Study Consortium towards Dr. Friedman’s salary. Within the past two years, The Epilepsy Study Consortium received payments for research services performed by Dr. Friedman from: Alterity, Baergic, Biogen, BioXcell, Cerevel, Cerebral, Jannsen, Lundbeck, Neurocrine, SK Life Science, and Xenon. He has also served as a paid consultant for Neurelis Pharmaceuticals and Receptor Life Sciences. He has received research support from NINDS, CDC, Epitel, and Neuropace unrelated to this study. He holds equity interests in Neuroview Technology. He received royalty income from Oxford University Press. Sasha Devore receives salary support from the National Institutes of Health, Department of Defense, and the Templeton World Charity Foundation unrelated to this study.
Data Availability
The data that support the findings of this study are available in the public repositories listed in the methods, supplemental files, and from the authors upon reasonable request.
References
- 1.Varadkar S, Bien CG, Kruse CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. The Lancet Neurology. Feb 2014;13(2):195–205. doi: 10.1016/s1474-4422(13)70260-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardo CA, Vining EP, Guo L, Skolasky RL, Carson BS, Freeman JM. The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia. May 2004;45(5):516–26. doi: 10.1111/j.0013-9580.2004.33103.x [DOI] [PubMed] [Google Scholar]
- 3.Bien CG, Bauer J, Deckwerth TL, et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol. Mar 2002;51(3):311–8. doi: 10.1002/ana.10100 [DOI] [PubMed] [Google Scholar]
- 4.Schwab N, Bien CG, Waschbisch A, et al. CD8+ T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain. May 2009;132(Pt 5):1236–46. doi: 10.1093/brain/awp003 [DOI] [PubMed] [Google Scholar]
- 5.Owens GC, Garcia AJ, Mochizuki AY, et al. Evidence for Innate and Adaptive Immune Responses in a Cohort of Intractable Pediatric Epilepsy Surgery Patients. Front Immunol. 2019;10:121. doi: 10.3389/fimmu.2019.00121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider-Hohendorf T, Mohan H, Bien CG, et al. CD8(+) T-cell pathogenicity in Rasmussen encephalitis elucidated by large-scale T-cell receptor sequencing. Nat Commun. Apr 04 2016;7:11153. doi: 10.1038/ncomms11153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tröscher AR, Wimmer I, Quemada-Garrido L, et al. Microglial nodules provide the environment for pathogenic T cells in human encephalitis. Acta Neuropathol. 04 2019;137(4):619–635. doi: 10.1007/s00401-019-01958-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owens GC, Huynh MN, Chang JW, et al. Differential expression of interferon-γ and chemokine genes distinguishes Rasmussen encephalitis from cortical dysplasia and provides evidence for an early Th1 immune response. J Neuroinflammation. May 02 2013;10:56. doi: 10.1186/1742-2094-10-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al Nimer F, Jelcic I, Kempf C, et al. Phenotypic and functional complexity of brain-infiltrating T cells in Rasmussen encephalitis. Neurol Neuroimmunol Neuroinflamm. Jan 2018;5(1):e419. doi: 10.1212/NXI.0000000000000419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dandekar S, Wijesuriya H, Geiger T, Hamm D, Mathern GW, Owens GC. Shared HLA Class I and II Alleles and Clonally Restricted Public and Private Brain-Infiltrating αβ T Cells in a Cohort of Rasmussen Encephalitis Surgery Patients. Front Immunol. 2016;7:608. doi: 10.3389/fimmu.2016.00608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ai J, Wang Y, Liu D, et al. Genetic Factors in Rasmussen’s Encephalitis Characterized by Whole-Exome Sequencing. Front Neurosci. 2021;15:744429. doi: 10.3389/fnins.2021.744429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain. Mar 2005;128(Pt 3):454–71. doi: 10.1093/brain/awh415 [DOI] [PubMed] [Google Scholar]
- 13.Olson HE, Lechpammer M, Prabhu SP, et al. Clinical application and evaluation of the Bien diagnostic criteria for Rasmussen encephalitis. Epilepsia. Oct 2013;54(10):1753–60. doi: 10.1111/epi.12334 [DOI] [PubMed] [Google Scholar]
- 14.Hoch D, Novakovic B, Cvitic S, Saffery R, Desoye G, Majali-Martinez A. Sex matters: XIST and DDX3Y gene expression as a tool to determine fetal sex in human first trimester placenta. Placenta. 08 2020;97:68–70. doi: 10.1016/j.placenta.2020.06.016 [DOI] [PubMed] [Google Scholar]
- 15.Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. February 22 2021;13(1):31. doi: 10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. Aug 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. Jan 01 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. Apr 2014;30(7):923–30. doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 19.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramírez F, Ryan DP, Grüning B, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. July 08 2016;44(W1):W160–5. doi: 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. May 2015;16:169. doi: 10.1186/s12859-015-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pires G, Leitner D, Drummond E, et al. Proteomic differences in the hippocampus and cortex of epilepsy brain tissue. Brain Commun. 2021;3(2):fcab021. doi: 10.1093/braincomms/fcab021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leitner DF, Mills JD, Pires G, et al. Proteomics and Transcriptomics of the Hippocampus and Cortex in SUDEP and High-Risk SUDEP Patients. Neurology. Apr 2021;doi: 10.1212/WNL.0000000000011999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lake BB, Chen S, Sos BC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 01 2018;36(1):70–80. doi: 10.1038/nbt.4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nirmal AJ, Regan T, Shih BB, Hume DA, Sims AH, Freeman TC. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol Res. 11 2018;6(11):1388–1400. doi: 10.1158/2326-6066.CIR-18-0342 [DOI] [PubMed] [Google Scholar]
- 26.Doellinger J, Schneider A, Hoeller M, Lasch P. Sample Preparation by Easy Extraction and Digestion (SPEED) - A Universal, Rapid, and Detergent-free Protocol for Proteomics Based on Acid Extraction. Mol Cell Proteomics. 01 2020;19(1):209–222. doi: 10.1074/mcp.TIR119.001616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727–742. [PMC free article] [PubMed] [Google Scholar]
- 28.Binini N, Sancini G, Villa C, et al. Identification of two mutations in cis in the SCN1A gene in a family showing genetic epilepsy with febrile seizures plus (GEFS+) and idiopathic generalized epilepsy (IGE). Brain Res. Dec 15 2017;1677:26–32. doi: 10.1016/j.brainres.2017.09.023 [DOI] [PubMed] [Google Scholar]
- 29.Nabbout R, Gennaro E, Dalla Bernardina B, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. Jun 24 2003;60(12):1961–7. doi: 10.1212/01.wnl.0000069463.41870.2f [DOI] [PubMed] [Google Scholar]
- 30.Beaufrère A, Caruso S, Calderaro J, et al. Gene expression signature as a surrogate marker of microvascular invasion on routine hepatocellular carcinoma biopsies. J Hepatol. 02 2022;76(2):343–352. doi: 10.1016/j.jhep.2021.09.034 [DOI] [PubMed] [Google Scholar]
- 31.Shaib WL, Zakka K, Staley C, et al. Blood-Based Next-Generation Sequencing Analysis of Appendiceal Cancers. Oncologist. 05 2020;25(5):414–421. doi: 10.1634/theoncologist.2019-0558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayer R, Dandulakis M, Richards S, Roque DM, Staats PN. Malignant Sex Cord-Stromal Tumor, Not Otherwise Specified, Harboring FOXL2, p53, and TERT Promoter Mutations: Report of a Case. Int J Gynecol Pathol. Nov 2020;39(6):567–572. doi: 10.1097/PGP.0000000000000651 [DOI] [PubMed] [Google Scholar]
- 33.Nagelkerke SQ, Schmidt DE, de Haas M, Kuijpers TW. Genetic Variation in Low-To-Medium-Affinity Fcγ Receptors: Functional Consequences, Disease Associations, and Opportunities for Personalized Medicine. Front Immunol. 2019;10:2237. doi: 10.3389/fimmu.2019.02237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. Dec 2013;45(12):1452–8. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douillard V, Castelli EC, Mack SJ, et al. Approaching Genetics Through the MHC Lens: Tools and Methods for HLA Research. Front Genet. 2021;12:774916. doi: 10.3389/fgene.2021.774916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vezzani A, Lang B, Aronica E. Immunity and Inflammation in Epilepsy. Cold Spring Harb Perspect Med. Dec 18 2015;6(2):a022699. doi: 10.1101/cshperspect.a022699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramaswamy V, Walsh JG, Sinclair DB, et al. Inflammasome induction in Rasmussen’s encephalitis: cortical and associated white matter pathogenesis. J Neuroinflammation. Dec 13 2013;10:152. doi: 10.1186/1742-2094-10-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guelfi S, Botia JA, Thom M, et al. Transcriptomic and genetic analyses reveal potential causal drivers for intractable partial epilepsy. Brain. Jun 2019;142(6):1616–1630. doi: 10.1093/brain/awz074 [DOI] [PubMed] [Google Scholar]
- 39.Mills JD, van Vliet EA, Chen BJ, et al. Coding and non-coding transcriptome of mesial temporal lobe epilepsy: Critical role of small non-coding RNAs. Neurobiol Dis. Feb 2020;134:104612. doi: 10.1016/j.nbd.2019.104612 [DOI] [PubMed] [Google Scholar]
- 40.Kushwah R, Hu J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology. Aug 2011;133(4):409–19. doi: 10.1111/j.1365-2567.2011.03457.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chester C, Fritsch K, Kohrt HE. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front Immunol. 2015;6:601. doi: 10.3389/fimmu.2015.00601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrow AD, Martin CJ, Colonna M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol. 2019;10:909. doi: 10.3389/fimmu.2019.00909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas R, Yang X. NK-DC Crosstalk in Immunity to Microbial Infection. J Immunol Res. 2016;2016:6374379. doi: 10.1155/2016/6374379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walzer T, Dalod M, Vivier E, Zitvogel L. Natural killer cell-dendritic cell crosstalk in the initiation of immune responses. Expert Opin Biol Ther. Sep 2005;5 Suppl 1:S49–59. doi: 10.1517/14712598.5.1.s49 [DOI] [PubMed] [Google Scholar]
- 45.Ludewig P, Gallizioli M, Urra X, et al. Dendritic cells in brain diseases. Biochim Biophys Acta. 03 2016;1862(3):352–67. doi: 10.1016/j.bbadis.2015.11.003 [DOI] [PubMed] [Google Scholar]
- 46.Darling TK, Lamb TJ. Emerging Roles for Eph Receptors and Ephrin Ligands in Immunity. Front Immunol. 2019;10:1473. doi: 10.3389/fimmu.2019.01473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark IC, Gutiérrez-Vázquez C, Wheeler MA, et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science. April 23 2021;372(6540)doi: 10.1126/science.abf1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Liberto G, Pantelyushin S, Kreutzfeldt M, et al. Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell. October 04 2018;175(2):458–471.e19. doi: 10.1016/j.cell.2018.07.049 [DOI] [PubMed] [Google Scholar]
- 49.Melzer N, Meuth SG, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J. Nov 2009;23(11):3659–73. doi: 10.1096/fj.09-136200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the public repositories listed in the methods, supplemental files, and from the authors upon reasonable request.