

Abstract
We offer a new biogenetic proposal for the origin of the complex alkaloid alstonlarsine A, via rearrangement of the Strychnos alkaloids alstolucines B and F. Further, we provide evidence of the chemical feasibility of this proposal via the facile conversion of synthetic alstolucines into alstonlarsine A via a short, efficient sequence of N-methylation, β-elimination, and a cascade 1,7-hydride shift/Mannich cyclization. We believe that this is the first biogenetic proposal involving the “tert-amino effect”, a hydride-shift-based internal redox trigger of a Mannich cyclization. A further interesting feature of the cascade is that its stereochemical outcome likely originates in conformational preferences during the hydride shift.
Keywords: alkaloids, biosynthesis, internal redox, total synthesis, Mannich reaction
Graphical Abstract
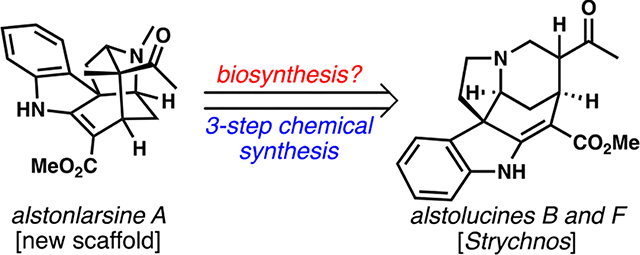
Alstonlarsine A, an indole monoterpene alkaloid isolated from Alstonia plants, was prepared by a rearrangement of other Alstonia alkaloids, alstolucines B and F. The successful process was patterned after a revised biogenetic proposal that features the first example of the “tert-amino effect” in alkaloid biogenesis.
The indole monoterpene alkaloids comprise a huge number of biogenetically related polycyclic compounds that commonly derive from tryptamine and seco-loganin (ultimately from the monoterpene geraniol), via the intermediacy of strictosidine.[1] This understanding took its first form as prescient hypotheses by Thomas and Wenkert.[2,3] Further evidence was gathered by groups of Arigoni, Battersby, Scott, and Kutney to unearth the common origins of many Corynanthé, Iboga and Strychnos alkaloids.[4] A very rewarding aspect of both biochemical and chemical work in this area is the opportunity to increase the understanding of the biosynthetic relationships among this vast collection of complex molecules. Not surprisingly, biomimetic synthesis has oftentimes yielded particularly attractive solutions to the chemical preparation of these compounds,[5] and stunning recent progress has made synthetic biology an equally promising approach to complex naturally occurring alkaloids and analogues.[6]
Alstonlarsine A (1, Figure 1) is a recently described hexacyclic indole monoterpene alkaloid that shows modest DRAK-2 inhibitory activity in vitro.[7] In their description of the isolation from Alstonia scholaris, structural characterization, and biological evaluation of alstonlarsine A, Li, Gao, Yue, and co-workers also put forth a biogenetic proposal for the formation of this new alkaloid via multi-stage rearrangement of N(4)-demethylalstogustine (2),[8] a Strychnos alkaloid that is also known in different diastereomeric forms.[9] This situation mirrored the one that we found ourselves in several years ago, when our work with the alsmaphorazine alkaloids[10] led us to propose a revision of their biogenesis from the Strychnos alkaloids alstolucines B and F (3a/b, Figure 2).[11] We posited that alsmaphorazine B (4) was likely to arise via oxidation steps and a particularly interesting cascade rearrangement of 3a/b. Notably, each alkaloid shown in Figures 1 and 2 can be found in species in the genus Alstonia, as could several of the intermediates en route to alsmaphorazine B[11,12] and along the proposed path to alstonlarsine A.[8]
Figure 1.

Probable origin of alstonlarsine A’s novel ring system from a Strychnos alkaloid precursor
Figure 2.
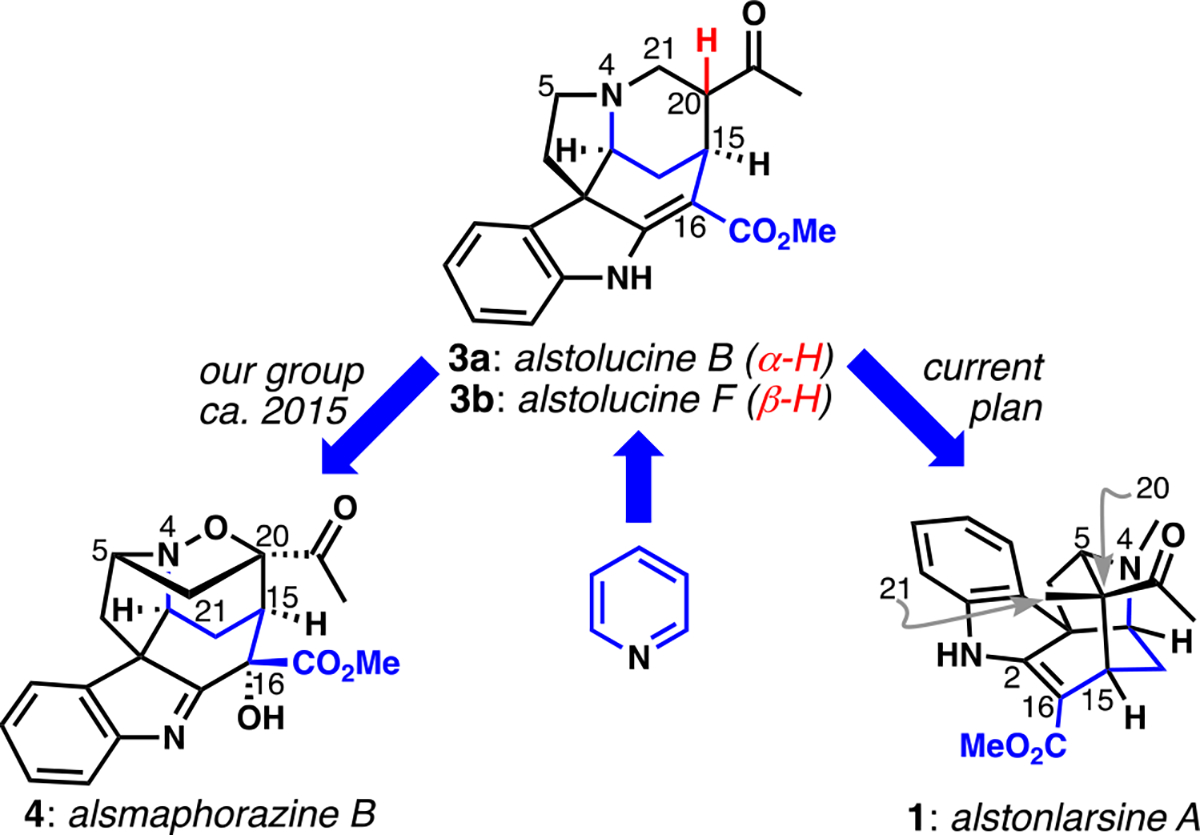
We previously converted synthetic Strychnos alkaloids alstolucines B and F, which we made from pyridine, into alsmaphorazine B via oxidation and rearrangement. Here, we aim to implement a different rearrangement to generate alstonlarsine A.
In our previous work on alsmaphorazine B,[11] we operated on a synthetic mixture of the known alkaloids alstolucine B and F, which are C20-epimeric ketones one oxidation state higher than 2.[12] N-Oxidation of the mixed epimers permitted elimination via an E1cB mechanism to a presumed enone/hydroxylamine intermediate; the latter functional group was intercepted by molecular oxygen, giving a reactive nitrone/alkene pair for intramolecular dipolar cycloaddition (this sequence is shown in the Supporting Information). This short protocol rearranged the common Strychnos scaffold into that of the alsmaphorazines, and required only C16-oxidation to complete the synthesis. Furthermore, this sequence was based upon our proposed revision of the biogenesis for the alsmaphorazine alkaloids, put forth as an alternative to the initial proposal of Morita and colleagues.[10]
The inspiration for our current study came from the realization that a different rearrangement sequence of the C20-epimers alstolucines B and F might quickly deliver alstonlarsine A, which has been the subject of two very recent, attractive total syntheses.[13] If this plan were reduced to practice, it would provide support for a revised biogenetic proposal by establishing the chemical feasibility of its key rearrangement sequence, which is shown in Scheme 1. N-Methylation of alstolucines B/F (3a/b) would provide an impetus for elimination via an E1cB mechanism to give enone 5. We considered that this reaction might be thermodynamically disfavored (as was the hydroxylamine elimination in our previous work[11]). In that case, we would need to intercept the potentially transient enone intermediate via an internal redox process[14] accomplished via a relatively rare 1,7-shift of hydride;[15] the resulting enolate/iminium ion pair 6 should then undergo a Mannich reaction to establish the bridged polycyclic scaffold of the alkaloid target. A particularly interesting part of this proposal comes from the enforced proximity of the reactive functional groups that should both increase the likelihood of the internal redox event and impose stereochemical control on the ensuing Mannich ring closure.
Scheme 1.
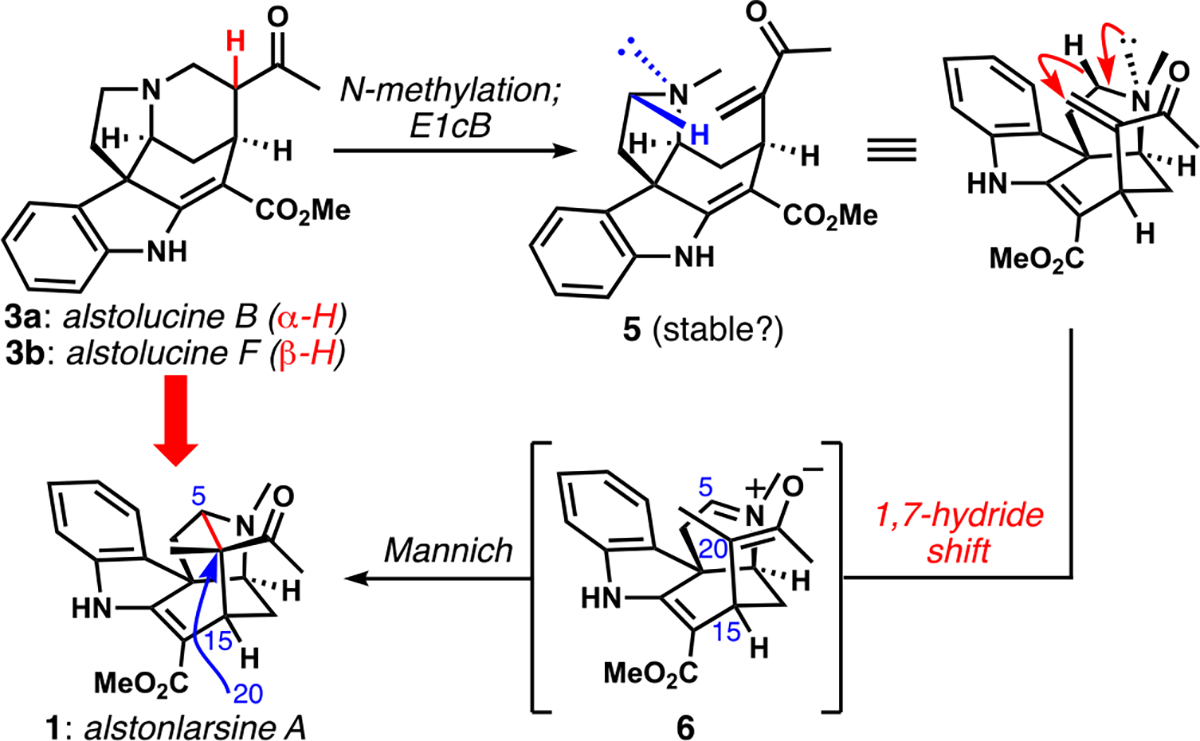
Proposed biogenesis of alstonlarsine A from alstolucines B or F
To our knowledge, reaction cascades of hydride shifts to electron-deficient alkenes that trigger subsequent Mannich reactions are unprecedented in alkaloid biosynthesis. However, these reactions have been successfully applied in many contexts for efficient C–C bond construction.[14] In synthetic settings, these types of reactions have been called Reinhoudt reactions,[14b,16] which are a specific type of amine-driven, hydride-shift-initiated rearrangements found under the unusual umbrella term “the tert-amino effect”.[14] Lewis acids or amines are often essential promoters of this useful reactivity by LUMO-lowering of the acceptor alkene. Successful hydride transfer typically requires close proximity of an electron rich C–H bond (most often α to an amine) to the electron-deficient alkene, and therefore the most frequently encountered processes involve 1,4- and 1,5-shifts. In the instance at hand, an unusual 1,7-shift[15] would be needed; however, a hand-held model of the proposed enone (5) suggested near-perfect alignment of the C5 C–H bond with the electrophilic enone β-carbon when held in the presumably higher energy conformation shown in Scheme 1 (N-methyl group on concave face providing appropriate alignments of the unshared electron pair and the C–H bond for transfer to the proximal electrophilic carbon). Because we could efficiently access racemic alstolucines B and F on the basis of our previous synthesis of alsmaphorazine B,[11] we were able to test this hypothesis.
To initiate our investigations, pure samples of alstolucines B and F (3a and 3b, Scheme 2) were separately treated with excess iodomethane to afford their methiodide salts (7a and b), which were isolated in high yield and purity by crystallization. To effect the desired E1cB reaction, each epimer was heated in a mixture of aqueous NaOH and THF. The only observable product in each case was desired enone 5, which was sufficiently pure after extraction; thus this enone was obtained in >90% from 3a or 3b without chromatography. In the hope of initiating the desired hydride shift/Mannich cascade, this material was warmed in the presence of Sc(OTf)3, which is well-precedented as a promoter of this type of transformation.[14] Perhaps unsurprisingly, conjugate addition was observed, thus reforming the pentacyclic scaffold in the form of a pair of C20-epimeric N-methylammonium triflate salts 8. The structural assignments of these products were supported by their reversion to enone 5 after treatment with base and heat.
Scheme 2.
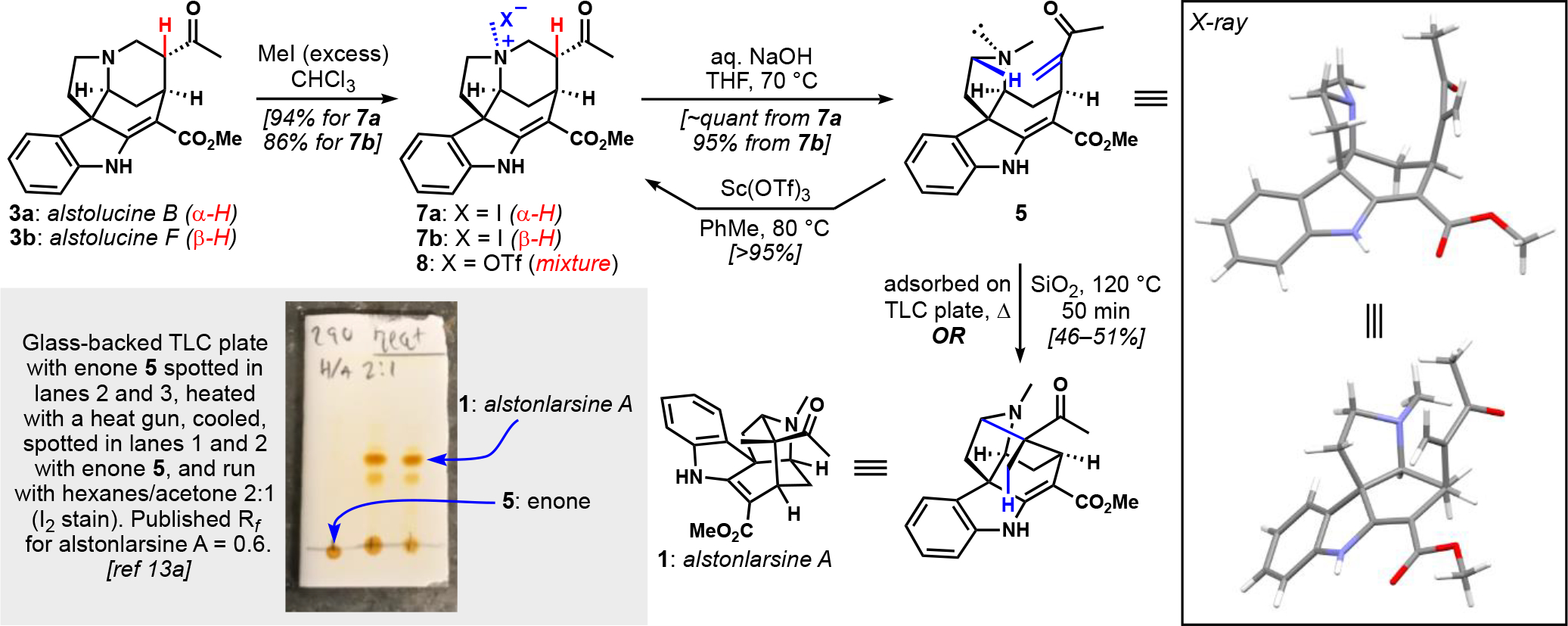
Synthesis of alstonlarsine A from alstolucines B and F
In addition to varying Lewis and protic acidic conditions—which were uniformly unsuccessful[17]—we considered triggering the rearrangement via heating enone 5 while adsorbed on silica gel. This approach was inspired by alkaloid interconversion reactions on silica gel conducted by Scott.[18] Two specific cases seemed noteworthy: (1) the conversion of dihydrostemmadenine acetate to pseudocatharanthine and its dihydro derivative, and (2) the generation of vincadifformine from stemmadenine acetate. In both cases, deep-seated rearrangements were observed that required elementary oxidation state changes that might be explained by disproportionations or internal redox chemistry. Though no through-space hydride shifts are implicated in either case, we thought that that these conditions might mediate such an event between the well-positioned functional groups in 5.
To this end, enone 5 was adsorbed onto a silica-gel thin layer chromatography (TLC) plate and heated briefly using a heat gun (Scheme 2, inset). After cooling the plate and eluting the crude material using a standard eluent, a major new spot was observed; the observed Rf value was consistent with the literature report for alstonlarsine A.[13a] The reaction was then optimized qualitatively using the surface of a hot plate with an infrared thermometer to heat the TLC plates to specific temperatures. Consumption of 5 over time could be estimated by development of the cooled TLC plate. On this basis, preparative conditions were identified: 5 was adsorbed onto silica gel and heated in an oil bath as a dry powder with stirring in a vial or flask; alstonlarsine A was produced in 46–51% yield after purification. Approximately 20 mg of alstonlarsine A was generated in a single preparative experiment. Importantly, control experiments involving heating neat 5 to 120 °C without silica gel did not yield alstonlarsine. The specific characteristics of silica that are responsible for the observed reactivity are not understood, but could involve activation of the enone, as well as partial sequestration of the nitrogen atom by H-bonding, thus slowing down the aza-Michael addition. We note that heating quaternary ammonium salts 7a or 7b adsorbed on silica gel does not lead to alstonlarsine A, suggesting that aza-Michael ring closure is not occurring to a significant extent in the key thermal reaction.
Three minor products, each formed in ~5% amounts, are observed in the key reaction, each with the same molecular mass as enone 5 and alstonlarsine A. A less polar compound that is barely visible in the TLC in Scheme 2 has many of the NMR spectral features of alstonlarsine A, and might well be its C20 epimer; it was isolated in such small quantities that full characterization was not possible. Assuming the possibility of rotation about the C15–C20 bond, only one conformation (presumably similar to that shown in the X-ray structure) of 5 orients the electrophilic enone β-carbon in proximity to the nucleophilic hydride on C5. After hydride shift, it seems reasonable to expect that Mannich recombination of the reactive enolate/iminium ion pair would be sufficiently rapid to largely outcompete rotation about the C15–C20 bond, thus leading nearly exclusively to the naturally configured C20 center of alstonlarsine A. Assuming that this less polar compound is the C20-epimer, we estimate the Mannich reaction diastereoselectivity to be ≥10:1. The two other compounds that were isolated correspond to the diffuse, minor spot visible on the TLC plate just below alstonlarsine A. These products are clearly related, and appear to be significantly rearranged products, as the aromatic region shows signatures of 2,3-disubstituted indoles. At this stage, we are unable to establish their identity.
While not yet successfully implemented in the laboratory, we recognize that amine catalysis of this cascade (via enamine-triggered elimination, iminium-ion-induced hydride shift, and enamine/iminium ion Mannich ring closure) would be more plausibly biomimetic conditions for this alkaloid interconversion. Efforts to effect this transformation under such milder, biologically relevant conditions are underway.[17]
We have established an efficient synthesis of the complex alkaloid alstonlarsine A via a plausibly biomimetic conversion of alstolucines B and F to this target. Given our previous synthesis of these alkaloids from pyridine, our total synthesis of alstonlarsine A proceeds via a longest linear sequence of 15 steps, and with an overall yield of about 10%, which compares favorably to the recent syntheses of this alkaloid.[13,19] Our demonstration of the chemical feasibility of this new biogenetic hypothesis provides circumstantial evidence that such a hydride shift/Mannich cascade features in the biosynthesis of alstonlarsine A. Taken together with our work on alsmaphorazine B, this sequence shows how complex rearrangements can be driven by the in situ formation of complementary pairs of reactive functional groups that would be difficult to generate in a stepwise manner. One of the most fascinating outcomes of our work on chemical investigations into the provenance of both the alsmaphorazines and alstonlarsine A is the realization that these two structurally incongruous alkaloids are in fact likely to be very closely biogenetically related. Finally, we note that—should our proposal be confirmed by rigorous biosynthetic experiments—this would appear to be the first instance of an internal redox/Mannich cascade in alkaloid biosynthesis.
Supplementary Material
Acknowledgements
This work was funded by the National Science Foundation (CHE-2102480), and A.Y.H. was supported by an NIH NRSA Postdoctoral Fellowship (F32 GM103058).
Footnotes
Dedicated to the memory of Prof. Rodrigo Andrade.
References
- [1].O’Connor SE, Maresh JJ, Nat. Prod. Rep. 2006, 23, 532–547. [DOI] [PubMed] [Google Scholar]
- [2].Thomas R, Tetrahedron Lett. 1961, 2, 544–553. [Google Scholar]
- [3].Wenkert E, J. Am. Chem. Soc. 1962, 84, 98–102. [Google Scholar]
- [4].(a) Goeggel H, Arigoni D, Chem. Commun. (London) 1965, 538–539; [Google Scholar]; (b) Battersby AR, Pure Appl. Chem. 1967, 14, 117–136; [DOI] [PubMed] [Google Scholar]; (c) Battersby AR, Byrne JC, Kapil RS, Martin JA, Payne TG, Arigoni D, Loew P, Chem. Commun. (London) 1968, 951–953; [Google Scholar]; (d) Kutney JP; Cretney WJ; Hadfield JR; Hall ES; Nelson VR; Wigfield DC J. Am. Chem. Soc. 1968, 90, 3566–3567; [DOI] [PubMed] [Google Scholar]; (e) Kutney JP; Ehret C; Nelson VR; Wigfield DCJ Am. Chem. Soc. 1968, 90, 5929–5930. [DOI] [PubMed] [Google Scholar]; (f) Scott published prolifically in this area, and some of his group’s key insights are reviewed in: Scott AI Acc. Chem. Res. 1970, 3, 151–157; [Google Scholar]
- [5].(a) For some key examples, see: Kuehne ME; Roland DM; Hafter RJ Org. Chem. 1978, 43, 3705–3710; [Google Scholar]; (b) Kuehne ME; Podhorez DE; Mulamba T; Bornmann WG J. Org. Chem. 1987, 52, 347–353; [Google Scholar]; (c) Carroll WA; Grieco PA J. Am. Chem. Soc. 1993, 115, 1164–1165; [Google Scholar]; (d) Tietze LF; Zhou Y Angew. Chem. Int. Ed 1999, 38, 2045–2047; [DOI] [PubMed] [Google Scholar]; (e) Ito M; Clark CW; Mortimore M; Goh JB; Martin SF J. Am. Chem. Soc. 2001, 123, 8003–8010; [DOI] [PubMed] [Google Scholar]
- [6].(a) For some recent achievements in the alkaloid field, see: Galanie S, Thodey K, Trenchard IJ, Filsinger Interrrante M, Smolke CD, Science 2015, 349, 1095–1100; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nett RS, Sattely ES, J. Am. Chem. Soc. 2021, 143, 19454–19465; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dudley QM, Jo S, Guerrero DAS, Chhetry M, Smedley MA, Harwood WA, Sherden NH, O’Connor SE, Caputi L, Patron NJ, Commun. Biol. 2022, 5, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhu X-X, Fan Y-Y, Xu L, Liu Q-F, Wu J-P, Li J-Y, Li J, Gao K, Yue J-M, Org. Lett. 2019, 21, 1471–1474. [DOI] [PubMed] [Google Scholar]
- [8].Hu W, Zhu J, Hesse M, Planta. Med. 1989, 55, 463–466. [DOI] [PubMed] [Google Scholar]
- [9].Gan L-S, Yang S-P, Wu Y, Ding J, Yue J-M, J. Nat. Prod. 2006, 69, 18–22. [DOI] [PubMed] [Google Scholar]
- [10].Koyama K, Hirasawa Y, Nugroho AE, Hosoya T, Hoe TC, Chan K-L, Morita H, Org. Lett. 2010, 12, 4188–4191. [DOI] [PubMed] [Google Scholar]
- [11].a) Hong AY, Vanderwal CD, J. Am. Chem. Soc. 2015, 137, 7306–7309; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hong AY, Vanderwal CD, Tetrahedron 2017, 73, 4160–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tan S-J, Low Y-Y, Choo Y-M, Abdullah Z, Etoh T, Hayashi M, Komiyama K, Kam T-S, J. Nat. Prod. 2010, 73, 1891–1897. [DOI] [PubMed] [Google Scholar]
- [13].a) Yao J-J, Ding R, Chen X, Zhai H, J. Am. Chem. Soc. 2022, 144, 14396–14402; [DOI] [PubMed] [Google Scholar]; b) Ferjancic Z, Kukuruzar A, Bihelovic F, Angew. Chem. Int. Ed. 2022, 61, e202210297. [DOI] [PubMed] [Google Scholar]
- [14].a) The “tert-amino effect” is a term seeming first coined by Meth-Cohn and Suschitzky to describe the overall transformation of hydride-shift-induced cyclizations of o-substituted tert-anilines, wherein the group ortho is electron-deficient and thus provides impetus for the amine-driven shift of the hydride: Meth-Cohn O; Suschitzky H Adv. Heterocycl. Chem. 1972, 14, 211–278, [Google Scholar]; b) For more recent reviews of internal redox processes via hydride shift mechanisms, see: Peng B, Maulide N, Eur. J. Org. Chem. 2013, 19, 13274–13287; [DOI] [PubMed] [Google Scholar]; c) Platanova AY, Glukhareva TV, ZImovets OA, Morzherin YY, Chem. Heterocyclic Comp. 2013, 49, 357–385; [Google Scholar]; (d) Haibach MC, Seidel D, Angew. Chem. Int. Ed. 2014, 53, 5010–5036. [DOI] [PubMed] [Google Scholar]
- [15].a) For transformations via a proposed 1,7-hydride shift, see Polonka-Bálint Á, Saraceno C, Ludányi K, Bényei A, Mátyus P, Synlett 2008, 2008, 2846–2850; [Google Scholar]; b) Dunkel P, Túrós G, Bényei A, Ludányi K, Mátyus P, Tetrahedron 2010, 66, 2331–2339; [Google Scholar]; c) Földi ÁA, Ludányi K, Bényei AC, Mátyus P, Synlett 2010, 2010, 2109–2113; [Google Scholar]; d) Cui L, Ye L, Zhang L, Chem. Commun. 2010, 46, 3351–3353; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nishide K, Shigeta Y, Obata K, Node M, J. Am. Chem. Soc. 1996, 118, 13103–13104; [Google Scholar]; f) Wicker G, Schoch R, Paradies J, Org. Lett. 2021, 23, 3626–3630; [DOI] [PubMed] [Google Scholar]; g) Kataoka M, Otawa Y, Ido N, Mori K, Org. Lett. 2019, 21, 9334–9338. [DOI] [PubMed] [Google Scholar]
- [16].Reinhoudt studied this type of process extensively, and reviewed much of his work in: Verboom W, Reinhoudt DN, Recl. Trav. Chim. Pays-Bas 1990, 109, 311–324. [Google Scholar]
- [17].See the Supporting Information for other conditions we have used in attempts to generate alstonlarsine A from enone 5, including catalysis by amines and ammonium salts.
- [18].Scott AI, Bioorg. Chem. 1974, 3, 398–429. [Google Scholar]
- [19].(a) Taking Andrade’s six-step synthesis of (±)-akuammicine into account, we can arrive at a 12-step longest linear sequence for (±)-alstonlarsine A: Sirasani G, Paul T, Dougherty W Jr., Kassel S, Andrade RB, J. Org. Chem. 2010, 75, 3529–3532. [DOI] [PubMed] [Google Scholar]; (b) Teijaro CN, Munagala S, Zhao S, Sirasani G, Kokkonda P, Malofeeva EV, Hopper-Borge E, Andrade RB, J. Med. Chem. 2014, 57, 10383–10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.