

Abstract
Purpose:
We sought to determine whether sequencing analysis of circulating cell-free (cf)DNA in prospectively accrued endometrial cancer (EC) patients captures the mutational repertoire of the primary lesion and allows for disease monitoring.
Design:
Peripheral blood was prospectively collected from 44 newly diagnosed EC patients over a 24-month period (i.e., baseline, post-surgery, every six months after). DNA from the primary ECs was subjected to targeted next-generation sequencing (NGS) of 468 cancer-related genes, and cfDNA to a high-depth NGS assay of 129 genes with molecular barcoding. Sequencing data were analyzed using validated bioinformatics methods.
Results:
cfDNA levels correlated with surgical stage in ECs, with higher levels of cfDNA being present in advanced stage disease. Mutations in cfDNA at baseline were detected pre-operatively in 8 of 36 (22%) patients with sequencing data, all of whom were diagnosed with advanced stage disease, high tumor volume and/or aggressive histologic type. Of the 38 somatic mutations identified in the primary tumors also present in the cfDNA assay, 35 (92%) and 38 (100%) were detected at baseline and follow-up, respectively. In six patients with recurrent disease, changes in circulating tumor (ct)DNA fraction/ variant allele fractions in cfDNA during follow-up closely mirrored disease progression and therapy response, with a lead time over clinically-detected recurrence in two cases. The presence of ctDNA at baseline (p<0.001) or post-surgery (p=0.014) was significantly associated with reduced progression-free survival.
Conclusions:
cfDNA sequencing analysis in EC patients at diagnosis has prognostic value and serial post-surgery cfDNA analysis enables disease and treatment response monitoring.
Keywords: cfDNA, sequencing, disease monitoring, mutation profiling, recurrence
INTRODUCTION
Endometrial cancer (EC) is the fourth most common female malignancy in the United States with more than 65,000 new cases estimated in 2022 (1). Despite advances in therapy, over the course of 2013–2017, uterine cancer death rates have increased more than any other type of cancer among women (2), and when compared to 1975, the overall 5-year relative survival for EC has decreased (3). EC is histologically, clinically and molecularly heterogeneous (4). The majority of low-risk histology, early-stage disease can be treated with primary surgery and carries a favorable prognosis (5), whereas serous carcinomas and carcinosarcomas, representing only about 15% of all ECs, account for the majority of deaths. In these high-risk ECs, the efficacy of standard therapeutic regimens (i.e. paclitaxel/ carboplatin) (6,7), is limited, and novel targeted therapies are emerging (8). Other than radiologic imaging and serum CA125 level assessment in some patients, currently there is no non-invasive form of treatment response or disease progression monitoring available for patients with EC.
Sequencing analysis of cell-free DNA (cfDNA) has emerged as a potential biomarker for early cancer detection, diagnosis, disease monitoring, and prognostication (9–11), and has been well studied in patients with early and advanced disease in a number of cancer types, including breast, colorectal and lung cancer (12–14). Unlike those solid tumors, distant metastatic disease in EC is not common (approximately 4%) (15). To date, the role of cfDNA sequencing analysis in women with EC is less defined. Small gene panels assessed by digital or quantitative PCR have identified circulating cell-free tumor (ct)DNA in subsets of patients with EC, often those with high-risk disease (16–21); these studies were retrospective, however, and only provided limited longitudinal information. Next generation sequencing (NGS) of hotspot amplicons of CTNNB1, KRAS, PTEN and PIK3CA in plasma sampled at hysterectomy from patients with endometrioid EC revealed mutations in 33% of patients, and found to be associated with advanced stage, deep myometrial/vascular invasion, and primary tumor size (22).
In this proof-of-principle study, we sought to define whether i) the presence of cfDNA is associated with the histologic subtype or disease stage in newly diagnosed EC patients, ii) whether sequencing analysis of longitudinal cfDNA samples can be used for disease monitoring in this disease and iii) whether the presence of ctDNA in plasma derived from EC patients is associated with disease progression or recurrence. To address these aims, we performed a retrospective analysis of prospectively collected plasma samples from patients with newly diagnosed EC using a high-sensitivity NGS-based mutation profiling assay.
MATERIAL AND METHODS
Patient selection
In this prospective proof-of-principle study, 44 biopsy-confirmed newly diagnosed EC patients consented to an institutional research protocol between 09/2017 and 07/2018 were included (Supplementary Figure S1, Consort Diagram). Rather than a consecutive series, cases representative of all histologic types and FIGO stages were included to assess the presence of mutations in cfDNA according to histologic type and stage. All surgical specimens were subjected to a central pathology review by a dedicated gynecologic pathologist (L.H.E.) to i) define the histologic type following the World Health Organization of Female Genital Tumors (23), ii) assess lymphovascular invasion, iii) confirm the presence of isolated tumor cells (ITCs) in the lymph nodes, and iv) confirm the FIGO staging. Clinical data, including demographic information, surgical staging, chemotherapy, radiation treatment, imaging, and follow-up were extracted from the electronic medical records from the date of patient consent. This study was approved by Memorial Sloan Kettering Cancer Center’s (MSK’s) institutional review board (IRB), and written informed consent was obtained from all patients. This study was conducted in accordance with the Declaration of Helsinki.
Liquid biopsies
Peripheral blood samples were collected from each patient in two Streck Cell-Free DNA Blood Collection Tubes (Streck, La Vista, NE). Samples were collected prior to surgery, at the post-operative visit (median 22 days post-surgery, range 9–57 days), and at 6, 12, 18, and 24 months. Blood was processed and cfDNA extracted using the QIAsymphony SP system (Qiagen), quantified, and stored following validated standard operating procedures at MSKs cfDNA extraction laboratory in the Department of Pathology and Laboratory Medicine, as previously described (24).
MRI-based FIGO staging and tumor volume analysis
Pretreatment magnetic resonance imaging (MRI) scans were obtained on a 3 Tesla magnet (GE Healthcare, Chicago IL) in accordance with guidance from the European Society of Urogenital Radiology (ESUR) (25). A board-certified radiologist (Y.L.) reviewed all MR images for the presence of tumor and, if present, assigned a preliminary MRI-based FIGO stage following the FIGO classification system, and according to the rules laid out in the ESUR guidelines (25,26). Briefly, tumor was considered present if MR images demonstrated an intermediate T2 signal intensity (SI) lesion with diffusion restriction (high SI on high b-value DWI and low SI on ADC map), and hypoenhancement compared to myometrium on later phase DCE. The MRI-based FIGO stages are detailed in the Supplementary Methods. In addition to the above qualitative evaluation, two radiologists (J.N. and Y.L.) manually segmented every lesion on each tumor-containing slice using AquariusNet (Terarecon, Foster City, USA) to automatically generate total tumor volume for each patient.
Tumor and cfDNA NGS sequencing analysis
Primary ECs underwent clinical tumor-normal targeted DNA NGS of 468 cancer-related genes using MSK-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) (27,28). NGS was performed on the primary tumor specimen in all but one case; for CD23, NGS was performed on a diagnostic biopsy (Supplementary Table S1). In addition, DNA was extracted from microdissected extra-uterine tumor sites from 6 cases who presented with stage III or stage IV disease and subjected separately to MSK-IMPACT sequencing at MSK’s Integrated Genomics Operation (IGO), as described (29). The median depth of coverage for tumor samples was 618x (range 101x-1030x) and for normal samples was 504x (range 325x-729x). MSK-IMPACT sequencing data were analyzed using previously described bioinformatics approaches (27,30). Somatic mutations identified in the primary tumor or extra-uterine site from a given patient were interrogated in the matched respective primary tumor/ extra-uterine site by manual inspection of BAM files using mpileup files (SAMtools mpileup; v1.2 htslib 1.2.1) (SAMTOOLS, RRID:SCR_002105) (31), as previously described (30). Primary ECs were stratified into The Cancer Genome Atlas (TCGA) molecular subtypes (32), as previously described (28).
For each patient, cfDNA obtained at baseline and post-surgery was subjected to MSK-ACCESS (Analysis of Circulating cfDNA to Examine Somatic Status), a high-depth NGS assay with molecular barcoding technology for the detection of very low frequency somatic alterations in 129 genes, a subset of the 468 MSK-IMPACT genes (33). Of the 44 ECs, 36 had a pre- and/or post-operative blood draw and the required plasma cfDNA input of 10–20ng cfDNA for MSK-ACCESS sequencing (Supplementary Figure S1 and Table S1). In addition, longitudinal cfDNA samples from 25 patients with sufficient cfDNA and/or detectable mutations at baseline or post-surgery and/or disease progression were also subjected to MSK-ACCESS sequencing (Supplementary Table S1). cfDNA samples were sequenced to a median raw coverage of 25,611x (range 13,295 – 58,743x); post collapsing, the median duplex coverage was 1,340x (range 212x – 2,839x). MSK-ACCESS raw sequence data were demultiplexed and processed as previously described (33) (Supplementary Methods). Variant calling was performed in a matched tumor-informed manner (“genotyping”) using Waltz (https://github.com/mskcc/Waltz), and required at least 2 duplex consensus reads, comprising both strands of DNA, to call a somatic single nucleotide variant (SNV) or short insertion and deletion (indel) at a site known to be altered in the matched tumor sample from a given patient, as previously described (33). Mutation-specific ctDNA fraction values were computed for all somatic mutations determined to be clonal in the tissue sample using MSK-IMPACT and covered in the MSK-ACCESS liquid biopsy panel (Supplementary Methods).
Statistical analysis
Comparisons between cfDNA concentrations and clinicopathologic parameters were performed using the Wilcoxon rank sum test. Progression-free survival (PFS) was defined from the date of surgery to the time of recurrence/progression or death or last follow-up whichever occurred first. The median PFS and PFS rates were estimated applying the Kaplan-Meier method. The association between somatic mutation in pre- and post-surgery cfDNA with PFS was examined using Log-rank test with permutation (34). To accommodate the time dependent nature of post-surgery cfDNA mutation, a landmark analysis was performed using the landmark time of 51 days (or 1.68 months) which is the longest time interval between surgery date and post-surgery cfDNA measurement date in this cohort. A p-value of p<0.05 was considered statistically significant. All statistical analyses were performed using R version 4.1.1 (https://cran.r-project.org/) (CRAN, RRID:SCR_003005).
DATA AVAILABLITY
Clinicopathologic information and the somatic mutations identified in the primary tumors and cfDNA are available in Supplementary Tables S1–S3. Targeted sequencing data that support the findings of this study is available at cBioPortal [www.cbioportal.org; Endometrial Carcinoma cfDNA (MSK, Clin Cancer Res 2022)].
RESULTS
EC patient population and association with cfDNA
In this study, 77% (34/44) of EC patients had FIGO stage I disease and 23% (10/44) had advanced-stage disease at diagnosis (stages III and IV; Table 1). As expected, ECs of endometrioid histology were the most common (24/44, 55%), followed by serous carcinoma (7/44; 16%) and carcinosarcoma (6/44; 14%; Table 1). All but one EC patient underwent primary surgery; half of the patients (21/43; 49%) received no further treatment, and the remaining patients were treated with either radiation and/or chemotherapy following surgery (Supplementary Table S1). Despite a subset of the 18- and 24-month timepoints occurring during the COVID-19 pandemic, 229 of the intended 258 (89%) longitudinal blood draws were collected. Median length of clinical follow-up was 33 months (range 0.3–44), with 11 patients being diagnosed with recurrent or persistent disease (Supplementary Table S1).
Table 1:
Histologic type, stage, tumor volume and cfDNA concentration of endometrial cancer patients included this study.
| n (%) | Mean tumor volume cm3 (Std Dev) | MSK-IMPACT sequencing primary tumor n (%) | Mean cfDNA concentration pre-op/baseline ng/ml (Std Dev) | MSK-ACCESS sequencing cfDNA pre-op/baseline n (%) | |
|---|---|---|---|---|---|
| Surgical stage | 44 | 42 | 33 | ||
| Stage IA | 30* (68%) | - | 28* (67%) | 2.68 (1.94) | 19* (58%) |
| Stage IB | 4* (9%) | - | 4* (10%) | 4.05 (2.29) | 4* (12%) |
| Stage II | 0 (0%) | - | 0 (0%) | - | 0 (0%) |
| Stage III | 8 (18%) | - | 8 (19%) | 4.78 (2.56) | 8 (24%) |
| Stage IV | 2 (5%) | - | 2 (5%) | 11.61 (12.74) | 2 (6%) |
| MRI stage | 37 | 105.2 (446) | 35 | 24 | |
| No measurable disease | 7 (21%) | 0 (0) | 5 (14%) | 3.03 (1.76) | 4 (17%) |
| Stage IA | 18 (53%) | 14.4 (26) | 18 (51%) | 2.91 (1.89) | 11 (46%) |
| Stage IB | 3 (9%) | 42.2 (47) | 3 (9%) | 6.01 (3.82) | 3 (13%) |
| Stage II | 1 (3%) | 12.6 (NA) | 1 (3%) | 3.10 (NA) | 1 (4%) |
| Stage III | 3 (9%) | 171.7 (128) | 3 (9%) | 4.06 (2.91) | 3 (13%) |
| Stage IV | 2 (6%) | 1332.5 (1804) | 2 (6%) | 11.61 (12.74) | 2 (8%) |
| Histologic type | 44 | 42 | 33 | ||
| Endometrioid | 24 (55%) | 28.7 (73) | 23 (55%) | 2.90 (1.69) | 16 (48%) |
| Serous | 7 (16%) | 16.0 (27) | 7 (17%) | 4.01 (2.45) | 6 (18%) |
| Carcinosarcoma | 6 (15%) | 671.6 (1291) | 6 (14%) | 5.52 (7.63) | 5 (15%) |
| High-grade carcinoma | 4 (9%) | 13.8 (20) | 3 (7%) | 3.58 (2.21) | 3 (9%) |
| Mixed | 1 (2%) | 7.7 (NA) | 1 (2%) | 7.47 (NA) | 1 (3%) |
| De-differentiated | 1 (2%) | 95.4 (NA) | 1 (2%) | 8.89 (NA) | 1 (3%) |
| Adenosarcoma | 1 (2%) | 111.0 (NA) | 1 (2%) | 2.07 (NA) | 1 (3%) |
One stage IA and two stage IB cases had isolated tumor cells in the lymph nodes on pathology. cfDNA: cell-free DNA; NA, not available; Std Dev, standard deviation.
Median cfDNA concentration in pre-operative cfDNA samples across cases was 2.71 ng/mL (range 0.66–20.62; Supplementary Table S1). We found EC patients with stage III/IV disease at diagnosis to have significantly higher amounts of cfDNA compared to those with stage I disease (Wilcoxon rank sum test, p=0.028; Figure 1A). In addition to surgical stage, MRI stage (25) was assigned based on a pre-operative MRI in 34/44 (77%) patients, which aligned with final surgical stage in 29/34 patients (Table 1, Supplementary Table S1). cfDNA concentrations were found to be numerically higher in histologic types associated with an aggressive clinical behavior, including carcinosarcoma and high-grade carcinomas of no specific type, however this association was not statistically significant likely due to the small sample size in some of the groups (p>0.1; Figure 1B).
Figure 1. Baseline characteristics of newly diagnosed endometrial cancers.
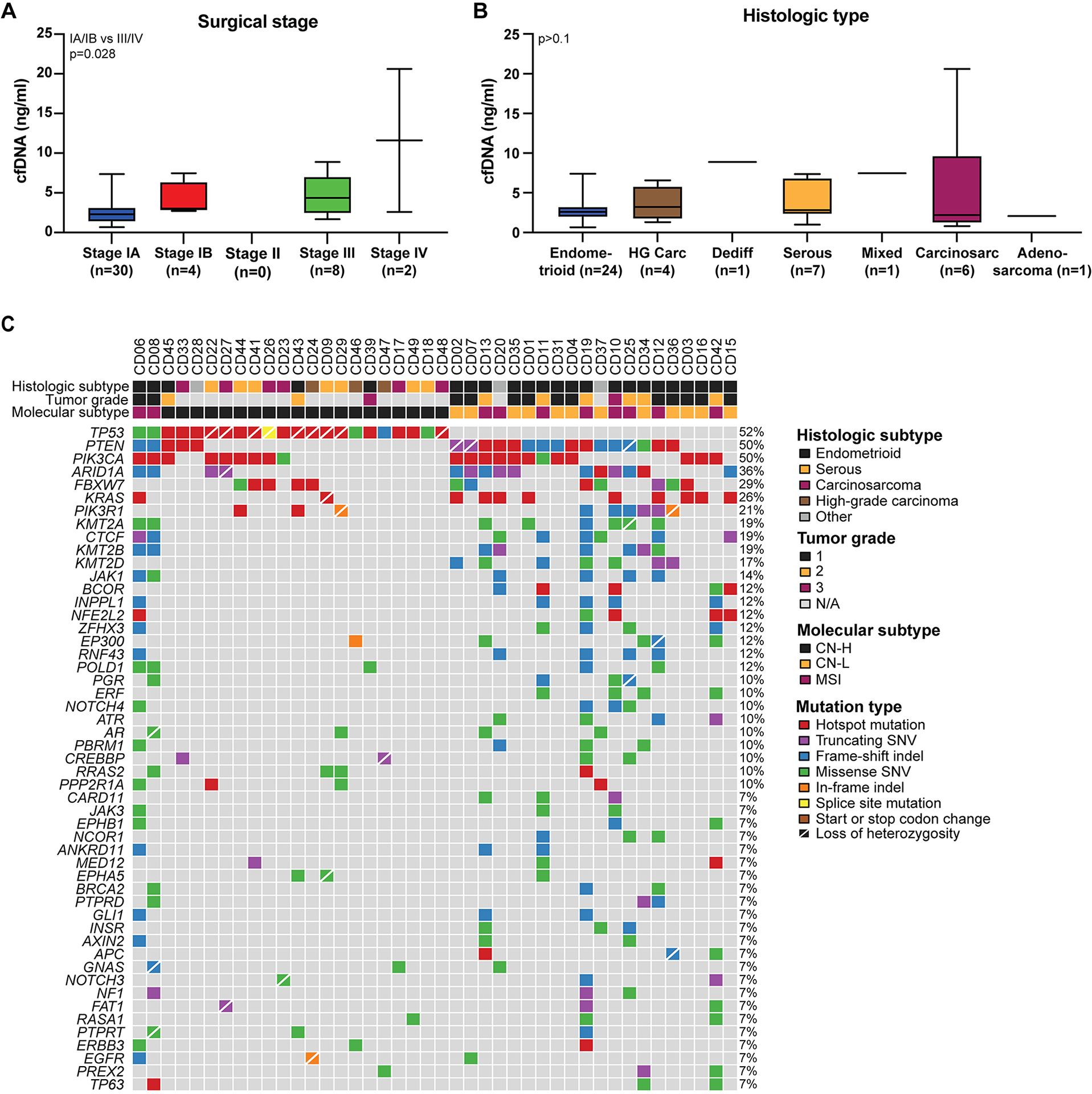
A, cfDNA concentrations in ng/ml plasma at baseline in newly diagnosed endometrial cancers according to surgical stage. Wilcoxon rank sum test. B, cfDNA concentrations in ng/ml plasma at baseline in newly diagnosed endometrial cancers according to histologic type. HG Carc, high-grade carcinoma; Dediff, dedifferentiated; Carcinosarc, carcinosarcoma. Wilcoxon rank sum test was used for comparisons. C, somatic mutations identified in primary newly diagnosed endometrial cancers included in this study. Mutation types are color coded according to the legend. Information on the histologic subtype, tumor grade of endometrioid endometrial cancers, and molecular subtype are provided on top.
The primary ECs of 42 patients were subjected to clinical tumor-normal FDA-authorized MSK-IMPACT sequencing of 468 cancer-related genes; for two cases there was no/ insufficient tumor for molecular profiling (Supplementary Figure S1). A median of 7.5 non-synonymous mutations were detected (range 1 – 92; Supplementary Table S2). Consistent with previous reports (32), the most commonly mutated genes were TP53 (52%), PTEN (50%), PIK3CA (50%), ARID1A (36%), and FBXW7 (29%; Figure 1C, Supplementary Table S2). Of all 42 cases included, ten ECs (24%) were of microsatellite instable (MSI) molecular subtype, 12 (29%) of copy number-low molecular subtype and 20 (48%) of copy number-high molecular subtype (Figure 1C). Of the 23 endometrioid ECs, 11 (48%) were of copy number-low, 9 (39%) of MSI, and 3 (13%) of copy number-high molecular subtype (Figure 1C).
Landscape of somatic mutations in baseline cfDNA
We next sought to define whether the landscape of somatic mutations in baseline/ pre-operative cfDNA would be representative of that identified in the respective primary ECs. A high-depth UMI-based sequencing assay (MSK-ACCESS) revealed the baseline presence of tumor-derived mutations in cfDNA in eight of 36 cases subjected to MSK-ACCESS sequencing (22%), six of which had stage III or stage IV disease (Figure 2, Supplementary Figures S1 and S2, Supplementary Table S1). ctDNA was also detected in a stage 1A high-grade adenosarcoma and a stage 1B high-grade mixed endometrioid/ serous carcinoma with isolated tumor cells (ITCs) and lymphovascular space invasion (Figure 2, Supplementary Figure S2), however none of the low-grade stage I endometrioid EC patients had detectable mutations in their pre-operative cfDNA. Of these 8 ECs with ctDNA being present in plasma at baseline, 6 displayed lymphovascular space invasion (LVSI), which was not significantly different from ECs without detectable ctDNA at baseline (5/25; Fisher’s exact test, p=0.137; Supplementary Table S1). When defining the cancer cell fractions of the mutations identified (i.e., bioinformatically inferred percentage of tumor cells harboring the mutation in a given sample), we observed that all mutations that were clonal in the primary tumor and present in the MSK-ACCESS assay were also detected in baseline/ pre-operative cfDNA (Figure 2, Supplementary Figure S2). The only exception was case CD28, a stage IB mixed EC with ITCs, where a clonal CDKN2A E61Pfs*54 and a TERT promoter C228T mutation present in the primary tumor were not detected in the plasma at baseline, but were detected at later time points (see below), whereas a subclonal TP53 mutation was (Figure 2, Supplementary Figure S2).
Figure 2. Mutations detected in cfDNA at baseline of newly diagnosed endometrial cancers.

In eight patients with newly diagnosed endometrial cancer, mutations present in the primary tumor were detected in plasma at baseline using a high-sensitivity sequencing assay (MSK-ACCESS). For each case, information on the histologic type, surgical stage, MRI-based tumor burden/ volume and circulating tumor (ctDNA) fraction is provided on top. The cancer cell fractions (CCFs) of the somatic mutations identified in the primary tumor (T) and extra-uterine sites are shown on the left, and the mutation types in the primary tumor (T), extra-uterine sites and cfDNA (cf) on the right, color-coded according to the legend. Only genes/ mutations that are present in both, the tumor sequencing assay (MSK-IMPACT) and the cfDNA sequencing assay (MSK-ACCESS) are shown (see Supplementary Figure S2 for all alterations). FT, fallopian tube; ITCs, isolated tumor cells in the lymph nodes; LN, left hypogastric lymph node; LLN1, left external iliac sentinel lymph node; LLN2, right exterior iliac sentinel lymph node; MP, mesenteric implant; SLN1, left exterior iliac sentinel lymph node; SLN2, obturator sentinel lymph node.
In four stage III/IV cases (i.e., CD20, CD22, CD26 and CD36) with detectable mutations in the cfDNA at baseline, in addition to the primary EC, DNA from extra-uterine sites, including (sentinel) lymph nodes, peritoneal implants and fallopian tube, was extracted and subjected to MSK-IMPACT sequencing. Heterogeneity between the primary tumor and extra-uterine sites was observed, with mutations identified in the primary EC not being present in the respective lymph nodes, whereas some lymph nodes/ peritoneal implants harbored additional mutations that were not detected in the respective primary lesions (Supplementary Figure S2). In case CD20, the primary tumor, the left exterior iliac sentinel lymph node (SLN) and obturator SLN shared a set of mutations, including an ARID1A truncating mutation and PIK3CA and KRAS hotspot mutations that were subclonal in the primary tumor but were found to become clonal in the sentinel lymph nodes (Figure 2; Supplementary Figure S2). A clonal PTEN mutation and RNF43, TSC1 and SMARCA4 frameshift mutations were, however, only shared between the primary EC and left exterior iliac SLN but were absent in the obturator SLN. In turn, AGO2 and FOXA1 missense mutations were restricted to the two SLNs. In case CD22, the primary EC and the mesenteric implant and three (sentinel) lymph nodes shared hotspot PIK3CA, TP53 and PPP2R1A mutations. An ARID1A truncating mutation was present in the primary lesion and three of the four extra-uterine sites as well as in cfDNA (Figure 2; Supplementary Figure S2). Whereas the primary EC and the fallopian tube lesion of case CD36 shared all somatic mutations identified, in case CD26, while FBXW7, NTRK3 and PIK3CA mutations were shared between all sites and cfDNA, a different PIK3CA hotspot mutation and a TP53 splice site mutation were restricted to the primary tumor. All mutations, however, were detected in the pre-surgery cfDNA. Sequencing analysis of the liquid biopsy at baseline was generally more representative of the repertoire of somatic mutation in the primary EC than that of the (sentinel) lymph nodes (Figure 2).
The ctDNA fraction in plasma obtained at baseline varied and was between 0.7% and 2.0% in the majority of cases (i.e., CD11, CD20, CD26, CD28, CD36 and CD37; Figure 2, Supplementary Table S3). The ctDNA fraction in pre-operative plasma was elevated in two cases, however. In CD22, a stage IVB endometrial serous carcinoma, the ctDNA fraction was 5.8%, and in CD23, a stage IVB carcinosarcoma with a very high tumor burden (2,608cm3) the ctDNA fraction was 74.6%; both patients died from their disease 31 and 30 months after the baseline sample, respectively (Figure 2; Supplementary Tables S1 and S3).
Mutation tracking in serial cfDNA samples during follow-up
Next, we sought to define whether cfDNA sequencing of serial plasma DNA samples could be employed for disease monitoring including the assessment of treatment response and identification of early disease recurrence. For this, blood samples were collected every 6 months during follow-up; for 25 patients with either recurrent disease and/or detectable post-surgery ctDNA in plasma, the longitudinal cfDNA samples were subjected to MSK-ACCESS sequencing. In six of these 25 EC patients (24%), ctDNA was detected in the serial blood samples (Supplementary Table S1). The change in the mutant allele fraction (MAFs) of the mutations identified as well as of the ctDNA fraction during follow-up closely followed disease progression and therapy response (Figures 3–5, Supplementary Figures S3 and S4).
Figure 3. Longitudinal disease monitoring by sequencing of plasma-derived cfDNA samples from patients CD20 and CD26 with newly diagnosed endometrial cancer.
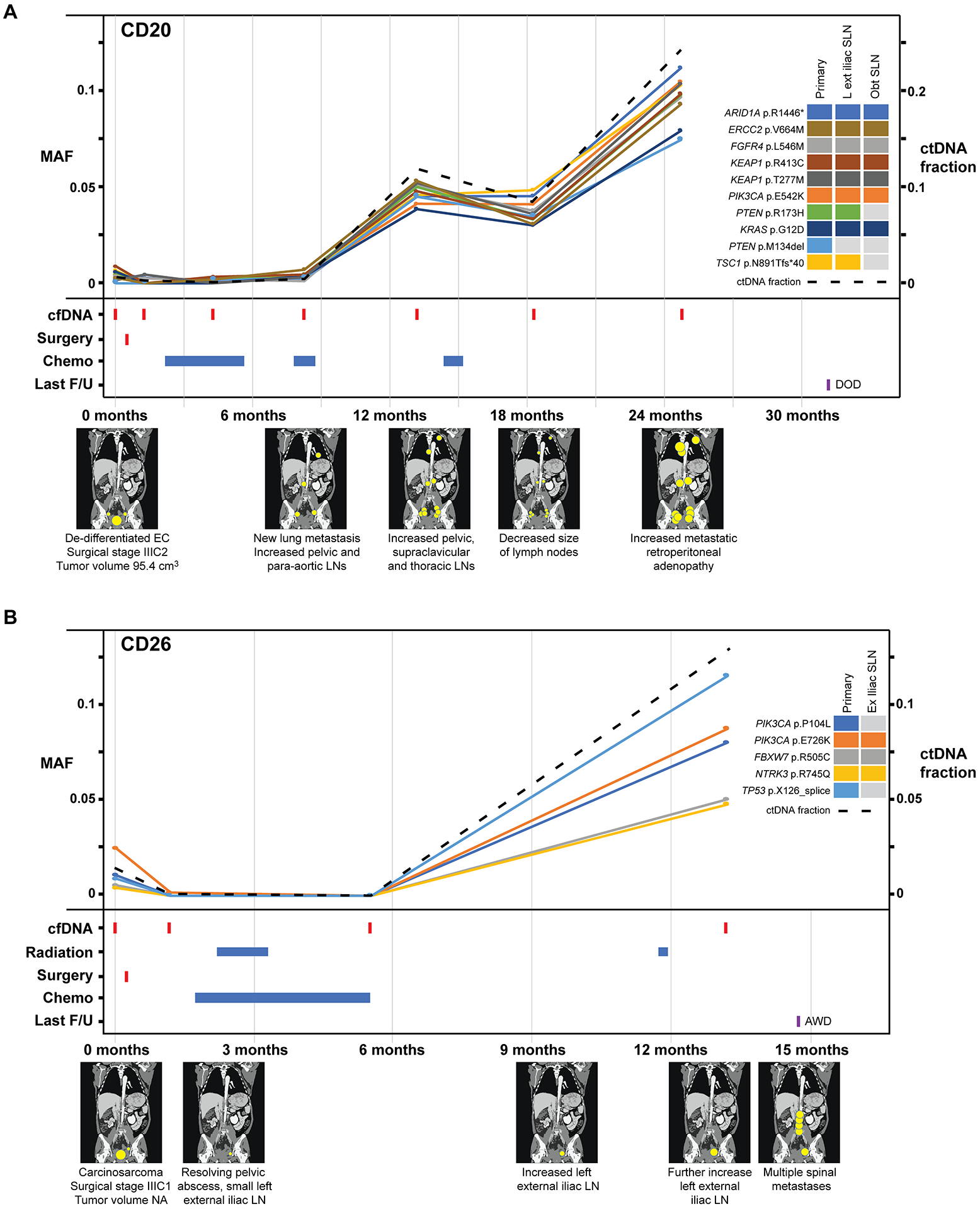
A, Case CD20, B, Case CD26 with detectable pre-operative circulating tumor (ct) DNA and corresponding graphs of mutation allele frequencies (MAFs) according to the time of blood collection, treatment timeline and a pictographic representation of disease burden. MAFs of mutation in plasma DNA (left y-axis) extracted from peripheral blood samples obtained at baseline, post-surgery and serially every 6 months during follow-up, color-coded according to the legend. The ctDNA fraction (right y-axis) is shown by a dashed line. Below the graph, information about surgery, radiation therapy, chemotherapy, last follow-up, and schematic radiographic representations of disease burden are shown. AWD, alive with disease; DOD, dead of disease; EC, endometrial cancer; F/U, follow-up; MAF, mutant allele fraction.
Figure 5. Association of baseline ctDNA and disease progression.
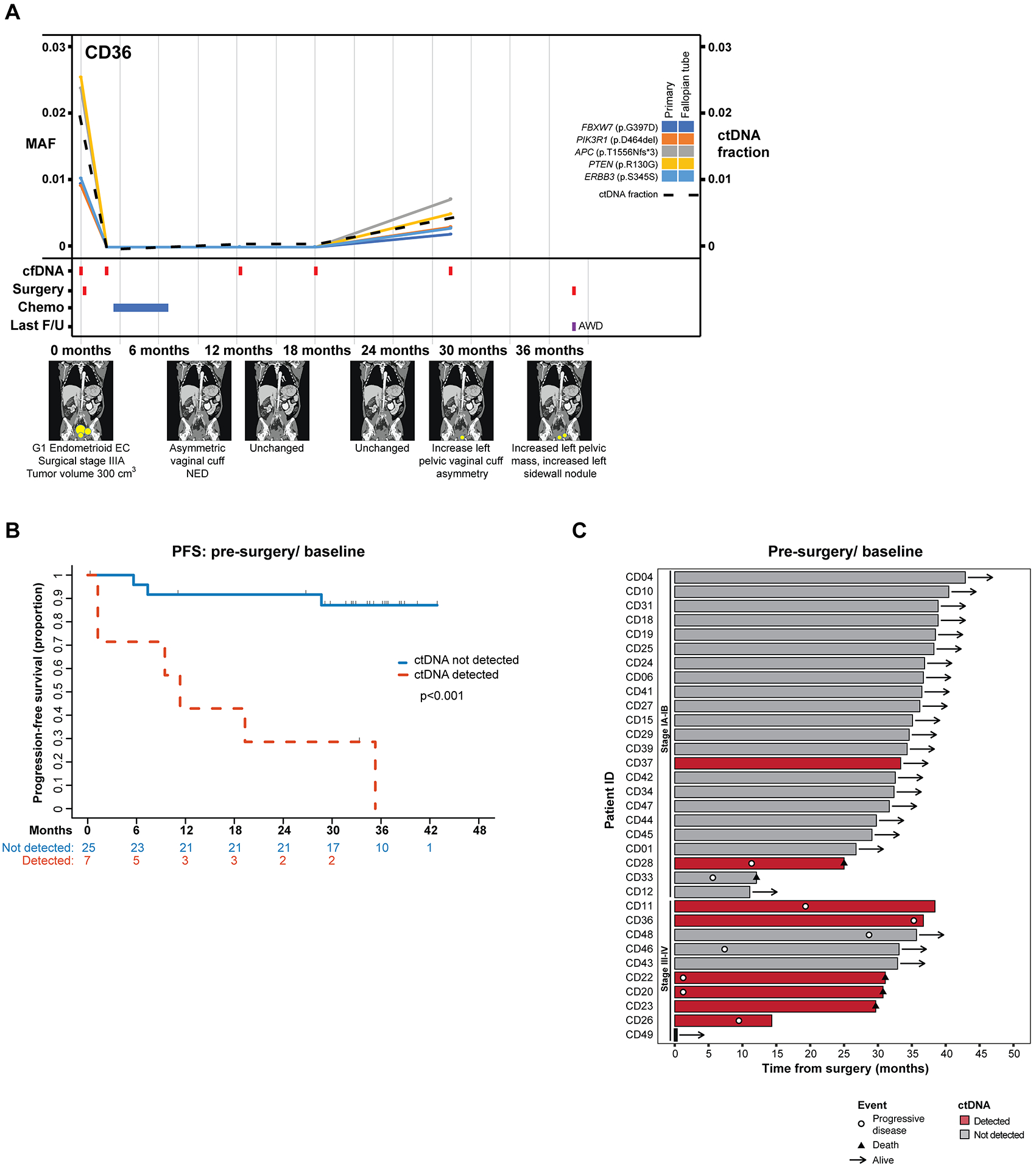
A, Longitudinal disease monitoring of case CD36. MAFs of mutation in plasma DNA (left y-axis) extracted from peripheral blood samples obtained at baseline, post-surgery and serially every 6 months during follow-up, color-coded according to the legend. The ctDNA fraction (right y-axis) is shown by a dashed line. Below the graph, information about surgery, chemotherapy, last follow-up, and schematic radiographic representations of disease burden are shown. AWD, alive with disease; F/U, follow-up, MAF, mutant allele fraction. B, Progression-free survival according to the detection of ctDNA in the baseline/ pre-surgery plasma. P value determined by log rank test. C, Swimmer plot depicting disease progression in newly diagnosed endometrial cancer patients according to the detection of ctDNA at baseline/ pre-surgery and stage.
In case CD20, a stage IIIC2 dedifferentiated carcinoma, a marked increase in the MAFs was observed following the second course of chemotherapy (i.e., 8–9 months post diagnosis). Imaging at that time revealed increased pelvic, supraclavicular and thoracic lymph nodes (Figure 3A). The overall ctDNA fraction and the allele fractions of the 10 mutations detected in plasma decreased during another course of chemotherapy and a clinical response with decreased size of lymph nodes was observed at 18 months. Mirroring the steep increase in MAFs/ ctDNA fraction between 18 and 24 months, imaging at 24 months revealed increased metastatic retroperitoneal adenopathy throughout (Figure 3A). Similarly, in case CD26, a stage IIIC1 carcinosarcoma, tumor was cleared during radiation and chemotherapy post-surgery with almost undetectable levels of tumor mutations in cfDNA at 6 months. Following chemotherapy, at the 12 months blood draw, a marked increase in the MAFs of all five mutations detected in the cfDNA, including two PIK3CA and a TP53 mutation, was observed, which was consistent with the increased tumor burden observed in this patient, including increase in lymph node sizes at 13 months and multiple spinal metastases at 15 months (Figure 3B).
In patient CD22, a stage IVB serous carcinoma, a sharp reduction in the MAFs of all four somatic mutations (i.e., ARID1A, PIK3CA, TP53 and PPP2R1A mutations) was detected post-surgery and chemotherapy. At 12 months, ctDNA levels increased, consistent with new lung nodules and increased perirectal metastases observed by imaging (Figure 4A). Despite the administration of 3 courses of chemotherapy over the next 12 months, the levels of ctDNA continued to rise; this lack of response aligned with the imaging data, which showed an increase in tumor burden and new splenic nodules (Figure 4A; Supplementary Figure S3). In case CD28, a stage IB grade 3 mixed endometrioid/ serous carcinoma with ITCs, only 3 of the 5 mutations (i.e., two PTEN and a TP53 mutation) present in the primary tumor were detected in ctDNA at baseline (Figure 2). Following the first course of chemotherapy plus radiation, an increase in the ctDNA fraction was observed. At 6 months, also the TERT promoter mutation and CDKN2A p.E61Pfs*54 mutation not detected at baseline were now identified in plasma, whereas there was no evidence of disease by imaging (Figure 4B). A sharp increase in MAFs were observed for six months before the detection of new pulmonary nodules on imaging at 12 months. Also in patient CD28 the MAFs of the mutations kept rising despite additional courses of chemotherapy and radiation (Figure 4B). This lack of response to chemo/ radiation therapy was also found in the imaging studies, which showed increased disease volume and new distant metastases at 12, 18 and 24 months (Figure 4B; Supplementary Figure S3). Whilst all five primary tumor mutations (i.e., FBXW7, PIK3R1, APC, PTEN and ERBB3 mutations) were identified in plasma at baseline in CD36, a patient with stage IIIA grade 1 endometrioid EC, these remained undetectable following surgery and chemotherapy for 18 months, and the patient was rendered disease-free (Figure 5A). We did observe an increase in the MAFs of the mutations at the 30-month blood draw, without any change in tumor burden on imaging until 36 month when the patient was noted to have an increased pelvic mass and sidewall nodule and underwent surgery for the removal of these lesions (Figure 5A). Whilst exploratory and further studies would be required to confirm these findings, these data provide evidence demonstrating that the detection of ctDNA changes can provide a lead time over the radiologically and/or clinically detected recurrence in EC patients.
Figure 4. Longitudinal disease monitoring by sequencing of plasma-derived cfDNA samples from patients CD22 and CD28 with newly diagnosed endometrial cancer and comparison with serum CA125 levels.
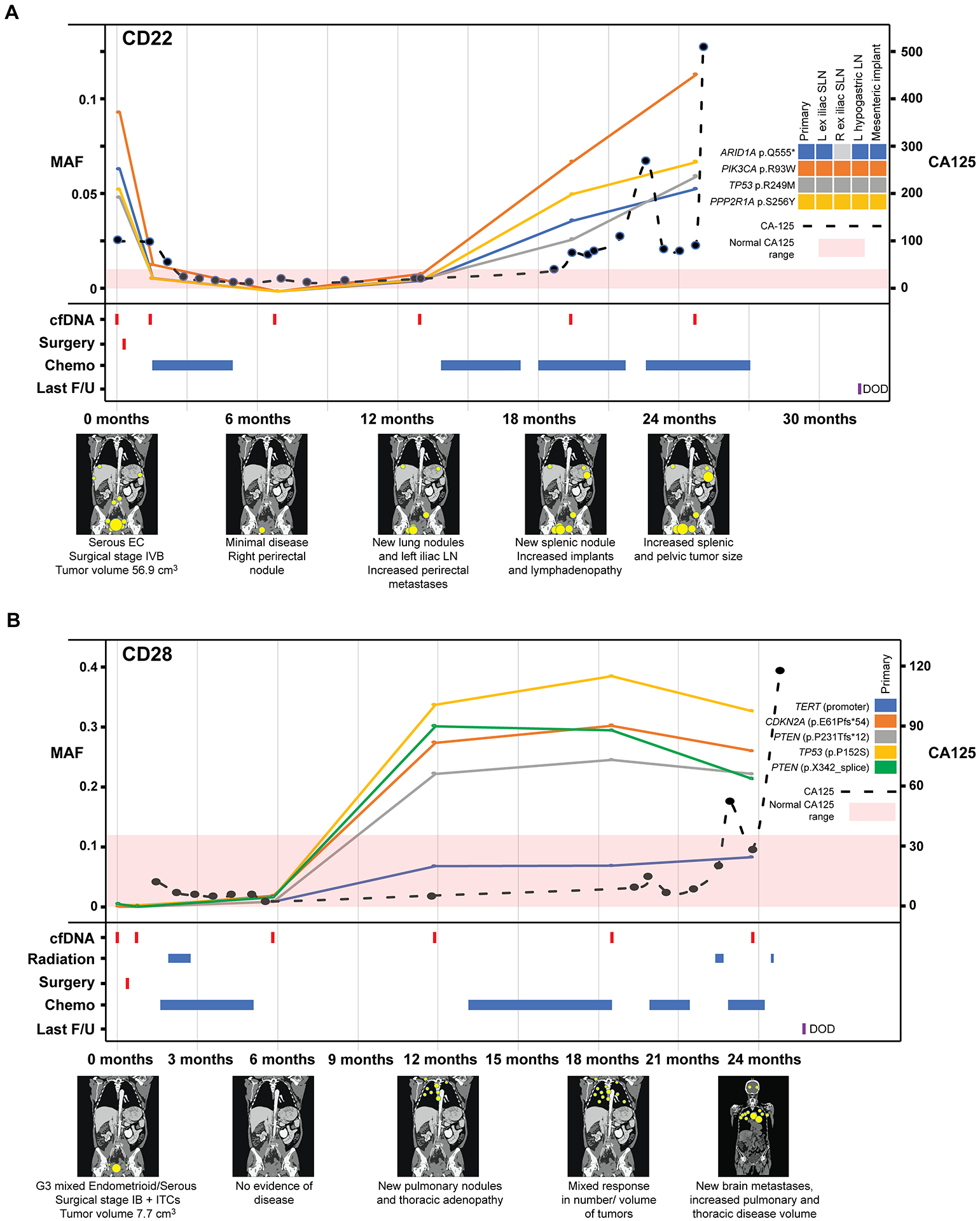
A, Case CD22, B, Case CD28 with detectable pre-operative circulating tumor (ct) DNA and corresponding graphs of mutation allele frequencies (MAFs) according to the time of blood collection, treatment timeline and a pictographic representation of disease burden. MAFs of mutation in plasma DNA (left y-axis) extracted from peripheral blood samples obtained at baseline, post-surgery and serially every 6 months during follow-up, color-coded according to the legend. The serum CA125 levels (right y-axis) in U/ml are shown by a dashed line, with <35 U/mL CA125 being the normal range. Below the graph, information about surgery, radiation therapy, chemotherapy, last follow-up, and schematic radiographic representations of disease burden are shown. DOD, dead of disease; EC, endometrial cancer; F/U, follow-up; ITCs, isolated tumor cells in the lymph nodes; MAF, mutant allele fraction; ND, not determined.
In contrast to ovarian cancer, the role of the tumor antigen CA125 for surveillance in EC is less well established. For two of the ECs with detectable mutations in the cfDNA during follow-up, regular CA125 measurements were obtained. In both CD22 and CD28, elevated CA125 levels were detected, however only 5.8 months and 17 months after the increase in MAFs of tumor-derived mutations in cfDNA (Figures 4A and B). This exploratory finding suggests that the detection of ctDNA in plasma may be more sensitive than the CA125 levels in serum of EC patients, warranting further studies.
ctDNA detection and prediction of progression-free survival
We examined whether the detection of ctDNA at different time points would have the potential to predict recurrences in this series of newly diagnosed ECs. Of the eight patients with detectable ctDNA mutations at baseline, all but one (CD37, adenosarcoma) recurred within the time of follow-up (median 33 months, range 0.3–44; Supplementary Table S1). The four EC patients with detectable mutations in cfDNA immediately post-surgery displayed recurrent or persistent disease following surgery (Supplementary Tables S1 and S3). Furthermore, for CD33, a stage IA carcinosarcoma, while no mutations were detected in the baseline/ pre-surgery blood sample, we observed an increase in the MAFs of the mutations at 6 months, the only longitudinal sample, which mirrored the disease recurrence observed by imaging (Supplementary Figure S4 and Table S3). We found that the presence of ctDNA at baseline/ pre-operatively in this series of newly diagnosed ECs was significantly associated with progression-free survival (p<0.001; Hazard ratio (HR) 11.14 (95% confidence interval (CI) 2.72–45.59); Figures 5B and C). Due to the limited number of events, a meaningful multivariate analysis could not be performed. Finally, we performed a landmark analysis showing that the detection of ctDNA immediately post-surgery also to be significantly associated with disease progression (p=0.014; HR 15.56 (95% CI 2.16–112.16), Supplementary Figure S5), however, the number of cases with measurable ctDNA after surgery in this cohort was small.
DISCUSSION
Our findings demonstrate that the amount of cfDNA present in plasma correlates with surgical stage in newly diagnosed EC patients, and that the detection of mutations in cfDNA using a high-sensitivity high-depth liquid biopsy sequencing assay was almost exclusively restricted to EC patients with advanced disease and more aggressive histology types. We have also shown that mutations detected in plasma/ peripheral blood of EC patients are representative of those present in the primary tumor, with 35/38 (92%) of the primary tumor mutations identified in the cfDNA at diagnosis. Whilst there was limited genetic heterogeneity between the primary ECs and extra-uterine sites/ metastases analyzed by MSK-IMPACT sequencing, the mutations detected in cfDNA captured also those present in the extra-uterine sites. Our study provides evidence that ctDNA detection is not only feasible in patients with EC but also that ctDNA, levels closely mirror treatment response and disease progression. Finally, we demonstrate that the detection of mutations in cfDNA, both pre- and post-surgery, correlates with disease progression in women with newly diagnosed EC.
Our study illustrates that even using a high-sensitivity high-depth UMI-based sequencing assay, ctDNA is detected only in a subset of patients with EC and is associated with stage and histologic type (although not significant for histology, probably due to low numbers). Akin to other cancer types (35,36), we found that cfDNA analysis may be useful in the setting of disease monitoring and detection of minimal residual disease (MRD) to predict disease recurrence. Furthermore, although we and others have previously shown that in early breast cancer serial sampling during follow-up was required for accurate MRD detection (14,37–39), in EC, the analysis of even a single blood sample taken before surgery offered information on disease recurrence. In contrast to breast or ovarian cancer, ECs are characterized by a high number of recurrently mutated cancer genes (4,40). Therefore, sequencing panels targeting cancer-related genes rather than personalized tumor-specific assays are well suited for longitudinal cfDNA-based disease monitoring. At the individual mutation level, the MSK-ACCESS assay employed in this study detects through de novo and tumor-informed analysis high confidence mutations at 0.5% and 0.1%, respectively (33). With the ongoing development of assays for MRD detection following curative-intent treatment and during the surveillance period for relapse, which often target larger numbers of mutations and thereby increase sensitivity for ctDNA (11), future studies using ultra-sensitive assays, in particular for EC patients with low-grade low-stage disease, are warranted.
Shedding of tumor DNA from cancer cells has been shown to be highly variable (12,41), and the fraction of ctDNA in plasma in the ECs studied here was frequently undetectable in early-stage disease or low-grade histologies using a deep sequencing method. ECs with high-risk histologic types, however, including carcinosarcoma and serous ECs, had higher ctDNA fractions in plasma. In addition to disease burden, these higher ctDNA fractions may be due to higher levels of processes that play a role in cfDNA release from cells such as proliferation, apoptosis, and necrosis (42).
Case CD37 included in this study was, following review of the resection specimen, an adenosarcoma rather than an endometrial carcinoma. The differential diagnosis of adenosarcoma includes benign and malignant endometrial conditions, including carcinosarcomas, and the clinical presentation may mimic that of EC with abnormal uterine bleeding (43). Uterine adenosarcomas are a genetically heterogeneous group, and recurrent DICER1, FGFR2 and TP53 mutations, MDM2/CDK4/HMGA2 and TERT amplification as well as NCOA2/3 gene fusions, amongst others, have been reported in these tumors (43,44). The primary adenosarcoma CD37 in this study was found to harbor 12 somatic mutations, including pathogenic DICER1, FGFR2, PTEN and ARID1A mutations (Supplementary Figure S2). Notably, the 3 mutations covered by the MSK-ACCESS assay were detected in the cfDNA at baseline (Figure 2), providing evidence to suggest that cfDNA analysis may be used at least for a subset of uterine sarcomas. The diagnostic performance of liquid biopsy in sarcoma in general has been reported to vary and to be dependent on tumor type (45). Larger studies to demonstrate the prognostic value and clinical utility of cfDNA assays in uterine sarcoma are warranted.
Our study has several limitations. Despite the prospective accrual of patients with EC, the number of patients when stratified according to stage or histologic type is small, and PFS analyses are based on limited number of events. Furthermore, due to the COVID-19 pandemic, 11% of samples could not be collected during the follow-up period as initially planned. Nevertheless, our findings provide a proof-of-principle of the role of cfDNA detection in prognostication and disease monitoring in EC.
It is currently still unclear which early-stage EC patients would benefit from adjuvant therapy after surgery with radiation, chemotherapy or both (6,46–48). In addition, recurrent EC can still be cured with salvage therapy if it is caught before wide-spread metastasis occurs (49). Currently, the role of ITCs in the evaluation of risk of recurrence in EC is also unanswered (50). Given these challenges, cfDNA analysis could benefit not only those EC patients with advanced disease for early detection of recurrence but could also prevent women the unnecessary morbidity of adjuvant therapy. Based on our findings, larger, prospective studies are warranted to incorporate cfDNA analysis as a monitoring tool and to guide additional therapies in EC patients at high risk for recurrence.
In conclusion, we have shown here that ctDNA mutation detection in plasma from newly diagnosed EC patients is not only feasible but has prognostic value and enables disease and treatment response monitoring.
Supplementary Material
TRANSLATIONAL RELEVANCE.
The use of minimally invasive methods for risk stratification, therapy response monitoring, and recurrence detection may impact treatment decision-making and outcomes for patients with endometrial cancer (EC), the leading cause of gynecologic cancer mortality. We demonstrate in EC patients that the detection of circulating tumor (ct)DNA at baseline or immediately post-surgery allowed for the identification of patients with high-risk of disease progression. Further, analyses of longitudinally collected plasma samples revealed that changes in ctDNA fraction mirrored the disease course and therapeutic response and had lead time compared to CT imaging in a subset of EC patients. This proof-of-principle study paves the way for future work assessing the clinical utility of ctDNA-based risk stratification and disease monitoring. Future studies are warranted to define whether EC patients with advanced disease who have changes in ctDNA fraction during surveillance would benefit from immediate therapeutic intervention.
ACKNOWLEDGMENTS/ FUNDING
Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748). B. Weigelt and J.S. Reis-Filho are funded in part by Breast Cancer Research Foundation and by the National Cancer Institute/National Institutes of Health (P50 CA247749 01). B. Weigelt is also funded in part by Cycle for Survival grants.
Disclosures:
N.R. Abu-Rustum reports institutional grants from Stryker/Novadaq, Olympus and GRAIL, outside the submitted work. B. Weigelt reports ad hoc membership of Repare Therapeutics, outside the scope of the submitted work. J.S. Reis-Filho reports receiving personal/consultancy fees from Goldman Sachs, REPARE Therapeutics, Paige.AI and Eli Lilly, membership of the scientific advisory boards of VolitionRx, REPARE Therapeutics, Paige.AI and Personalis, membership of the Board of Directors of Grupo Oncoclinicas, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech and InVicro, outside the scope of this study. M.M. Leitao Jr reports institutional research funds from KCI/ Acelity, advisory boards for JnJ/ Ethicon and Takeda, and ad hoc speaker for Intuitive Surgical, Inc, outside the current work. D. Zamarin reports institutional grants from Genentech, AstraZeneca, and Plexxikon, as well as personal fees from Genentech, AstraZeneca, Xencor, Memgen, Takeda, Synthekine, Immunos, and Calidi Biotherapeutics, outside of the submitted work. D. Zamarin is also an inventor on a patent related to the use of oncolytic Newcastle Disease Virus for cancer therapy. C.F. Friedman reports institutional grants from Genentech, Bristol-Meyers Squibb, Daiichi, and Merck, as well as personal fees from Bristol-Meyers Squibb and Seagen, and uncompensated scientific advisory board work for Merck and Genentech; these are all outside the scope of the submitted work. C. Aghajanian has received research grants from Abbvie, Clovis, Genentech, and Astra Zeneca and served on advisory boards for Abbvie, AstraZeneca/Merck, Eisai/Merck, Mersana Therapeutics, Repare Therapeutics, and Roche/Genentech. Y. Lakhman reports serving as a consultant of Calyx Clinical Trial Solutions, outside the submitted work. The remaining authors have no conflicts of interest to disclose.
REFERENCES:
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020;126:2225–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J Natl Cancer Inst 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol 2014;15:e268–78. [DOI] [PubMed] [Google Scholar]
- 5.Wright JD, Barrena Medel NI, Sehouli J, Fujiwara K, Herzog TJ. Contemporary management of endometrial cancer. Lancet 2012;379:1352–60. [DOI] [PubMed] [Google Scholar]
- 6.de Boer SM, Powell ME, Mileshkin L, Katsaros D, Bessette P, Haie-Meder C, et al. Adjuvant chemoradiotherapy versus radiotherapy alone for women with high-risk endometrial cancer (PORTEC-3): final results of an international, open-label, multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller DS, Filiaci VL, Mannel RS, Cohn DE, Matsumoto T, Tewari KS, et al. Carboplatin and Paclitaxel for Advanced Endometrial Cancer: Final Overall Survival and Adverse Event Analysis of a Phase III Trial (NRG Oncology/GOG0209). J Clin Oncol 2020;38:3841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Q, Peng H, Qi X, Wu M, Zhao X. Targeted therapies in gynecological cancers: a comprehensive review of clinical evidence. Signal Transduct Target Ther 2020;5:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan YY, Guo QR, Wang FH, Adhikari R, Zhu ZY, Zhang HY, et al. Cell-Free DNA: Hope and Potential Application in Cancer. Front Cell Dev Biol 2021;9:639233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weigelt B, Comino-Mendez I, de Bruijn I, Tian L, Meisel JL, Garcia-Murillas I, et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin Cancer Res 2017;23:6708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan JCM, Mughal TI, Razavi P, Dawson SJ, Moss EL, Govindan R, et al. Liquid biopsies for residual disease and recurrence. Med (N Y) 2021;2:1292–313. [DOI] [PubMed] [Google Scholar]
- 12.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hench IB, Hench J, Tolnay M. Liquid Biopsy in Clinical Management of Breast, Lung, and Colorectal Cancer. Front Med (Lausanne) 2018;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015;7:302ra133. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Sun L, Zhang Y, Cai S. Patterns of distant metastases in patients with endometrial carcinoma: A SEER population-based analysis. Journal of Clinical Oncology 2019;37:e17109–e. [Google Scholar]
- 16.Feng W, Jia N, Jiao H, Chen J, Chen Y, Zhang Y, et al. Circulating tumor DNA as a prognostic marker in high-risk endometrial cancer. J Transl Med 2021;19:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shintani D, Hihara T, Ogasawara A, Sato S, Yabuno A, Tai K, et al. Tumor-related mutations in cell-free DNA in pre-operative plasma as a prognostic indicator of recurrence in endometrial cancer. Int J Gynecol Cancer 2020;30:1340–6. [DOI] [PubMed] [Google Scholar]
- 18.Casas-Arozamena C, Diaz E, Moiola CP, Alonso-Alconada L, Ferreiros A, Abalo A, et al. Genomic Profiling of Uterine Aspirates and cfDNA as an Integrative Liquid Biopsy Strategy in Endometrial Cancer. J Clin Med 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayo-de-Las-Casas C, Velasco A, Sanchez D, Martinez-Bueno A, Garzon-Ibanez M, Gatius S, et al. Detection of somatic mutations in peritoneal lavages and plasma of endometrial cancer patients: A proof-of-concept study. Int J Cancer 2020;147:277–84. [DOI] [PubMed] [Google Scholar]
- 20.Cicchillitti L, Corrado G, De Angeli M, Mancini E, Baiocco E, Patrizi L, et al. Circulating cell-free DNA content as blood based biomarker in endometrial cancer. Oncotarget 2017;8:115230–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grassi T, Harris FR, Smadbeck JB, Murphy SJ, Block MS, Multinu F, et al. Personalized tumor-specific DNA junctions to detect circulating tumor in patients with endometrial cancer. PLoS One 2021;16:e0252390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolivar AM, Luthra R, Mehrotra M, Chen W, Barkoh BA, Hu P, et al. Targeted next-generation sequencing of endometrial cancer and matched circulating tumor DNA: identification of plasma-based, tumor-associated mutations in early stage patients. Mod Pathol 2019;32:405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.WHO Classification of Tumours Editorial Board. Female Genital Tumours. Lyon: IARC; 2020. [Google Scholar]
- 24.Kothari P, Marass F, Yang JL, Stewart CM, Stephens D, Patel J, et al. Cell-free DNA profiling in retinoblastoma patients with advanced intraocular disease: An MSKCC experience. Cancer Med 2020;9:6093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nougaret S, Horta M, Sala E, Lakhman Y, Thomassin-Naggara I, Kido A, et al. Endometrial Cancer MRI staging: Updated Guidelines of the European Society of Urogenital Radiology. Eur Radiol 2019;29:792–805. [DOI] [PubMed] [Google Scholar]
- 26.Amant F, Mirza MR, Koskas M, Creutzberg CL. Cancer of the corpus uteri. Int J Gynaecol Obstet 2018;143 Suppl 2:37–50. [DOI] [PubMed] [Google Scholar]
- 27.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Momeni-Boroujeni A, Dahoud W, Vanderbilt CM, Chiang S, Murali R, Rios-Doria EV, et al. Clinicopathologic and Genomic Analysis of TP53-Mutated Endometrial Carcinomas. Clin Cancer Res 2021;27:2613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SH, Da Cruz Paula A, Basili T, Dopeso H, Bi R, Pareja F, et al. Identification of recurrent FHL2-GLI2 oncogenic fusion in sclerosing stromal tumors of the ovary. Nat Commun 2020;11:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.da Silva EM, Fix DJ, Sebastiao APM, Selenica P, Ferrando L, Kim SH, et al. Mesonephric and mesonephric-like carcinomas of the female genital tract: molecular characterization including cases with mixed histology and matched metastases. Mod Pathol 2021;34:1570–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brannon AR, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun 2021;12:3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heller G, Venkatraman ES. Resampling procedures to compare two survival distributions in the presence of right-censored data. Biometrics 1996;52:1204–13. [Google Scholar]
- 35.Moding EJ, Nabet BY, Alizadeh AA, Diehn M. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discov 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alix-Panabieres C, Pantel K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov 2021;11:858–73. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Murillas I, Chopra N, Comino-Mendez I, Beaney M, Tovey H, Cutts RJ, et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol 2019;5:1473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coombes RC, Page K, Salari R, Hastings RK, Armstrong A, Ahmed S, et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin Cancer Res 2019;25:4255–63. [DOI] [PubMed] [Google Scholar]
- 39.Chedid J, Allam S, Chamseddine N, Bou Zerdan M, El Nakib C, Assi HI. Role of circulating tumor DNA and circulating tumor cells in breast cancer: History and updates. SAGE Open Med 2022;10:20503121221077838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018;33:690–705 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 2005;102:16368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 43.Parra-Herran C, Howitt BE. Uterine Mesenchymal Tumors: Update on Classification, Staging, and Molecular Features. Surg Pathol Clin 2019;12:363–96. [DOI] [PubMed] [Google Scholar]
- 44.Piscuoglio S, Burke KA, Ng CK, Papanastasiou AD, Geyer FC, Macedo GS, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol 2016;238:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Laan P, van Houdt WJ, van den Broek D, Steeghs N, van der Graaf WTA. Liquid Biopsies in Sarcoma Clinical Practice: Where Do We Stand? Biomedicines 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keys HM, Roberts JA, Brunetto VL, Zaino RJ, Spirtos NM, Bloss JD, et al. A phase III trial of surgery with or without adjunctive external pelvic radiation therapy in intermediate risk endometrial adenocarcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2004;92:744–51. [DOI] [PubMed] [Google Scholar]
- 47.Creutzberg CL, Nout RA, Lybeert ML, Warlam-Rodenhuis CC, Jobsen JJ, Mens JW, et al. Fifteen-year radiotherapy outcomes of the randomized PORTEC-1 trial for endometrial carcinoma. Int J Radiat Oncol Biol Phys 2011;81:e631–8. [DOI] [PubMed] [Google Scholar]
- 48.Wortman BG, Creutzberg CL, Putter H, Jurgenliemk-Schulz IM, Jobsen JJ, Lutgens L, et al. Ten-year results of the PORTEC-2 trial for high-intermediate risk endometrial carcinoma: improving patient selection for adjuvant therapy. Br J Cancer 2018;119:1067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Francis SR, Ager BJ, Do OA, Huang YJ, Soisson AP, Dodson MK, et al. Recurrent early stage endometrial cancer: Patterns of recurrence and results of salvage therapy. Gynecol Oncol 2019;154:38–44. [DOI] [PubMed] [Google Scholar]
- 50.Ghoniem K, Larish AM, Dinoi G, Zhou XC, Alhilli M, Wallace S, et al. Oncologic outcomes of endometrial cancer in patients with low-volume metastasis in the sentinel lymph nodes: An international multi-institutional study. Gynecol Oncol 2021;162:590–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.