

ABSTRACT
Background
Spinal cord damage is a hallmark of Friedreich's ataxia (FRDA), but its progression and clinical correlates remain unclear.
Objective
The objective of this study was to perform a characterization of cervical spinal cord structural damage in a large multisite FRDA cohort.
Methods
We performed a cross‐sectional analysis of cervical spinal cord (C1–C4) cross‐sectional area (CSA) and eccentricity using magnetic resonance imaging data from eight sites within the ENIGMA‐Ataxia initiative, including 256 individuals with FRDA and 223 age‐ and sex‐matched control subjects. Correlations and subgroup analyses within the FRDA cohort were undertaken based on disease duration, ataxia severity, and onset age.
Results
Individuals with FRDA, relative to control subjects, had significantly reduced CSA at all examined levels, with large effect sizes (d > 2.1) and significant correlations with disease severity (r < −0.4). Similarly, we found significantly increased eccentricity (d > 1.2), but without significant clinical correlations. Subgroup analyses showed that CSA and eccentricity are abnormal at all disease stages. However, although CSA appears to decrease progressively, eccentricity remains stable over time.
Conclusions
Previous research has shown that increased eccentricity reflects dorsal column (DC) damage, while decreased CSA reflects either DC or corticospinal tract (CST) damage, or both. Hence our data support the hypothesis that damage to the DC and damage to CST follow distinct courses in FRDA: developmental abnormalities likely define the DC, while CST alterations may be both developmental and degenerative. These results provide new insights about FRDA pathogenesis and indicate that CSA of the cervical spinal cord should be investigated further as a potential biomarker of disease progression. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Keywords: Friedreich's ataxia, MRI, spinal cord, ENIGMA‐ataxia, SCT
Friedreich's ataxia (FRDA) is a neurogenetic disease caused by GAA expansions or point mutations in the first intron of the FXN gene, 1 leading to lower levels of the protein frataxin and resulting in mitochondrial dysfunction and neurodegeneration. 2 FRDA is the most common autosomal recessive ataxia worldwide. 2 The first symptoms typically begin in late childhood or adolescence and are characterized by slowly progressive ataxia and sensory abnormalities. 2 , 3 A smaller subset of individuals manifest symptoms after the age of 25 years and are known as individuals with late‐onset FRDA (LOFA). 4 , 5 These individuals are clinically characterized by slower disease progression and milder nonneurological symptoms. 6
Pathology studies in FRDA show that structural damage affects both the central and the peripheral nervous systems. 7 , 8 In fact, the spinal cord, dorsal root ganglia, and dentate nucleus of the cerebellum are the main targets of damage in the disease. 7 Magnetic resonance imaging (MRI)‐based studies have confirmed such findings and, beyond that, have shown structural damage in the cerebellum, brainstem, cerebellar peduncles, and motor cortex. 9
In recent years, there has been renewed interest in assessing spinal cord damage using noninvasive MRI in FRDA. 10 , 11 , 12 , 13 , 14 Quantitative structural neuroimaging studies have shown atrophy and anteroposterior flattening in affected subjects, particularly at cervical and thoracic levels. 10 , 11 , 14 Using diffusion tensor imaging, Hernandez et al 13 and Joers et al 14 also reported on microstructural changes in the corticospinal tracts and dorsal columns of the cervical spinal cord in individuals with FRDA. Using magnetic resonance spectroscopy, Joers et al 14 reported on large neurochemical changes in the spinal cord in FRDA. In all these studies, the authors were able to find significant correlations between spinal cord MRI metrics and disease severity. Thus, in vivo imaging is well aligned with histological evidence that spinal cord compromise plays a major role in the pathophysiology of FRDA.
Several aspects of spinal cord changes in people with FRDA remain unclear. It is not yet established how spinal cord morphometric abnormalities—atrophy and flattening—change along the disease course. Moreover, differences may exist in the magnitude, progression, and association with clinical variables of spinal cord damage in pediatric versus adult patients and in individuals with early versus late symptom onset. These are relevant issues, not only to understand the underlying biology of the disorder but also to uncover potential imaging‐based biomarkers.
Prior neuroimaging studies have generally relied on modest sample sizes from single sites, limiting opportunities to provide robust disease characterization, reliable effect size (ES) estimates, and subgroup analyses. The ENIGMA‐Ataxia working group is an international collaboration that aggregates MRI data from individuals with ataxias. This consortium offers a unique opportunity to enlarge cohort sizes and to accomplish more detailed analyses in rare diseases, such as FRDA. 15 Hence the main goal of this study was to perform a comprehensive evaluation of cervical spinal cord damage in FRDA using a large dataset collected within the ENIGMA‐Ataxia group. We sought to characterize the pattern of damage and how it evolves across disease subgroups, stratified according to the time from onset and the magnitude of disease severity.
Subjects and Methods
Participants and Data
We performed a retrospective cross‐sectional analysis of data from eight sites in the ENIGMA‐Ataxia working group, totaling 256 patients with molecular confirmation of FRDA and 223 age‐ and sex‐matched nonataxic control subjects (Table 1, Supporting Information Table S S1). Disease duration and age at symptom onset were recorded for all participants with FRDA, and disease severity was quantified using one of the following validated clinical scales: the Friedreich's Ataxia Rating Scale (FARS), 16 , 17 the modified FARS (mFARS), 18 or the Scale for Assessment and Rating of Ataxia (SARA). 19 All diagnoses of FRDA were genetically confirmed at all sites, but individual‐level GAA repeat information was not consistently available because of local reporting procedures or data privacy considerations. To assess the cervical spinal cord, we used high‐resolution T1‐weighted MRIs covering the brain and upper cervical vertebrae acquired on 3‐T clinical scanners with spatial resolution not inferior to 1 mm isotropic (Supporting Information Table S2). Subjects with FRDA and control subjects from each site underwent MRI scans using the same scanner and protocol.
TABLE 1.
Demographics data for all sites
| Sites | Average age, y (range) | Sex, n | Average GAA1 | Average GAA2 | Average onset age, y | Average disease duration, y | Disease severity | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Scale | Average | Normalized scale | ||||||
| Aachen (n = 32) | 36 ± 12 (19–59) | 16 | 16 | 497 ± 224 | 819 ± 218 | 17 ± 8 | 20 ± 9 | SARA | 20 ± 9 | 0.486 ± 0.227 |
| Campinas (n = 83) | 30 ± 13 (7–66) | 31 | 52 | 1026 ± 267 | 869 ± 215 | 18 ± 9 | 12 ± 10 | FARS | 55 ± 22 | 0.440 ± 0.178 |
| Conegliano (n = 46) | 24 ± 12 (8–51) | 18 | 21 | 671 ± 179 | – | 12 ± 7 | 13 ± 9 | SARA | 18 ± 8 | 0.441 ± 0.199 |
| Essen (n = 15) | 44 ± 11 (26–60) | 6 | 9 | 415 ± 292 | 648 ± 306 | 17 ± 10 | 23 ± 9 | SARA | 24 ± 4 | 0.598 ± 0.107 |
| Melbourne1 (n = 22) | 39 ± 14 (22–63) | 11 | 11 | 532 ± 234 | 908 ± 222 | 21 ± 9 | 18 ± 10 | FARS | 84 ± 29 | 0.667 ± 0.226 |
| Melbourne2 (n = 14) | 30 ± 9 (18–49) | 8 | 6 | 604 ± 206 | 870 ± 186 | 15 ± 4 | 14 ± 7 | mFARS | 47 ± 22 | 0.526 ± 0.164 |
| Minnesota (n = 26) | 19 ± 7 (10–35) | 14 | 12 | 598 ± 184 | 960 ± 213 | 14 ± 5 | 6 ± 4 | FARS a | 43 ± 14 | 0.341 ± 0.111 |
| Tubingen (n = 14) | 32 ± 11 (18–53) | 10 | 4 | – | – | 18 ± 9 | 14 ± 7 | SARA | 18 ± 8 | 0.447 ± 0.208 |
| Total (N = 252) | 30 ± 13 (7–66) | 114 | 131 | 639 ± 286 | 640 ± 421 | 17 ± 9 | 14 ± 10 | – | – | 0.473 ± 0.201 |
For the healthy control subjects demographics data, see Supporting Information Table S S1.
Abbreviations: SARA, Scale for Assessment and Rating of Ataxia; FARS, Friedreich's Ataxia Rating Scale; mFARS, modified Friedreich's Ataxia Rating Scale.
Maximum score is 117.
This study was approved by the Institutional Review Board of each site, and all participants provided written informed consent.
Image Processing
Data processing was undertaken using harmonized protocols developed by the ENIGMA‐Ataxia consortium (http://enigma.ini.usc.edu/ongoing/enigma-ataxia/), based on publicly available and well‐validated software toolboxes. 20
To measure the cross‐sectional area (CSA) and eccentricity, we employed the Spinal Cord Toolbox (SCT) version 4.2.2, an open‐source software package specifically designed to process spinal cord multimodal MRI data. 20 In brief, automatic segmentation of the cervical spinal cord was conducted using a deep learning algorithm, 21 and if deemed necessary after visual inspection, the segmentations were manually corrected. Next, the C2 and C3 vertebral levels were manually marked at the posterior tip of the vertebral discs, which enabled the registration of subject images to a standardized template of the spinal cord and brainstem (the PAM50 template). 22 , 23 , 24 Lastly, the mean CSA and eccentricity were computed at each of the C1 to C4 vertebrae after correcting for the curvature of the spine. The CSA is quantified by the number of pixels in the set of axial slices defining each vertebral level of the segmented spinal cord, reported in millimeters squared. Eccentricity is computed by fitting an ellipse to each axial spinal slice and determining the deviation (ie, flattening) of the ellipse relative to a perfect circle. Mathematically, such a measure characterizes the shape of the spinal cord cross‐section defined as the square root of 1 − (d/D), 2 where d and D are respectively defined as the smallest and largest diameter of the ellipse. Values closer to 1 indicate an anteroposterior flattening of the spinal cord. We assessed only the upper cervical spinal cord because we used MRIs centered on the brain with limited spinal cord coverage (Fig. 1). Because the spinal cord coverage was slightly different across individuals due to head size variability or field‐of‐view placement during data acquisition, different sample sizes were available for each vertebral level we examined (control subjects: C1 = 223, C2 = 223, C3 = 215, and C4 = 170; patients: C1 = 252, C2 = 252, C3 = 237, and C4 = 170).
FIG 1.
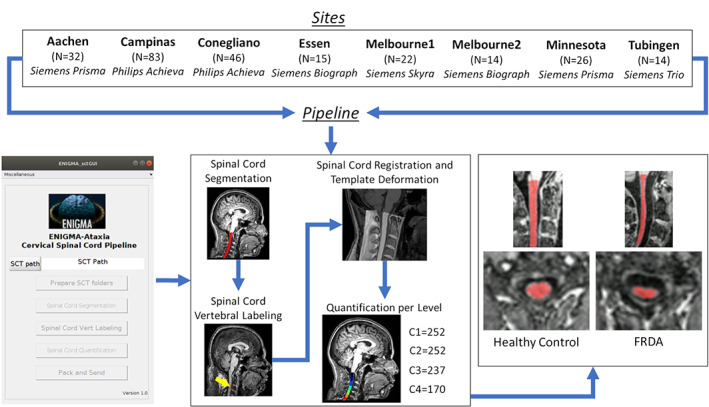
Study design and imaging processing pipeline. For healthy control subject numbers, see Supporting Information Table S S1. N is the number of patients with Friedreich's ataxia (FRDA) enrolled by each site, and C1, C2, C3, and C4 are the sample sizes available for each vertebral level assessed. SCT, Spinal Cord Toolbox. [Color figure can be viewed at wileyonlinelibrary.com]
Statistical Analysis
Overall FRDA Versus Control Comparison
We compared CSA and eccentricity at each vertebral level from C1 to C4 in all individuals with FRDA relative to the age‐ and sex‐matched control cohort using analyses of covariance with age, sex, and site as covariates of no interest. There is no systematic relationship between spinal cord CSA or eccentricity and brain/head size 25 ; therefore, brain volume was not included as a covariate. We corrected for multiple comparisons using Bonferroni adjustment of statistical significance thresholds. ESs of statistically significant results were computed as follows (Cohen's d):
| (1) |
| (2) |
where μ 1 and μ 2 are the mean values for the control and FRDA groups, respectively; s pooled is the pooled standard deviation; n 1 and n 2 are the number of subjects in each group; and s 1 and s 2 are the respective group standard deviations. We considered ES values of 0.2 as small, 0.5 as moderate, 0.8 as large, and >1.2 as very large, according to established convention. 26
Clinical Correlation Analyses
To assess correlations between spinal cord morphometric data (CSA and eccentricity) and clinical parameters (disease duration and disease severity), we used the Pearson correlation coefficient. Before performing the analyses, we first adjusted the data to account for site, age, and sex effects using a linear model. Multiple clinical rating scales were used to assess disease severity across the sites (FARS, mFARS, and SARA). There was a high correlation between SARA and FARS total neurological scores (r = 0.860 and P < 0.0001) in our participants for whom both scales were collected at the same time (Conegliano: n = 28, age = 23.7 ± 11.4 years, 19 males/23 females, SARA = 17.5 ± 8.0, FARS = 57.1 ± 20.4; Melbourne1: n = 31, age = 36.6 ± 13.0 years, 17 males/14 females, SARA = 19.3 ± 8.6, FARS = 81.1 ± 28.4; Minnesota: n = 11, age = 18.1 ± 7.7 years, 6 males/5 females, SARA = 9.4 ± 2.7, FARS = 40.7 ± 9.7), which is in agreement with comparable previous work from Bürk et al 27 (r = 0.953, P < 0.0001). To accomplish a direct pooled analysis, we therefore created a normalized disease severity variable by dividing the disease severity scores by the respective maximum value of the respective scale; eg, disease severity measures quantified using FARS were divided by 125 (except by Minnesota site, maximum score of 117), mFARS scores were divided by 93, and SARA measures were divided by 40. We note that these scales likely have slightly different psychometric properties (eg, differing ceiling and floor effects) 18 , 28 ; thus, although this normalization approach is strongly supported by the very high interscale correlations, we acknowledge that there is not a precise 1‐to‐1 correspondence in their absolute or relative scores. However, our goal is not to establish absolute harmonization across scales or investigate detailed symptom expression and progression, but rather to test for general trends between overall ataxia severity and spinal cord structure. To corroborate the results obtained using this normalization and data pooling approach, we also performed secondary correlation analyses separately for subjects only with FARS and only with SARA.
Clinical Subtype Comparison
We also analyzed spinal cord damage in two subgroups that merit special attention in FRDA. First, we examined pediatric patients (age < 18 years; n = 40, mean age = 13.3 ± 2.5 years, 18 males/22 females, mean disease duration = 4.8 ± 2.3 years, normalized disease severity = 0.379 ± 0.124) relative to an age/sex‐matched control group (n = 26, mean age = 14.0 ± 2.4 years, 12 males/14 females; Supporting Information Table S3) and to a disease duration–matched adult FRDA group (n = 30, mean age = 26.7 ± 9.5 years, 17 males/13 females, mean disease duration = 5.6 ± 2.7 years, normalized disease severity = 0.295 ± 0.134). Next, we examined individuals with LOFA (first onset of symptoms at age > 25 years; n = 45, mean age = 45.6 ± 8.8 years, 25 males/20 females, mean disease duration = 14.6 ± 8.8 years, normalized disease severity = 0.394 ± 0.189) first relative to an age/sex‐matched control group (n = 42, mean age = 45.4 ± 9.3 years, 22 males/20 females; Supporting Information Table S3), second to an age/sex‐matched classical onset FRDA group (n = 43, mean age = 39.2 ± 9.3 years, 11 males/32 females, mean disease duration = 24.3 ± 9.2 years, normalized disease severity = 0.666 ± 0.172), and finally to a disease duration–matched classical onset FRDA group (n = 35, mean age = 28.7 ± 10.4 years, 15 males/20 females, mean disease duration = 14.2 ± 8.9 years, normalized disease severity = 0.546 ± 0.215).
In both analyses, between‐group comparisons of spinal cord measures (CSA and eccentricity) and correlations with clinical parameters (disease duration and severity) were undertaken as described earlier.
Disease Staging
To supplement the clinical correlations described earlier, provide a clearer picture of disease staging and progression, and allow for direct quantitative and qualitative comparisons between disease subgroups and healthy control data, we also undertook a categorical division of the data. Five subgroups (DD1–DD5) were defined according to disease duration (time since first symptom expression) at the time of each participant's scan: <5, 5–10, 11–15, 16–20, and >20 years, respectively. Four subgroups (DS1–DS4) were also defined according to the normalized disease severity at the time of each participant's scan: <0.25, 0.26–0.50, 0.51–0.75, and >0.75, respectively. These divisions do not represent clinically determined cutoffs but rather provide an intuitive means of quantitatively assessing and reporting changes in ESs with disease progression.
Each subgroup was first compared with a nonataxic control cohort matched by age, sex, and site. Subsequently, we compared each subgroup with the earliest (DD1) or least severe (DS1) subgroup to assess evidence for progressive degeneration independent of early/presymptomatic effects. Similar to the statistical approach used in the general comparison, we used analysis of covariance to assess each group's differences, using age, sex, and site as covariates, and used Bonferroni correction to adjust for multiple comparisons.
Data Sharing
All code and data processing instructions are available online at: https://github.com/Harding-Lab/enigma-ataxia.
Results
Overall FRDA Versus Control Comparison
Participants with FRDA relative to control subjects had significantly reduced CSA at all vertebral levels (Fig. 2A) with very large ESs (C1 ES = 2.6, C2 ES = 2.6, C3 ES = 2.3, C4 ES = 2.1). Similarly, we found significantly increased eccentricity at all vertebral levels (Fig. 2A), also with very large ESs (C1 ES = 1.2, C2 ES = 1.4, C3 ES = 1.3, C4 ES = 1.4), although substantially smaller in comparison with CSA. In addition, the spinal cord growth curve, ie, the plot of spinal cord CSA versus age, showed distinct patterns in the control group (C1: r = −0.050, P = 0.999; C2: r = −0.045, P = 0.999; C3: r = −0.068, P = 0.999; C4: r = −0.039, P = 0.999) and CSA remains stable over the entire life span, whereas in individuals with FRDA (C1: r = −0.247, P < 0.001; C2: r = −0.216, P = 0.003; C3: r = −0.227, P = 0.002; C4: r = −0.244, P = 0.006), CSA appears to show a progressive decline with age (Fig. 3A).
FIG 2.
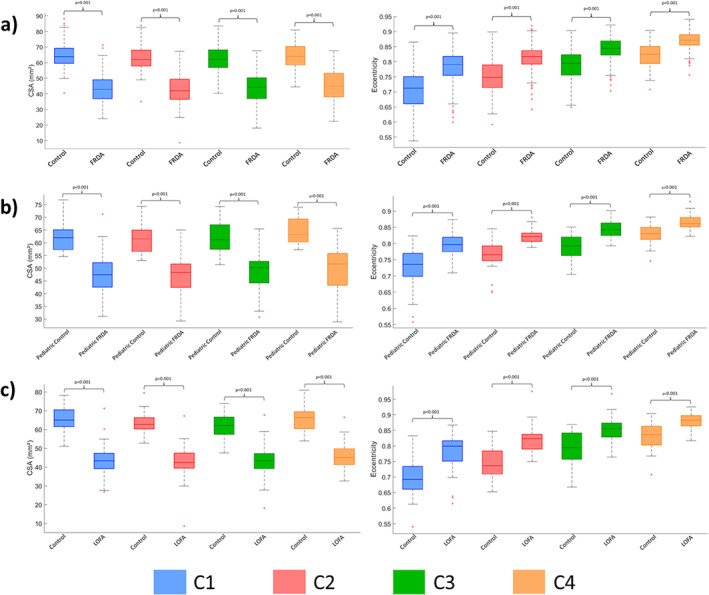
Boxplots displaying group differences at each spinal cord segment, C1 to C4, for the total cohort. (A) All patients with Friedreich's ataxia (FRDA) versus all matched control subjects. (B) Children (age < 18 years) with FRDA versus matched control subjects. (C) Individuals with late‐onset Friedreich ataxia (LOFA) versus matched control subjects. CSA, cross‐sectional area. [Color figure can be viewed at wileyonlinelibrary.com]
FIG 3.
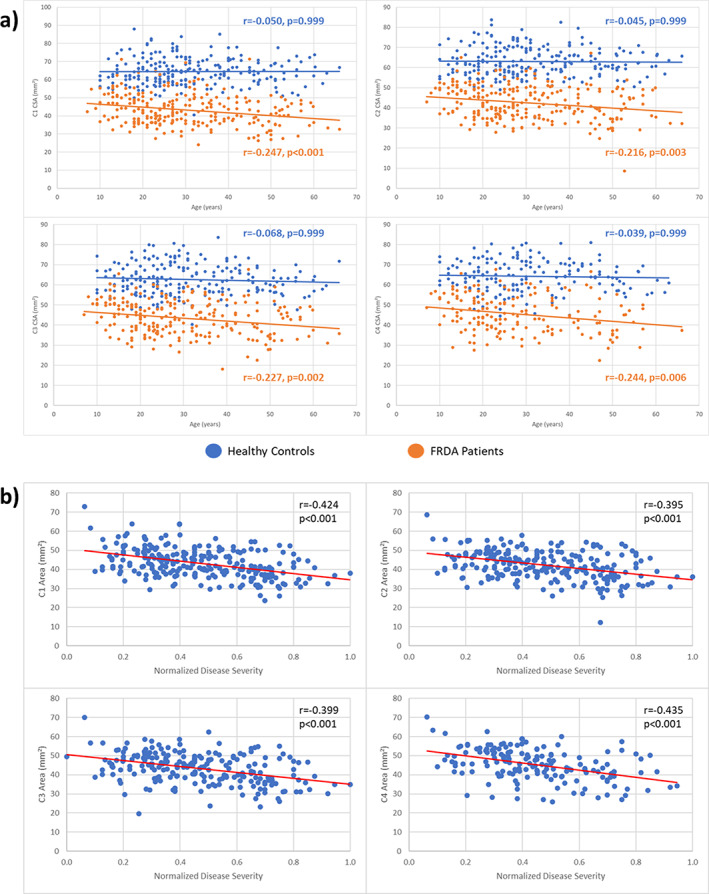
(A) Plot of spinal cord cross‐sectional area (CSA) versus age in patients and control subjects for all assessed vertebral levels. (B) Significant correlations between normalized disease severity and CSA in individuals with Friedreich's ataxia (FRDA) at vertebral levels C1, C2, C3, and C4. [Color figure can be viewed at wileyonlinelibrary.com]
In Supporting Information Figure S1, we also present the site‐specific between‐group effects, demonstrating largely consistent results irrespective of scan site and protocol.
Clinical Correlation Analysis
We found significant correlations between the normalized disease severity or ataxia duration and CSA at all vertebral levels assessed (C1–C4) (Fig. 3B) after Bonferroni adjustment for multiple comparisons (normalized disease severity—C1: r = −0.424, P < 0.001; C2: r = −0.395, P < 0.001; C3: r = −0.399, P < 0.001; C4: r = −0.435, P < 0.001; ataxia duration—C1: r = −0.174, P = 0.006; C2: r = −0.146, P = 0.044; C3: r = −0.164, P = 0.026; C4: r = −0.237, P = 0.004). In contrast, we did not find any significant correlations between eccentricity and normalized disease severity or ataxia duration. Secondary analysis of clinical correlations separately for subjects with FARS and again for those subjects with SARA produced comparable results (Supporting Information Table S4).
Comparison of Clinical Subtypes
Children with FRDA showed abnormal CSA and eccentricity when compared with matched nonataxic control subjects (Fig. 2B) with very large ESs (CSA: C1 ES = 1.7, C2 ES = 2.1, C3 ES = 2.0, C4 ES = 2.1; eccentricity: C1 ES = 1.3, C2 ES = 1.8, C3 ES = 1.8, C4 ES = 1.5). Differences relative to adults with FRDA (with classical onset age) did not reach statistical significance (Supporting Information Table S5). We did not find significant correlations between spinal cord measures and clinical variables in the pediatric cohort (Supporting Information Table S6).
Individuals with LOFA also had very large ESs when compared with matched nonataxic control subjects (Fig. 2C) (CSA: C1 ES = 3.0, C2 ES = 2.9, C3 ES = 2.6, C4 ES = 2.3; eccentricity: C1 ES = 1.4, C2 ES = 1.7, C3 ES = 1.4, C4 ES = 1.2). Differences relative to adults with classical FRDA did not show statistical significance. Significant correlations were evident between CSA and normalized disease severity for all vertebral levels assessed, except for C4, after Bonferroni correction (C1: r = −0.385, P < 0.010; C2: r = −0.371, P = 0.026; C3: r = −0.357, P = 0.035).
Disease Staging
Disease Duration
The subgroup analyses based on disease duration showed that CSA and eccentricity are already abnormal in the earliest stages of the disease, with significant differences relative to controls in all subgroups (Fig. 4, Supporting Information Table S7). In addition, we found significantly reduced CSA when DD3 (10–15 years postsymptom duration), DD4 (15–20 years duration), and DD5 (20+ years duration) were compared with DD1 (<5 years duration) at all vertebral levels (Supporting Information Table S8). In contrast, eccentricity remained stable across the subgroups.
FIG 4.

Results showing the progressive atrophy of the cervical spinal cord area (cross‐sectional area [CSA]) (A, B) and eccentricity (C, D) in participants with Friedreich's ataxia (FRDA) and healthy control subjects. (A, C) Subgroups based on disease duration (DD). (B, D) Subgroups based on disease severity (DS). To the healthy control subjects, the measures represent the mean cervical spinal cord area or eccentricity; error bars represent standard error of the mean. Subgroups are based on disease duration: DD1, time from ataxia onset <5 years; DD2, time from ataxia onset between 5 and 10 years; DD3, time from ataxia onset between 10 and 15 years; DD4, time from ataxia onset between 15 and 20 years; DD5, time from ataxia onset >20 years. Subgroups are based on disease severity: DS1, normalized disease severity <0.25; DS2, normalized disease severity between 0.26 and 0.50; DS3, normalized disease severity between 0.51 and 0.75; DS4, normalized disease severity >0.75. [Color figure can be viewed at wileyonlinelibrary.com]
Disease Severity
The subgroup analyses based on disease severity showed similar results. Abnormalities in CSA and eccentricity are observable in patients with normalized disease severity <0.25, with significant effects relative to controls in all subgroups (Fig. 4, Supporting Information Table S9). We also found significantly reduced CSA when DS3 (normalized disease severity 0.51–0.75) and DS4 (severity > 0.75) were compared with DS1 (severity < 0.25) at all vertebral levels. Meanwhile, DS2 (severity 0.26–0.50) showed reduced CSA, relative to DS1, for only C1 and C2; eccentricity remained stable across the subgroups (Supporting Information Table S10).
Discussion
Spinal cord damage has been recognized as a hallmark of FRDA since Nikolaus Friedreich's first reports and confirmed in more recent histology and neuroimaging studies. 7 , 10 , 11 , 12 , 13 , 14 , 29 In this study, we performed a harmonized and reliable retrospective cross‐sectional analysis of cervical spinal cord structure using MRI data from a large multisite cohort. We report significant and substantial CSA reduction in individuals with FRDA at all vertebral levels examined, relative to nonataxic individuals, and significant correlations with disease severity scores. Eccentricity differences were also pronounced in this cohort relative to controls, but ES values were smaller than for CSA, and no significant clinical correlations were observed. Subgroup analyses based on disease duration and severity showed that CSA and eccentricity are already abnormal in the early stages of the disease and that CSA likely declines with disease progression, whereas eccentricity remains stable. Taken together, CSA emerges as a potential MRI biomarker candidate for clinical tracking in FRDA.
Our results are consistent with previous MRI‐based studies that found cervical spinal cord atrophy and anteroposterior flattening in FRDA. 10 , 11 , 12 , 13 , 14 , 30 Post mortem studies indicate that the pathological correlates of these findings are severe depletion of myelinated fibers in the dorsal columns, dorsal spinocerebellar, and lateral corticospinal tracts. 29 These findings are also consistent with a single‐site prospective study that showed a decrease in CSA over time in individuals with FRDA in an early‐stage cohort, with no decrease over time in eccentricity. 14
Prior studies undertaken in other spinal cord diseases help us understand the pathological underpinnings of changes in CSA and eccentricity. 31 , 32 , 33 , 34 Indeed, different patterns emerge when one compares diseases characterized by predominant/exclusive lateral column involvement (eg, amyotrophic lateral sclerosis, pure subtypes of hereditary spastic paraplegia) versus diseases with predominant/exclusive dorsal column involvement (eg, acquired sensory neuronopathies). 31 , 32 , 33 CSA reduction is evident in both groups, but eccentricity increase is reported only in the latter. 31 Therefore, eccentricity can be considered a surrogate MRI marker for dorsal column damage, whereas CSA may be related to abnormal integrity in both lateral and dorsal columns. Using this conceptual framework, relevant insights can be inferred from our results. The stability of eccentricity alongside decreasing CSA across FRDA stages (based on duration or severity) suggests that the corticospinal tract and dorsal columns follow distinct mechanisms and time courses of damage in the disease. Corticospinal tract damage is most consistent with a combination of abnormal developmental and progressive degenerative processes, as shown by both early (already seen in the pediatric subgroup) and progressive CSA abnormalities. In contrast, dorsal column abnormalities, assessed by eccentricity, may be related to early maldevelopment but remain stable along the entire disease course, at least from the point of first symptom expression. However, extrapolation of these results beyond the cervical spinal cord must be approached with caution. Indeed, a whole‐spine study in a small FRDA cohort reported similar findings in the cervical cord but also observed significant correlations between disease duration and eccentricity in thoracic regions. 11
Our assumption that dorsal column damage is largely neurodevelopmental is in line with neuropathological reports from Koeppen et al. 29 These authors suggest that the developmental failure of the dorsal root ganglia (DRGs) leads to the secondary hypoplasia of dorsal columns, because dorsal root ganglia are the source of myelinated fibers in the dorsal columns. Indeed, the autopsy of two young patients with FRDA showed that the neurons in the dorsal nuclei were severely reduced or absent, probably because of the lack of innervation from the dorsal root collaterals that occurs during the gestational period, 34 arguing in favor of a developmental failure. Furthermore, experiments using animal models provide evidence that frataxin plays a role in embryonic development. 35 , 36 Our imaging data suggest that CSA of individuals with FRDA, on average, reaches its maximum before 10 years of age and then starts to decrease, whereas healthy control subjects have higher CSA values relative to patients with FRDA even at the earliest disease stages and keep stable over time. A preceding MRI‐based study performed by Rezende and colleagues 12 found a very similar result.
Progressive neurodegeneration in the corticospinal tract is consistent with the hypothesis that pyramidal tract damage in FRDA arises from a dying‐back process. Neuropathological studies have found that the corticospinal tract is more affected in the spinal cord than in the brain, with the exception of the lack of Betz cells in the motor cortex. 37 , 38 Koeppen and Mazurkiewicz 7 also showed that spinal cord damage is more severe in thoracic levels compared with cervical regions. Previous neuroimaging studies also support such a concept. Indeed, a diffusion MRI‐based study showed that microstructural abnormalities were more robust in caudal levels, although clinical correlations were stronger at the upper levels of the corticospinal tract. 13 Rezende and colleagues 12 also reported motor cortex thinning only in adults and not in children with FRDA, alongside progressive damage in the cerebral corticospinal tract. This is in agreement with Harding and colleagues, 15 who proposed a disease staging schema for brain damage in FRDA. These authors highlight the progressive pattern of damage in the disease that begins in infratentorial structures and spreads to cortical structures in later disease stages. 15
From a clinical perspective, our data indicate that CSA at C1 level is a potential biomarker candidate because it showed the highest correlation coefficient with disease severity and the highest ES compared with control subjects. CSA at C1 level also had the highest ES in a recent single‐site longitudinal study. 14 However, this may not be the case for all FRDA stages or subphenotypes. For the pediatric cohort (age < 18 years), we did not find any significant correlations between CSA and normalized disease severity, whereas such associations were evident in the adult cohort. Although this observation may reflect statistical power because there were fewer pediatric individuals (n = 40) relative to adults (n = 159), a similar result was previously reported by Rezende and colleagues. 12 This relative decoupling of structure and function may reflect partial neural reserve, or the influence of parallel developmental and degenerative processes obscuring clear clinical associations in pediatric patients, relative to a pure degenerative profile in adults. Different neuroimaging biomarkers may also vary in their sensitivity to change at different disease stages, similar to what has been suggested for SCA3. 39 This hypothesis is supported by the proposed mechanism of corticospinal degeneration in FRDA, which appears to follow a dying‐back motor axonopathy. Lower levels of the spinal cord may therefore already be extensively impacted very early in the disease course and reach an early floor effect. Ongoing damage to the spinal corticospinal tract may therefore be more easily captured by MRI metrics at upper levels. At this point, prospective studies with pediatric and adult cohorts must be undertaken to confirm such hypotheses.
Notwithstanding the original contributions of this study, several limitations must be acknowledged. This is a cross‐sectional study, and many of the findings presented in this article must be confirmed by prospective longitudinal neuroimaging studies, particularly those enriched with a pediatric cohort. Our analyses were performed using T1‐weighted brain MRI, which is the most common and widely used MRI sequence for research and clinical practice. However, this confines our assessment to the upper portions of the cervical spinal cord and prevents investigation of whether different patterns of degeneration characterize different levels of the spinal cord. More targeted spinal cord imaging acquisitions would enable more detailed analyses, such as tract‐specific microstructural evaluation and individual assessment of white matter and gray matter regions. The use of different clinical scales across sites and general availability of only total scores (instead of individual subitems) also limit more extensive investigation of correlations between spinal cord damage and both overall disease severity and different symptom domains. In this study, we use a relatively blunt normalization approach to pool scores across different clinical scales. Future work modeling the relationship between different clinical scales (ie, SARA and FARS) would be beneficial to establish more specific conversion scores. Lastly, the small sample size of the pediatric and LOFA cohorts did not allow us to split them into subgroups to assess their disease evolution. Prospective natural history imaging studies (eg, TRACK‐FA; https://www.monash.edu/medicine/trackfa) will be key to addressing many of these limitations.
To conclude, our data support the hypothesis that damage to the spinal dorsal column and corticospinal tract follow distinct courses in the disease: developmental damage likely defines the former, while alterations in the latter may be both developmental and degenerative in origin. These results provide new insights about FRDA pathogenesis and indicate that spinal cord MRI may be a useful biomarker to track disease progression.
Author Roles
Conception and design of the study and methods: T.J.R.R., I.H.H., M.C.F., N.J., S.I.T., G.F.E., and P.M.T.
Acquisition and analysis of data: (Aachen) K.R., I.D., S.R., and J.B.S.; (Campinas) M.C.F., T.J.R.R., A.R.M.M., and F.C.; (Conegliano) A.M., F.A., G.P., and D.P.; (Essen) D.T., A.D., and S.L.G.; (Melbourne) N.G.‐K., I.H.H., L.A.C., M.D., and G.F.E.; (Minnesota) C.L., P.‐G.H., J.M.J., I.M.A., D.H.; (Tubingen) B.B., T.L., M.S., and L.S.
Analysis of multisite data and drafting of initial manuscript: T.J.R.R., I.H.H., and M.C.F.
Financial Disclosures
K.R., I.D., S.R., J.B.S., M.C.F., T.J.R.R., A.R.M.M., F.C., A.M., F.A., M.V., D.P., S.I.T., D.T., A.D., S.L.G., N.G.‐K., I.H.H., L.A.C., M.D., G.F.E., J.M.J., I.M.A., D.H., B.B., T.L., and S.I.T.: none.
P.M.T. and N.J. received a research grant from Biogen, Inc., unrelated to the topic of this manuscript. P.‐G.H. and C.L. received research grants from Minoryx Therapeutics. C.L. also received support by a research grant from Biogen Inc. for activities unrelated to the topic of this manuscript. L.S. served as an advisory board member for VICO Therapeutics. M.S. received consultancy honorary from Janssen Pharmaceuticals, Ionis Pharmaceuticals, and Orphazyme Pharmaceuticals, all unrelated to this manuscript.
Supporting information
Table S1 Demographics data for non‐ataxic individuals for each site.
Table S2 Scanner and imaging acquisition details for each site.
Table S3 Demographics data of pediatric and LOFA patients.
Table S4 Separated clinical correlations analysis for subjects with FARS only and SARA only. N is the sample size used for each clinical scale.
Table S5 Group differences between pediatric vs adult patients with FRDA at each spinal cord level, C1‐C4.
Table S6 Correlations between time from ataxia onset or normalized disease severity and CSA or eccentricity for the pediatric cohort.
Table S7 Results of ROI‐based analyses to assess CSA damage at each stage of FRDA (DD1: Time from ataxia onset <5 years, DD2: Time from ataxia onset between 5–10 years, DD3: Time from ataxia onset between 10–15 years, DD4: Time from ataxia onset between 15–20 years, DD5: Time from ataxia onset >20 years). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Table S8 Results of ROI‐based analyses to assess progressive CSA damage at each stage of FRDA relative to the subgroup small time from ataxia onset (DD1: Time from ataxia onset <5 years, DD2: Time from ataxia onset between 5–10 years, DD3: Time from ataxia onset between 10–15 years, DD4: Time from ataxia onset between 15–20 years, DD5: Time from ataxia onset >20 years). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Table S9 Results of ROI‐based analyses to assess CSA damage at each stage of FRDA (DS1: Normalized disease severity <0.25, DS2: Normalized disease severity between 0.26–0.50, DS3: Normalized disease severity between 0.51–0.75, DS4: Normalized disease severity >0.75). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Table S10 Results of ROI‐based analyses to assess progressive CSA damage at each stage of FRDA relative to the subgroup small normalized disease severity (DS1: Normalized disease severity <0.25, DS2: Normalized disease severity between 0.26–0.50, DS3: Normalized disease severity between 0.51–0.75, DS4: Normalized disease severity >0.75). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Figure S1 Site‐specific effect sizes for (a) cross‐sectional area (CSA) and (b) eccentricity for each vertebral level assessed.
Acknowledgments
The methods of harmonization and multisite data analysis elements of this work were supported by National Institutes of Health (NIH) Big Data to Knowledge (BD2K) program grant U54 EB020403 and grants from the Australian National Health and Medical Research Council (Fellowship 1106533, Grant 1184403). FARA (Friedreich's Ataxia Research Alliance, grant 92133) and FAPESP (São Paulo Research Foundation) also supported this study through CEPID/BRAINN (grant 2013/07559‐3), the German Research Foundation (DFG; DE 2516/1‐1 and TI 239/17‐1), and the European Joint Programme on Rare Diseases, under the EJP RD COFUND‐EJP N° 825575 as part of the PROSPAX consortium (to M.S. and D.T. via DFG). Center for Magnetic Resonance Research (CMRR) at the University of Minnesota was supported by NIH grants P41 EB027061 and P30 NS076408. P.‐G.H. and C.L. also acknowledge support by grants from the FARA, GoFAR, Ataxia UK, and the Bob Allison Ataxia Research Center. The funding agencies did not interfere with the design of the study, collection of data, or drafting of the manuscript.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
Data Availability Statement
All code and data processing instructions are available at https://github.com/Harding-Lab/enigma-ataxia.
References
- 1. Campuzano V, Montermini L, Molto MD, et al. Friedreich's ataxia: autosomal recessive disease caused by anintronic GAA triplet repeat expansion. Science 1996;271:1423–1427. [DOI] [PubMed] [Google Scholar]
- 2. Pandolfo M. Friedreich ataxia. Arch Neurol 2008;65:1296–1303. [DOI] [PubMed] [Google Scholar]
- 3. Reetz K, Dogan I, Hilgers RD, et al. Progression characteristics of the European Friedreich's ataxia consortium for translational studies (EFACTS): a 4‐year cohort study. Lancet Neurol 2021;20:362–372. [DOI] [PubMed] [Google Scholar]
- 4. De Michele G, Filla A, Cavalcanti F, et al. Late onset Friedreich's disease: clinical features and mapping of mutation to the FRDA locus. J Neurol Neurosurg Psychiatry 1994;57:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martinez AR, Moro A, Abrahao A, et al. Nonneurological involvement in late‐onset Friedreich ataxia (LOFA): exploring the phenotypes. Cerebellum 2017;16:253–256. [DOI] [PubMed] [Google Scholar]
- 6. Bhidayasiri R, Perlman SL, Pulst SM, Geschwind DH. Late‐onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol 2005;62:1865–1869. [DOI] [PubMed] [Google Scholar]
- 7. Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol 2013;72:78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harding IH, Lynch DR, Koeppen AH, Pandolfo M. Central nervous system therapeutic targets in Friedreich ataxia. Hum Gene Ther 2020;31(23–24):1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Öz G, Harding IH, Krahe J, Reetz K. MR imaging and spectroscopy in degenerative ataxias: toward multimodal, multisite, multistage monitoring of neurodegeneration. Curr Opin Neurol 2020;33:451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chevis CF, da Silva CB, D'Abreu A, Lopes‐Cendes I, Cendes F, Bergo FP, França MC Jr. Spinal cord atrophy correlates with disability in Friedreich's ataxia. Cerebellum 2013;12:43–47. [DOI] [PubMed] [Google Scholar]
- 11. Dogan I, Romanzetti S, Didszun C, et al. Structural characteristics of the central nervous system in Friedreich ataxia: an in vivo spinal cord and brain MRI study. J Neurol Neurosurg Psychiatry 2019;90:615–617. [DOI] [PubMed] [Google Scholar]
- 12. Rezende TJR, Martinez ARM, Faber I, et al. Developmental and neurodegenerative damage in Friedreich's ataxia. Eur J Neurol 2019;26:483–489. [DOI] [PubMed] [Google Scholar]
- 13. Hernandez ALCC, Rezende TJR, Martinez ARM, de Brito MR, França MC Jr. Tract‐specific spinal cord diffusion tensor imaging in Friedreich's ataxia. Mov Disord 2022;37:354–364. [DOI] [PubMed] [Google Scholar]
- 14. Joers JM, Adanyeguh IM, Deelchand DK, et al. Spinal cord MRI and MRS detect early‐stage alterations and disease progression in Friedreich ataxia. Brain Commun 2022;4:fcac246. 10.1093/braincomms/fcac246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harding IH, Chopra S, Arrigoni F, et al. Brain structure and degeneration staging in Friedreich ataxia: magnetic resonance imaging volumetrics from the ENIGMA‐ataxia working group. Ann Neurol 2021;90:570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramony SH, May W, Lynch D, et al. Measuring Friedreich ataxia: interrater reliability of a neurologic rating scale. Neurology 2005;12(64):1261–1262. [DOI] [PubMed] [Google Scholar]
- 17. Lynch DR, Farmer JM, Tsou AY, et al. Measuring Friedreich ataxia: complementary features of examination and performance measures. Neurology 2006;66(11):1711–1716. [DOI] [PubMed] [Google Scholar]
- 18. Rummey C, Corben LA, Delatycki MB, et al. Psychometric properties of the Friedreich ataxia rating scale. Neurol Genet 2019;29(5):371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmitz‐Hübsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66(11):1717–1720. 10.1212/01.wnl.0000219042.60538.92 Erratum in: Neurology. 2006 Jul 25;67(2):299. Fancellu, Roberto [added]. PMID: 16769946 [DOI] [PubMed] [Google Scholar]
- 20. De Leener B, Lévy S, Dupont SM, et al. SCT: spinal cord toolbox, an open‐source software for processing spinal cord MRI data. Neuroimage 2017;145:24–43. [DOI] [PubMed] [Google Scholar]
- 21. Gros C, De Leener B, Badji A, et al. Automatic segmentation of the spinal cord and intramedullary multiple sclerosis lesions with convolutional neural networks. Neuroimage 2019;184:901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ullmann E, Pelletier Paquette JF, Thong WE, Cohen‐Adad J. Automatic labeling of vertebral levels using a robust template‐based approach. Int J Biomed Imaging 2014;2014:719520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Leener B, Taso M, Fonov V, et al. Fully‐integrated T1, T2, T2*, white and gray matter atlases of the spinal cord. In: Proceedings of the International Society for Magnetic Resonance in Medicine 24th Annual Meeting and Exhibition, Singapore. May 7–8, 2016.
- 24. De Leener B, Fonov VS, Collins DL, et al. PAM50: unbiased multimodal template of the brainstem and spinal cord aligned with the ICBM152 space. Neuroimage 2018;165:170–179. [DOI] [PubMed] [Google Scholar]
- 25. Papinutto N, Asteggiano C, Bischof A, et al. Intersubject variability and normalization strategies for spinal cord Total cross‐sectional and gray matter areas. J Neuroimaging 2020;30(1):110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cohen J. Statistical Power Analysis for the Behavioral Sciences. New York, NY: Routledge Academic; 1988. [Google Scholar]
- 27. Bürk K, Mälzig U, Wolf S, et al. Comparison of three clinical rating scales in Friedreich ataxia (FRDA). Mov Disord 2009;24:1779–1784. [DOI] [PubMed] [Google Scholar]
- 28. Perez‐Lloret S, van de Warrenburg B, Rossi M, et al. Assessment of ataxia rating scales and cerebellar functional tests: critique and recommendations. Mov Disord 2021;36:283–297. [DOI] [PubMed] [Google Scholar]
- 29. Koeppen AH, Becker AB, Qian J, Feustel PJ. Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J Neuropathol Exp Neurol 2017;76:101–108. [DOI] [PubMed] [Google Scholar]
- 30. Mascalchi M, Bianchi A, Ciulli S, et al. Lower medulla hypoplasia in Friedreich ataxia: MR imaging confirmation 140 years later. J Neurol 2017;264:1526–1528. [DOI] [PubMed] [Google Scholar]
- 31. França MC Jr, D'Abreu A, Zanardi VA, Faria AV, Lopes‐Cendes I, Nucci A, Cendes F. MRI shows dorsal lesions and spinal cord atrophy in chronic sensory neuronopathies. J Neuroimaging 2008;18:168–172. [DOI] [PubMed] [Google Scholar]
- 32. Rezende TJ, de Albuquerque M, Lamas GM, et al. Multimodal MRI‐based study in patients with SPG4 mutations. PLoS One 2015;10:e0117666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Querin G, Bede P, El Mendili MM, et al. Presymptomatic spinal cord pathology in c9orf72 mutation carriers: a longitudinal neuroimaging study. Ann Neurol 2019;86:158–167. [DOI] [PubMed] [Google Scholar]
- 34. Pisharady PK, Eberly LE, Cheong I, et al. Tract‐specific analysis improves sensitivity of spinal cord diffusion MRI to cross‐sectional and longitudinal changes in amyotrophic lateral sclerosis. Commun Biol 2020;3(1):370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Altman J, Bayer SA. Development of the human spinal cord. An Interpretation Based on Experimental Studies in Animals. Oxford: Oxford University Press; 2001. [Google Scholar]
- 36. Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M. Frataxin shows developmentally regulated tissue‐specific expression in the mouse embryo. Neurobiol Dis 1997;4:103–113. [DOI] [PubMed] [Google Scholar]
- 37. Oppenheimer DR. Brain lesions in Friedreich's ataxia. Can J Neurol Sci 1979;6:173–176. [DOI] [PubMed] [Google Scholar]
- 38. Urich H, Norman RM, Lloyd OC. Suprasegmental lesions in Friedreich's ataxia. Confin Neurol 1957;17:360–371. [DOI] [PubMed] [Google Scholar]
- 39. Rezende TJR, de Paiva JLR, Martinez ARM, et al. Structural signature of SCA3: from presymptomatic to late disease stages. Ann Neurol 2018;84:401–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Demographics data for non‐ataxic individuals for each site.
Table S2 Scanner and imaging acquisition details for each site.
Table S3 Demographics data of pediatric and LOFA patients.
Table S4 Separated clinical correlations analysis for subjects with FARS only and SARA only. N is the sample size used for each clinical scale.
Table S5 Group differences between pediatric vs adult patients with FRDA at each spinal cord level, C1‐C4.
Table S6 Correlations between time from ataxia onset or normalized disease severity and CSA or eccentricity for the pediatric cohort.
Table S7 Results of ROI‐based analyses to assess CSA damage at each stage of FRDA (DD1: Time from ataxia onset <5 years, DD2: Time from ataxia onset between 5–10 years, DD3: Time from ataxia onset between 10–15 years, DD4: Time from ataxia onset between 15–20 years, DD5: Time from ataxia onset >20 years). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Table S8 Results of ROI‐based analyses to assess progressive CSA damage at each stage of FRDA relative to the subgroup small time from ataxia onset (DD1: Time from ataxia onset <5 years, DD2: Time from ataxia onset between 5–10 years, DD3: Time from ataxia onset between 10–15 years, DD4: Time from ataxia onset between 15–20 years, DD5: Time from ataxia onset >20 years). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Table S9 Results of ROI‐based analyses to assess CSA damage at each stage of FRDA (DS1: Normalized disease severity <0.25, DS2: Normalized disease severity between 0.26–0.50, DS3: Normalized disease severity between 0.51–0.75, DS4: Normalized disease severity >0.75). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Table S10 Results of ROI‐based analyses to assess progressive CSA damage at each stage of FRDA relative to the subgroup small normalized disease severity (DS1: Normalized disease severity <0.25, DS2: Normalized disease severity between 0.26–0.50, DS3: Normalized disease severity between 0.51–0.75, DS4: Normalized disease severity >0.75). All comparisons are significant, i.e., P < 0.001 after Bonferroni correction.
Figure S1 Site‐specific effect sizes for (a) cross‐sectional area (CSA) and (b) eccentricity for each vertebral level assessed.
Data Availability Statement
All code and data processing instructions are available at https://github.com/Harding-Lab/enigma-ataxia.