

Abstract
Background:
Hydrogen sulfide (H2S) is a critical endogenous signaling molecule that exerts protective effects in the setting of heart failure (HF). Cystathionine γ-lyase (CSE), one of three H2S producing enzyme, is predominantly localized in the vascular endothelium. The interaction between the endothelial CSE – H2S axis and endothelial-mesenchymal transition (EndoMT), an important pathological process contributing to the formation of fibrosis, has yet to be investigated.
Methods:
Endothelial cell (EC) specific CSE knockout (EC-CSE KO) and EC-CSE overexpressing (EC-CSE Tg) mice were subjected to transverse aortic constriction (TAC) to induce HF with reduced ejection fraction (HFrEF). Cardiac function, vascular reactivity, and treadmill exercise capacity following TAC were measured to determine the severity of HF. Histological and gene expression analysis were performed to investigate changes in cardiac fibrosis and EndoMT activation.
Results:
EC-CSE KO mice exhibited increased EndoMT and reduced nitric oxide (NO) bioavailability in the myocardium which was associated with increased cardiac fibrosis, impaired cardiac and vascular function, and worsened exercise performance. In contrast, genetic overexpression of CSE in endothelial cells led to increased myocardial NO, decreased EndoMT and cardiac fibrosis, preserved cardiac and endothelial function, and improved exercise capacity.
Conclusions:
Our data demonstrate that endothelial CSE modulates endothelial-mesenchymal transition and ameliorate the severity of pressure overload – induced heart failure, in part, through NO-related mechanisms. These data further suggest that endothelium-derived H2S is a potential therapeutic for the treatment of heart failure with reduced ejection fraction.
Keywords: Hydrogen sulfide, Endothelial function, Heart Failure, Fibrosis, Endothelial-mesenchymal transition
Graphical Abstract

INTRODUCTION
Cardiovascular disease is the leading cause of death world-wide, accounting for approximately 18.6 million deaths in 20191, 2. Though mortality rates associated with acute myocardial infarction, valvular and congenital heart disease, hypertension, and arrhythmias have fallen dramatically, heart failure (HF) is the exception3. Despite considerable progress in the treatment of HF, its incidence and economic burden are steadily increasing. By 2030, the prevalence of HF is expected to increase by 46% and cost will increase by 127% to $69.8 billion4. Therefore, new therapeutic measures are urgently needed. Today, there are no approved treatments that stimulate repair, regeneration and recovery of heart function after HF ensues.
Endothelial cells (ECs) are a critical component of the cardiovascular system that are responsible for modulating blood flow and vascular permeability, nitric oxide (NO) production, regulation of global redox status, and maintenance of thromboresistance5, 6. Endothelial dysfunction is characterized by impaired endothelial nitric oxide synthase (eNOS) activity, reduction in NO production, and imbalanced redox state, all of which are commonly seen in HF patients6. Moreover, dysfunctional ECs are now recognized to play an important role in contributing to the progression of pathological fibrosis. In the setting of systemic inflammation, dysfunctional ECs have been shown to undergo transformation into a mesenchymal-like phenotype, referred to as endothelial-mesenchymal transition (EndoMT)7–9. The phenotypic switch of ECs into EndoMT have been shown to mediate fibrosis directly by increasing the number of pro-fibrotic mesenchymal-like cells, including fibroblasts and myofibroblasts, smooth muscle cells, and osteoblasts9. The contribution of EndoMT to initiating the fibrotic response and to the pathological progression of fibrosis has been observed in a variety of cardiovascular including atherosclerosis, valvular disease, pulmonary hypertension, and cardiac fibrosis10, 11. Previous studies have shown that EndoMT contributes to cardiac fibrosis12–14. While myofibroblasts are still considered to be the main contributors to fibrosis, the critical role of EndoMT in myocardial fibrosis is increasingly being recognized. As HF is characterized by profound myocardial fibrosis, targeting EndoMT may represent a novel, anti-fibrotic therapeutic strategy.
Hydrogen sulfide (H2S), produced endogenously by three enzymes, cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST), has been recognized as an important physiological signaling molecule modulating cardiovascular homeostasis15–17. H2S exerts several protective actions in the cardiovascular system, including vasodilation as well as potent antioxidant and anti-inflammatory properties15, 17–22. CSE is predominantly localized to the vascular endothelium where it has been shown to mediate smooth muscle relaxation and subsequent vasodilation23. We previously reported that genetic deletion of CSE specifically in endothelium leads to reduced NO bioavailability, impaired vascular relaxation, and impaired exercise capacity24. Conversely, genetic overexpression of CSE in endothelial cells improves endothelial function and attenuates myocardial infarction following myocardial ischemia reperfusion injury24. Others have also reported endothelial CSE derived H2S preserves endothelial function, balance redox status and reduce membrane lipid peroxidation in endothelium, and ultimately attenuates atherogenic response25, 26. Taken together, these data suggest a critical role of endothelial CSE-derived H2S in cardiovascular homeostasis and acute cardioprotection. However, the roles of endothelial CSE-derived H2S in chronic heart failure has yet to be investigated.
In the current study, we aimed to investigate the roles of endothelial cell CSE-derived H2S in the setting of pressure overload induced heart failure with reduced ejection fraction. Genetic modified mouse models of endothelial specific CSE knockout (EC-CSE KO) or overexpression (EC-CSE Tg) were generated and subjected to heart failure with reduced ejection fraction. We investigated the severity of the heart failure, endothelial function, exercise capacity, cardiac fibrosis, and levels of EndoMT.
MATERIALS and METHODS
Data Availability.
Detailed Materials and Methods and Major Resources Table are available in the Online Supplemental Material. The data that support the findings of this study, experimental materials, and analytic methods are available from the corresponding author upon reasonable request.
RESULTS
EC-CSE KO Mice Exhibit Exacerbated Severity of Pressure Overload Induced Heart Failure.
EC-CSE KO mice displayed a significant decrease in LV posterior wall thickness at 9 weeks post-TAC; however, it was similar to controls at all other timepoints (Figure 2A). LV chamber dimensions at end-diastole was significantly greater in EC-CSE KO mice starting 9 weeks post TAC through 15 weeks when compared to controls (Figures 2B). LV chamber enlargement was associated with a significant reduction in LV ejection fraction in EC-CSE KO mice compared to controls starting 6 weeks through 12 weeks post TAC procedure (Figure 2C). Despite similar LV wall thickness, LV chamber size, and LVEF in EC-CSE KO and control mice at the 21-week end point, invasive hemodynamic measurements revealed a moderate yet significant increase in LV end-diastolic pressure as well as relaxation constant Tau (Figure 2D, E). Moreover, circulating BNP, a clinically used biomarker for assessing the severity of HF, was significantly elevated in EC-CSE KO mice as compared to control mice (Figure 2F) at 21 weeks post TAC.
Figure 2: Exacerbated pressure overload HFrEF in EC-CSE KO mice.

(A) LV posterior wall thickness, (B) LV end-diastolic diameters and (C) LV ejection fraction throughout the 21 weeks study for EC-CSE KO and control mice. (D) LV end-diastolic pressure, (E) LV relaxation constant Tau, (F) Circulating BNP, (G) aortic vascular reactivity to ACh, (H) aortic vascular reactivity to SNP, and (I) Treadmill running distance in EC-CSE KO and control mice at 21 weeks post TAC. Circled number inside bars indicates samples size. Data in (A) - (C) and (G) - (H) are analyzed with 2-way ANOVA; other data are analyzed with unpaired student t-test. Data are presented as mean ± SD.
To determine whether vascular function was altered in the absence of CSE in endothelial cells, we measured endothelial function in isolated thoracic aortic ring segments. At the 21-week end point, EC-CSE KO mice exhibited significantly impaired endothelial cell-dependent vasorelaxation responses to ACh compared to controls (Figure 2G). There were no differences detected in endothelial cell-independent relaxation between EC-CSE KO and control mice (Figure 2H). Endothelial-dependent vascular dysfunction in EC-CSE KO mice was further evidenced by exercise intolerance with a significant reduction in treadmill running distance at 21 weeks post TAC compared to controls (Figure 2I).
Genetic Deletion of Endothelial cell CSE Results in Increased Myocardial Fibrosis via Exacerbating EndoMT.
Next, we evaluated the effects of EC-CSE KO on cardiac fibrosis with Masson’s trichrome staining at 10- and 21-weeks post TAC. Representative photomicrographs of heart sections are shown in Figure 3A and B, Figure S1A and B. Selective genetic deletion of CSE only in endothelial cells significantly increased overall total cardiac fibrosis (Figure 3C and S1C). Interestingly, the significantly higher levels of total fibrosis observed in EC-CSE KO hearts was driven almost exclusively by the increased vascular fibrotic response. At 10- and 21-weeks following TAC, perivascular fibrosis was significantly increased in EC-CSE KO hearts compared to controls (Figure 3B and D, S1B and D). Furthermore, EC-CSE KO resulted in increased transcription of pro-fibrotic genes including collagen 1a1, collagen 3a1, IL-6, and fibronectin 1. Interestingly, TGFβ, a secreted inflammatory protein that promotes activation of myofibroblasts and EndoMT, was not elevated in EC-CSE KO mice at 21 weeks post TAC (Figure 3E).
Figure 3: Increased cardiac fibrosis in EC-CSE KO mice at 21 weeks post TAC.

Best illustrative microphotography of (A) cardiac fibrosis in interstitial area and (B) perivascular area. Total fibrosis (C) and perivascular fibrosis (D) quantified. (E) Relative fold changes in cardiac mRNA levels of a panel of fibrosis-related genes in EC-CSE KO and control mice. Circled number inside bars indicates samples size. Data are analyzed with unpaired student t-test and presented as mean ± SD. Scale bar indicates 50 μm.
Following the observation of increased total and perivascular fibrosis in EC-CSE KO mice, we next evaluated the level of EndoMT by immunohistochemical staining of vimentin and Von Willebrand Factor at 21 weeks post TAC. Representative photomicrographs of heart sections are shown in Figure 4A. There were significantly more vimentin and VWF double-positive cells in EC-CSE KO hearts compared to control (Figure 4B). We then measured the transcriptional levels of EndoMT markers (αSMA, vimentin) and activators (Snai1, Slug, Twist) in the heart. While αSMA and vimentin were significantly elevated, Snai1, Slug, and Twist were similar in EC-CSE KO mice compared to controls at 21 weeks post TAC (Figure 4D). Interestingly, at an earlier timepoint (6-week), all of the markers and activators for EndoMT were significantly elevated (Figure 4C), indicating that the process of EndoMT is more likely to occur at the early stage of HF induced by pressure overload. Moreover, the bioavailability of NO, an established EndoMT inhibitor, were reduced in circulation and hearts of EC-CSE KO mice at 21-week following TAC (Figures 4E and F).
Figure 4: Overactivation of EndoMT in EC-CSE KO mice.

(A) Best illustrative microphotography of heart sections stained for Vimentin (Red), VWF (green), and Dapi (blue). Vimentin+/VWF+ double positive cells displayed yellow color and quantified in (B). Relative fold changes in cardiac mRNA levels of a panel of EndoMT-related genes at 6 weeks post TAC (C) and 21 weeks post TAC (D) in EC-CSE KO and control mice. Circled number inside bars indicates samples size. Data are analyzed with unpaired student t-test and presented as mean ± SD. Scale bar indicates 50 μm.
Endothelial Cell Overexpression of CSE Ameliorates Pressure Overload-Induced Heart Failure.
Building on our results showing profound vascular dysfunction and increased EndoMT in hearts when CSE is absent in endothelial cells, we investigated whether overexpression of CSE in endothelial cells could protect the heart when subjected to TAC-induced heart failure and if this protection involved EndoMT. Additionally, in a subset of EC-CSE Tg mice, we temporarily suppressed overexpression of EC-CSE with doxycycline diet for the first 10 weeks of the 21-week study, allowing us to evaluate the therapeutic potential of delayed EC-CSE gene therapy after heart failure has ensued. When constitutively overexpressed, EC-CSE Tg mice were protected against adverse cardiac remodeling as evidenced by stable LV wall thickness and LV chamber dimension (Figure 5A and B), which translated into largely preserved LV ejection fraction (Figure 5C). When overexpression of CSE in the endothelium was delayed until 10 weeks post-TAC, eccentric hypertrophic remodeling and LV chamber dilation were significantly attenuated at the later timepoints and LVEF was preserved at 18- and 21-weeks post-TAC (Figure 5A to C). Similarly, both constitutive and delayed EC-CSE Tg improved LV hemodynamics, evidenced by significantly reduced LVEDP and Tau when compared to control mice (Figure 5D and E). Circulating BNP, a measure of cardiac enlargement and HF severity, was also lower in both constitutive and delayed EC-CSE Tg mice (Figure 5F).
Figure 5: Attenuated severity of pressure overload HFrEF in EC-CSE Tg mice.

(A) LV posterior wall thickness at diastole, (B) LV end-diastolic diameters, and (C) LV ejection fraction throughout the 21 weeks study for constitutive EC-CSE Tg, 10-week-delayed EC-CSE Tg, and control mice. (D) LV end-diastolic pressure, (E) LV relaxation constant Tau (F) Circulating BNP, (G) aortic vascular reactivity to ACh (H) aortic vascular reactivity to SNP, and (I) Treadmill running distance for constitutive EC-CSE Tg, 10-week-delayed EC-CSE Tg, and control mice at 21 weeks post TAC. Circled number inside bars indicates samples size. Data in (A) - (C) and (G) - (H) are analyzed with 2-way ANOVA; other data are analyzed with one-way ANOVA. Data are presented as mean ± SD.
Next, we evaluated vascular endothelial function as well as the exercise performance in both constitutive and delayed EC-CSE Tg mice as compared to control mice. At 21 weeks post-TAC, both constitutive and 10-week-delayed EC-CSE Tg mice displayed significantly better endothelial-dependent vasorelaxation responses to ACh while all groups responded similarly to SNP, indicating that EC-CSE overexpression, either constitutive or delayed, preserve endothelial function in pressure overload heart failure (Figure 5G and H). Exercise intolerance in control mice observed at 21 weeks post TAC was markedly attenuated in constitutive EC-CSE Tg mice. Exercise intolerance was also attenuated in mice in which EC-CSE overexpression was delayed until 10 weeks post-TAC, but not to the same extent as the constitutive overexpression (Figure 5I).
Genetic Overexpression of Endothelial Cell CSE Attenuated EndoMT and Cardiac Fibrosis.
We next evaluated the effects of EC-CSE Tg on cardiac fibrosis with Masson’s trichrome staining at 10- and 21- weeks post-TAC. Representative photomicrographs of heart sections are shown in Figure 6A and B, Figure S2A and B. Genetic overexpression of endothelial cell CSE, either constitutively or after the onset of heart failure, significantly reduced the total fibrotic area as well as the perivascular fibrosis in the heart. EC-CSE Tg therapy starting at 10 weeks following TAC also significantly reduced myocardial fibrosis but to a lesser extent when compared to constitutive EC-CSE expression mice (Figure 6C and D). Interestingly, at 10 weeks post TAC, constitutive EC-CSE Tg significantly lower perivascular fibrotic response but failed to reduce the total fibrosis (Figure S2) Furthermore, both constitutive and 10-week-delayed EC-CSE Tg led to decreased amount of transcription of pro-fibrotic genes such as collagen 1a1, collagen 3a1, and IL6 at 21 weeks post TAC (Figure 6E). Notably, TGFβ and fibronectin 1 were significantly reduced only in mice with constitutive overexpression of EC-CSE Tg but not in the mice in which EC-CSE overexpression was delayed (Figure 6E).
Figure 6: Reduced cardiac fibrosis in EC-CSE Tg mice at 21 weeks post TAC.

Best illustrative microphotography of (A) cardiac fibrosis in interstitial area and (B) perivascular area; Total fibrosis (C) and perivascular fibrosis (D) quantified; (E) Relative fold changes in cardiac mRNA levels of a panel of fibrosis-related genes for constitutive EC-CSE Tg, 10-week-delayed EC-CSE Tg, and control mice. Circled number inside bars indicates samples size. Data are analyzed with one-way ANOVA and presented as mean ± SD. Scale bar indicates 50 μm.
We next evaluated the level of EndoMT in our model of HFrEF by utilizing immunohistochemical staining of vimentin and Von Willebrand Factor at the 21-week end point of the study. Representative photomicrographs of heart sections are shown in Figure 7A. Vimentin and VWF double-positive cells were counted, and a significant reduction of such double-positive cells was observed in both constitutive and 10-week-delayed EC-CSE Tg mice when compared to control mice Figure 7B. Further, we measured the transcriptional levels of markers (αSMA, vimentin) and activators (Snai1, Slug, Twist) in the heart. At 6 weeks post-TAC, an early stage of the pressure overload HF progression, αSMA, Vimentin, Snai1, Slug, and Twist were all significantly reduced transcriptionally in constitutive EC-CSE Tg mice when compared to control mice (Figure 7C). Since the overexpression was under suppression by doxycycline, we did not observe any differences between 10-week-delayed EC-CSE Tg and control mice (Figure 7C). At 21-week end point, αSMA, vimentin, Snai1 and Slug remained reduced transcriptionally in constitutive EC-CSE Tg mice while no difference was observed in Twist (Figure 7D). Interestingly, although CSE overexpression was suppressed until 10 weeks post-TAC, we did observe a significantly reduction in vimentin expression in delayed EC-CSE Tg mice, which corresponded to reduced Vimentin+/VWF+ double positive cells (Figure 7B and D). However, this observation may not be attributed to expression levels of Snai1, Slug, and Twist, as no difference was found between 10-week-delayed EC-CSE Tg and control mice at either 6- or 21-weeks post TAC (Figure 7D). Similarly, we also evaluated the NO bioavailability in EC-CSE Tg mice at 21 weeks following TAC. We observed significant elevated circulating nitrite levels only in constitutive EC-CSE Tg mice but not 10-wk-delayed EC-CSE Tg mice (Figure 7E). However, myocardial nitrite levels were significantly increased in myocardial tissues from both constitutive and 10-wk-delayed EC-CSE Tg mice (Figure 7F).
Figure 7: Downregulation of EndoMT in EC-CSE Tg mice.
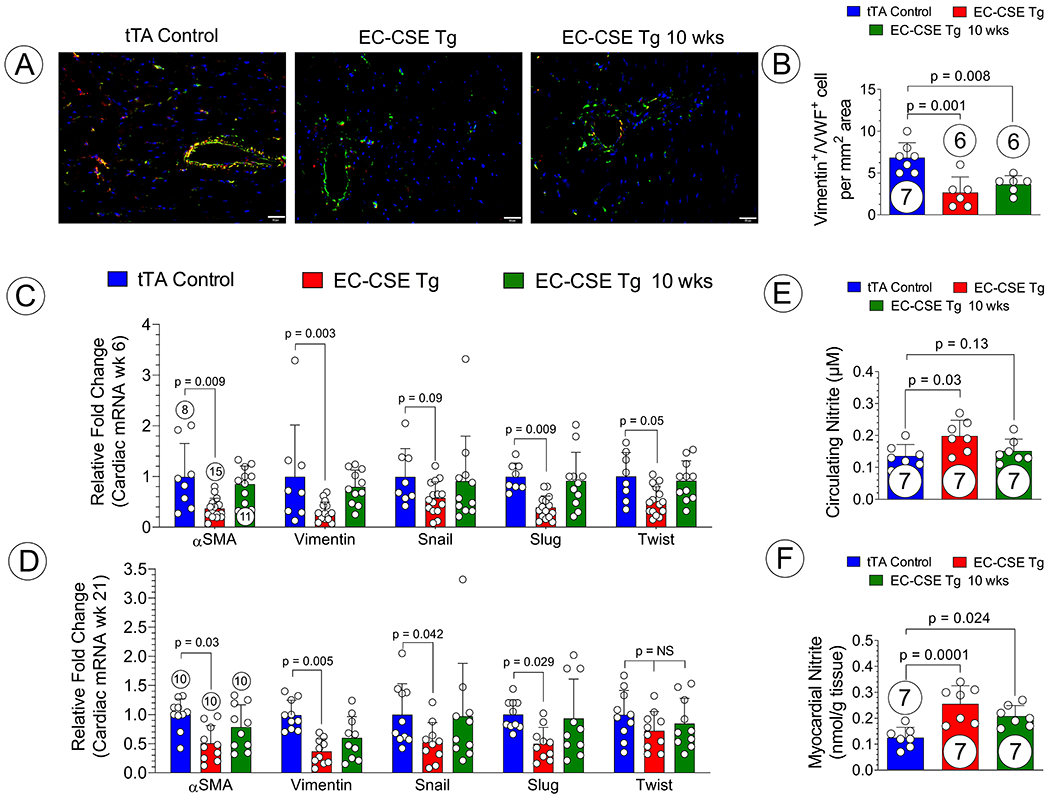
(A) Best illustrative microphotography of heart sections stained for Vimentin (Red), VWF (green), and Dapi (blue); Vimentin+/VWF+ double positive cells displayed yellow color and quantified in (B); Relative fold changes in cardiac mRNA levels of a panel of EndoMT-related genes at 6 weeks post TAC (C) and 21 weeks post TAC (D) for constitutive EC-CSE Tg, 10-week-delayed EC-CSE Tg, and control mice. Circled number inside bars indicates samples size. Data are analyzed with one-way ANOVA and presented as mean ± SD. Scale bar indicates 50 μm.
Endothelial Cell CSE/H2S Reduced Expression of EndoMT Markers in vitro.
To validate the observation that EndoMT was enhanced in EC-CSE KO while reduced in EC-CSE Tg hearts we investigated the mRNA expression levels of critical EndoMT markers in primary endothelial cells isolated from either wild-type control or EC-CSE KO mice under baseline condition, following TGF-β exposure, or following administration of a potent hydrogen sulfide donor (i.e., JK-1). Under baseline condition, endothelial cells lacking CSE exhibited significantly elevated mRNA levels of Snai1 and Twist as compared to control endothelial cells (Figure S3A). Following TGF-β treatment, ECs with CSE KO displayed a 2.2 fold increase in Vimentin, 4.7 and 4.9-fold increase in Snai1 and Snai2, respectively, a 30-fold increase in Twist, as well as a 95-fold increase in Col1a1 (Figure S3B). More importantly, with the treatment of an H2S donor, JK-1, the increased expression of Snai1 and Col1a1 were significantly blunted (Figure S3C).
Genetic Modulation of Endothelial Cell CSE Did Not Alter Systemic Blood Pressure.
To better understand the systemic effects of modulating EC-CSE and its H2S production, radio-telemetry systems were utilized to record blood pressure and heart rates in EC-CSE KO, EC-CSE Tg mice, and the respective littermate control mice. We observed no significant difference between EC-CSE KO, EC-CSE Tg, and the respective littermate control mice in terms of systolic, diastolic, mean arterial blood pressure at baseline or after subcutaneous AngII infusion (Figures S4A to C and S5A to C). Similarly, there wasn’t any difference observed in conscious heart rate in EC-CSE mutant as compared to the control mice (Figures S4D and S5D).
Pharmacological H2S Supplementation Reversed HF Severity in EC-CSE KO Mice.
Additionally, a well-studied experimental H2S donor, JK-1 (0.2 mg/kg/day), were administered via i.p. injection in a subset of EC-CSE KO mice following TAC to evaluate the effects of pharmacological H2S supplementation20, 27. H2S supplementation resulted in attenuated hypertrophic response as LVPWd were significantly lower at 3wk- and 6wk- post TAC while thicker in 21wk- post TAC in EC-CSE KO mice that received JK-1 treatment (Figure S6A). LV chamber dimensions and ejection fraction were also restored in EC-CSE KO mice that received JK-1 treatment (Figures S6B – C). Additionally, JK-1 therapy significantly lowered LV end-diastolic pressure, TAU, and circulating BNP levels (Figures S6D – F) while improving and aortic vascular reactivity to acetylcholine (Figure S6G) and exercise performance (Figure S6I).
DISCUSSION
It is well appreciated that H2S plays a critical beneficial role in the pathogenesis of heart failure19–21, 28. Global CSE knockout mice subjected to TAC procedure developed exacerbated cardiac adverse remodeling and reduced function, accompanied by increased oxidative stress, compromised eNOS coupling and function, and reduced NO bioavailability18, 21, 28. H2S donors have been shown to ameliorates the severity of pressure overload induced heart failure by modulating vascular tone, promoting angiogenesis, reducing inflammation, attenuating oxidative stress, and downregulating apoptosis18, 21, 28. These studies have laid the foundation for the development of H2S based therapy for the treatment of heart failure.
However, to date, the effects of H2S in the pathophysiological process of heart failure have been investigated primarily by using pharmacological donors or inhibitors of H2S, or by using gene targeted mice with global overexpression or deletion of H2S producing enzymes. Such “lack-of-cell-type-selectivity” approaches significantly limits our understanding of the effects of H2S in specific cell populations and certain cell-specific pathological process, such as endothelial cells. More recently, studies utilizing EC-CSE knockout mice greatly advanced our knowledge in the roles of EC-CSE in the pathogenesis of atherosclerosis and demonstrated the capability of EC-CSE derived H2S in modifying protein structure and function25, 29. In the current study, we investigated endothelial cell-specific CSE genetic knockout or overexpression mouse models to define the effects of endothelial cell-specific H2S production on EndoMT in the setting of pressure overload-induced heart failure, and determine if the severity of HFrEF is affected by endothelium-derived H2S. We observed significantly compromised cardiac structure and function in mice with endothelial cell-specific CSE deletion, as evidenced by elevated LV chamber size as well as reduced LV ejection fraction at 6-, 9-, and 12-weeks post-TAC, respectively. Interestingly, these worsened cardiac adverse remodeling were no longer displayed by EC-CSE KO mice at later time points, indicating that EC-CSE KO accelerates the progression of pressure overload-induced cardiac remodeling and failure but does not exacerbate echocardiographic parameters in the late stages of the disease process. Despite the enhanced early pathological cardiac remodeling in EC-CSE KO mice, LVEF was not significantly worse at 21 weeks post-TAC when compared to the control group. Nevertheless, we did observe that invasive LV hemodynamic parameters such as LVEDP and Tau were significantly worsened and vascular relaxation to ACh was also significantly reduced in EC-CSE KO mice when compared to control. In combination, these alterations in cardiac pressure and vascular function ultimately translated into significantly reduced treadmill exercise performance in EC-CSE KO mice. These data suggest that endothelial cell CSE-derived H2S limits the severity of heart failure and genetic deletion of endothelial cell CSE exacerbates pressure overload-induced HFrEF. In contrast, when CSE is overexpressed constitutively or in a delayed manner in ECs, we observed attenuated cardiac adverse remodeling and dysfunction, significantly improved endothelial responses to acetylcholine, and ultimately improved exercise performance as compared to control mice. More importantly, all symptoms of HFrEF were attenuated even when the EC-CSE overexpression was delayed until 10 weeks post TAC, at which time HFrEF had already been ensued. Pharmacological supplementation of H2S also halted the progression of HFrEF in a manner that’s similar to constitutive EC-CSE overexpression following TAC. Taken together, these observations provide the first evidence that endothelial-derived H2S plays a role in the pathogenesis of and may be a therapeutic target for pressure overload HFrEF.
We also observed increased fibrotic response in myocardial tissues following CSE deletion but attenuated cardiac fibrosis in mice with CSE overexpressed ECs. Given the endothelial specific nature of these CSE mutant mouse models, and pressure overload heart failure is induced by long-standing systolic hypertension, we decided to investigate cardiac perivascular fibrosis. Significantly increased perivascular fibrosis was observed in EC-CSE KO mice when compared to control, while both constitutive and delayed-activated EC-CSE overexpression reduced the amount of perivascular fibrosis. Mechanistically, we investigated the endothelial-mesenchymal transition phenomenon in these mice. Co-staining of vimentin and von Willebrand factor identified the transitioning ECs in the heart. We observed a significant elevation in vimentin+/VWF+ double positive cells in EC-CSE KO hearts while EC-CSE Tg hearts displayed significantly reduced numbers of such double positive cells. Additionally, we investigated the mRNA expressional levels of a panel of EndoMT marker genes in primary endothelial cells isolated from either control or EC-CSE KO mice in vitro. Interestingly, under baseline condition, we observed significantly elevated Snai1 and Twist mRNA, indicating that genetic deletion of CSE might predispose ECs to EndoMT process. Following TGF-β exposure, CSE deficient endothelium expressed elevated levels of Vimentin, Snai1, Snai2, and Twist, suggesting an enhanced EndoMT phenotype. JK-1 treatment partially rescued such phenotype. These data provide the first evidence that EC-CSE derived H2S may preserve the cellular signature of ECs, preventing them from entering the endothelial-mesenchymal transition.
The precise contribution of EndoMT in the development of myocardial fibrosis in the adult heart remains controversial7, 9. The initial lineage-tracing studies using constitutive endothelial Tie1-Cre approach were limited by the non-specific mis-labeling of embryonic EC - derived mesenchymal cells12–14, 30, 31. More recent fate-mapping studies have suggested that de novo EndoMT plays limited roles in cardiac fibrosis formation in adult injured heart32, 33. In the current study, we utilized co-staining of vimentin and VWF to identify ECs that are potentially transitioning into mesenchymal-like cells. We observed a significantly elevation in vimentin+/VWF+ double positive cells, which is associated with increased total cardiac fibrosis and especially cardiac perivascular fibrosis. Although we cannot draw a definite conclusion that EndoMT is the major contributor to cardiac fibrosis in these mice, we believe that under the stress of systemic pressure overload such as long-standing hypertension, ECs in the blood vessels are more likely to undergo EndoMT and contribute to perivascular fibrosis. Besides the direct effects on perivascular fibrosis, further studies are warranted to investigate the paracrine effects of EC-CSE derived H2S on nearby quiescent fibroblasts. EC-CSE derived H2S may be a paracrine factor that inhibits the activation and transformation of surrounding quiescent fibroblasts into myofibroblasts, leading to attenuated cardiac fibrosis. On the other hand, we speculate that under other stress stimuli such as myocardial infarction, the activation of EndoMT may not be as profound, and contributes less to the scar formation and overall cardiac fibrotic response. Future studies are required to elucidate the role of EC-CSE derived H2S in EndoMT in the setting of myocardial infarction and subsequent HFrEF. Further, it is worth noted that although studies have suggested that CSE/H2S plays an important role in BP regulation, genetic modulation of CSE specifically in endothelial cells did not appear to affect systemic blood pressure, despite improving aortic vascular reactivity to acetylcholine treatment ex vivo34–36. We observed no difference in SBP, DBP, and MAP between EC-CSE KO, EC-CSE Tg and the respective littermate controls at baseline or after subcutaneous AngII infusion. However, we previously reported that at baseline condition, EC-CSE deletion led to dampened aortic vascular reactivity to acetylcholine and poor exercise performance while EC-CSE overexpressing mice displayed the opposite, indicating that modifying H2S production in endothelial cell likely result in some degree of systemic ramifications such as altered local blood flow, oxidative stress, and inflammatory state, which could contribute to the cardiovascular phenotypes of the EC-CSE KO or Tg mice in the current study24.
In the efforts of understanding the underlying mechanisms by which EC-CSE derived H2S modulates EndoMT, we performed a gene expression panel analysis on key markers and activators such as Snai1, Slug, and Twist, of EndoMT in EC-CSE KO, constitutive and delayed EC-CSE Tg, and their littermate control mice. In EC-CSE KO mice, we observed elevated markers for EndoMT such as αSMA and Vimentin at 6 weeks post-TAC, the early stage of the pathogenesis of pressure overload HFrEF, as well as at 21 weeks post-TAC, the late stage of the disease model as compared to control. Interestingly, we observed elevation of activators of EndoMT such as Snai1, Slug, and Twist only at the early stage, but not the late stage of the pressure overload HFrEF model. Similarly, in EC-CSE Tg mice we observed reductions in the mRNA levels of both the markers and activators of EndoMT during the early stages of heart failure, while only the EndoMT markers were reduced during the late stages of heart failure. These data suggest that the EndoMT pathological process may occur in the early stage of the pressure overload HFrEF due to the increase of Snai1, Slug, and Twist, which are modulated by endothelial-derived H2S. However, when we activated the EC-CSE Tg at 10 weeks post TAC, which has passed the early stage of our protocol, EC-CSE overexpression still suppressed the EndoMT evidenced by reduction in vimentin+/VWF+ double positive cells, via a Snai1/Slug/Twist – independent mechanism. Additionally, it has been reported that eNOS – NO pathway exerts inhibitory effects on EndoMT31, 37–39. Given the profound eNOS-activating and NO-promoting effects of endothelial CSE24, 40, and indeed we observed significant reduction in nitrite levels in both the circulation and myocardial tissues in EC-CSE KO mice while enhanced NO bioavailability in EC-CSE Tg mice, it is plausible that endothelial derived H2S modulate EndoMT, in part, through regulating eNOS activity and NO production21, 22, 28. These data further confirmed not only the importance of EC-CSE in preserving normal function and cellular signature, but also the crosstalk between gaseous molecules such as NO and H2S within the endothelium. Additionally, EC-CSE/H2S - eNOS/NO crosstalk modulates coronary blood flow, which may in turn, impact the formation of cardiac fibrosis41, 42. Additionally, it has been recently established that CSE - derived H2S determines the cellular s-sulfhydrome which modifies the functions of important transcription regulators in the EC25, 29. To fully decipher the underlying mechanisms by which endothelial CSE - derived H2S modulates EndoMT in different stages of heart failure, a more comprehensive time-course and gain- or loss- of function studies with the use of advanced proteomic/sulfhydromic technologies are warranted.
In summary, this is the first study using tissue/cell specific gene modulation to address the role of endothelial cell specific H2S generation in the setting of pressure overload - induced heart failure. Our results provide evidence that endothelial CSE derived H2S mediates a variety of beneficial effects and ameliorates the severity of heart failure. Moreover, we demonstrated that H2S produced in endothelial cells is critical in maintaining cellular signature of the endothelium and preventing endothelial-mesenchymal transition under the stress environment of pressure overload, in part, through regulating eNOS activity and NO bioavailability. Modulation of endogenous endothelial H2S production may be a potential therapeutic strategy for the treatment of heart failure.
Supplementary Material
Figure 1: Study Timeline.
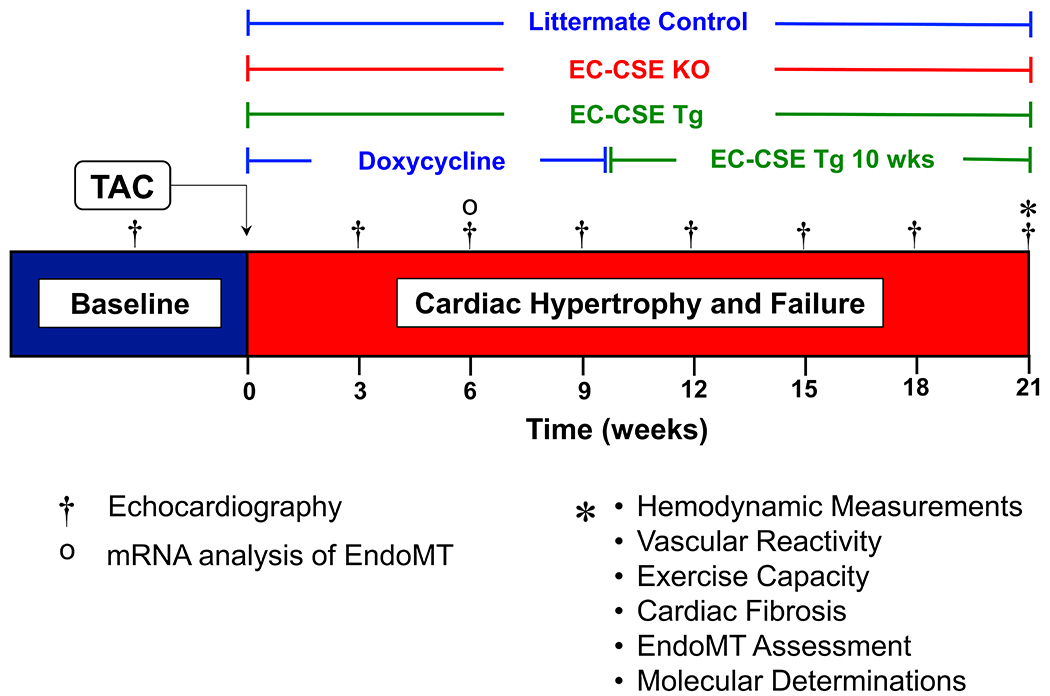
Echocardiography was performed prior to the induction of HFrEF by TAC procedure then once every 3 weeks for a duration of 21 weeks. Invasive hemodynamic, vascular reactivity, exercise capacity, cardiac fibrosis, EndoMT activation, and other molecular determination were performed at 21 weeks post TAC. A separate set of mice were sacrificed at 6 weeks post TAC to assess early stage EndoMT activation.
Novelty and Significance.
What is Known?
Hydrogen sulfide (H2S) is an endogenous physiological signaling molecule that exerts protective effects in the cardiovascular system.
Endothelial-derived H2S produced by cystathionine γ lyase (CSE) preserves endothelial cell function, promotes exercise capacity, and protects against myocardial ischemia/reperfusion injury.
Endothelial CSE/H2S mediated sulfhydration delays atherogenesis in a murine model of atherosclerosis.
What New Information Does This Article Contribute?
Endothelial specific CSE KO resulted in increased cardiac remodeling, impaired vascular reactivity, and worsened left ventricular function in a transverse aortic constriction (TAC) - induced heart failure model.
Endothelial specific CSE KO resulted in increased expression of key regulatory genes in the Endothelial-Mesenchymal transition pathway, including Snai, Slug, and Twist, leading to elevated cardiac interstitial and perivascular fibrosis in a TAC - induced heart failure model.
Endothelial specific CSE overexpression suppressed the expression of Snai, Slug, and Twist, leading to a blunted cardiac fibrotic response, and correction of cardiac and vascular dysfunction following pressure overload heart failure.
Endothelial protective effects by H2S have been previously shown. In our study, we investigate the role of endothelial cell (EC) specific CSE in pressure overload - induced heart failure by utilizing EC specific CSE knockout or overexpressing mouse models. Our data demonstrates that endothelial cells with CSE genetic knockout are more prone to undergo endothelial-mesenchymal transition when exposed to pressure overload or TGF-β treatment, ultimately contributing to more severe heart failure phenotypes including elevation in cardiac fibrosis, accelerated decline in LV ejection fraction, increased LV remodeling and filling pressure, impaired vascular reactivity, and significant exercise intolerance. In contrast, CSE overexpression or H2S supplementation in endothelial cells inhibits the endothelial-mesenchymal transition process and corrected the deteriorating phenotypes of EC-CSE KO mice following TAC. These data suggest a key regulatory role of EC-CSE derived H2S in maintaining the cellular signature of ECs in pathological settings. Modulation of endothelium-derived H2S production may represent a novel therapeutic strategy for the treatment of heart failure with reduced ejection fraction and, more broadly, cardiovascular diseases involving endothelial-mesenchymal transition.
SOURCES OF FUNDING
This work was supported by Grants from NIH National Heart, Lung, and Blood Institute (R01 HL146098, R01 HL146514, R01 HL137711) to David J Lefer, American Heart Association Postdoctoral Grant (20POST35200075) to Zhen Li, American Heart Association Postdoctoral Grant (18POST34020143) to Thomas E. Sharp III, the German Research Foundation (SFB 815) to Josef Pfeilschifter, and the Hellenic Foundation for Research and Innovation under the “First Call for Research Projects to support Faculty Members and Researchers and the procurement of high-cost research equipment grant” (HFRI-FM17-886) to Dr. Papapetropoulos.
CONFLICT OF INTEREST DISCLOSURES
Drs. David Lefer and Andreas Papapetropoulos serve as scientific consultant for Sulfagenix Inc, a company focusing on developing H2S-based therapy for clinical use. Dr. Lefer also has stock in NovoMedix and SAJE Pharma. Dr. Papapetropoulos has received a grant from Antibe Therapeutics outside the submitted work. Other authors declare no conflict of interests.
Nonstandard Abbreviations and Acronyms
- 3-MST
3-mercaptopyruvate sulfurtransferase
- ACh
acetylcholine
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- ECs
endothelial cells
- EC50
half maximal effective concentration
- EC-CSE KO
endothelial cell specific CSE knockout
- EC-CSE Tg
endothelial cell specific CSE transgenic
- EDD
end-diastolic dimension
- EDP
end-diastolic pressure
- EF
ejection fraction
- EndoMT
endothelial-mesenchymal transition
- eNOS
endothelial nitric oxide synthase
- H2S
hydrogen sulfide
- LV
left ventricle
- NO
nitric oxide
- PWd
posterior wall thickness at diastole
- SNP
sodium nitroprusside
- TAC
transverse aortic constriction
- TetO
tetracycline operator sequence
- VWF
Von Willebrand factor
REFERENCES
- 1.Bozkurt B, Hershberger RE, Butler J, Grady KL, Heidenreich PA, Isler ML, Kirklin JK and Weintraub WS. 2021 ACC/AHA Key Data Elements and Definitions for Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Clinical Data Standards for Heart Failure). Circ Cardiovasc Qual Outcomes. 2021:HCQ0000000000000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, VanWagner LB, Wang NY, Tsao CW, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143:e254–e743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nabel EG and Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. [DOI] [PubMed] [Google Scholar]
- 4.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention and Stroke C. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alem MM. Endothelial Dysfunction in Chronic Heart Failure: Assessment, Findings, Significance, and Potential Therapeutic Targets. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuchi C, Tritto I, Carluccio E, Mattei C, Cattadori G and Ambrosio G. Role of endothelial dysfunction in heart failure. Heart Fail Rev. 2020;25:21–30. [DOI] [PubMed] [Google Scholar]
- 7.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G and Baker AH. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bischoff J Endothelial-to-Mesenchymal Transition. Circ Res. 2019;124:1163–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Lui KO and Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15:445–456. [DOI] [PubMed] [Google Scholar]
- 10.Jackson AO, Zhang J, Jiang Z and Yin K. Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases. Trends Cardiovasc Med. 2017;27:383–393. [DOI] [PubMed] [Google Scholar]
- 11.Gong H, Lyu X, Wang Q, Hu M and Zhang X. Endothelial to mesenchymal transition in the cardiovascular system. Life Sci. 2017;184:95–102. [DOI] [PubMed] [Google Scholar]
- 12.Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M and Hirata K. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–18. [DOI] [PubMed] [Google Scholar]
- 13.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S and Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–61. [DOI] [PubMed] [Google Scholar]
- 14.Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, Brewer AC, Zhang M and Shah AM. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. 2014;63:2734–41. [DOI] [PubMed] [Google Scholar]
- 15.Polhemus DJ and Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res. 2014;114:730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabil O and Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal. 2014;20:770–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Polhemus DJ and Lefer DJ. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ Res. 2018;123:590–600. [DOI] [PubMed] [Google Scholar]
- 18.LaPenna KB, Polhemus DJ, Doiron JE, Hidalgo HA, Li Z and Lefer DJ. Hydrogen Sulfide as a Potential Therapy for Heart Failure-Past, Present, and Future. Antioxidants (Basel). 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polhemus D, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ and Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail. 2013;6:1077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Organ CL, Kang J, Polhemus DJ, Trivedi RK, Sharp TE 3rd, Jenkins JS, Tao YX, Xian M and Lefer DJ. Hydrogen Sulfide Attenuates Renin Angiotensin and Aldosterone Pathological Signaling to Preserve Kidney Function and Improve Exercise Tolerance in Heart Failure. JACC Basic Transl Sci. 2018;3:796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R and Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci U S A. 2014;111:3182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bibli SI, Szabo C, Chatzianastasiou A, Luck B, Zukunft S, Fleming I and Papapetropoulos A. Hydrogen Sulfide Preserves Endothelial Nitric Oxide Synthase Function by Inhibiting Proline-Rich Kinase 2: Implications for Cardiomyocyte Survival and Cardioprotection. Mol Pharmacol. 2017;92:718–730. [DOI] [PubMed] [Google Scholar]
- 23.Zhao W and Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;283:H474–80. [DOI] [PubMed] [Google Scholar]
- 24.Xia H, Li Z, Sharp TE 3rd, Polhemus DJ, Carnal J, Moles KH, Tao YX, Elrod J, Pfeilschifter J, Beck KF and Lefer DJ. Endothelial Cell Cystathionine gamma-Lyase Expression Level Modulates Exercise Capacity, Vascular Function, and Myocardial Ischemia Reperfusion Injury. J Am Heart Assoc. 2020;9:e017544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bibli SI, Hu J, Sigala F, Wittig I, Heidler J, Zukunft S, Tsilimigras DI, Randriamboavonjy V, Wittig J, Kojonazarov B, Schurmann C, Siragusa M, Siuda D, Luck B, Abdel Malik R, Filis KA, Zografos G, Chen C, Wang DW, Pfeilschifter J, Brandes RP, Szabo C, Papapetropoulos A and Fleming I. Cystathionine gamma Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation. 2019;139:101–114. [DOI] [PubMed] [Google Scholar]
- 26.Bibli SI, Hu J, Leisegang MS, Wittig J, Zukunft S, Kapasakalidi A, Fisslthaler B, Tsilimigras D, Zografos G, Filis K, Brandes RP, Papapetropoulos A, Sigala F and Fleming I. Shear stress regulates cystathionine gamma lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation. Redox Biol. 2020;28:101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang J, Li Z, Organ CL, Park CM, Yang CT, Pacheco A, Wang D, Lefer DJ and Xian M. pH-Controlled Hydrogen Sulfide Release for Myocardial Ischemia-Reperfusion Injury. J Am Chem Soc. 2016;138:6336–9. [DOI] [PubMed] [Google Scholar]
- 28.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr., Gojon G Jr., Wang R, Karusula N, Nicholson CK, Calvert JW and Lefer DJ. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bibli SI, Hu J, Looso M, Weigert A, Ratiu C, Wittig J, Drekolia MK, Tombor L, Randriamboavonjy V, Leisegang MS, Goymann P, Delgado Lagos F, Fisslthaler B, Zukunft S, Kyselova A, Justo AFO, Heidler J, Tsilimigras D, Brandes RP, Dimmeler S, Papapetropoulos A, Knapp S, Offermanns S, Wittig I, Nishimura SL, Sigala F and Fleming I. Mapping the Endothelial Cell S-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation. 2021;143:935–948. [DOI] [PubMed] [Google Scholar]
- 30.Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, Mammoto A, Chen J, Ariaans M, Francis SE, Van der Heiden K, Ridger V and Evans PC. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep. 2017;7:3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH and Thum T. Transforming growth factor-beta-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol. 2012;32:361–9. [DOI] [PubMed] [Google Scholar]
- 32.Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Muller AM, Volz KS, Tang Z, Red-Horse K and Ardehali R. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115:625–35. [DOI] [PubMed] [Google Scholar]
- 33.Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J and Evans SM. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cui C, Fan J, Zeng Q, Cai J, Chen Y, Chen Z, Wang W, Li SY, Cui Q, Yang J, Tang C, Xu G, Cai J and Geng B. CD4(+) T-Cell Endogenous Cystathionine gamma Lyase-Hydrogen Sulfide Attenuates Hypertension by Sulfhydrating Liver Kinase B1 to Promote T Regulatory Cell Differentiation and Proliferation. Circulation. 2020;142:1752–1769. [DOI] [PubMed] [Google Scholar]
- 35.Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacos E, Buhimschi IA, Buhimschi CS and Ahmed A. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation. 2013;127:2514–22. [DOI] [PubMed] [Google Scholar]
- 36.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH and Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo Y, Li P, Bledsoe G, Yang ZR, Chao L and Chao J. Kallistatin inhibits TGF-beta-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp Cell Res. 2015;337:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smeda M, Kieronska A, Adamski MG, Proniewski B, Sternak M, Mohaissen T, Przyborowski K, Derszniak K, Kaczor D, Stojak M, Buczek E, Jasztal A, Wietrzyk J and Chlopicki S. Nitric oxide deficiency and endothelial-mesenchymal transition of pulmonary endothelium in the progression of 4T1 metastatic breast cancer in mice. Breast Cancer Res. 2018;20:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Han X, Tang J, Long X and Wang X. Salidroside inhibits endothelialmesenchymal transition via the KLF4/eNOS signaling pathway. Mol Med Rep. 2021;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altaany Z, Yang G and Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. 2013;17:879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai Z, Aoki T, Fukumoto Y and Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol. 2012;60:416–21. [DOI] [PubMed] [Google Scholar]
- 42.Casalini ED, Goodwill AG, Owen MK, Moberly SP, Berwick ZC and Tune JD. Contribution of hydrogen sulfide to the control of coronary blood flow. Microcirculation. 2014;21:104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knowlton AA and Lee AR. Estrogen and the cardiovascular system. Pharmacol Ther. 2012;135:54–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Organ CL, Li Z, Sharp TE 3rd, Polhemus DJ, Gupta N, Goodchild TT, Tang WHW, Hazen SL and Lefer DJ. Nonlethal Inhibition of Gut Microbial Trimethylamine N-oxide Production Improves Cardiac Function and Remodeling in a Murine Model of Heart Failure. J Am Heart Assoc. 2020;9:e016223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bradley JM, Spaletra P, Li Z, Sharp TE 3rd, Goodchild TT, Corral LG, Fung L, Chan KWH, Sullivan RW, Swindlehurst CA and Lefer DJ. A novel fibroblast activation inhibitor attenuates left ventricular remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol. 2018;315:H563–H570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conchinha NV, Sokol L, Teuwen LA, Veys K, Dumas SJ, Meta E, Garcia-Caballero M, Geldhof V, Chen R, Treps L, Borri M, de Zeeuw P, Falkenberg KD, Dubois C, Parys M, de Rooij L, Rohlenova K, Goveia J, Schoonjans L, Dewerchin M, Eelen G, Li X, Kalucka J and Carmeliet P. Protocols for endothelial cell isolation from mouse tissues: brain, choroid, lung, and muscle. STAR Protoc. 2021;2:100508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia H, Sriramula S, Chhabra KH and Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113:1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed Materials and Methods and Major Resources Table are available in the Online Supplemental Material. The data that support the findings of this study, experimental materials, and analytic methods are available from the corresponding author upon reasonable request.