

Abstract
Aromatic interactions are commonly involved in the assembly of naturally occurring building blocks, and these interactions can be replicated in an artificial setting to produce functional materials. Here we describe a colorimetric biosensor using co-assembly experiments with plasmonic gold and surfactant-like peptides (SLPs) spanning a wide range of aromatic residues, polar stretches, and interfacial affinities. The SLPs programmed in DDD–(ZZ)x–FFPC self-assemble into higher-order structures in response to a protease and subsequently modulate the colloidal dispersity of gold leading to a colorimetric readout. Results show the strong aggregation propensity of the FFPC tail without polar DDD head. The SLPs were specific to the target protease, i.e., Mpro, a biomarker for SARS-CoV-2. This system is a simple and visual tool that senses Mpro in phosphate buffer, exhaled breath condensate, and saliva with detection limits of 15.7, 20.8, and 26.1 nM, respectively. These results may have value in designing other protease testing methods.
Keywords: main protease, colorimetric test, peptide amphiphile, aromatic interactions, saliva
Graphical Abstract
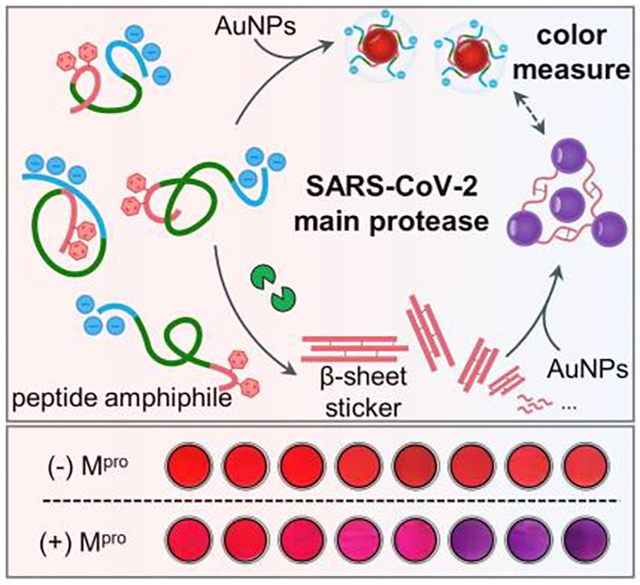
A colorimetric probe for the SARS-CoV-2 protease is reported by mediating the interplay of peptide amphiphiles and gold nanoparticles via aromatic force. This probe showed good performance in exhaled breath and saliva, in favor of designing amphiphilic peptides for plasmonic coupling in complex media.
Introduction
The FF dipeptide and its resulting β-sheet assemblies are most studied due to their unique mechanical, optical, and electrical properties.[1] For example, the core section of β-amyloid fibrils implicated in neurodegenerative pathologies has been identified as the sequence of KLVFF.[1b, 2] The PFF sequence has the greatest aggregation propensity of all combinatorial tripeptides. Gazit et al. showed that PFF tripeptide assembles into unique helical-like β-sheets that are about 2- to 3-fold stiffer than those formed by the FF motif.[3] They provided mechanistic insights into how aromatic residues in PFF units result in ordering and directional growth via zipper π–π stacking. As the fundamental understanding of peptide assembly evolves, increasingly sophisticated materials and applications have emerged, e.g., by complementing the self-assembly peptide (protease-responsive regions) with plasmonic nanoparticles (optical properties).[4] For instance, colorimetric measure based on plasmonic coupling offers affordable, equipment-free, and end-user deliverable point-of-care diagnostics for disease biomarkers (here, protease) analogous to the impact of lateral flow assays.
The exact nature of the interactions driving interparticle organization spans a range of covalent[5] and non-covalent modalities (e.g., electrostatic,[6] H-bonding,[7] hydrophobic and/or aromatic[8], and specific recognition[9]). For example, peptides consisting of divalent Cys (C) [i.e., C–(ZZ)x–C, where –(ZZ)x– is arbitrary amino acid] have been extensively applied to measure metalloproteinase, caspase-3, and furin in combination with gold nanoparticles (AuNPs) via covalent Au–S bond.[5b, 10] Alternatively, zwitterionic peptides [e.g., D3−(ZZ)x−R2] of switchable electrophoretic properties can induce phase transformation of charged AuNPs via Coulomb interactions, which has been further validated for detection of a viral protease, metalloproteinase, and phosphatase.[4d, 6a, 6b, 11] The key question raised by the above peptide designs concerns their functional loss in complex milieu due to oxidation and charge scavengers. Peptide functions under these conditions, however, are possible when the controlled assemblies are imparted by aromatic residues, a characteristic met by surfactant-like peptides (SLPs) deployed in a considerable body of work for theragnostic applications.[8, 12] Yet, the SLP design suffers from fine tuning the amphiphilicity balance and complex substrate post-modifications, e.g., conjugation of unnatural aromatic moieties and/or phosphorylation of side chains.
Here, we rationally designed simple SLPs with a general formula of DDD–(ZZ)x–FFPC to assemble metal nanostructures via aromatic-aromatic interactions. The SLPs use only natural occurring residues and include (i) a FFPC sticker tail, (ii) a protease responsive module, and (iii) a triple DDD stretch. Note that the sticker domain is engineered with a single Cys to interface with AuNPs. We then investigated the effect of mutating the sequence in different domains (e.g., aromatic residues, polar stretches, and interfacial affinities) on the colloidal dispersity and optical responses. We validated the system for colorimetric sensing of SARS-CoV-2 main protease (Mpro, or nsp5/3CLpro) in saliva. Mpro processes the viral polypeptides into functional proteins and is therefore a key enzyme for diagnostics and therapeutics (e.g., Pfizer’s Paxlovid).[13] Our results revealed that FF di-homopeptide is an indispensable domain that induces assembly of AuNPs in aqueous environment. The protic polar head was the most effective moiety to disturb the self-assembly of peptide amphiphile and thus restore the good colloidal dispersity of AuNPs. Overall, we determined that the detection limit of Mpro is 15 nM in the biological milieu (e.g., doped exhaled breath and saliva) with good specificity. Considering the broad interest in rapid diagnosis and mass surveillance of COVID-19, these findings have important implications for the development of portable biological sensors for SARS-CoV-2 proteases.
Results and Discussion
We introduced and tested the peptide amphiphiles (also referred to as SLPs) and then used them with AuNPs for colorimetric sensing of Mpro. Mpro is chosen as the target because of its essential role in protein maturation for viral proliferation.[13] We hypothesized that proteolysis of the monomeric SLPs would lead to sulfhydryl-rich supramolecular assemblies via aromatic stacking and subsequently flocculate colloidal gold, thus producing a color change (Figure 1a). The amphiphilic SLPs were rationally designed to encompass three functional domains composed of (i) an N-terminal polar head made of three Asp residues (DDD) for promoting the colloidal stability, (ii) an Mpro cleavage sequence consisting of TSAVLQ↓SG, and (iii) a C-terminal sticker tail for co-assembling the peptide and colloidal gold. The sticker domain contains a FFP motif as a strong β-sheet forming sequence and a Cys residue for sequestering surface gold via aromatic-aromatic interactions and dative bonds, respectively.[3, 5b, 14] The sticker tail is capped by a charged polar DDD stretch, and hydration of these protic groups would compromise aromatic stacking and thus break self-organization of the intact peptide to a low degree. This modular design can be reconfigured for other proteases.
Figure 1.
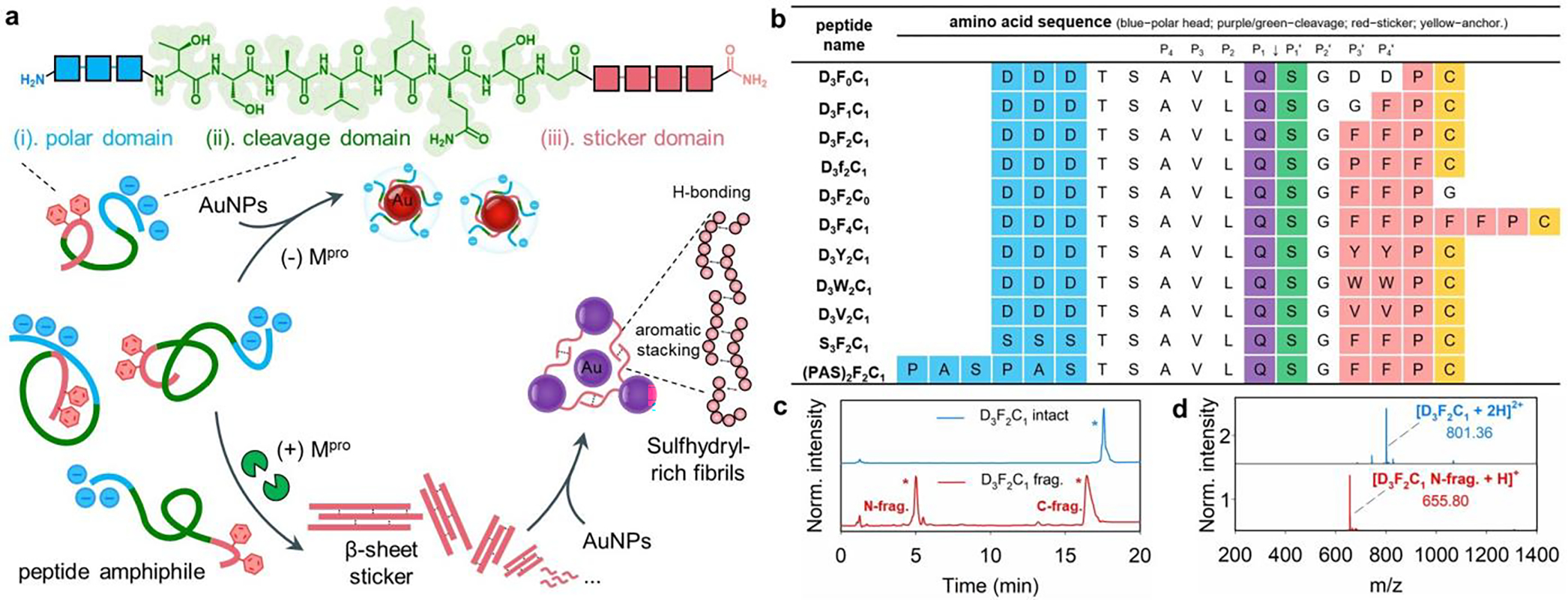
Library of surfactant-like peptides (SLPs) studied here for colorimetric assays. (a) Chemical structures of modular peptide amphiphiles have an aromatic amino acid sticker tethered by a polar head of increasing hydrophilicity; an Mpro cleavage sequence is in the center. The schematic illustrates the peptide self-assembly and subsequent co-assembly with plasmonic nanoparticle in the presence of Mpro. The intact peptides produce β-sheet structures rich in sulfhydryl groups after proteolysis, which favors plasmonic coupling via aromatic-stacking/hydrogen-bonding. (b) Synthetic peptide sequences spanning a wide range of aromatic residues, polar stretches, and interfacial affinities. The amino acids on the polar head, cleavage site, and sticker tail are color coded. Mpro cleaves the peptide at Q↓S (i.e., P1 and P1’ site). (c,d) HPLC and ESI-MS data confirm that Mpro cleaves the representative D3F2C1 peptide between Q and S. Peaks with * are the intact peptide (blue); the fragments are in red. The specificity constant (kcat/KM) for D3F2C1 substrate by Mpro is 4,178.6 M−1.s−1.
All SLP sequences contain the well-defined Mpro recognition motif, TSAVLQSG, where cleavage occurs between Q and S (Q↓S).[13a, 15] The sequence of amino acids on the sticker tail and polar head may affect the self-organization propensity and subsequent biological responses. Therefore, we synthesized several analogs, where the number of F residues varied or were substituted with other hydrophobic amino acids such as Y, W, and V. The charged DDD stretch is replaced by a neutral SSS or (PAS)2 segment;[16] see peptide library in Figure 1b. For example, Figure 1c shows the HPLC profile of a representative SLP synthesized following the above modular design: DDDTSAVLQ↓SGFFPC (named D3F2C1). The Mpro cleavage of D3F2C1 peptide is confirmed by HPLC and ESI-MS, which proteolytically liberated SGFFPC fragment (Figure 1d). The specificity constant (kcat/KM) for this customized D3F2C1 by Mpro was determined using a synthetic fluorogenic substrate (Cy3-D3F2C1-Cy5.5; See Figure S3) and was 4,178.6 M−1.s−1, which is close to that of the well-defined Mpro substrate (i.e., 4,650.8 M−1.s−1 for TSAVLQ↓SGF). These values agree with a previous work and a slight discrepancy could from the active fraction of protease used (here, 72.6%).[17] Detailed kinetic analysis and assay conditions were provided in Supporting Information, Section 3. The other peptide analog cleavage is also provided in Figure S2. Indeed, such a cleavage at the C-terminal Q is rarely seen for mammalian proteases except for kallikrein-3 that is solely expressed in the prostate.[18] Hence this cleavage site is highly unique to viral proteases.
Figure 2a shows that SLPs (600 μM) copolymerize noncovalently to form supramolecular fibrils in buffer (pH 8.0) after Mpro proteolysis. The intact SLPs were completely soluble without secondary structure formation. A close comparison of the TEM images suggests that the ordered fibrils were mostly formed by FFP-bearing fragments, i.e., the other fragments of Mpro break down and contain one- or zero-Phe with little-to-no aggregation. In particular, the SGFFPC hexapeptide [i.e., proteolytically liberated from D3F2C1, S3F2C1, and (PAS)2F2C1] clearly yielded linear fibrils—typical units from which supramolecular materials can be formed.[14] The long and thin nanofibrils displayed a width of ~5 nm and a length extending a few micrometers, which agree with the fiber thickness of reported FF-formed nanotubes.[2, 19]
Figure 2.
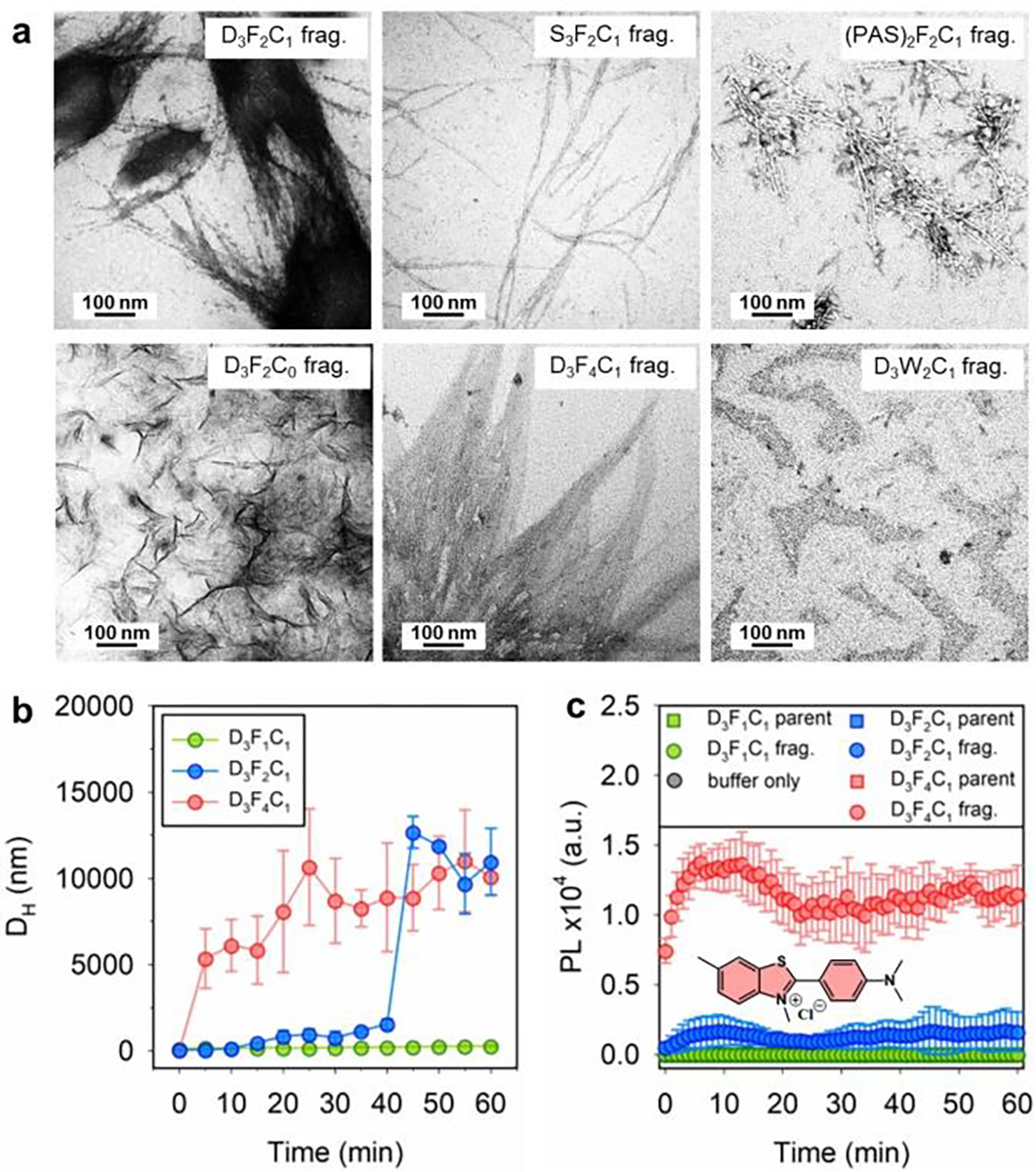
(a) TEM micrographs of the stained SLP fragments prepared by incubating the corresponding intact peptide (600 μM) with Mpro (200 nM) and aging for 48 h. The SGFFPC and SGFFPFFPC form a network of ordered nanofibrils. (b) Hydrodynamic size (DH) change of the D3FnC1 peptide (n = 1, 2, 4, at 600 μM) when incubated with Mpro (200 nM) at 37 °C. The time of initializing macroscopic aggregation in D3F4C1 and D3F2C1 solutions is about 5 and 40 min, respectively. While the D3F1C1 produced no secondary structure under the tested conditions. (c) ThT kinetic experiment showing fluorescence intensity at 485 nm for 50 μM ThT incubated with 300 μM peptides (e.g., D3F1C1, D3F2C1, D3F4C1, and their aged proteolytic products), as recorded over 1 h. The negative control consisted of buffer only. Error bars = standard errors (n = 3). Inset shows the dye structure.
In addition, the versatile network of nanofibrils resulting from the proteolytic D3F2C1, S3F2C1, and (PAS)2F2C1 peptide indicates that the environment or the counter fragments in which the peptide self-assembly takes place may affect the final secondary structure.[1b] For example, the mixture of D3F2C1 fragments showed a hierarchical self-assembly process from the proto-fibers to multi-fibrous alignment bundles (i.e., branched spindle in Figure 2a, top left), which is attributed to the fiber–fiber interactions at the aromatic zipper interfaces.[3, 20] The segments from S3F2C1 and (PAS)2F2C1 peptides had more broken fibril stripes with a tubular structure as seen by the densely-stained parallel sides with a hollow center (i.e., Figure 2a, top middle and right). Interestingly, the SGFFPG hexapeptide lacking a cysteine sulfhydryl (i.e., derived from D3F2C0) produced amorphous sheet-like structures without nanofibrils. This is not unusual: The introduction of a thiol group into the FF unit has been reported to modify the assembles morphology.[14] Notably, increasing the Phe amino acids in the sticker tail (i.e., FFPFFPC from D3F4C1) promoted large peptide aggregates (see Figure 2a, bottom middle). To this end we also tested an SLP bearing the PFF tripeptide initially proposed by Tuttle and Gazit et al. (i.e., named D3f2C1) as opposed to the FFP used in D3F2C1. We found less-to-no Mpro cleavage, thus implying that substitution of Phe with Pro at the P3’ site significantly dysregulates the specificity between SLP/Mpro (Figure S2d).[3, 21] More TEM images in the zoom-out view are provided in Figure S5. The electron microscopy data are further corroborated with molecular dynamics simulation (Figure S6) and optical microscopy data (Figure S7) to elucidate the morphological difference of SLPs before/after proteolysis.
Next, the Mpro-activated assembly kinetics of SLPs (i.e., D3F1C1 vs D3F2C1 vs D3F4C1) were evaluated through hydrodynamic size analysis using DLS. Figure 2b summarizes the time-dependent size profile of each peptide: Both D3F2C1 and D3F4C1 (600 μM, pH 8.0) showed the formation of peptide aggregates while D3F1C1 peptide had no size change indicating that a di-homo FF is required to induce peptide aggregation. A higher number of Phe in SLPs resulted in fast and large aggregation (e.g., 40 min for D3F2C1 and 5 min for D3F4C1).
A previous study reported that the PFF tripeptide exhibits unexpected α-helical intermediates and hierarchically assemblies into a supramolecular structure exerting helical-like amyloid β-sheet.[3] To examine the structural arrangement of the formed peptide fibers we applied a thioflavin-T (ThT) binding assay—an amyloid-specific fluorescent dye.[22] Staining the β-type fibers with ThT resulted in high fluorescence levels, thus establishing their amyloidogenic nature. Figure 2c shows the emission kinetics of ThT-staining on the SGGFPC, SGFFPC, and SGFFPFFPC fibers in the suspension. Except for the SGGFPC solution, the suspension of SGFFPC and SGFFPFFPC yielded an intense and characteristic fluorescence peak at 485 nm in the stable plateau regime, thus resembling a characteristic kinetic profile of amyloid aggregates. Control experiments used the corresponding intact SLPs in buffer and showed no fluorescence enhancement.
The modular SLPs were next used to modulate the colloidal dispersity and thus sample color to measure Mpro. Recent studies by our group and others have illustrated that the nanoparticle surface ligand is important during such experiments.[5b, 23] Typically, compact and labile capping ligands (e.g., derivatives of carboxylate, hydroxyl, and phosphine[5b]) cause AuNPs to undergo rapid and intense color changes during assays. Thus, we used citrate-AuNPs by the Turkevich method and diphenylphosphinobenzene-3-sulfonate (DPPS)-modified AuNPs as the color indicators (TEM size = 13.1 ± 1.3 nm). More characterizations of ligand exchange and colloidal gold such as TEM, DLS, UV/Vis, and FT-IR are provided in Figures S9,10.
We first validated the colorimetric assay by incubating citrate-AuNPs with a representative D3F2C1 SLP and its pre-cleaved fragments (e.g., SGFFPC). Then, the sensing capability of a series of D3F2C1 peptide analogs was compared in colorimetric assays, which allowed us to investigate the effect of mutating peptide modules and therefore the altered SLP scaffold’s properties on co-assembly with colloidal gold. Figure 3a shows the time-lapsed progression of absorption profiles of a SLP/AuNP mixture. The nanoparticles remained stable with intact D3F2C1 (cSLP = 1.5 μM), but D3F2C1 fragments aggregated AuNPs in 30 min and led to a pronounced bathochromic shift in the SPR peak from 520 nm to 600 nm. We defined a ratiometric signal, Abs600/Abs520, to quantify the aggregation level and color change (Figure 3b,c). This color change increases with SLP concentration and incubation time. TEM (Figures 3d,e and S10), DLS (Figure 3f) and multispectral advanced nanoparticle tracking analysis (MANTA,[22b, 24] Figure 3g,h) further confirm the formation of nanoparticle aggregates in the presence of D3F2C1 fragments.
Figure 3.
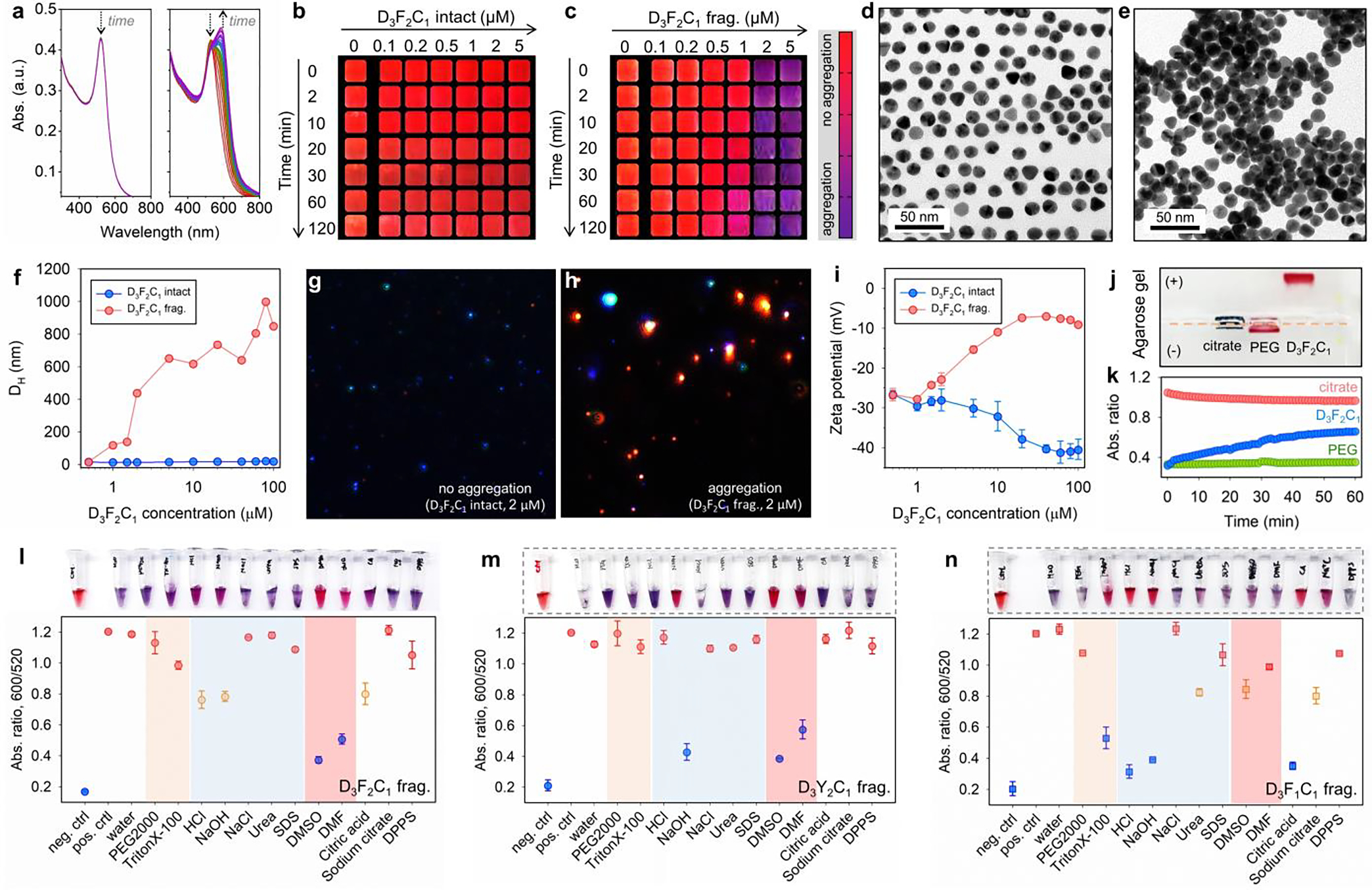
Mpro-induced color change using the modular peptide amphiphiles and colloidal gold (citrate). (a) The time progression of optical absorption of AuNPs (3.4 nM, 100 μL) when incubated with D3F2C1 intact (left) and its fragments (right, c = 1.5 μM). The curves were recorded every 1 min for 30 min. Arrows designate sizable optical changes at 520 and 600 nm. (b-c) The concentration- and time-dependent color evolution of AuNPs (3.4 nM, 100 μL) in the presence of intact D3F2C1 and its pre-cleaved fragments. These are cropped images with a color bar where purple represents particle flocculation. See also TEM images of AuNPs (3.4 nM, 8 μL) when mixed with D3F2C1 intact (d, monolayer) and its fragments (e, heterogeneous stacking). (f) DLS profiles of AuNPs (3.4 nM, 100 μL) incubated with D3F2C1 intact (blue) and its fragments (red) of 0 – 100 μM. (g-h) View of MANTA[22b] size measurement shows that AuNPs (c = 0.2 – 0.6 nM) scatter blue light with D3F2C1 intact (2.0 μM), whereas the fragment-induced colloidal aggregates scatter red light. (i) Zeta potential of AuNPs (3.4 nM, 100 μL) when incubated with increasing concentrations of D3F2C1 intact (blue, reduced from −26.0 to −40.4 mV) and its fragments (red, increased from −26.6 to −9.0 mV). Error bars represent triplicate measurements for one sample. (j) The agarose gel (0.7% w/v) electrophoresis image collected from citrate-AuNPs only, AuNPs incubated with HS-PEG2k-OCH3, and intact D3F2C1 (from left to right). Samples were prepared using the AuNPs (~15 nM, 40 μL) mixed with glycerol (10 μL). Note that TBE buffer (1×) promotes instant aggregation of citrate-AuNPs. (k) Ca2+ cation (20 mM)-modulated dispersity of citrate (red), PEGylated (green), and D3F2C1-capped AuNPs (blue). The colloidal dispersity is quantified by ratiometric signal, i.e., Abs600/Abs520. The DDD stretch negatively charges the surfaces and promotes colloidal stability via electrostatic double repulsion. (l-n) White-light image (top) and quantified reversal color change (bottom) of the gold pellet in different surfactant solutions (10 mM, 100 μL) or solvents (100 μL). Panel l indicates an aromatic stacking-driven co-assembly of D3F2C1 fragments and AuNPs. The gold pellet is prepared by aggregating AuNPs (3.4 nM, 100 μL) with D3F2C1/D3Y2C1/D3F1C1 fragments (at 600 μM, 30 μL). Error bars = standard deviations (n = 3).
The size increases commensurate with the surface potential shifting from −26.6 to −9.0 mV (Figure 3i), presumably due to the cysteine sulfhydryl displacing the native citrate anions. The intact D3F2C1 SLP did not produce a size change. Nonetheless, the surface potential of AuNPs mixed with intact D3F2C1 decreased by −14.4 mV due to the triple acidic aspartate from the coordinated peptide. Agarose gel imaging shows that the AuNPs incubated with intact D3F2C1 migrated toward the anode, thus confirming the presence of carboxylates from the chemisorbed peptide via Au-S bond (referred to as D3F2C1-AuNP, Figure 3j). We further titrated the D3F2C1-AuNPs with 20 mM of CaCl2 where the Ca2+ can electrostatically crosslink the negatively charged colloids following the Schulze-Hardy rule (Figure 3k).[6a, 6b] Under the same conditions, this cation additive had no effect on the neutral PEGylated nanoparticles (i.e., control). These combined findings suggest that the inclusion of charged DDD residues in a modular SLP can stabilize colloids via electrostatic repulsion, thus retaining red color upon addition of intact SLPs.
The aromatic interaction-guided peptide/AuNP co-assembly was further probed with reversibility experiments employing several different solvents and surfactants (10 mM). In Figure 3l, the gold pellet (3.4 nM, 100 μL) clustered by SGFFPC (600 μM, 30 μL) readily disassociated and returned to a red color in organic solvents including dimethyl sulfoxide (DMSO) and dimethylformamide (DMF). This suggests an aromatic stacking-driven co-assembly.[25] In addition, shifting the pH to either 3 or 11 (e.g., by 10 mM HCl/NaOH) partially recovered the colloidal gold (~40% efficacy). This is likely due to: (i) a high concentration of H+/OH− competes with NH···O=C hydrogen-bonds in the polymerized peptide aggregates, and (ii) H+ protonates the N-terminal amino at low pH or OH− deprotonates the native citrate residues at high pH, thus restoring interparticle electrostatic repulsions.[26] A similar trend was observed for the gold aggregates prepared by SGYYPC (600 μM, 30 μL) under the same testing conditions (Figure 3m). Likewise, the use of HCl/citric acid/NaOH additives disassembled the co-aggregates of SGGFPC/AuNPs with a high color recovery (~80% efficacy, Figure 3n). Despite this, SGGFPC/AuNPs co-assemblies redispersed in non-ionic Triton X-100 detergent rather than DMSO or DMF, indicating a large fraction of hydrophobic interactions with scarce π-stacking. This also agrees with the notion that a di-homo FF is the key block for promoting aromatic stacking and directional growth, whereas SGGFPC only gave rise to the amorphous structure without secondary arrangement under TEM.[3] Note that in all the above scenarios, the addition of SDS surfactant cannot recover any of those co-assemblies implying a lack of electrostatic interactions.[6b, 24]
We then determined the dynamic range for Mpro detection based on the proposed SLP/AuNP system. We limited our experiments and descriptions using DPPS-AuNPs for the sake of improved colloidal stability and sensing performance, however, data on citrate-AuNPs are also provided in Figures S13,14. Experimentally, the dynamic range is extracted by recording the optical signals (Abs600/Abs520 at 3 h) after addition of DPPS-AuNPs (3.4 nM, 100 μL) to each SLPs and its corresponding fragments of varying concentrations (i.e., 0 – 100 μM). As shown in Figure 4a,b, the pre-digested D3F0C1 and D3F1C1 fragments yielded no co-assembly with DPPS-AuNPs under tested conditions due to the weak-or-no aromatic interaction. The D3F2C1 and D3F4C1 fragments induced a noticeable color change in buffer and sizeable optical signals starting from 36.6 and 16.5 μM, respectively (Figure 4c,d). Notably, the curves obtained from the intact and fragmented D3F2C0 lacked thiol groups and were inseparable, thus revealing that a di-homo FF element cannot solely bridge aromatic-rich ligands (e.g., DPPS and tannic acid) via π-stacking (Figure 4e); this is likely uncoupled by the strong hydration layer created by the ligand shell. Therefore, inclusion of a cystine sulfhydryl in the sticker domain is a key consideration for tight-locking aromatic interactions at bio-nano interfaces.[5b, 25b]
Figure 4.
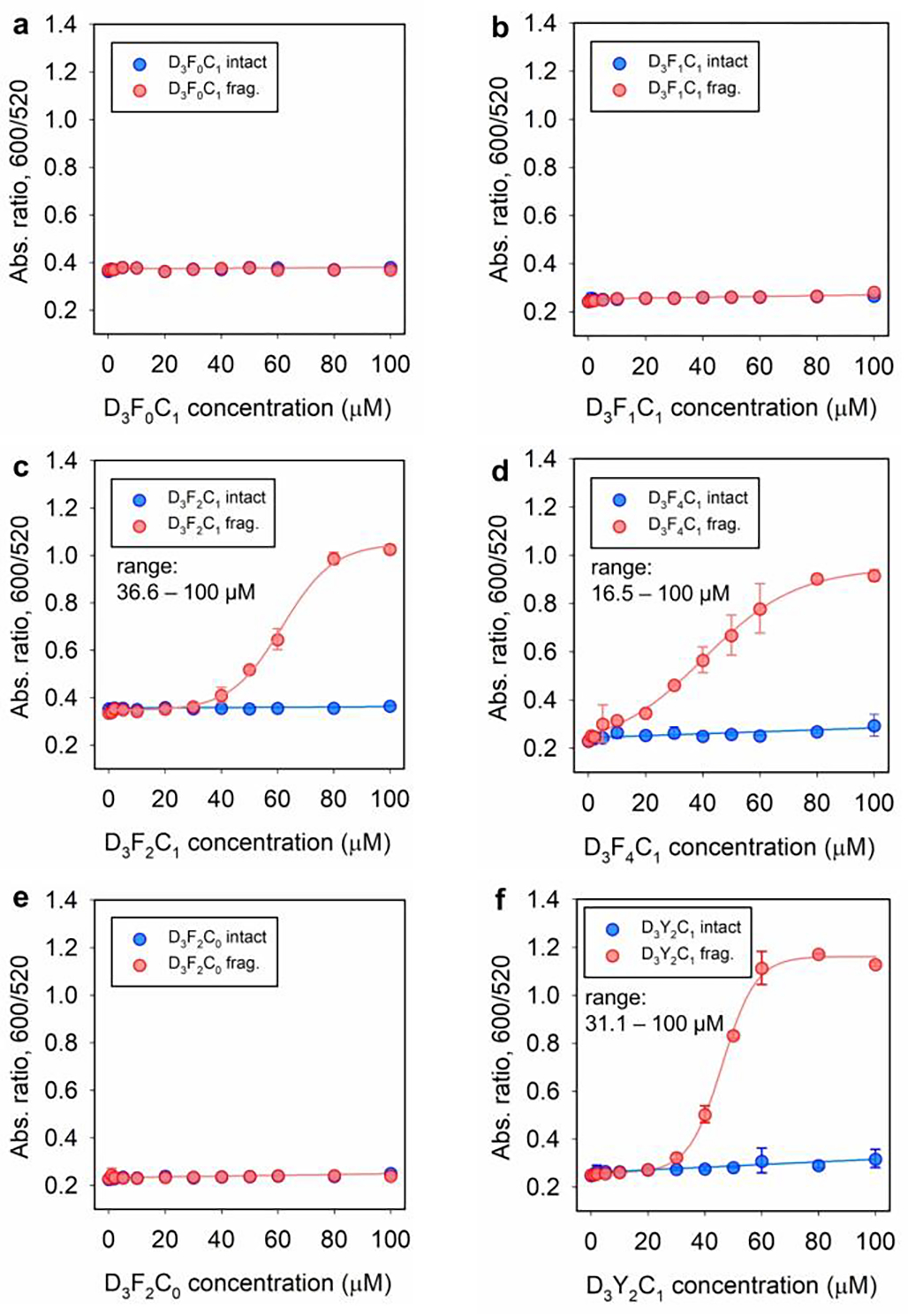
Dynamic range of peptide. Ratiometric signal (i.e., Abs600/Abs520 at 3 h readout time) recorded from DPPS-AuNPs (2.8 nM, 120 μL) incubated with various amount of D3F0C1 intact (a), D3F1C1 intact (b), D3F2C1 intact (c), D3F4C1 intact (d), D3F2C0 intact (e), D3Y2C1 intact (f), and their corresponding proteolytic fragments (red). The charged polar head, di-homo aromatic amino acid, and cysteine are indispensable modules for peptide amphiphiles to colorimetrically measure protease. The determined lower limit of dynamic range for D3F2C1, D3F4C1, and D3Y2C1 SLP is 36.6, 16.5, and 31.1 μM, respectively. Error bars = standard deviations (n = 2).
Furthermore, we found that substitution of FF with other hydrophobic residues (i.e., Y, W, and V; that is, D3Y2C1, D3W2C1, and D3V2C1) also results in SLPs with particle co-assembly capability after proteolysis, albeit at different aggregation levels. For instance, the dynamic range for D3Y2C1 and D3W2C1 is 31.1 – 100 and 30.6 – 100 μM, respectively (see Figures 4f and S15,16), which are comparable to that of D3F2C1 peptide with the highest aggregation reached. The D3V2C1 fragments imposed a slow aggregation on DPPS-AuNPs and thus there were only mild color changes (Figure S15h). We then ranked the propensity of these aggregating moieties in the order of: FFPFFP > FFP ≈ YYP ≈ WWP > VVP >> GFP. Interestingly, despite the exquisite self-assembled structures by neutral SLPs [e.g., from S3F2C1 and (PAS)2F2C1, Figure 2a], these peptides exhibit stabilizing deficiencies for nanoparticles regardless of Mpro addition (Figure S13). We thus conclude that the steric hindrance provided by the aprotic polar SSS or PASPAS head cannot compensate for the sticker domain of aromatic aggregation, thus failing to preserve good colloidal stability. In practice, the proteolysis of D3F4C1 produced macroscopic aggregation and subsequently led to an opaque gel that impeded proteolysis and particle aggregation. Therefore, we chose a combination of D3F2C1 peptide (at 100 μM) and DPPS-AuNPs (3.4 nM, 100 μL) for the next Mpro sensing.
Release of the viral protease to the respiratory fluids such as saliva (upper airway) and exhaled breath (lower airway) could potentially occur in SARS-CoV-2-infected patients.[27] This motivated us to study the limit of detection (LoD) of Mpro in biological milieu including phosphate buffer (PB, 10 mM, pH 8.0), exhaled breath condensate (EBC), saliva (50% dilution), and human plasma (50% dilution). Note that DPPS-AuNP itself was stable in all tested media (Figure S17). The stepwise assay was performed by first incubating the D3F2C1 peptide (100 μM) with various concentrations (i.e., 0 – 100 nM) of Mpro doped in the above media in a 40 μL volume for 3 h. Then, the DPPS-AuNPs (2.4 nM) were added as a readout at 3 h. We defined 6 h as the total assay time, but a continuous measurement could also be conducted with improved detection limits. The overall dithiothreitol (DTT) level in the assay was maintained at ~2 μM for Mpro stabilization. All concentrations are with respect to 120 μL volume; more details are provided in Experimental Section.
Figure 5a plots the ratiometric signal (Abs600/Abs520) against Mpro concentration using DPPS-AuNPs. High concentrations of Mpro activated more particle aggregation and rapid color change and vice versa. Accordingly, the LoD for Mpro was determined to be 25.5 nM in phosphate buffer, 25.3 nM in the EBC matrix, and 68.3 nM in saliva. These values decreased to 15.7 nM in phosphate buffer, 20.8 nM in the EBC matrix, and 26.1 nM in saliva when using citrate-AuNPs as the color reporter (Figure 5b). Notably, the turn-on signals in saliva indicates that co-assembly of SLP/AuNP is not affected by any charge scavengers. Nonetheless, no color change was recorded in human plasma regardless of the type of colloidal gold used.[5b, 6b] The clinically relevant level of Mpro in respiratory droplets from COVID-positive patients is still unclear, but the Mpro LoDs of our system reach low nanomolar concentrations that are similar to other reported fluorogenic probes yet with a simple visual readout.[15, 28] Transformation of the above aromatic-driven sensing systems to a lateral flow device requires stringent supporting substrates as many aromatic-rich materials (e.g., polyester fibers) would dysregulate the co-assembly process.[29] Consecutively, we also confirmed that the enzymatic SLPs cleavage and colloidal aggregation could not take place in situ; this is presumably because the surface-docked SLP lacks a spacer domain. Thus, proteolysis is sterically prevented at the bio-nano interface (Figure S17e).[30]
Figure 5.
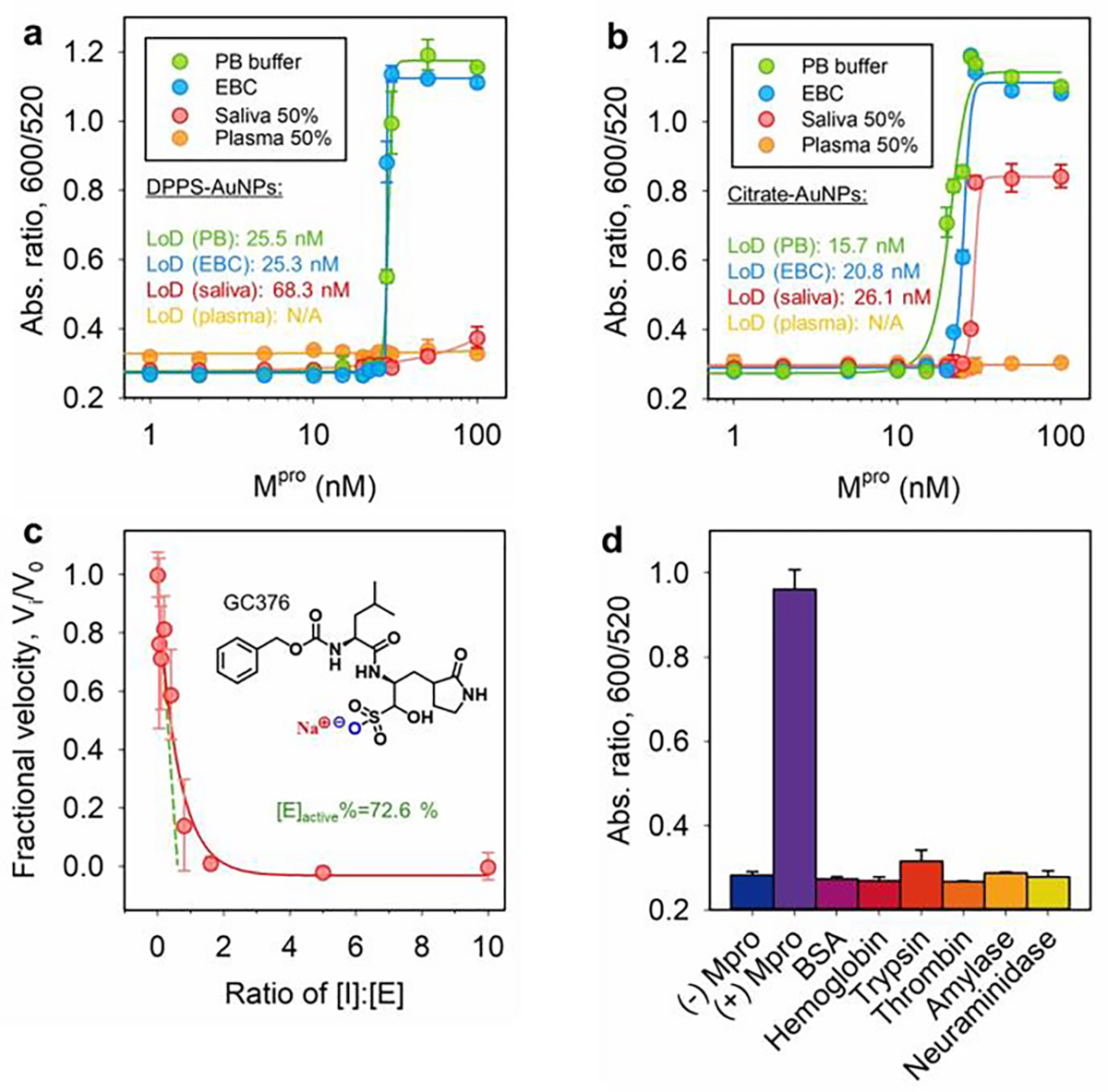
Sensitivity and specificity test. Ratiometric absorbance as a function of Mpro concentration: The D3F2C1 substrate (100 μM), DPPS-AuNPs (2.4 nM), and a 6 h assay time are employed in (a), while citrate-AuNPs (2.4 nM) and a 3 h 10 min assay time are used in (b). The LoDs are shown and the linear region is provided in Figure S17.[31] Error bar = standard deviation (n = 2). (c) Inhibition curve collected by titrating Mpro (50 nM) with varying amount of GC376 in the presence of D3F2C1 substrate (100 μM): % active Mpro = [I]/[E] at the x-intercept (see dash green line, 72.6%).[32] Inset is the chemical structure of inhibitor GC376. Error bar = standard deviation (n = 2). (d) Sensor performance interfered by other mammalian proteins (50 nM), including bovine serum albumin (BSA), hemoglobin, trypsin, thrombin, α-amylase (50 U/mL), and neuraminidase (5 U/mL). Samples with and without Mpro were the controls.
Lastly, a competitive inhibitor (GC376) for Mpro was used to gain insights into the enzymatic role of the protease in colorimetric assays. Mpro (50 nM) was incubated with an increasing molarity of GC376 (i.e., 0 – 500 nM) for 10 min prior to mixing with D3F2C1 substrate (100 μM) for another 30 min. As expected, following addition of DPPS-AuNPs the aggregation kinetic was slowed down due to the inactivation of Mpro-GC376 complexes. Examination of the ratiometric signals at 3 h yields a typical inhibitor titration curve, suggesting a positive correlation between colloidal aggregation and protease activity (Figure 5c). The inhibitor itself does not affect the colloidal dispersity. In addition, the extrapolated line in Figure 5c indicates that the amount of active Mpro was 36.3 nM (or 72.6% active of the stock assuming a tight-binding kinetic).[32] We further cross-tested the effect of several related proteins on our sensing system such as bovine serum albumin (BSA), hemoglobin, trypsin (i.e., cleaves at the C-terminus of Arg or Lys), thrombin (i.e., cleaves the Arg–Gly bond in fibrinogen), salivary α-amylase (i.e., digests α-1,4-glucosidic bonds in starch[29]), and viral neuraminidase (i.e., high levels in influenza virus[33]). Figure 5d shows that only the positive control (i.e., 50 nM Mpro) yielded a prominent optical signal due to the liberated SGFFPC sticker. There was limited signal activation from other off-target mammalian proteins (e.g., BSA and hemoglobin) and enzymes (e.g., α-amylase, thrombin, trypsin, neuraminidase). This highlights the remarkable specificity of our sensor to the SARS-CoV-2 protease, which could be used to non-invasively monitor a wide range of clinical-relevant samples in dynamic conditions.
Conclusion
In summary, a peptide amphiphile of universal formula, DDD–(ZZ)x–FFPC, was established to modulate the colloidal dispersity of AuNPs via aromatic-aromatic interactions. This peptide used only natural occurring amino acids with fine-tuned amphiphilicity, which showed dramatic discrepancy in aggregation propensity in the presence of target protease (here, SARS-CoV-2 Mpro). Several surfactant-like peptide derivatives with altered modular domains (e.g., different polar heads and nonpolar stickers) were synthesized and compared in terms of their value in colorimetric sensing. In particular, we found that the intact peptides bearing protic DDD domain stabilize the best for the pristine or DPPS-AuNPs through electrostatic repulsions, comparing to that of the aprotic polar heads such as SSS and PAS. Proteolysis of the intact SLP yields a highly aggregative fragment that co-assembles AuNPs and changes the dispersion color. We ranked the aggregation propensity of the sticker domain in the order of FFPFFP > FFP ≈ YYP ≈ WWP > VVP >> GFP. By quantifying the color with a measurable absorbance ratio (Abs600/Abs520), the sensor involving D3F2C1 peptide showed good performance in complex media including PB, EBC, and saliva with an LoD of 15.7, 20.8, and 26.1 nM, respectively. Inhibitor assays confirmed the critical role of Mpro in mediating the color changes. We also cross-tested the responsiveness of the SLP/AuNP sensing system to other related proteins and enzymes and found no nonspecific and off-site activation. This SLP/AuNP-based sensor does not require bioconjugations or sophisticated instrumentations, and hence offers easy integration into qualitative diagnostic kits.
Supplementary Material
Acknowledgements
The authors thank internal funding from the UC Office of the President (R00RG2515) and the National Institutes of Health (R01 DE031114; R21 AG065776-S1; R21 AI157957) for financial support. This work was also supported by National Science Foundation (DMR-2011924) via equipment in the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC). M.N.C. was supported by NIT under T32 CA15391. M.R. acknowledges the Wallonie-Bruxelles International (WBI) of the Fédération Wallonie-Bruxelles for financial support. The electron microscopy work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of University of California San Diego, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-1542148). The MANTA analysis work was supported by the National Institutes of Health (S10 OD023555). The computational work for this article was fully carried out on National Supercomputing Centre, Singapore (https://www.nscc.sg).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Gazit E, Chem. Soc. Rev. 2007, 36, 1263–1269; [DOI] [PubMed] [Google Scholar]; b) Levin A, Hakala TA, Schnaider L, Bernardes GJL, Gazit E, Knowles TPJ, Nat. Rev. Chem. 2020, 4, 615–634; [Google Scholar]; c) Lampel A, Chem 2020, 6, 1222–1236. [Google Scholar]
- [2].Sheehan F, Sementa D, Jain A, Kumar M, Tayarani-Najjaran M, Kroiss D, Ulijn RV, Chem. Rev. 2021, 121, 13869–13914. [DOI] [PubMed] [Google Scholar]
- [3].Bera S, Mondal S, Xue B, Shimon LJW, Cao Y, Gazit E, Nat. Mater. 2019, 18, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Saha K, Agasti SS, Kim C, Li X, Rotello VM, Chem. Rev. 2012, 112, 2739–2779; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nowinski AK, Sun F, White AD, Keefe AJ, Jiang S, J. Am. Chem. Soc. 2012, 134, 6000–6005; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xie X, Xu W, Liu X, Acc. Chem. Res. 2012, 45, 1511–1520; [DOI] [PubMed] [Google Scholar]; d) Huang RH, Nayeem N, He Y, Morales J, Graham D, Klajn R, Contel M, O’Brien S, Ulijn RV, Adv. Mater. 2022, 34, 2104962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Ruan S, Xiao W, Hu C, Zhang H, Rao J, Wang S, Wang X, He Q, Gao H, ACS Appl. Mater. Interfaces 2017, 9, 20348–20360; [DOI] [PubMed] [Google Scholar]; b) Jin Z, Yeung J, Zhou J, Cheng Y, Li Y, Mantri Y, He T, Yim W, Xu M, Wu Z, Fajtova P, Creyer MN, Moore C, Fu L, Penny WF, O’Donoghue AJ, Jokerst JV, Chem. Mater. 2022, 34, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Bian T, Gardin A, Gemen J, Houben L, Perego C, Lee B, Elad N, Chu Z, Pavan GM, Klajn R, Nat. Chem. 2021, 13, 940–949; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jin Z, Mantri Y, Retout M, Cheng Y, Zhou J, Jorns A, Fajtova P, Yim W, Moore C, Xu M, Creyer MN, Borum RM, Zhou J, Wu Z, He T, Penny WF, O’Donoghue AJ, Jokerst JV, Angew. Chem. Int. Ed. 2022, 61, e202112995; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Feng Y, Liu G, La M, Liu L, Molecules 2022, 27, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ, Nature 1996, 382, 607–609; [DOI] [PubMed] [Google Scholar]; b) Stevens MM, Flynn NT, Wang C, Tirrell DA, Langer R, Adv. Mater. 2004, 16, 915–918; [Google Scholar]; c) Yang K, Liu Y, Wang Y, Ren Q, Guo H, Matson JB, Chen X, Nie Z, Biomaterials 2019, 223, 119460; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhou W, Hu K, Kwee S, Tang L, Wang Z, Xia J, Li X, Anal. Chem. 2020, 92, 2739–2747. [DOI] [PubMed] [Google Scholar]
- [8].Laromaine A, Koh L, Murugesan M, Ulijn RV, Stevens MM, J. Am. Chem. Soc. 2007, 129, 4156–4157. [DOI] [PubMed] [Google Scholar]
- [9].a) Gupta S, Andresen H, Ghadiali JE, Stevens MM, Small 2010, 6, 1509–1513; [DOI] [PubMed] [Google Scholar]; b) Piao JY, Chung DS, Analyst 2012, 137, 2669–2673; [DOI] [PubMed] [Google Scholar]; c) Liu P, Yang X, Sun S, Wang Q, Wang K, Huang J, Liu J, He L, Anal. Chem. 2013, 85, 7689–7695. [DOI] [PubMed] [Google Scholar]
- [10].a) Guarise C, Pasquato L, De Filippis V, Scrimin P, Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 3978–3982; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pan Y, Guo M, Nie Z, Huang Y, Peng Y, Liu A, Qing M, Yao S, Chem. Commun. 2012, 48, 997–999; [DOI] [PubMed] [Google Scholar]; c) Chen P, Selegård R, Aili D, Liedberg B, Nanoscale 2013, 5, 8973–8976; [DOI] [PubMed] [Google Scholar]; d) Liu X, Zhang Q, Knoll W, Liedberg B, Wang Y, Adv. Mater. 2020, 32, 2000866; [DOI] [PubMed] [Google Scholar]; e) Creyer MN, Jin Z, Moore C, Yim W, Zhou J, Jokerst JV, ACS Appl. Mater. Interfaces 2021, 13, 45236–45243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Choi Y, Ho N-H, Tung C-H, Angew. Chem. Int. Ed. 2007, 46, 707–709. [DOI] [PubMed] [Google Scholar]
- [12].a) Ma M, Kuang Y, Gao Y, Zhang Y, Gao P, Xu B, J. Am. Chem. Soc. 2010, 132, 2719–2728; [DOI] [PubMed] [Google Scholar]; b) Song Z, Han Z, Lv S, Chen C, Chen L, Yin L, Cheng J, Chem. Soc. Rev. 2017, 46, 6570–6599; [DOI] [PubMed] [Google Scholar]; c) Feng Z, Wang H, Xu B, J. Am. Chem. Soc. 2018, 140, 16433–16437; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hu Y, Miao Y, Zhang J, Chen Y, Qiu L, Lin J, Ye D, Nano Lett. 2021, 21, 10377–10385; [DOI] [PubMed] [Google Scholar]; e) Wang C, Du W, Wu C, Dan S, Sun M, Zhang T, Wang B, Yuan Y, Liang G, Angew. Chem. Int. Ed. 2022, 61, e202114766. [DOI] [PubMed] [Google Scholar]
- [13].a) Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Yang H, Nature 2020, 582, 289–293; [DOI] [PubMed] [Google Scholar]; b) Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, Boras B, Cardin RD, Carlo A, Zhu Y, Science 2021, 374, 1586–1593. [DOI] [PubMed] [Google Scholar]
- [14].Reches M, Gazit E, Nano Lett. 2004, 4, 581–585. [Google Scholar]
- [15].Cheng Y, Borum RM, Clark AE, Jin Z, Moore C, Fajtová P, O’Donoghue AJ, Carlin AF, Jokerst JV, Angew. Chem. Int. Ed. 2022, 61, e202113617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Abbas M, Lipiński WP, Nakashima KK, Huck WTS, Spruijt E, Nat. Chem. 2021, 13, 1046–1054; [DOI] [PubMed] [Google Scholar]; b) Brito A, Dave D, Lampel A, Castro VIB, Kroiss D, Reis RL, Tuttle T, Ulijn RV, Pires RA, Pashkuleva I, J. Am. Chem. Soc. 2021, 143, 19703–19710; [DOI] [PubMed] [Google Scholar]; c) Zhang L, Li Y, Mu G, Yang L, Ren C, Wang Z, Guo Q, Liu J, Yang C, Anal. Chem. 2022, 94, 2236–2243; [DOI] [PubMed] [Google Scholar]; d) Yu H, Palazzolo JS, Zhou J, Hu Y, Niego B. e., Pan S, Ju Y, Wang T-Y, Lin Z, Hagemeyer CE, Caruso F, ACS Appl. Mater. Interfaces 2022, 14, 3740–3751. [DOI] [PubMed] [Google Scholar]
- [17].Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R, Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vol. 2022, MEROPS database.
- [19].Ma X, Zhao Y, He C, Zhou X, Qi H, Wang Y, Chen C, Wang D, Li J, Ke Y, Wang J, Xu H, Nano Lett. 2021, 21, 10199–10207. [DOI] [PubMed] [Google Scholar]
- [20].Wu B, Zhao S, Yang X, Zhou L, Ma Y, Zhang H, Li W, Wang H, ACS Nano 2022, 16, 4126–4138. [DOI] [PubMed] [Google Scholar]
- [21].a) Shan YF, Xu GJ, Acta. Biochim. Biophys. Sin. 2005, 37, 807–813; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Frederix PWJM, Scott GG, Abul-Haija YM, Kalafatovic D, Pappas CG, Javid N, Hunt NT, Ulijn RV, Tuttle T, Nat. Chem. 2015, 7, 30–37. [DOI] [PubMed] [Google Scholar]
- [22].a) Xue C, Lin TY, Chang D, Guo Z, Soc R. Open Sci. 2017, 4, 160696; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moore C, Wing R, Pham T, Jokerst JV, Anal. Chem. 2020, 92, 11590–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Chen P, Liu X, Goyal G, Tran NT, Shing Ho JC, Wang Y, Aili D, Liedberg B, Anal. Chem. 2018, 90, 4916–4924; [DOI] [PubMed] [Google Scholar]; b) Jin Z, Sugiyama Y, Zhang C, Palui G, Xin Y, Du L, Wang S, Dridi N, Mattoussi H, Chem. Mater. 2020, 32, 7469–7483. [Google Scholar]
- [24].Retout M, Mantri Y, Jin Z, Zhou J, Noël G, Donovan B, Yim W, Jokerst JV, ACS Nano 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Zhou J, Lin Z, Penna M, Pan S, Ju Y, Li S, Han Y, Chen J, Lin G, Richardson JJ, Yarovsky I, Caruso F, Nat. Commun. 2020, 11, 4804; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou J, Creyer MN, Chen A, Yim W, Lafleur RPM, He T, Lin Z, Xu M, Abbasi P, Wu J, Pascal TA, Caruso F, Jokerst JV, J. Am. Chem. Soc. 2021, 143, 12138–12144; [DOI] [PubMed] [Google Scholar]; c) Yim W, Takemura K, Zhou J, Zhou J, Jin Z, Borum RM, Xu M, Cheng Y, He T, Penny W, Miller BR, Jokerst JV, ACS Nano 2022, 16, 683–693; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wu Z, Zhou J, Nkanga CI, Jin Z, He T, Borum RM, Yim W, Zhou J, Cheng Y, Xu M, Steinmetz NF, Jokerst JV, ACS Appl. Mater. Interfaces 2022, 14, 13692–13702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tang C, Smith AM, Collins RF, Ulijn RV, Saiani A, Langmuir 2009, 25, 9447–9453. [DOI] [PubMed] [Google Scholar]
- [27].Huang N, Pérez P, Kato T, Mikami Y, Okuda K, Gilmore RC, Conde CD, Gasmi B, Stein S, Byrd KM, Nat. Med. 2021, 27, 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Martínez-Fleta P, Alfranca A, González-Álvaro I, Casasnovas JM, Fernández-Soto D, Esteso G, Cáceres-Martell Y, Gardeta S, López-Sanz C, Valés-Gómez M, J. Immunol. Res. 2020, 205, 3130–3140. [DOI] [PubMed] [Google Scholar]
- [29].Jin Z, Jorns A, Yim W, Wing R, Mantri Y, Zhou J, Zhou J, Wu Z, Moore C, Penny WF, Jokerst JV, Anal. Chem. 2021, 93, 11025–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Kwon EJ, Dudani JS, Bhatia SN, Nat. Biomed. Eng. 2017, 1, 0054; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Loynachan CN, Soleimany AP, Dudani JS, Lin Y, Najer A, Bekdemir A, Chen Q, Bhatia SN, Stevens MM, Nat. Nanotechnol. 2019, 14, 883–890; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) [Google Scholar]; Jin Z, Ling C, Li Y, Zhou J, Li K, Yim W, Yeung J, Chang Y-C, He T, Cheng Y, Fajtová P, Retout M, O’Donoghue AJ, Jokerst JV, Nano Lett. 2022. [DOI] [PubMed] [Google Scholar]
- [31].Armbruster DA, Pry T, Clin. Biochem. Rev. 2008, 29 Suppl 1, S49–S52. [PMC free article] [PubMed] [Google Scholar]
- [32].Hurst DR, Schwartz MA, Ghaffari MA, Jin Y, Tschesche H, Fields GB, Sang Q-XA, Biochem. J. 2004, 377, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang X, Dhawane AN, Sweeney J, He Y, Vasireddi M, Iyer SS, Angew. Chem. Int. Ed. 2015, 54, 5929–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.