

Abstract
Congenital disorders of glycosylation (CDG) are a group of heterogeneous inherited metabolic disorders affecting posttranslational protein modification. DDOST-CDG, caused by biallelic pathogenic variants in DDOST which encodes dolichyl-diphospho-oligosaccharide-protein glycosyltransferase, a subunit of N-glycosylation oligosaccharyltransferase (OST) complex, is an ultra-rare condition that has been described in two patients only. The main clinical features in the two reported patients include profound developmental delay, failure to thrive, and hypotonia. In addition, both patients had abnormal transferrin glycosylation. Here we report an 18-year-old male who presented with moderate developmental delay, progressive opsoclonus, myoclonus, ataxia, tremor, and dystonia. Biochemical studies by carbohydrate deficient transferrin analysis showed a type I CDG pattern. Exome sequencing identified compound heterozygous variants in DDOST: a maternally inherited variant, c.1142dupT (p.Leu381Phefs*11), and a paternally inherited variant, c.661T>C (p.Ser221Pro). Plasma N-glycan profiling showed mildly increased small high mannose glycans including Man0–5GlcNAc2, a pattern consistent with what was previously reported in DDOST-CDG or defects in other subunits of OST complex. Western blot analysis on patient’s fibroblasts revealed decreased expression of DDOST and reduced intracellular N-glycosylation, as evident by the biomarkers ICAM-1 and LAMP2. Our study highlights the clinical variability, expands the clinical and biochemical phenotypes, and describes new genotype, which all are essential for diagnosing and managing patients with DDOST-CDG.
Introduction
Congenital disorders of glycosylation (CDG) are a group of clinically and biochemically heterogeneous inherited conditions affecting posttranslational protein modification and causing abnormal glycosylation of several types of macromolecules including glycoproteins, glycolipids, and proteoglycans 1–3. Over 175 genetic disorders have been identified across several glycosylation pathways, and the list of conditions is ever growing with the identification of new genes implicated in the pathogenesis of these disorders, accelerated by the advent of next-generation sequencing 3,4. Although the clinical spectrum is variable, impaired glycosylation of different macromolecules can result in overlapping multisystemic manifestations that often begin in infancy, and may include failure to thrive, developmental delay, neurologic abnormalities, hypoglycemia, hepatocellular injury, protein losing enteropathy, dermatologic manifestations, ocular anomalies, and skeletal anomalies 5–7.
CDG involving N-linked protein glycosylation are the most common CDG types. They are caused by defects in the synthesis of glycans that are based on the common high-mannose core motif, or in their attachment to asparagine residues in protein chains 7–9. N-linked glycosylation is a complex multistep process including three main steps: formation of nucleotide-linked sugars in the cytosol, assembly in the endoplasmic reticulum (ER) and transferring to the nascent proteins, and processing/remodeling of the glycan molecule on glycoproteins in the Golgi apparatus.
Type I CDGs are caused by defects that block glycan assembly before the high-mannose glycan core is formed and transferred for processing and trimming in the Golgi – that encompasses both cytosolic and ER glycan synthesis. Type II CDGs are caused by defects in Golgi processing of glycans. A CDG type I biochemical pattern of carbohydrate-deficient transferrin (CDT) is characterized by a decrease in fully-sialylated transferrin (Tf) glycoforms (i.e., a decrease in tetrasialotransferrin) with a loss of full glycan antenna units at a time, leading to an incrase in disialotransferrin and monosialotransferrin 10. Conversely, a type II pattern is characterized by abnormally sialylated glycan antennas that are not necessarily reduced in abundance, but that contain less terminal sialic acid residues than mature glycan antennas, leading to not only a decrease in tetrasialotransferrin but also an increase of the aberrant glycans trisialotransferrin and monosialotransferrin, as well as a minor increase in pentasialotransferrin. By plasma N-glycan profiling, type I CDG often presents with abnormal high-mannose glycan pattern with increases of small high-mannose glycans from intermediate glycan assembling steps, while truncated complex glycans are often increased in type II CDG 11.
DDOST (MIM 602202) is localized in chromosomal region 1p36.1 and encodes dolichyl-diphosphooligosaccharide-protein glycosyltransferase (DDOST, EC 2.4.1.119). The encoded protein is a 48 kDa subunit of the N-glycosylation oligosaccharyltransferase (OST) complex, which catalyzes the transfer of a high mannose oligosaccharide from a dolichol-linked oligosaccharide donor onto the asparagine acceptor site within an Asn-X-Ser/Thr consensus motif in nascent polypeptide chains across the ER membrane 12,13. To date, only two patients have been reported with a disease attributable to biallelic variants in DDOST 13,14.
Here we describe a third patient with an emerging form of an ultra-rare DDOST-CDG (previously known as CDG-Ir), associated with intellectual disability, progressive opsoclonus, myoclonus, ataxia, tremor, and dystonia. Exome sequencing demonstrated compound heterozygous variants in the DDOST gene, which confirmed to be in trans by parental segregation analysis. Exome results supported by abnormal glycosylation pattern and a fitting clinical phenotype, led to the diagnosis of DDOST-CDG.
Material and Methods
Case Description
The proband is an 18-year-old male of Northern European ancestry who presented with developmental delay/intellectual disability and a movement disorder. He was born at 37 weeks of gestation via C-section to a then 28-year-old mother. His birth weight was 2.8 kg (38%ile) and his birth length was 49.5 cm (70%ile). Pregnancy and perinatal period were unremarkable. Developmental delay was first noted at age 2; the patient began walking at 12–18 months of age, and was toilet trained at 2 years of age. He could copy a line or circle at 5 years, and button or zip clothing at 8 years. He was noted to have a short attention span and required speech therapy. He was noted to have recurrent sinopulmonary infections. He was first brought to the attention of pediatric neurology at the age of 7 years, and was diagnosed with essential tremor, ataxia, and a learning disorder. At the time, his brain magnetic resonance imaging (MRI) was normal. Tremor, ataxia and chorea were progressing and became more noticeable by the age of 12 years when he was evaluated by our facilities. Repeat brain MRI at the age of 12 years was unchanged and electroencephalogram was also normal. His weight was at the 66th percentile, his height was at the 83rd percentile and his occipitofrontal circumference (OFC) was at the 41st percentile. He had deep set eyes, overbite and high foot arches but otherwise was nondysmorphic (Figure 1A, B). Neurological findings include opsoclonus, generalized myoclonus worsened with exertion, generalized dystonia, tremor at rest and action (Supplementary Video), as well as abnormal cerebellar signs and gait.
Figure 1.
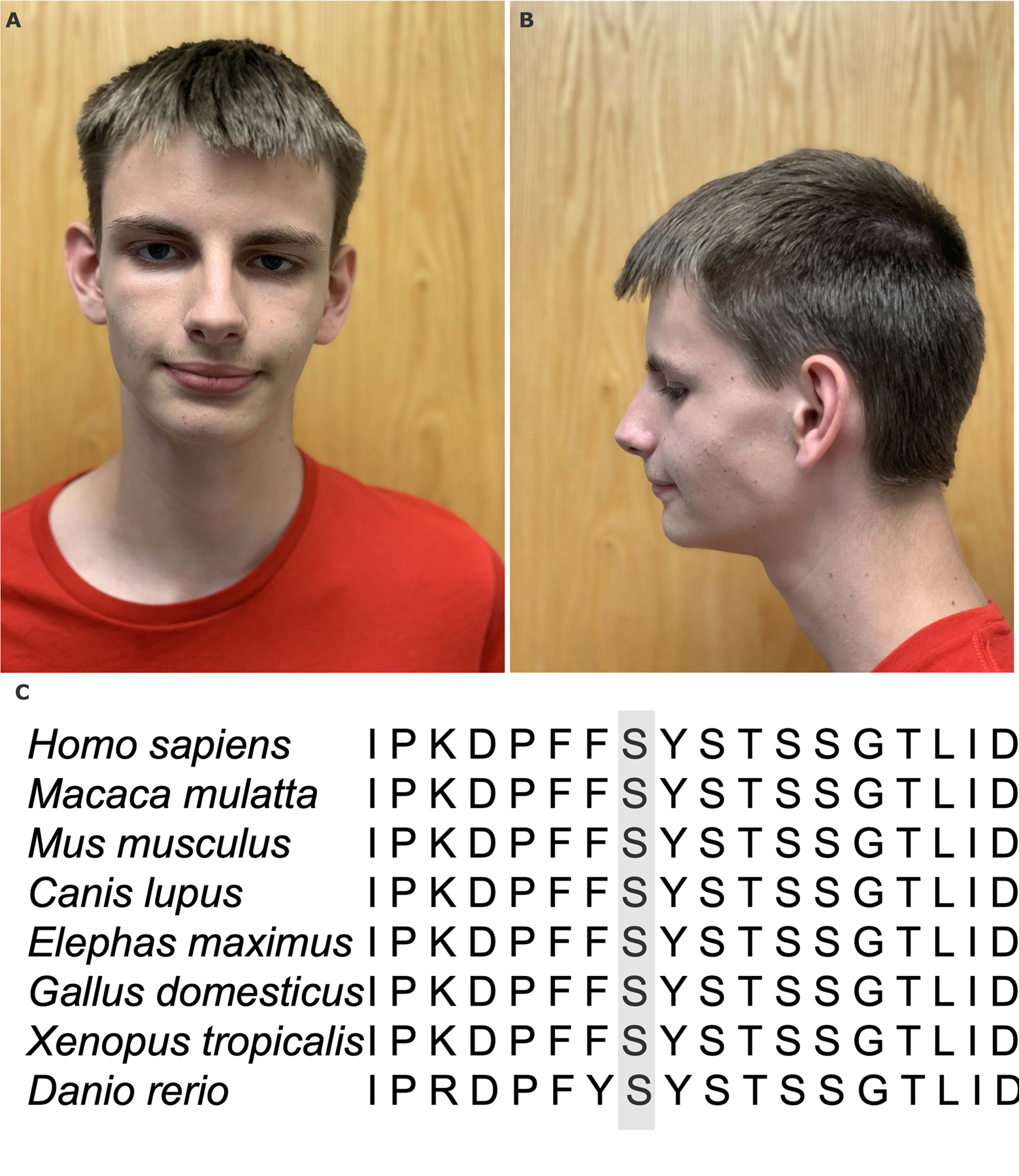
The proband with DDOST-CDG exhibits mild non-specific facial dysmorphism (A). The residue Ser221 is highly conserved among species (B).
The proband was started on gabapentin for tremor but was discontinued due to excessive sleepiness. At his evaluation at 18 years of age, his weight was at the 21st percentile, height was at the 60th percentile and OFC was at the 50th percentile. His neurological examination appeared to be stable without significant progression.
Laboratory workup, including renal and hepatic function panels as well as endocrinologic and hematologic studies, was normal except for decreased antithrombin activity (66%−79%; normal reference 80–125%). Biochemical studies including ceruloplasmin, fractionated metanephrines, lipid profile, free thyroxine, thyroid stimulating hormone, vitamin E, creatine kinase, cerebrospinal fluid neurotransmitter metabolite analysis, tetrahydrobiopterin, neopterin, and 5-methyltetrahydrofolate, acylcarnitine profile, serum amino acids, lactate and pyruvate, ammonia, urine organic acid analysis, urine purine and pyrimidine levels were normal. Serum carbohydrate deficient transferrin performed by Mayo Clinic Laboratories (Rochester, MN) revealed elevated ratios of mono-oligo/di-oligosaccharide, and a-oligo/di-oligosaccharide, suggestive of a diagnosis of type I CDG (Table 1). Subsequent enzyme testing for phosphomannomutase (PMM, EC 5.4.2.8) and mannosephosphate isomerase (MPI, EC 5.3.1.8), as indicated by usual diagnostic algorithms upon detection of a type I CDG on CDT 15, was normal. Chromosomal microarray analysis was negative.
Table 1.
Serum carbohydrate deficient transferrin analysis in the proband
| Component | Reference Range | Proband |
|---|---|---|
| Mono-oligo/di-oligosaccharide ratio | ≤0.06 | 0.65 |
| A-oligo/di-oligosaccharide ratio | ≤0.011 | 0.087 |
| Tri-sialo/di-oligosaccharide ratio | ≤0.05 | 0.04 |
Molecular Analysis
Trio clinical exome sequencing performed by GeneDx (Gaithersburg, MD). The variants were confirmed by Sanger sequencing. Variant annotation and analysis were performed using company’s pipeline. In silico analysis of the effect of missense variants was performed by various bioinformatic tools including Combined Annotation Dependent Depletion (CADD) 16, Rare exome variant ensemble learner (Revel) 17, VARITY, Sorting Intolerant From Tolerant (SIFT) 18, and MutationTaster2 (MT) 19. The variants were classified according to the American College of Medical Genetics and Genomics (ACMG) recommendations 20.
Glycoprotein studies
To evaluate the glycosylation status of secreted glycoproteins, we studied the N-glycan profile of total plasma glycoproteins from the eight individuals using our recently described and clinically validated quantitative N-glycan assay 11. Carbohydrate transferrin testing (CDT) was performed using a previously reported mass spectrometry approach (LC-ESI-TOF/MS) 21.
Western blot analysis
Western blot analysis was performed on whole fibroblast extracts of the proband and controls using a rabbit polyclonal DDOST antibody (D6820 Sigma), a mouse monoclonal anti-ICAM-1/CD54 antibody (G5 Santa Cruz), a mouse monoclonal anti-LAMP2 antibody (DSHB Hybridoma Product H4B4), and a mouse monoclonal GAPDH antibody (MA5–15738 Thermo Fisher) as previously described 13.
Results
Next generation sequencing
Trio exome sequencing identified two novel heterozygous variants in DDOST with trans phase confirmed by parental targeted sequencing. The maternally inherited variant was designated as c.1142dupT (p.Leu381Phefs*11) and the paternally inherited variant was designated c.661T>C (p.Ser221Pro). The variant c.1142dupT is a frameshift change that leads to a the creation of a stop codon after 11 codons; this variant is predicted to cause loss of function through protein truncation, or more likely, nonsense-mediated mRNA decay (NMD). Based on the American College of Medical Genetics, this maternally inherited 1-bp duplication variant was classified as pathogenic (PVS1, PM2 and PP3).
The paternally inherited variant, c.661T>C, leads to a substitution of serine (a small polar amino acid) for proline (a cyclic nonpolar iminoacid), at a residue located in the hinge region between two beta-strands of the transmembrane domain, likely disrupting the angulation of the subsequent beta-strand. This amino acid is conserved from Homo sapiens to Dario rerus (Figure 1C). In silico analysis predicted a deleterious effect on the protein function (Table 2). This variant has not been reported in gnomAD. Given that it was detected in trans with a pathogenic variant and the biochemical phenotype is specific for this disorder, as well as evolutionary, protein structure, and formal in silico analyses, and its absence from controls, this variant was classified as likely pathogenic (PM2, PM3, PP3, PP4). Clinical exome re-analysis after two years did not identify any further diagnostic information or variants.
Table 2.
In silico prediction and ACMG classification of the missense variant
| c.661T>C (p.Ser221Pro) | |
|---|---|
| Revel | Deleterious (low) (0.56) |
| Varity | Deleterious (0.89) |
| SIFT | Damaging (0) |
| MT | Deleterious (1) |
| CADD score | 20.2 |
| gnomAD frequency | Absent |
| ACMG Classification | PM2, PM3, PP3, PP4 |
| Interpretation | Likely pathogenic |
Abnormal plasma N-glycan profiles in DDOST-CDG
We found mild increases of N-linked small high mannose species including Man0–5GlcNAc2 were detected in our patient, along with increased Man4/9, Man5/9 and Man7/9 ratios (Table 3). Similar to what was reported before in a known DDOST-CDG patient, Man4, Man5, Man 4/9 and Man5/9 ratio are increased in our patient 11. In addition, Man0, Man1, Man2, Man3 and Man7/9 ratio are also increased in our patient, suggesting a possible shift in substrate specificity of the OST complex in DDOST-CDG. Unlike other type I CDG that have been previously reported, the ratios between these very small high-mannose glycans, in particular, Man3/4 ratio, is normal.
Table 3.
Plasma N-glycan profiles in DDOST-CDG
| N-Glycan species (% Total glycan) |
Proband | Normal Control | Known DDOST | Reference Range | |
|---|---|---|---|---|---|
| Low | High | ||||
| Man 0 | 0.08 | 0.04 | 0.06 | 0.01 | 0.07 |
| Man 1* | 0.12 | 0.07 | 0.11 | 0.01 | 0.10 |
| Man 2 | 0.25 | 0.13 | 0.28 | 0.00 | 0.24 |
| Man 3 | 0.44 | 0.26 | 0.66 | 0.02 | 0.42 |
| Man 4 | 1.25 | 0.7 | 1.94 | 0.22 | 1.20 |
| Man 5 | 3.18 | 1.91 | 5.15 | 0.81 | 3.13 |
| Man 6 | 2.76 | 2.36 | 4.70 | 0.92 | 3.11 |
| Man 7 | 0.90 | 0.65 | 1.14 | 0.24 | 1.09 |
| Man 8 | 0.93 | 0.73 | 1.05 | 0.35 | 1.33 |
| Man 9 | 0.77 | 0.76 | 1.43 | 0.41 | 1.18 |
| Neu5Ac2Hex5HexNAc4 (Di-sialo) (Disialo biantennary) | 35.62 | 44 | 27.18 | 25.55 | 55.21 |
| Man 3/4 | 0.35 | 0.37 | 0.34 | 0.21 | 0.39 |
| Man 4/9 | 1.62 | 0.92 | 1.36 | 0.44 | 1.35 |
| Man 5/9 | 4.13 | 2.51 | 3.60 | 1.33 | 3.70 |
| Man 7/9 | 1.17 | 0.86 | 0.80 | 0.50 | 1.07 |
N-glycan changes that are shared between proband and previously tested known DDOST case are shown in bold.
Western blot analysis revealed decreased DDOST expression of patient’s fibroblasts
To evaluate the level and activity of DDOST in the proband, we performed Western blot analysis (Figure 2). The proband was found to have significantly decreased expression of DDOST when compared to controls. ICAM-1 levels were also reduced in the proband’s fibroblasts compared to controls. LAMP2 Western blot showed a decrease in this protein’s molecular weight compared to control, with an increased spread of lower-molecular weight positivity.
Figure 2.

Western blot analysis of whole cell extracts from control (GM-00038, GM-05565, GM-09503) and patient (CDG-0462) fibroblasts showing the expression of DDOST, ICAM1, LAMP2 and the loading control GAPDH.
Discussion
OST, a multi-subunit protein complex, is essential part of the glycosylation machinery in the ER and responsible for the en bloc transfer of dolichol-linked oligosaccharides to nascent proteins. Two types of OST complexes were identified in human: OST-A and OST-B 22,23. Both types share six subunits including ribophorin-I, ribophorin-II, DDOST, TMEM258, DAD1, and OST4. OST-A contains the catalytic subunit STT3A in addition to two complex-specific accessory subunits, KRTCAP2 and OSTC, while OST-B contains the catalytic subunit STTB and either TUSC3 or MAGT1 22. Human genetic disorders were so far identified in association with DDOST, STT3A, STT3B, TUSC3, OSTC and MAGT1. Biallelic pathogenic variants in TUSC3 have been associated with non-syndromic moderate-to-severe intellectual disability 24,25, while mutations in DDOST, STT3A, STT3B, and MAGT1 were associated with various types of CDGs 13,14,26–28. STT3A-CDG is inherited in both autosomal dominant and recessive fashions 27,29,30, while MAGT1-CGD is inherited in X-linked recessive manner 26. The patients with OST complex disorders exhibit overlapping but nonspecific clinical pictures including severe developmental delay, intellectual disability, and failure to gain adequate weight.
There have been two previous reports of patients with pathogenic variants in DDOST 13,14 (Table 4). The first patient presented at six months of age with failure to thrive, developmental delay, oromotor dysfunction, recurrent ear infections, and gastroesophageal reflux disease. At one year of age, the patient started exhibiting hypotonia, external strabismus, and mild to moderate liver dysfunction. The second patient exhibited feeding difficulties soon after birth and presented at six months of age for evaluation of developmental delay, hypotonia, failure to thrive, dysmorphic features, strabismus, severe myopia and astigmatism. He also exhibited dysmorphic features. The developmental milestones of previously reported patients appeared more severely affected when compared to the proband in this report. The first patient did not walk until three years of age, while the second patient was able to walk with support at 18 months of age. At five years old, the first patient’s fine motor skills were at the 26-month level and gross motor skills were at the 16-month level. Upon evaluation at four years of age, the second patient’s developmental milestones were at 11 months for gross motor skills, 10 months for fine motor skills, and seven months for language skills. Brain MRI of the first patient demonstrated disordered myelination. Interestingly, the second patient’s brain MRI as well as the proband’s were normal, indicating that a degree of developmental disabilities may not directly correlate with abnormalities of brain imaging. Two episodes of tremor in extremities with no loss of consciousness was reported in the first patient, although it did not appear to be persistent or part of a movement disorder as in the patient in this report, where the predominant neurological phenotype was tremor, ataxia, dystonia, and chorea.
Table 4.
Comparison of clinical characteristics of our patient and previously reported patient
| Characteristic | Patient A (our patient) | Pi et al. | Jones et al. |
|---|---|---|---|
| Age of Onset | 2 years | 6 months | 6 months |
| Genotype | c.1142dupT (p.Leu381Phefs*11) c.661T>C (p.Ser221Pro) |
c.1187G>A (p.Arg396Gln) | c.1265_1286del22 (p.Ile422Thrfs*7) c.650G>A (p.Gly217Asp) |
| Presenting features | Developmental delay age, essential tremor and ataxia | Strabismus, hypotonia, failure to thrive, developmental delay | Failure to thrive, oromotor dysfunction, developmental delay |
| Developmental delay | Moderate | Severe | Severe |
| Hypotonia | - | + | + |
| Brain MRI | Normal | Normal | Hypomyelination |
| Movement disorder | Chorea, episodic tremor, ataxia | - | -* |
| Eye Findings | Strabismus | Strabismus; high myopia; astigmatism |
Strabismus |
| Failure to gain adequate weight | - | + | + |
| Facial dysmorphism | Deep set eyes, overbite | Broad forehead, brow ptosis, depressed nasal tip, thick lip, and low-set cup ears | N/A |
| Coagulation | Decreased anti-thrombin III activity | - | Decreased anti-thrombin II, factor XI, protein C, and protein S |
| Liver dysfunction | - | - | + |
| Recurrent infections | Sinopulmonary | - | Ear |
| Abnormal carbohydrate deficient transferrin | + | + | + |
Patient had two episodes of tremor in extremities at 7 years of age, which apparently resolved
Hyperkinetic movement disorders (HMD) are characterized by excessive, abnormal, and involuntary movement. Five major types of HMD were described: tremor, chorea, dystonia, myoclonus and tics. Various types of HMD has been found to be associated with CDGs. Previous study revealed that approximately 14% (8 out of 56) of patients with CDGs exhibited one or more symptoms of HMD 31. Four out of the 8 patients with HMD had PMM2-CDG and the remainder 4 had one of the following diseases each: ALG-6CDG, ALG8-CDG, COG5-CDG and unknown type of CDG-I. The onset of abnormal movement varies from infantile to adulthood, while its progression is usually slow or stable. Abnormal movements became more noticeable with mental or physical stress, similar to the patient in this report. These data suggest that CDGs should be considered as part of the differential diagnosis in patients presenting with HMD.
It is generally believed that dystonia involves dysfunction of the direct pathway (caudate nucleus, putamen → globus pallidus interna), while chorea involves dysfunction of the indirect pathway (caudate nucleus, putamen → globus pallidus externa → substantia nigra → globus pallidus interna). Both pathways eventually reach the thalamus and then the cortex. The thalamus is an important part of the brain that integrates motor and sensory information and is involved in motor control 32. Lesions of the thalamus have been reported to cause various involuntary movement disorders, including dystonia, athetosis, chorea, and action tremor 33. Although this case did not show any structural lesions on MRI, it is possible that microstructural or functional changes have occurred and caused symptoms associated with dystonia and chorea 34. It would be beneficial if more precise brain imaging studies could be performed in the future to investigate possible microstructural changes in CDG patients with HMD.
As in our patient, serum transferrin analysis demonstrated a pattern consistent with type 1 glycosylation defect in which both monoglycosylated and aglycosylated transferrin glycoforms were markedly elevated. Jones et al. also performed functional studies on patient’s fibroblasts using a combination of immunoblot analysis and markers of glycosylation and confirmed protein hypoglycosylation 13. Both our patient and the previously reported patient also had evidence of some degree of reduction of antithrombin III activity 13. This is unsurprising as many pro- and anti- coagulant proteins are glycosylated, and glycosylation defects are expected to affect protein stability and function. Altered levels of these proteins are observed in the plasma of individuals with CDG 35–38. In contrast to our patient, the previously reported patient was also found to have reduced factor XI, protein C, and protein S, as well as evidence of liver dysfunction, none of which were seen in our patient. The liver dysfunction may have contributed to the more severe abnormalities of the coagulation profile observed in the previously reported patient. It is also possible that broad clinical differences in patients’ phenotypes can be explained by hitherto unknown genotype-phenotype associations, or genetic and non-genetic modifiers.
The first reported patient with DDOST-CDG was found to harbor a 22 bp deletion in exon 11, namely c.1265_1286del22 (p.Ile422Thrfs*7) creating a stop codon 7 amino acids downstream and produces a protein with a 29 amino acid C-terminal truncation, and a heterozygous missense variant, c.650G>A (p.Gly217Asp) in DDOST 13. Family studies subsequently confirmed that the variants were in trans. The second patient harbors a homozygous variant, designated as c.1187G>A (p.Arg396Gln), inherited from both parents 14. The proband, similar to the first reported patient, harbors a frameshift variant c.1142dupT (p.Leu381Phefs*11) and a missense variant c.661T>C (p.Ser221Pro). Western blot analysis revealed significant reduction of DDOST protein level in the proband’s fibroblasts. The variant c.1142dupT is predicted to cause haploinsufficiency through nonsense-mediated mRNA decay (NMD). To analyse intracellular glycosylation, we performed Western blotting of ICAM-1 and LAMP2. ICAM-1 is an ubiquitous structural protein, and its localization and stability rely on normal glycosylation. LAMP2 is a lysosomal membrane protein which requires normal glycosylation for adequate insertion in the lysosomal membrane. In patient reported here, ICAM-1 levels were quantitatively decreased, while LAMP2 levels displayed a lower molecular weight with an increased spread which is thought to represent aberrant glycoforms of this protein. Together, these two abnormalities are consistent with previously published patterns of patients with N-linked congenital disorders of glycosylation 39,40.
To date, none of the reported patients were found to have homozygous or compound heterozygous truncating variants. In mouse model, homozygous knock-out of mouse homologue of human DDOST is lethal 41. It is possible that complete loss of DDOST function leads to severe developmental defects causing the embryonic lethality. The severity of clinical phenotype may be inversely correlated with the residual enzymatic activity. As previously mentioned, the proband in our study has a less severe phenotype (moderate developmental delay/intellectual disability, lack of hypotonia and failure to thrive) than the two previously reported patients. Given that the first variant likely causes complete loss-of-function, p.Ser221Pro may represent a hypomorphic allele – a similar molecular pathogenesis occurs in the most frequent CDG type, PMM2-CDG 42.
This is the second time that we have reported plasma N-glycan changes in a DDOST-CDG patient. We previously reported changes in high mannose species in the plasma from DDOST-CDG and STT3B-CDG as part of our method validation 11. At that time, de-identified known positive samples were provided to us by a reference lab from which clinical information cannot be retrieved. In this report, we describe detailed N-glycan changes that showed increases of small high mannose species from Man0-5GlcNAc2. The canonical substrate for OST complex is Glc3Man9GlcNAc2, which is subsequently trimmed to Man9GlcNAc2 and Man8GlcNAc2 after the glycans are transferred to the nascent proteins. The increase of Man7/9 ratio or Man7 and Man9 abundance in DDOST-CDG suggests potential impairment of substrate specificity of OST complex. Alternatively, Glc3Man5GlcNAc2 can also be transferred onto nascent proteins 43. The increases of Man4, Man5 abundance along with increased Man5/9 and Man4/9 ratios in our cohort suggests the alternative pathway is probably upregulated. However without extended high mannose glycans that are greater than Man5, a major glycoprotein folding quality control and ER response to unfolding stresses will be lost 44, which is predicted to be pathogenic. Interestingly, we also observed mild increases of Man0–3GlcNA2 in our proband. Man0–3GlcNA2 are intermediate glycans with very low abundance in normal plasma, and have been reported in multiple type I CDGs including PMM2-CDG, ALG3-CDG and ALG9-CDG. Since the high mannose assembling pathway is not impaired in DDOST-CDG, we suspect that the mild increases of these intermediate glycans are likely from impairment of substrate specificity of OST complex.Consistently with this, unlike other type I CDGs in high mannose assembling, the ratios between these intermediate glycans, such as Man3/4, is normal in DDOST-CDG. Thus the changes of N-glycans in DDOST-CDG have an interesting pattern that is different from other type I CDG and may be used to provide biochemical confirmation for this condition. Nonetheless, this is a single case report and more future studies are warranted.
Given the compelling evidence of DDOST function in glycosylation elucidated by previous publications, as well as the fitting clinical and biochemical phenotype in our patient and the evolutionary, in silico, and populational frequency analyses, we postulate that the reported variants in our patient are indeed disease-causing. Our study expands phenotypic and genotypic spectra of DDOST-CDG. Given the limited information available on DDOST-CDG patients, larger cohorts are needed to establish genotype-phenotype correlations and prognostic features.
Supplementary Material
Acknowledgements
We thank the patient and his families for their cooperation with this article.
Details of funding
Research reported in this publication was supported by the Rocket Fund and the National Institutes of Health grants R01DK99551 (HF).
Footnotes
Conflict of Interest
Ibrahim Elsharkawi, Parith Wongkittichote, Earnest James Paul Daniel, Rodrigo Tzovenos Starosta, Keisuke Ueda, Bobby G. Ng, Hudson H. Freeze, Miao He, Marwan Shinawi declare that they have no conflict of interest.
Details of ethics approval:
The guardian of patients signed a consent form for publication approved by Washington University IRB (Media Authorization for the Use and Disclosure of Protected Health Information).
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from the patient for being included in the study.
Availability of data and materials Not applicable
Animal Rights
This article does not contain any studies with human or animal subjects performed by the any of the authors.
References
- 1.Gilfix BM. Congenital disorders of glycosylation and the challenge of rare diseases. Hum Mutat 08 2019;40(8):1010–1012. doi: 10.1002/humu.23829 [DOI] [PubMed] [Google Scholar]
- 2.Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet Feb 06 2014;94(2):161–75. doi: 10.1016/j.ajhg.2013.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng BG, Freeze HH. Perspectives on Glycosylation and Its Congenital Disorders. Trends Genet 06 2018;34(6):466–476. doi: 10.1016/j.tig.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson MP, Matthijs G. The evolving genetic landscape of congenital disorders of glycosylation. Biochim Biophys Acta Gen Subj 11 2021;1865(11):129976. doi: 10.1016/j.bbagen.2021.129976 [DOI] [PubMed] [Google Scholar]
- 5.Rymen D, Jaeken J. Skin manifestations in CDG. J Inherit Metab Dis Sep 2014;37(5):699–708. doi: 10.1007/s10545-014-9678-7 [DOI] [PubMed] [Google Scholar]
- 6.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 04 1993;3(2):97–130. doi: 10.1093/glycob/3.2.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grunewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation: a review. Pediatr Res Nov 2002;52(5):618–24. doi: 10.1203/00006450-200211000-00003 [DOI] [PubMed] [Google Scholar]
- 8.Grünewald S. Congenital disorders of glycosylation: rapidly enlarging group of (neuro)metabolic disorders. Early Hum Dev Dec 2007;83(12):825–30. doi: 10.1016/j.earlhumdev.2007.09.016 [DOI] [PubMed] [Google Scholar]
- 9.Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet 2001;2:129–51. doi: 10.1146/annurev.genom.2.1.129 [DOI] [PubMed] [Google Scholar]
- 10.Kingma HA, van der Sluijs FH, Heiner-Fokkema MR. Fast screening of N-glycosylation disorders by sialotransferrin profiling with capillary zone electrophoresis. Ann Clin Biochem Nov 2018;55(6):693–701. doi: 10.1177/0004563218779609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Li X, Edmondson A, et al. Increased Clinical Sensitivity and Specificity of Plasma Protein. Clin Chem 05 2019;65(5):653–663. doi: 10.1373/clinchem.2018.296780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamagata T, Tsuru T, Momoi MY, et al. Genome organization of human 48-kDa oligosaccharyltransferase (DDOST). Genomics Nov 01 1997;45(3):535–40. doi: 10.1006/geno.1997.4966 [DOI] [PubMed] [Google Scholar]
- 13.Jones MA, Ng BG, Bhide S, et al. DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am J Hum Genet Feb 10 2012;90(2):363–8. doi: 10.1016/j.ajhg.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pi S, Gong J, Xiao W, Xiao B, Mao X, Long H. The second DDOST-CDG patient with lactose intolerance, developmental delay, and situs inversus totalis. J Hum Genet Feb 2022;67(2):103–106. doi: 10.1038/s10038-021-00974-2 [DOI] [PubMed] [Google Scholar]
- 15.Francisco R, Marques-da-Silva D, Brasil S, et al. The challenge of CDG diagnosis. Mol Genet Metab 01 2019;126(1):1–5. doi: 10.1016/j.ymgme.2018.11.003 [DOI] [PubMed] [Google Scholar]
- 16.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 01 August 2019;47(D1):D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet Oct 06 2016;99(4):877–885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc Jan 2016;11(1):1–9. doi: 10.1038/nprot.2015.123 [DOI] [PubMed] [Google Scholar]
- 19.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods Apr 2014;11(4):361–2. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med May 2015;17(5):405–24. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacey JM, Bergen HR, Magera MJ, Naylor S, O’Brien JF. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem Mar 2001;47(3):513–8. [PubMed] [Google Scholar]
- 22.Harada Y, Ohkawa Y, Kizuka Y, Taniguchi N. Oligosaccharyltransferase: A Gatekeeper of Health and Tumor Progression. Int J Mol Sci Dec 02 2019;20(23)doi: 10.3390/ijms20236074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohorko E, Glockshuber R, Aebi M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J Inherit Metab Dis Aug 2011;34(4):869–78. doi: 10.1007/s10545-011-9337-1 [DOI] [PubMed] [Google Scholar]
- 24.Al-Amri A, Saegh AA, Al-Mamari W, et al. Homozygous single base deletion in TUSC3 causes intellectual disability with developmental delay in an Omani family. Am J Med Genet A 07 2016;170(7):1826–31. doi: 10.1002/ajmg.a.37690 [DOI] [PubMed] [Google Scholar]
- 25.Garshasbi M, Kahrizi K, Hosseini M, et al. A novel nonsense mutation in TUSC3 is responsible for non-syndromic autosomal recessive mental retardation in a consanguineous Iranian family. Am J Med Genet A Aug 2011;155A(8):1976–80. doi: 10.1002/ajmg.a.34077 [DOI] [PubMed] [Google Scholar]
- 26.Matsuda-Lennikov M, Biancalana M, Zou J, et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in. J Biol Chem September 13 2019;294(37):13638–13656. doi: 10.1074/jbc.RA119.008903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shrimal S, Ng BG, Losfeld ME, Gilmore R, Freeze HH. Mutations in STT3A and STT3B cause two congenital disorders of glycosylation. Hum Mol Genet Nov 15 2013;22(22):4638–45. doi: 10.1093/hmg/ddt312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryant EM, Millichap JJ, Spinelli E, et al. Oligosaccharyltransferase complex-congenital disorders of glycosylation: A novel congenital disorder of glycosylation. Am J Med Genet A 06 2020;182(6):1460–1465. doi: 10.1002/ajmg.a.61553 [DOI] [PubMed] [Google Scholar]
- 29.Wilson MP, Garanto A, Pinto E Vairo F, et al. Active site variants in STT3A cause a dominant type I congenital disorder of glycosylation with neuromusculoskeletal findings. Am J Hum Genet November 04 2021;108(11):2130–2144. doi: 10.1016/j.ajhg.2021.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh A, Urquhart J, Daly S, et al. Phenotypic Heterogeneity in a Congenital Disorder of Glycosylation Caused by Mutations in STT3A. J Child Neurol 05 2017;32(6):560–565. doi: 10.1177/0883073817696816 [DOI] [PubMed] [Google Scholar]
- 31.Mostile G, Barone R, Nicoletti A, et al. Hyperkinetic movement disorders in congenital disorders of glycosylation. Eur J Neurol 09 2019;26(9):1226–1234. doi: 10.1111/ene.14007 [DOI] [PubMed] [Google Scholar]
- 32.Bosch-Bouju C, Hyland BI, Parr-Brownlie LC. Motor thalamus integration of cortical, cerebellar and basal ganglia information: implications for normal and parkinsonian conditions. Front Comput Neurosci 2013;7:163. doi: 10.3389/fncom.2013.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JS. Delayed onset mixed involuntary movements after thalamic stroke: clinical, radiological and pathophysiological findings. Brain Feb 2001;124(Pt 2):299–309. doi: 10.1093/brain/124.2.299 [DOI] [PubMed] [Google Scholar]
- 34.Blaser SI, Steinlin M, Al-Maawali A, Yoon G. The Pediatric Cerebellum in Inherited Neurodegenerative Disorders: A Pattern-recognition Approach. Neuroimaging Clin N Am Aug 2016;26(3):373–416. doi: 10.1016/j.nic.2016.03.007 [DOI] [PubMed] [Google Scholar]
- 35.Thiel C, Schwarz M, Peng J, et al. A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. J Biol Chem Jun 20 2003;278(25):22498–505. doi: 10.1074/jbc.M302850200 [DOI] [PubMed] [Google Scholar]
- 36.Schwarz M, Thiel C, Lübbehusen J, et al. Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylation type Ik. Am J Hum Genet Mar 2004;74(3):472–81. doi: 10.1086/382492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters V, Penzien JM, Reiter G, et al. Congenital disorder of glycosylation IId (CDG-IId) -- a new entity: clinical presentation with Dandy-Walker malformation and myopathy. Neuropediatrics Feb 2002;33(1):27–32. doi: 10.1055/s-2002-23597 [DOI] [PubMed] [Google Scholar]
- 38.Dupré T, Vuillaumier-Barrot S, Chantret I, et al. Guanosine diphosphate-mannose:GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. J Med Genet Nov 2010;47(11):729–35. doi: 10.1136/jmg.2009.072504 [DOI] [PubMed] [Google Scholar]
- 39.He P, Srikrishna G, Freeze HH. N-glycosylation deficiency reduces ICAM-1 induction and impairs inflammatory response. Glycobiology Apr 2014;24(4):392–8. doi: 10.1093/glycob/cwu006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrer A, Starosta RT, Ranatunga W, et al. Fetal glycosylation defect due to ALG3 and COG5 variants detected via amniocentesis: Complex glycosylation defect with embryonic lethal phenotype. Mol Genet Metab 12 2020;131(4):424–429. doi: 10.1016/j.ymgme.2020.11.003 [DOI] [PubMed] [Google Scholar]
- 41.Dickinson ME, Flenniken AM, Ji X, et al. High-throughput discovery of novel developmental phenotypes. Nature September 22 2016;537(7621):508–514. doi: 10.1038/nature19356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schollen E, Kjaergaard S, Legius E, Schwartz M, Matthijs G. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). Eur J Hum Genet May 2000;8(5):367–71. doi: 10.1038/sj.ejhg.5200470 [DOI] [PubMed] [Google Scholar]
- 43.Polla DL, Edmondson AC, Duvet S, et al. Bi-allelic variants in the ER quality-control mannosidase gene EDEM3 cause a congenital disorder of glycosylation. Am J Hum Genet July 01 2021;108(7):1342–1349. doi: 10.1016/j.ajhg.2021.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shang J, Körner C, Freeze H, Lehrman MA. Extension of lipid-linked oligosaccharides is a high-priority aspect of the unfolded protein response: endoplasmic reticulum stress in Type I congenital disorder of glycosylation fibroblasts. Glycobiology May 2002;12(5):307–17. doi: 10.1093/glycob/12.5.307 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.