

Abstract
BLOC1S1 is a common component of BLOC and BORC multiprotein complexes which play distinct roles in endosome and lysosome biology. Recent human mutations in BLOC1S1 associate with juvenile leukodystrophy. As leukodystrophy is linked to perturbed lysosomal lipid storage we explored whether BLOC1S1 itself modulates this biology. Given the central role of the liver in lipid storage, our investigations were performed in hepatocyte specific liver bloc1s1 knockout (LKO) mice and in human hepatocyte-like lines (HLCs) derived from inducible pluripotential stem cells (iPSCs) from a juvenile leukodystrophy subject’s with bloc1s1 mutations and from isogenic corrected iPSCs. Here we show that hepatocyte lipid stores are diminished in parallel with increased lysosomal content, increased lysosomal lipid uptake and lipolysis in LKO mice. The lysosomal lipolysis program was independent of macro- and chaperone-mediated lipophagy but dependent on cellular lysosome content. In parallel, genetic induction of lysosomal biogenesis in a transformed hepatocyte cell line replicated depletion of intracellular lipid stores. Interestingly bloc1s1 mutant and isogenic corrected HLCs both showed normal lysosomal enzyme activity. However, relative to the isogenic corrected HLCs, mutant bloc1s1 HLCs showed reduced lysosomal content and increased lipid storage. Together these data show distinct phenotypes in human mutant HLCs compared to murine knockout cells. At the same time, human blcs1s1 mutation and murine hepatocyte bloc1s1 depletion disrupt lysosome content and the cellular lipid storage. These data support that BLOC1S1 modulates lysosome content and lipid handling independent of autophagy and show that lysosomal lipolysis is dependent on the cellular content of functional lysosomes.
Keywords: BLOC1S1, Lysosome, Lysosomal lipolysis, Hepatic Lipid droplets
1. Introduction
Based on diverse roles of BLOC1S1 as a component of numerous multiprotein complexes, it has been designated with alternate nomenclatures, including BLOS1, GCN5L1 and BORCS1 [1]. BLOC1S1/GCN5L1 was firstly identified as a homolog to the nuclear acetyltransferase KAT2A/GCN5 and was found to contribute to lysine residue acetylation of intracellular proteins [2–5]. Protein interaction and functional characterization studies identified that BLOC1S1/BORCS1 was a component of the BLOC and BORC super-complexes which modulate endo-lysosome biology, including conferring roles in endosome, vesicular and lysosome positioning [6–8], in vesicular trafficking to lysosomes [9], in lysosomal reformation [10], and in endosomal maturation and function [9; 11]. In parallel BLOC1S1/GCN5L1 was been linked to acetyl-modification of mitochondrial metabolic proteins linked to the regulation of amino acid and fatty acid metabolism [12–14], electron transfer chain[2], mitochondrial biogenesis and mitophagy programs [15; 16] and to modulate distinct retrograde mitochondrial signal transduction pathways [17; 18].
Mutations in BLOC1S1 have recently been found to associate with leukodystrophy and neurodegeneration [19], diseases linked with endosomal-lysosome dysfunction [20; 21] and/or disrupted lysosomal lipid storage [22]. Although whether BLOC1S1 mutations linked to leukodystrophy results from manifestation of endosomal-lysosome dysfunction function or altered lysosomal lipid handling remains unknown.
The role of the endosome-lysosome function in lipid handling is extensive with roles in lipid droplet (LD) directed supply of neutral lipid for autophagosome membrane biogenesis, in LD catabolism via chaperone-mediated autophagic programs including both macro- and micro-lipophagy [23] and by direct LD lysosome initiated lipolysis [24]. To date, the role of BLOC1S1 in lipid handling has exclusively been explored in the liver in response to acute high fat-feeding. In that study hepatic BLOC1S1/GCN5L1 knockout mice were protected from high fat diet-induced hepatosteatosis with a marked reduction in LD accumulation.[13] However, LD biology was not directly explored, rather BLOC1S1/GCN5L1 was found to augment mitochondrial fatty acid oxidation (FAO) and thereby increase fat consumption [13]. However, given that BLOC1S1 also modulates endosome-lysosome biology and its emerging link with leukodystrophy its direct role in LD catabolism warrants exploration.
To explore the biology of BLOC1S1 in LD biology we studied hepatic bloc1s1/gcn5l1 knockout (LKO) mice. As background to this biology, multiple interacting mechanisms function in the catabolism of LD’s, via hepatic cytosolic lipase and adipocyte triglyceride lipase (ATGL) induced lipolysis [25], via macro- or micro-lipophagy and/or by direct lysosomal lipolysis [26; 27]. Other distinct mechanisms are operational including LD mediated production of VLDL [28]. Given the emerging roles of BLOC1S1 on the regulation of endosome/lysosome biology, we initially focused on the endosome/lysosomal regulated lipolytic programs.
In this study we show that when bloc1s1 is genetically depleted in the liver, hepatic lipid content is reduced due to the enhanced lysosomal lipolysis independent of autophagic or chaperone mediated lipophagy. This induction of lysosomal lipolysis in response to the absence of BLOC1S1 was dependent on an increased number of functional lysosomes. Interestingly, in a human hepatocyte-like cells derived from induced pluripotential stem cells (iPSCs) from a patient with biallelic complex heterozygous mutations in bloc1s1, the levels of BLOC1S1 were increased in parallel with a reduction in lysosomal content, reduced lysosomal lipolysis and increased intracellular lipid content. Together these data show that the levels and function of BLOC1S1 dictate lysosomal content and the extent of lysosomal lipolysis in the liver.
2. Materials and Methods
All chemicals were purchased from Sigma-Aldrich (now Merck) unless otherwise stated.
2.1. Animal Studies.
Breeding and husbandry of bloc1s1 liver knockout mice and controls as described [10]. Animal studies were approved by the NHLBI Animal Care and Use Committee in accordance with the National Institutes of Health guide for the care and use of laboratory animals (8th Edition, revised 2011). Males and female mice were used to extract primary hepatocytes for all experiments.
2.2. Cell Culture and Transfection.
Primary hepatocytes were isolated from chow fed mice at age of 8–12 weeks as described [29]. For overexpression studies, primary hepatocytes and murine hepatocyte cell line AML12 cells were transfected with Lipofectamine 2000 (Invitrogen, 11668019). For knockdown experiments, cells were transfected with 25 nM siRNAs (GE Dharmacon, ON-TARGETplus SMART-pool) or ON-TARGETplus Non-Targeting SMARTpool (GE Dharmacon, D0018101050) with Lipofectamine RNAiMAX Reagent (Invitrogen, 13778150) and cells analyzed 48 h after knockdown or overexpression transfections.
Hepatic-like cells (HLCs) were differentiated from iPSCs as described [30]. iPSCs were differentiated to definitive endoderm with STEMdiff Definitive Endoderm Kit (Stemcell, 05110). The iPSC cells were generated from a patient following ethical approval and signed consent as documented previously [31].
2.3. Adenovirus Production and Transfection.
RFP-LAMP1 overexpression adenoviruses were produced using Adeasy Adenoviral System (Agilent, 240009) [10]. TFEB(S211A) overexpression adenovirus was a gift [32]. Primary hepatocyte infection with adenovirus overexpressing empty vector (control), RFP-LAMP1 or TFEB(S211A) were transduced at 20 plaque-forming units/cell and analyzed 48 h later.
2.4. Immunoblot analysis and antibodies.
Livers tissue or hepatocytes were homogenized in RIPA buffer (Boston BioProducts, BP-115), 40 μg total protein was used for each sample. Antibodies: α-ACTB/β-Actin (Cell Signaling Technology, Cat 3700); α-LC3B (Sigma, L8918); α-TFEB (Bethyl labs, A303–673A); α-BLOC1S1 (customized(Scott)); α-LAMP1 (Developmental Studies Hybridoma Bank, 1D4B or Abcam, ab24170); α-PLIN (Proteintech, 10694–1-AP); α-LAL (Abcam, ab154356); α-A1AT and α-albumin (R&D systems, MAB1268 and MAB1455); IRDye® 800CW Goat α-Mouse IgG (H + L), 800CW Goat α-Rabbit IgG (H + L), 680RD Goat α-Mouse IgG (H + L) and 680RD Goat α-Rabbit IgG (H + L) (LI-COR 926-32210, 926-32211, 926-68070 and 926-68071)
2.5. Lipid biology.
Liver frozen section or primary hepatocytes were fixed with 0.2% oil red O in 35% 2-propanol for 30 min, then imaging with fluorescent microscopy with excitation at 460–490nm. The cells then incubated with 10 min with isopropyl alcohol, which dissolved stained lipid droplet. The absorbance of the dye was measured at 420nm. Values were normalized to protein concentration.
Lipid was extracted from fasted mouse livers and triglyceride were quantified with a colorimetric kit from Pointe Scientific (T7532, T7531-SD) [26].
Palmitate oxidation was measured as our described. Values were normalized to protein concentration [13]. Lipid droplets were isolated from the liver of WT and LKO mice after 24 hours fasting [33]. The lipid protein was extracted with 1x LDS Sample Buffer and analyzed by immunoblotting.
2.6. Live Cell Imaging.
Primary hepatocytes were seeded on rat tail type I collagen (Sigma, C3867) coated μ-Slide 4 well glass (ibidi, 80426) slides. Twenty-four or forty-eight hours after transfection, cells were imaged using confocal laser-scanning microscope (LSM 880 or LSM 880 with Airyscan; CarlZeiss) fitted with a 63×, 1.4 numerical aperture (NA) objective in an environmental control system at 37°C in 5% CO2, and images were acquired continually. LysoTracker Deep Red (200nM) (Invitrogen, L12492) incubation for 30 min was used to label and image lysosomes. BODIPY 493/503 (1μg/ml) (Invitrogen, D3922) was incubated for 15 min before imaging for lipid staining. For lysosomal Cathepsin L assay, 20 μl Magic Red (ImmunoChemistry Technologies, 941) were incubated with hepatocytes at 37°C for 30 min. For BODIPY-C12 chase and lysosome retention, 7.5μm BODIPY FL C12 (Invitrogen, D3822) were incubated with cells for 2h and performed as described [34]. Figi ImageJ (NIH) was used with live imaging to quantify fluorescent intensity and fluorophore colocalization.
2.7. Statistical Analysis.
Results are displayed as the mean ± SEM. When the comparison of two groups was analyzed two-tailed unpaired Students t-test were used, alternatively, one way ANOVA for multiple comparisons followed by sidak multiple comparison testing was performed. A value of P<0.05 was considered statistically significant.
3. Results
3.1. BLOC1S liver knockout mice and primary hepatocytes have reduced cytosolic lipid stores
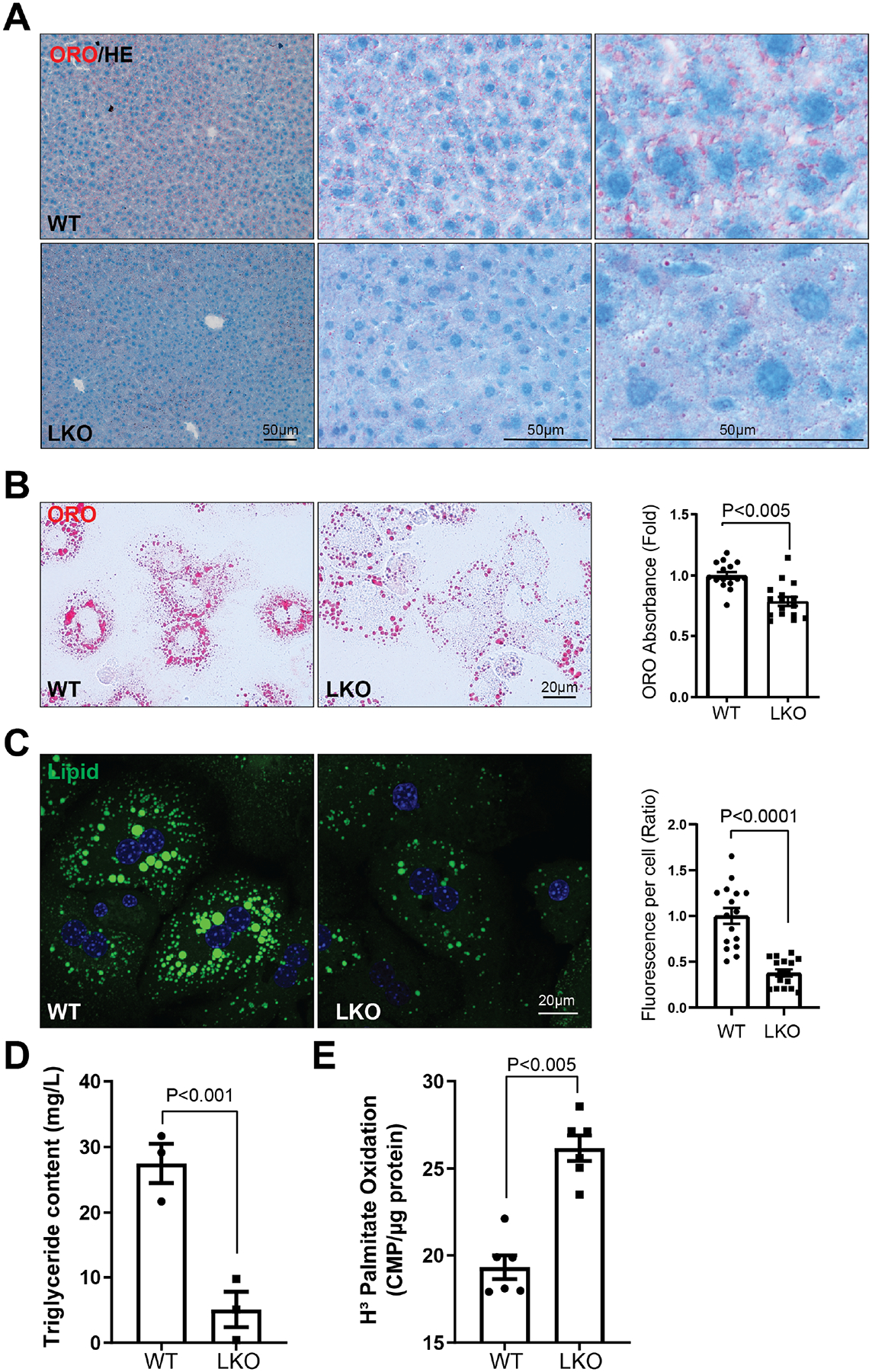
As bloc1s1/gcn5l1 liver knockout (LKO) mice are resistant to fat accumulation in response to high-fat feeding [13], we initially assessed baseline hepatocyte lipid storage. Levels of lipid was assessed using frozen liver sections and in primary hepatocytes stained with oil red O from control and LKO being fed on regular chow. Both tissue sections and primary cells showed reduced lipid stores in the absence of BLOC1S1 (Fig. 1, A and B). This was confirmed by fluorescent labelling of fat stores with the BODIPY fluorophore in primary hepatocytes (Fig. 1C). In parallel primary hepatocyte triglyceride content was markedly reduced in LKO hepatocytes (Fig. 1D).
Figure 1. BLOC1S1 liver knockout attenuates hepatic lipid content.
(A) Representative images show lipid content in frozen liver sections in WT and LKO mice with different magnification. Oil Red O and HE staining results are displayed. (B) Representative images of lipid droplets in WT and LKO primary hepatocytes by Oil Red O staining (left panel). Lipid were then extracted and quantified by their absorbance at 420nm (right panel). (C) BODIPY (Green) staining of lipid droplets in primary hepatocytes with the accompanying histograms showing quantitation. n > 10 cells from each group were analyzed. (D) Lipid was extracted from WT and LKO hepatocytes and triglyceride content quantified. (E) LKO primary hepatocytes show significantly increased palmitate oxidation rates relative to WT. Scale bars shown in image panels.
To see if the mechanism extends beyond putative fatty acid consumption, we explored numerous components of hepatic lipid handing. Fat uptake was not different between the genotypes as measured by the kinetics of dodecanoic acid fluorescent fatty acid substrate incorporation into hepatocytes (Supp. Fig. 1A). Moreover, there was no changes in transcript levels encoding proteins involved in fatty acid synthesis including sterol regulatory element binding protein 1 and fatty acid synthase (Supp. Fig. 1B) between genotypes. Interesting transcript levels of genes encoding cytosolic (hepatic lipase HL)) and LD (adipocyte triglyceride lipase (ATGL)) linked lipolysis and the canonical transcription factor (PPARα) linked with fatty acid oxidation were blunted in the absence of BLOC1S1 (Supp. Fig. 1C). Meanwhile, transcript levels of genes encoding VLDL biosynthesis enzymes were unchanged (Supp. Fig. 1D). Additionally, we confirmed the fatty acid oxidation, as measured by 3H-palmitate oxidation, was increased in primary LKO hepatocytes (Fig. 1E) [13].
3.2. The absence of BLOC1S1 augments lysosomal lipolysis
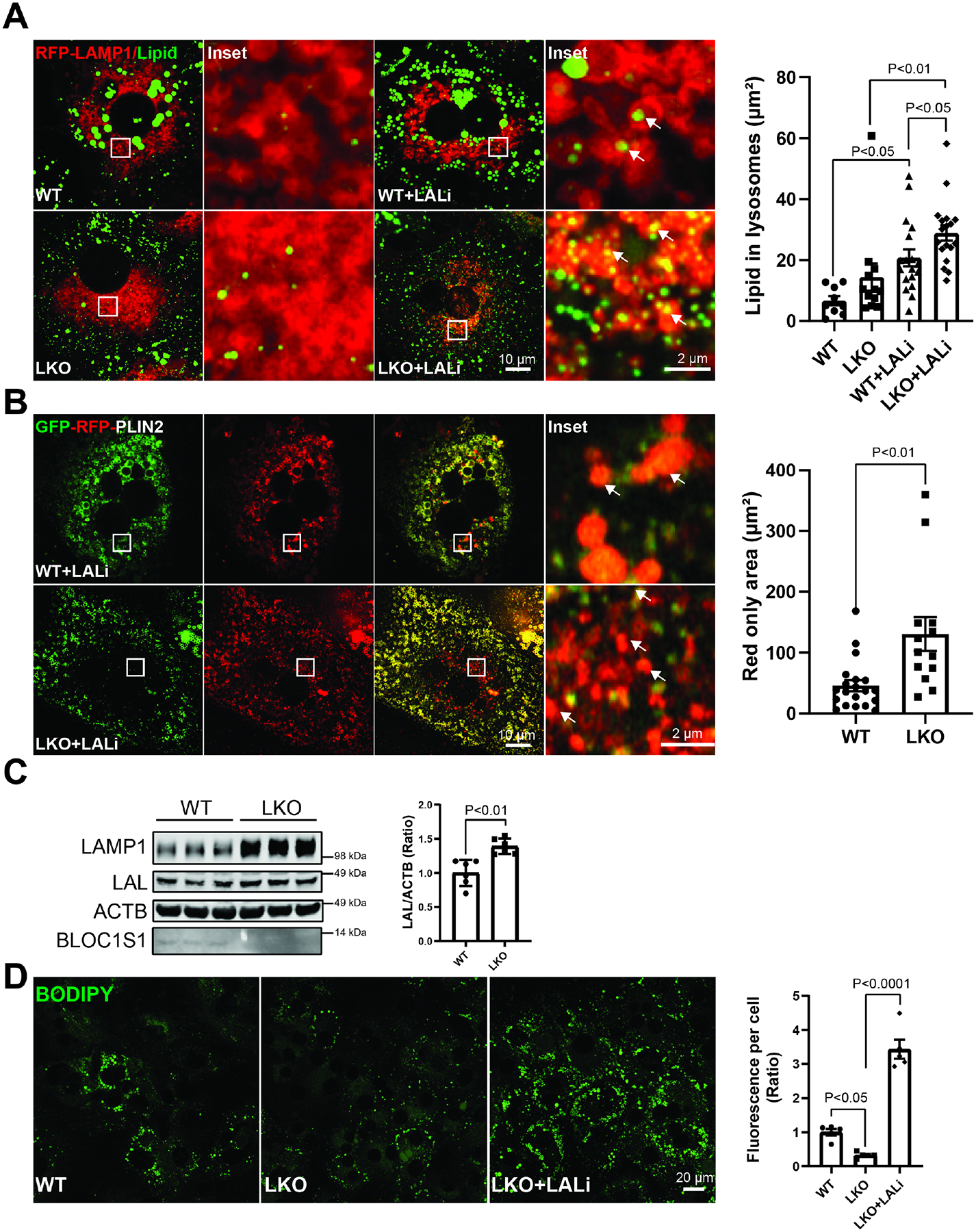
The finding that the LKO hepatocytes consume more fatty acids would be compatible with the catabolism of LD stores through modulation of lipolysis. At the same time, given the reduced transcript levels of ATGL and HL and our prior observation that BLOC1S1 hepatocytes exhibit an accumulation of lysosomes with sustained lysosome activity [10], we began to explore the role of lysosomal lipolysis in the BLOC1S1-deficient lipid storage depleted phenotype. To evaluate this, we measured lipid trafficking to lysosomes. Primary hepatocytes were infected with adenovirus expressing an RFP-labelled lysosomal membrane protein LAMP1, after 24 hours, lipids were stained with lipidspot dye, visualized by confocal microscopy in the presence or absence of 50 μM of the lysosomal acid lipase inhibitor Lalistat-2 (LALi) to slow lysosomal LD catabolism and preserve the number of lysosomes exhibiting ingested LDs [26; 35]. Here the overlapping-labelled lysosomal lipid accumulation indicate lysosomal structures containing lipid and were quantified as previous reported [26; 35]. LALi treatment increased the lysosomal lipid content in both WT and LKO hepatocytes (Fig. 2A). Interestingly, the accumulation of lysosomal lipid was augmented to a greater degree with LALi in LKO hepatocytes, supporting greater lysosomal lipid trafficking in the absence of BLOC1S1 (Fig. 2A). To further validate LD uptake and catabolism by lysosomes dual GFP- and RFP-labelled LD surface protein Perilipin-2 (PLIN2)[34] was transiently transfected into WT and LKO hepatocytes exposed to LALi. Lysosomal acidification of the pH-sensitive GFP-fluorophore resulting quenching resulting in augmented red-fluorescence of LD in lysosomes [26; 34]. Here too RFP signaling was augmented in LKO lysosomes supporting greater lysosomal lipid uptake and catabolism in the absence of BLOC1S1 (Fig. 2B). Lysosomal acid lipase (LAL), encoded by the lipase A (LIPA) gene in human, is the only known enzyme active at an acidic pH in lysosome to hydrolyze cholesteryl esters (CEs) and triglycerides. Consistently with excess functional lysosome in LKO hepatocytes, steady-state LAL levels were higher in LKO hepatocytes (Fig. 2C). Furthermore, pharmacologic inhibition of LAL by LALi dramatically restored lipid content in LKO hepatocytes (Fig. 2D).
Figure 2. BLOC1S1 deficiency increases lysosome-dependent lipid consumption.
(A) Confocal microcopy of lipid accumulation in LKO vs. WT lysosomes with/without lysosomal acid lipase inhibitor (LALi). The lysosomes labeled by RFP-LAMP1 (red) and lipid stained with BODIPY (green). The area of lipid inside lysosome was quantified in n > 15 cells from each group (histogram). (B) Live-cell confocal imaging of hepatocyte transiently expressing the RFP-GFP-PLIN2 reporter shows PLIN2 levels (red only) within lysosomes. The lysosomal PLIN2 (red signal) was quantified in n > 10 cells from each group (histogram). (C) Immunoblot analysis comparing hepatocyte LAL levels between groups. (D) Blocking the lipid degradation in lysosome by LALi restored lipid content in LKO hepatocytes. The relative fluorescence per cell was shown in the accompanying histograms. Scale bars shown in image panels.
3.3. Increased lysosome lipolysis in the absence of BLOC1S1 is independent of canonical lipophagy programs
To evaluate whether upstream membrane-dependent LD sequestration programs, i.e. macro- and microlipophagy or chaperone-mediated autophagy (CMA), additionally contribute to BLOC1S1 mediated control of LD catabolism we assayed whether these programs were perturbed in LKO hepatocytes. We initially explored whether disruption of autophagosome-lysosome fusion inhibitor, with the vacuolar H+-ATPase inhibitor Bafilomycin A1 (BafA1), would alter the accumulation of endogenous lipid in lysosomes in the presence of LALi. Confocal microscopy showed the BafA1 did not disrupt the accumulation of LDs in lysosomes in both genotypes as assessed by the colocalization of lipidspot (lipid staining) and lysotracker-633 (lysosome staining) [35] (Figure 3A). To trace the lipid trafficking to lysosomes upon uptake, fluorescent-labelled BODIPY-C12 fatty acid were then introduced to primary hepatocytes for 2 hours. Here lysosomal lipid accumulation (Lysotracker and C12 positive) was measured by colocalization. These studies were performed with the parallel siRNA knockdown of the macroautophagy mediator (ATG7) or of CMA mediator lysosome-associated membrane protein (LAMP2A) [36] in LKO hepatocytes. Depletion of these regulatory proteins (Supp Fig. 2A) did not impair accumulation of C12-bodipy in lysosomes (Fig. 3B). Recent data show the direct transfer of LD components into the lysosome for breakdown in hepatocytes [26]. To evaluate whether direct binding of lipid droplets (LD) to lysosomes, LDs and their binding proteins were purified from mice liver after a 24 hour fast using gradient centrifugation as described previously [33]. Immunoblotting shows excess LAMP1 binding to LDs in LKO mice (Fig. 3C). However, given increased lysosome content in LKO hepatocytes, the more directly lysosome-LD interaction in LKO hepatocytes may be due to the excess lysosomes. As we could not decrease the lysosome content in primary LKO hepatocytes to evaluate this concept, lysosome accumulation was replicated in AML12 hepatocytes via transfection of the lysosome biogenesis regulator TFEB [37]. Adenovirus mediated TFEB overexpression increased lysosomes as evident by increased LAMP1 and LC3-I levels (Supp. Fig. 2B). Here too, and mirroring the LKO hepatocytes, the triglycerides content was decreased with higher TFEB-induced lysosome content (Supp. Fig. 2C).
Figure 3. BLOC1S1 deficiency increases directly lysosome-lipid droplet interaction but not macroautophagy or chaperone-mediated lipophagy.
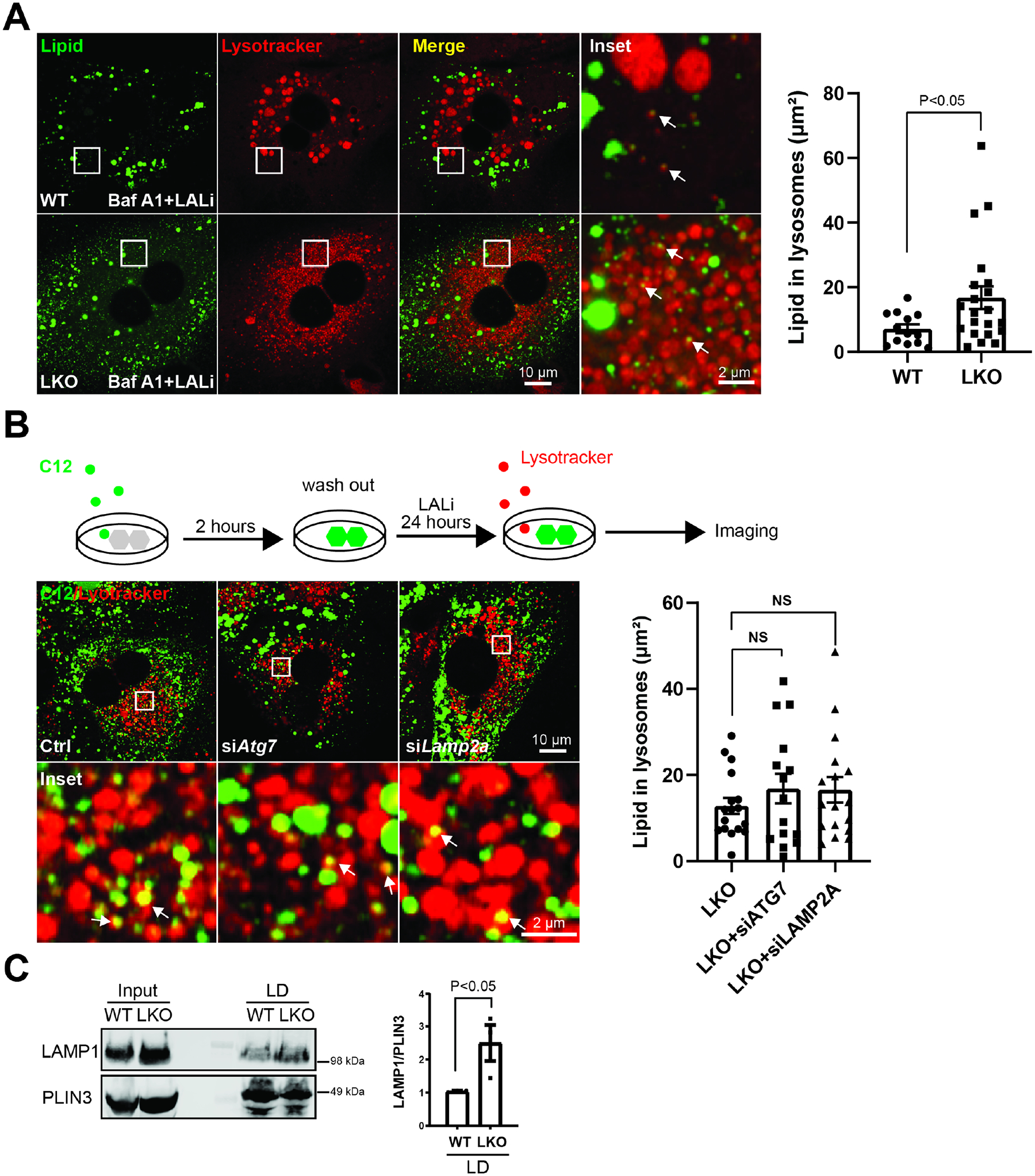
(A) Confocal microscopy to evaluate the effect of the disruption of autophagic flux by bafilomycin A1 to assess effect on LD accumulation in LALi treated primary hepatocytes in both groups. Lysosomes and lipid were stained with lysotracker (red) and BODIPY (green) respectively. The area of lipid inside lysosome were quantified in n > 15 cells from each group (B) Schematic diagram illustrates method to trace lysosome degradation of BODIPY labeled C12 long chain fatty acid in primary hepatocytes (upper panel). Confocal image following knockdown of macroautophagy (siATG7)/CMA machinery (siLAMP2A) to assess effect on lipid transfer to lysosomes in LKO hepatocytes (lower panel). Lysosomes were visualized by Lysotracker (red). The area of lipid inside lysosome was quantified in n > 15 cells from each group. (C) Lipid droplets were isolated from livers of WT and LKO mice after 24 hours fasting followed by immunoblot analysis of LAMP1 binding to lipid droplets. PLIN3 reflects the mass of LDs. Scale bars shown in image panels.
3.4. Lysosomal lipolysis is impaired in hepatocyte-like cells generated from a human iPSC line harboring the Bloc1s1 c.206A>C and c.359G>A mutations
A recent compound heterozygous mutations in bloc1s1 (BLOC1S1 c.206A>C and c.359G>A) has been identified in a patient with leukodystrophy. Inducible pluripotential stem cells (iPSCs) were generated from PMBCs from this patient [31]. In parallel, this bloc1s1 mutation iPSC line was employed to generate isogenic control lines to control for the genetic background [31]. These lines were designated as BLOC1S1-DM and BLOC1S1-DC respectively and successfully differentiated into hepatocyte-like cell (HLCs) which became more angular and displayed brighter junctures, suggesting cellular polarity and canaliculi formation (Fig. 4A). The validity of the hepatocyte phenotype was confirmed by the induction of expression of hepatic specific transcripts (Supp. Fig. 3A) and liver synthesized proteins (Supp. Fig. 3B) in the mutant and isogenic-corrected HLCs. In contrast to the LKO cells LAMP1 expression, which is a marker of lysosomal content, was diminished in BLOC1S1-DM and restored in the isogenic-control HLCs (Fig. 4B). In parallel, BLOC1S1-DM HLCs showed increased BLOC1S1 levels which were blunted following isogenic correction (Fig. 4B). At the same time, the Cathepsin L activity of these lysosomes was unchanged between genotypes (Fig. 4C), as was evident in control and LKO hepatocytes [10] (Fig. 4C). Interestingly, LD accumulation was significantly induced in the BLOC1S1-DM HLCs (Fig. 4D). These data suggest that a linear relationship exists between functional lysosome content and lysosomal lipolysis in hepatocytes, with an inverse relationship with LD accumulation.
Figure 4. Complex heterozygous BLOC1S1 mutants decreases lysosome biogenesis and results in more lipid accumulation in iPSCs-derived hepatic-like cells (HLCs).
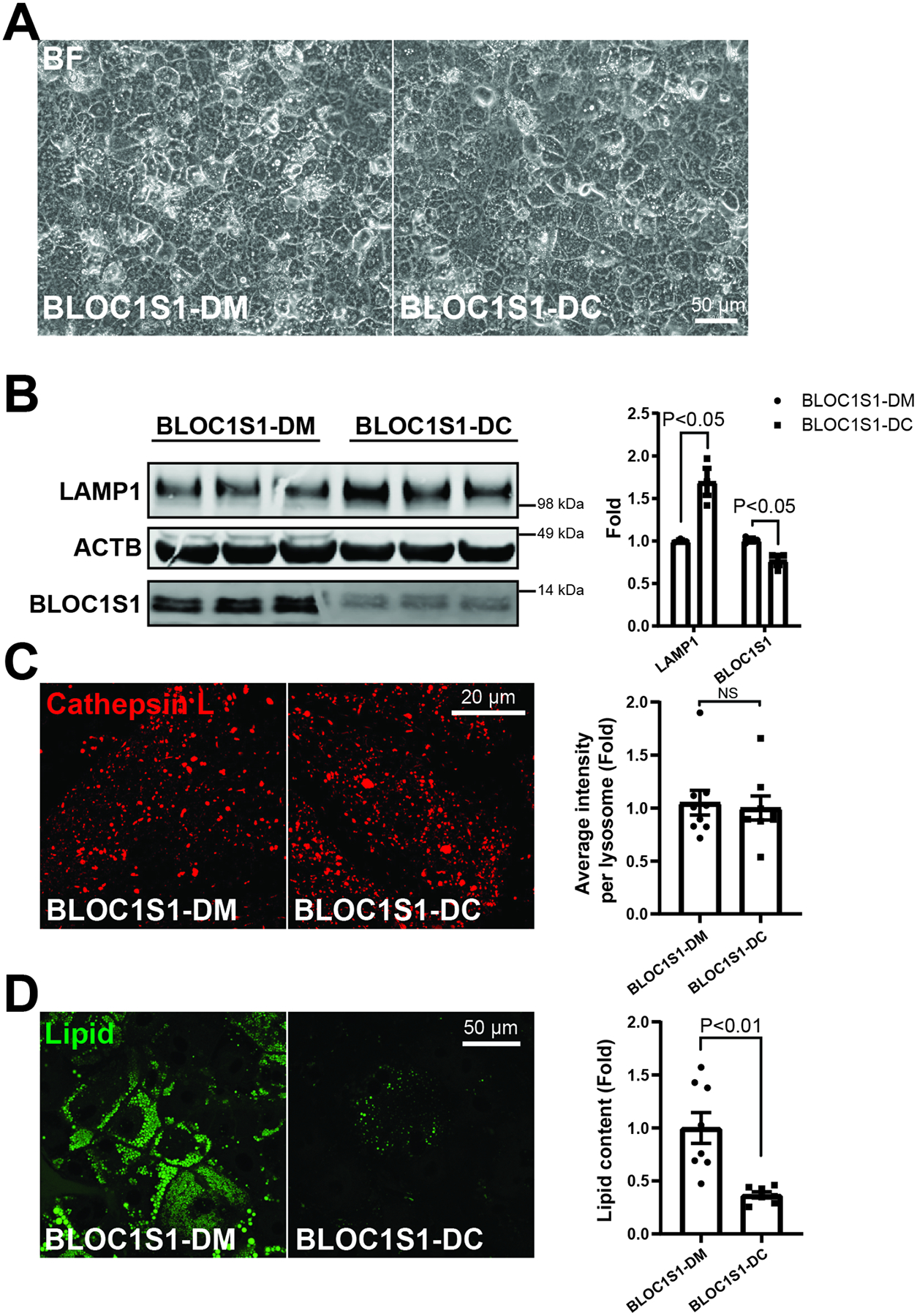
(A) Representative images show typical hepatic cell morphology in both BLOC1S1-DM and BLOC1S1-DC HLCs. (B) Immunoblot analysis of LAMP1 expression in heterozygous mutant and isogenic correct HLCs. (C) Confocal microscopy to evaluate lysosome activity with Cathepsin L activity staining in both HLC genotypes. GCN5L1 mutants (GCN5L1-DM) does not impair the CTSL activity of lysosome in HLCs. The relative fluorescence per cell and normalized to cellular lysosomal area was quantified in 10–12 cells per group (histogram). (D) BODIPY (Green) staining of lipid droplet in mutant an isogenic corrected HLCs with the accompanying histogram of quantitation of n > 10 cells per group. Scale bars shown in image panels.
4. Discussion:
In this report we show that the role of BLOC1S1 in hepatic endosome-lysosome biology extends to include a role in controlling lysosomal content and that the lysosomal content directly governs lysosomal lipolysis. This study also shows that excessive lysosome content orchestrates lysosomal lipolysis independent of autophagic or chaperone-mediated autophagy, and that this in turn depletes intracellular hepatocyte lipid deposits. Moreover, the studies in the human bloc1s1 mutation iPSC derived hepatocyte like cells further support the role of BLOC1S1 in controlling lysosome content and lipolysis, but also highlights the distinct phenotypes of genetic depletion in mouse models vs. point mutations in human disease.
The role of BLOC1S1 in the cytoplasm was initially linked with vacuolar organelle trafficking which included a requirement of BLOC1S1 for canonical lysosome positioning. This was mediated in part by the interaction of BLOC1S1 with alpha-tubulin acetyltransferase 1, and the nucleoporin RanBP2 to acetylate the microtubular network required to lysosome transport [4]. Lysosome positioning was also dependent on the interaction of BLOC1S1 with the BORC complex [6]. Subsequent data found that BLOC1S1 was essential for the recycling of lysosomes through the program of lysosomal reformation [10]. In that study BLOC1S1 enabled lysosomal reformation via its interaction with kinesin motor proteins and the actin cytoskeleton. At the same time, despite these disruptions of lysosome trafficking and recycling, the hepatic content of lysosomes was markedly increased by the genetic knockout of bloc1s1 and these lysosomes predominantly retained their functional capacity [10]. In this study we expand our understanding of the role of BLOC1S1 on lysosomal function where we show that this increase in lysosome content and activity augments lysosomal lipolysis to deplete hepatic intracellular lipid droplet accumulation. This preferential catabolism of lipids within lysosomes as a mass action effect was supported by the replication of this lipolysis following the genetic induction of lysosomal biogenesis and by the human BLOC1S1 mutant genetic correction effect, where the increase in lysosomal content diminished intracellular lipid content in iPSC-derived hepatocyte-like cells. Our findings are in line with the previous findings that exogenous overexpression of TFEB increase lysosomal biogenesis and lipid metabolism in mice liver [38].
The concept that lysosomes could promote lipolysis independent of lipid droplet catabolism or autophagic-dependent pathways was identified in the liver under nutrient limiting conditions using live cell microcopy [26]. There, direct lysosomes-LD interaction-dependent lipolysis was not affected by perturbations to crucial components of the canonical macro-autophagy machinery and occurred in the absence of double-membrane lipo-autophagosomes [34]. Consistently, in this manuscript we now demonstrate that, beside lysosome-lipid droplet interactions, the lysosome content in the liver also is important in driving lysosomal lipolysis and that the genetic induction of lysosomal biogenesis may be a strategy to ameliorate the fatty liver. Interestingly, even though lysosomes in LKO hepatocytes lost their centrifugal trafficking due to impaired KIF5B motor protein recruitment to lysosomes [6; 39], LD trafficking to lysosome appeared to be maintained (Figure 2A). Although the mechanism of directly lysosome-LD interaction is unknown, kiss-and-run events as well as prolonged interactions between LDs and lysosomes were observed [34]. Our findings indicate that KIF5B-dependent lysosome movement is not required for LD trafficking to lysosomes and that LDs may traffic to lysosomes independent of lysosomal cytosolic location.
Furthermore, this study expands the role of BLOC1S1 in controlling the disposition of fat stores in the liver. Prior work showed that BLOC1S1/GCN5L1 was a negative regulator of mitochondrial fatty acid oxidation, and it is feasible that the depletion of bloc1s1 may further orchestrate FAO via the generation of free fatty acids for uptake and utilization for catabolism within mitochondria. If this were operational it would mirror the effect of ATGL in LD’s which also facilitate fatty acid liberation for FAO [25]. It is interesting that in the LKO liver, ATGL and hepatic lipase transcript levels were diminished, which could be compatible with the LD enzymes being suppressed in response to excess lysosomal lipolysis in the absence of BLOC1S1. Furthermore, knockout of other subunits of BORC decreased LDs in HeLa cells [40], although in that scenario, there was no obvious difference in triglyceride levels between WT, BORC subunits KO and rescued HeLa cells after oleic acid supplementation [40]. In contrast, in this study we find that BLOC1S1 regulates TG in both murine hepatocytes and human iPSC-derived HLCs which indicates tissue specific regulation of BLOC1S1 on lipid metabolism.
This study also shows that the murine LKO phenotype is quite distinct from the compound heterozygous phenotype in human patient derived HLCs. This distinction is not uncommon comparing knockout mice to human mutations of the same gene and highlight the differences between genetic mutations of a protein that interacts with a multitude of proteins in different super-complexes [1; 10] compared to the depletion at the level of the protein. This concept also underscores the limitations of using the knockout mouse model to study the mutations in bloc1s1 linked with leukodystrophy. This concept is further complicated in the neuronal knockout of bloc1s1 in neurons by the nestin cre-recombinase is embryonically lethal [9]. Here, the generation of patient mutant iPSCs will be a useful model going forward to generate different neuronal cell lineages to explore if mutations in bloc1s1 confer detrimental effects via its role in lysosomal function, or via its effects on endosomal, autophagosome or mitochondrial functioning. Also, given the different phenotypes in the murine LKO model and the human heterozygous mutation in HLCs suggests that the functional consequences of this human mutation on lysosome biology will need to be explored at the structure-function and protein interaction level to determine how human mutations perse modulate endo-lysosomal biology.
In conclusion, this study shows that the repertoire of functions of BLOC1S1 in endo-lysosomal biology extends to controlling the intracellular hepatic lysosome content. Moreover, that the number of lysosomes within hepatocytes can moderate the degree of lysosomal lipolysis independent of lipophagy. Furthermore, the deletion of bloc1s1 diminished both baseline hepatic storage of lipids as shown in this study, and the attenuates the accumulation of fat in the liver in response to high fat feeding as shown previously [13]. Hence, the blunting of BLOC1S1 may be a therapeutic target in the management of fatty liver disease and/or alternatively the augmentation of lysosomal biogenesis itself could be exploited to ameliorate hepatosteatosis.
Supplementary Material
Highlights.
BLOC1S1 levels and genetic fidelity control hepatic lysosomal content
Hepatic functional lysosome content determines the rate of lysosomal lipolysis
Lysosomal lipolysis modulates hepatic lipid storage independent of autophagy
Acknowledgements.
We thank Dr. M.A. McNiven (Mayo Clinic) for the kind gift of GFP-RFP-PLIN2 plasmid and Dr. R. Puertollano (NHLBI) for TFEB(S211A) adenovirus; We thank the National Heart Lung and Blood Institute (NHLBI) Light Microscopy core for assistance on imaging acquisition and analysis, IPSC core for iPSC generation and technical support, and animal facility for animal maintenance.
Funding.
NHLBI Division of Intramural Research (MNS – ZIA-HL005102).
Abbreviations:
- ATGL
adipocyte triglyceride lipase
- BLOC1S1
biogenesis of lysosomal complex 1, subunit 1
- CMA
chaperone mediated autophagy
- FAO
fatty acid oxidation
- GCN5L1
general control non-depressible 5 like 1
- HLC
hepatocyte like cell
- iPSC
inducible pluripotent stem cell
- KIF5B
kinesin family member 5B
- LALi
lysosome acid lipase inhibitor
- LAMP1
lysosome associated membrane protein 1
- LD
lipid droplet
- LKO
liver knockout
- TFEB
transcription factor EB
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest.
No conflicts to declare.
Appendix A. Supplementary Data
Supplementary data to this article can be found online at…
Data availability
Data will be made available on request.
References:
- [1].Wu K, Scott I, Wang L, Thapa D, Sack MN, 2021. The emerging roles of GCN5L1 in mitochondrial and vacuolar organelle biology. Biochim Biophys Acta Gene Regul Mech 1864(2):194598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Scott I, Webster BR, Li JH, Sack MN, 2012. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J 443(3):655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Scott I, Wang L, Wu K, Thapa D, Sack MN, 2018. GCN5L1/BLOS1 Links Acetylation, Organelle Remodeling, and Metabolism. Trends Cell Biol 28(5):346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wu K, Wang L, Chen Y, Pirooznia M, Singh K, Walde S, et al. , 2018. GCN5L1 interacts with alphaTAT1 and RanBP2 to regulate hepatic alpha-tubulin acetylation and lysosome trafficking. J Cell Sci 131(22):pii: jcs221036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Donato V, Bonora M, Simoneschi D, Sartini D, Kudo Y, Saraf A, et al. , 2017. The TDH-GCN5L1-Fbxo15-KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat Cell Biol 19(4):341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pu J, Schindler C, Jia R, Jarnik M, Backlund P, Bonifacino JS, 2015. BORC, a multisubunit complex that regulates lysosome positioning. Dev Cell 33(2):176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Niwa S, Tao L, Lu SY, Liew GM, Feng W, Nachury MV, et al. , 2017. BORC Regulates the Axonal Transport of Synaptic Vesicle Precursors by Activating ARL-8. Curr Biol 27(17):2569–2578 e2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bae D, Moore KA, Mella JM, Hayashi SY, Hollien J, 2019. Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J Cell Biol 218(4):1118–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang A, He X, Zhang L, Yang L, Woodman P, Li W, 2014. Biogenesis of lysosome-related organelles complex-1 subunit 1 (BLOS1) interacts with sorting nexin 2 and the endosomal sorting complex required for transport-I (ESCRT-I) component TSG101 to mediate the sorting of epidermal growth factor receptor into endosomal compartments. J Biol Chem 289(42):29180–29194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu K, Seylani A, Wu J, Wu X, Bleck CKE, Sack MN, 2021. BLOC1S1/GCN5L1/BORCS1 is a critical mediator for the initiation of autolysosomal tubulation. Autophagy 17(11):3707–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].John Peter AT, Lachmann J, Rana M, Bunge M, Cabrera M, Ungermann C, 2013. The BLOC-1 complex promotes endosomal maturation by recruiting the Rab5 GTPase-activating protein Msb3. J Cell Biol 201(1):97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang L, Zhu L, Wu K, Chen Y, Lee DY, Gucek M, et al. , 2019. Mitochondrial General Control of Amino Acid Synthesis 5 Like 1 Regulates Glutaminolysis, Mammalian Target of Rapamycin Complex 1 Activity, and Murine Liver Regeneration. Hepatology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thapa D, Wu K, Stoner MW, Xie B, Zhang M, Manning JR, et al. , 2018. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial beta-oxidation enzyme HADHA. J Biol Chem 293(46):17676–17684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang T, Cui Y, Wu Y, Meng J, Han L, Zhang J, et al. , 2022. Mitochondrial GCN5L1 regulates glutaminase acetylation and hepatocellular carcinoma. Clin Transl Med 12(5):e852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Webster BR, Scott I, Traba J, Han K, Sack MN, 2014. Regulation of autophagy and mitophagy by nutrient availability and acetylation. Biochim Biophys Acta 1841(4):525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Scott I, Webster BR, Chan CK, Okonkwo JU, Han K, Sack MN, 2014. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem 289(5):2864–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang L, Scott I, Zhu L, Wu K, Han K, Chen Y, et al. , 2017. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat Commun 8(1):523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Manning JR, Thapa D, Zhang M, Stoner MW, Traba J, Corey C, et al. , 2019. Loss of GCN5L1 in cardiac cells disrupts glucose metabolism and promotes cell death via reduced Akt/mTORC2 signaling. Biochem J 476(12):1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bertoli-Avella AM, Kandaswamy KK, Khan S, Ordonez-Herrera N, Tripolszki K, Beetz C, et al. , 2021. Combining exome/genome sequencing with data repository analysis reveals novel gene-disease associations for a wide range of genetic disorders. Genet Med 23(8):1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ito Y, Hartley T, Baird S, Venkateswaran S, Simons C, Wolf NI, et al. , 2018. Lysosomal dysfunction in TMEM106B hypomyelinating leukodystrophy. Neurol Genet 4(6):e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Feltri ML, Weinstock NI, Favret J, Dhimal N, Wrabetz L, Shin D, 2021. Mechanisms of demyelination and neurodegeneration in globoid cell leukodystrophy. Glia 69(10):2309–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sevin C, Aubourg P, Cartier N, 2007. Enzyme, cell and gene-based therapies for metachromatic leukodystrophy. J Inherit Metab Dis 30(2):175–183. [DOI] [PubMed] [Google Scholar]
- [23].Drizyte-Miller K, Schott MB, McNiven MA, 2020. Lipid Droplet Contacts With Autophagosomes, Lysosomes, and Other Degradative Vesicles. Contact (Thousand Oaks) 3:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. , 2014. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15(9):846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG, 2011. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53(1):116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schulze RJ, Krueger EW, Weller SG, Johnson KM, Casey CA, Schott MB, et al. , 2020. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc Natl Acad Sci U S A 117(51):32443–32452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Grabner GF, Xie H, Schweiger M, Zechner R, 2021. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nat Metab 3(11):1445–1465. [DOI] [PubMed] [Google Scholar]
- [28].Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP, 2015. Hepatic lipid droplet biology: Getting to the root of fatty liver. Hepatology 62(3):964–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O, et al. , 2011. Wnt signaling regulates hepatic metabolism. Sci Signal 4(158):ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Du C, Feng Y, Qiu D, Xu Y, Pang M, Cai N, et al. , 2018. Highly efficient and expedited hepatic differentiation from human pluripotent stem cells by pure small-molecule cocktails. Stem Cell Res Ther 9(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu K, Takanohashi A, Woidill S, Seylani A, Helman G, Dias P, et al. , 2022. Generation of human induced pluripotential stem cells from individuals with complex heterozygous, isogenic corrected, and homozygous Bloc1s1 mutations. Stem Cell Res 64:102905. [DOI] [PubMed] [Google Scholar]
- [32].Martina JA, Chen Y, Gucek M, Puertollano R, 2012. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8(6):903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ding Y, Zhang S, Yang L, Na H, Zhang P, Zhang H, et al. , 2013. Isolating lipid droplets from multiple species. Nat Protoc 8(1):43–51. [DOI] [PubMed] [Google Scholar]
- [34].Schulze RJ, Krueger EW, Weller SG, Johnson KM, Casey CA, Schott MB, et al. , 2020. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proceedings of the National Academy of Sciences 117(51):32443–32452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, et al. , 2019. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol 218(10):3320–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM, 2006. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103(15):5805–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. , 2009. A gene network regulating lysosomal biogenesis and function. Science 325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [38].Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. , 2013. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15(6):647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu K, Wang L, Chen Y, Pirooznia M, Singh K, Walde S, et al. , 2018. GCN5L1 interacts with alphaTAT1 and RanBP2 to regulate hepatic alpha-tubulin acetylation and lysosome trafficking. J Cell Sci 131(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Anderson J, Walker G, Pu J, 2022. BORC-ARL8-HOPS ensemble is required for lysosomal cholesterol egress through NPC2. Mol Biol Cell 33(9):ar81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.