

Abstract
The combination of lipopolysaccharide (LPS) and hypoxia-ischemia (HI) has been used to model the brain injury sustained by sick pre-term infants in order to study the pathological conditions of diffuse white matter injury, which is a major cause of preterm morbidity. Prior studies have shown that the timing and dose of LPS administration will determine whether the injury is reduced or exacerbated. Here we show that administering a single injection of LPS (0.1 mg/kg) to postnatal-day-2 rat pups 14 hours before inducing HI effectively protects the brain from HI-associated damage. We show that the LPS-treated HI rat pups have significantly less histopathology compared to the saline-treated HI rat pups. Apoptotic deaths were dramatically curtailed in both the neocortex and white matter when evaluated at 2 days of recovery. Microglial activation was reduced when the percentage of CD68+/Iba1+ cells was quantified in the neocortex of the LPS-treated vs the saline-treated HI rat pups. One mechanism through which LPS pre-treatment appears to be preventing injury is through the AKT-endothelial nitric oxide synthase (eNOS) pathway as LPS induced an increase in both the expression and phosphorylation of eNOS. Altogether these data show that the neocortex, as well as the white matter sustain damage after HI at this timepoint in forebrain development and that acutely activating the immune system can protect the brain from brain injury.
Keywords: apoptosis, encephalopathy, inflammation, pre-term birth, cerebral blood flow
1. Introduction
Preterm birth is a major cause of neonatal mortality and permanent neurological disabilities, accounting for 17% of infant deaths every year in the United States [1]. It remains a severe public health problem with around 15 million infants worldwide that are born before 37 weeks of gestation annually. Approximately 40% of preterm infants develop cognitive and behavioral deficits [2, 3]. In the extremely preterm infant (born less than 28 weeks of gestation), evidence suggests that perinatal and postnatal inflammation exacerbates the extent of injury [3]. These inflammatory insults can induce focal necrosis in the deep white matter (WM) but more often produce diffuse white matter injury (dWMI) [4]. Recent studies investigating the conditions that trigger dWMI in rat models report that the combination of LPS-induced fetal inflammation and hypoxia produces white matter injury [5]. However, lipopolysaccharide preconditioning has been shown to have the opposite effect in other dual-hit models of perinatal brain injury [6, 7].
Lipopolysaccharide (LPS) is a large glycolipid found in the outer membrane of Gram-negative bacteria and it is widely used as an inflammatory agent. LPS is comprised of a hydrophobic lipid A, a core oligosaccharide and an O antigen. The hydrophobic anchor of LPS, lipid A, is detected by the innate immunity toll-like receptor 4 (TLR4) present on dendritic cells, macrophages and endothelial cells [8]. Upon detecting the lipid A of LPS, TLR4 triggers the biosynthesis of inflammatory mediators like tumor necrosis factor alpha (TNF-α) and interleukin 1β, which, in turn, activate adaptive immune responses and tissue inflammation. LPS has been shown previously to sensitize hypoxia-ischemia (HI) induced white matter injury through neuroinflammation induction [6]. However, other studies have shown that LPS can exert a neuroprotective effect [7]. Altogether, different results are obtained depending upon the LPS dose, timing, species and model.
Here, we investigated a dual-hit rat model in P2 rats to model injury sustained by sick preterm infants. We injected a single dose of 0.1 mg/kg LPS to both male and female Wistar rats on postnatal day 2, 14 hours prior to inducing systemic HI. The dose and the timing of the LPS injection had previously been shown to induce neuroinflammation in a dual-hit hypoxic model [5]. We used the Vannucci model to induce unilateral HI brain damage on P3 Wistar rat pups and evaluated the tissue damage in the ipsilateral neocortex and white matter. In addition, we quantified the overall level of apoptosis and microgliosis in both regions. To glean insights into the mechanisms through which systemic inflammation was affecting brain physiology, we measured mRNA levels of nitric oxide synthase 3 (Nos3), Insulin-like growth factor 1 (Igf1) and nuclear factor erythroid 2-related factor 2 (Nfe2l2 or Nrf2), as well as the protein levels of phosphorylated endothelial nitric oxide synthase (eNOS) and AKT kinase.
2. Materials and Methods
2.1. Rodents
All experiments were performed in accordance with research guidelines set forth by the Institutional Animal Care and Use Committee of Rutgers New Jersey Medical School and were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23) revised in 1996 and the ARRIVE guidelines. Timed pregnant Wistar rats at embryonic day 18 of gestation were purchased from Charles River Laboratories (Wilmington, MA, USA). Following delivery, litter sizes were adjusted to 12–13 pups per litter, and efforts were made to ensure the number of each sex and pup weights were equal and consistent. Animals were group housed and kept on a 12-h light/dark cycle with ad libitum access to food and autoclaved water. Rat pups were intraperitoneally injected with either single dose 0.1mg/kg lipopolysaccharide (LPS, Sigma, St. Louis, MO, Cat # L2880 Escherichia coli strain O55:B5) or same dose phosphate-buffered saline (PBS) at postnatal day 2, 14 hours before HI injury.
2.2. Neonatal HI brain injury
Cerebral HI in 3-day-old Wistar rat pups (P3, day of birth = P0; mean body mass = 7.07 g) as a model of late preterm injury was performed as previously described [9, 10]. Briefly, rats were anesthetized with isoflurane (3–4% induction, 1–2% maintenance) prior to right common carotid artery cauterization. A special effort was made to carefully isolate the carotid without damaging other structures contained within the carotid sheath (i.e., internal jugular vein and vagus nerve). The neck incision was sutured with 4–0 surgical silk. Following a one-hour recovery period, rats were exposed to 75 min of hypoxia in a humidified 8% oxygen/nitrogen balance. Sham rats were anesthetized and underwent isolation of the right common carotid without cauterization and then were exposed to hypoxia. Following hypoxia exposure, the pups were returned to their respective dam. At least 2 litters of rats were used for each experiment to reduce potential litter effects. Rats in each litter were randomly assigned to experimental groups after HI injury. Sample sizes per experiment were chosen to achieve sufficient statistical power with minimal numbers of animals based on pilot studies.
2.3. Brain Histology and Immunofluorescence
Two days after HI injury (P5), mice were deeply anesthetized with a mixture of ketamine (87.5 mg/kg) and xylazine (12.5 mg/kg) and then perfused with 3% paraformaldehyde (PFA). Whole brains were dehydrated in 70% ethanol and embedded into paraffin. A total of six sections per brain were used to evaluate the damage produced by the HI insults. Neocortex and white matter damages were assessed from sections taken 1.0 mm rostral to Bregma. All sections were stained with hematoxylin and eosin and imaged under 40X magnification using a Q-imaging mono 12-bit camera interfaced with iVision 4 scientific imaging software (Scanalytics, Rockville, MD, USA). Sections were labeled with random codes and evaluated by an investigator who was blinded to the identity of the samples. The whole-brain images were scanned with Keyence BZ-X710 All-in-One Fluorescent Microscope installed with BZ-X Viewer.
In situ end labeling (ISEL): Two days after HI injury, rats were deeply anesthetized with a mixture of ketamine (87.5 mg/kg) and xylazine (12.5 mg/kg) (or sodium pentobarbital) before intracardiac perfusion with 3% PFA in PBS. Brains were post-fixed overnight in 4% PFA/PBS, cryoprotected with 30% sucrose overnight and embedded in Tissue-Tek OCT matrix (Sakura Finetek, Torrance, CA, USA). Serial coronal sections of 25–30 μm thickness were taken through the 1.0 mm Bregma regions using a cryostat at −14 °C and mounted on slides. Sections were dehydrated and rehydrated in ethanol and water and incubated with 10 μM dNTP mix containing Digoxigenin-dUTP and 20 U/mL Klenow Fragment (Roche; Basel, Switzerland) at room temperature for 2 h. Digoxygenin-labeled nucleotides were detected using Anti-Digoxigenin-Fluorescein (sheep polyclonal, Roche, 1:75) incubated overnight at 4 °C. Images were collected by an investigator blinded to each group using an Olympus Provis fluorescent microscope. Images were captured using a Q-imaging mono 12-bit camera interfaced with iVision 4 scientific imaging software (Scanalytics, Rockville, MD, USA). Signal intensities were quantified using Fiji with plugins.
Two days (P5) after HI injury, mice were deeply anesthetized with ketamine/xylazine before intracardiac perfusion with 3% PFA in PBS. The brains were postfixed overnight and embedded in Tissue-Tek OCT matrix (Sakura Finetek, Torrance, CA, USA). Serial coronal sections 20 μm thick were taken through the 1.0mm Bregma using a cryostat cooled to −14 °C and mounted onto Superfrost+ slides. Sections were incubated with primary antibodies in 1% goat serum/0.05% Triton X-100/PBS at 4 °C overnight. Primary antibodies included: (1) anti-Iba-1 (rabbit polyclonal; Wako Chemicals USA, Cat. # 019–19,741; 1:250); (2) anti-CD68 (rat monoclonal; Bio-Rad, Cat. # MCA1957; 1:50). Secondary antibodies against the appropriate species were incubated for 2 h at room temperature. Secondary antibodies included: donkey anti-rabbit Cy5 (Cat. # 711–175-152); (2) Donkey anti-rat Alexa 488 (Cat. # 712–546-153) (all from J. Lin, et al. Experimental Neurology 330 (2020) 113324 2 Jackson ImmunoResearch Labs; 1:250). Sections were mounted in Prolong Gold Antifade with DAPI (ThermoFisher Scientific).
2.4. RNA extraction, reverse transcription and real-time quantitative PCR
Sixteen hours post intraperitoneal administration of either a single dose 0.1mg/kg of LPS or the same volume of PBS (vehicle) on postnatal day 2, the dorsal neopalium within the territory of the middle cerebral artery was extracted and flash frozen. The tissue was then pulverized using a micro-mortar and pestle and the RNA was extracted using the RNeasy Mini Kit (Qiagen, Cat # 74106). cDNA was reverse transcribed using the iScript™ cDNA Synthesis Kit (Bio-Rad, Cat # 1708891) and real-time quantitative polymerase chain reaction (qPCR) was performed using iTaq SYBR supermix (Bio-Rad, Cat # 172-5124) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). For all samples, the mRNA levels of Nos3, Igf1 and Nrf2 were normalized to the levels of the housekeeping gene Peptidylprolyl Isomerase A (Ppia). The primers for the different genes were purchased from Integrated DNA Technologies (Nos3: Rn.PT.58.6917365, Igf1: Rn.PT.58.37138478, Nfe2l2: Rn.PT.58.23835958, Ppia: Rn.PT.39a.22214830).
2.5. Western Blotting
Sixteen hours post intraperitoneal administration of LPS or PBS at postnatal day 2, the dorsal neopalium within the territory of the middle cerebral artery was extracted and flash frozen. The tissue was pulverized using a micro-mortar and pestle and then solubilized in lysis buffer (Thermo Scientific, Cat # 89900), supplemented with 1x Halt Protease/Phosphatase Inhibitor Cocktail (Thermo Scientific, Cat # 78440). Twenty micrograms of protein for each sample were loaded onto 4–12% Bis-Tris gels for SDS-PAGE. Protein levels were then quantified by Western blot using the following primary and secondary antibodies: p-eNOS (S1177) (Fisher Scientific, Cat. # BDB612393; 1/1000), p-AKT(S473) (Cell Signaling Cat. # 4060; 1/1000), t-AKT (Cell Signaling, Cat. # 9272; 1/1000), β-actin (Cell Signaling, Cat. # 5125; 1/1000), goat anti-rabbit HRP (Jackson ImmunoResearch Labs, Cat. # 111-035-003; 1/5000) and goat anti-mouse HRP (Jackson ImmunoResearch Labs, Cat. # 115-035-003; 1/5000). Three different total eNOS antibodies were tested on the rat lysates (Fisher Scientific, Cat. # BDB610297, Fisher Scientific, Cat # 610299 and Cell Signaling, Cat. # 32027), but none of these antibodies detected a band at the appropriate molecular weight for eNOS (140 kDa). Quantification was performed after normalizing each sample to β-actin.
2.6. Data Analysis and Statistics
Raw data from Image J/ Fiji analyses were imported into Prism 9 (GraphPad Software, La Jolla, CA, USA) for statistical analyses using unpaired t-test for comparison between 2 groups or ANOVA followed by Tukey’s post hoc when comparisons were made between more than 2 groups. For non-parametric analyses, results were analyzed using the Mann-Whitney test. Graphs were produced in Prism 9, and error bars denote standard error of the mean (SEM). Comparisons were interpreted as significant when associated with p < 0.05.
3. Results
3.1. LPS administered before HI surgery protects the neocortex and WM from HI-associated brain damage
To assess the effects of LPS on HI-associated brain damage, we injected Wistar rats with 0.1 mg/kg LPS 14 hours before HI on postnatal day 3 (Figure 1A). After perfusion, brain slices at 1.0 mm rostral to bregma were collected and stained with H&E to evaluate overall brain histopathology. HI induced significant brain tissue loss in the PBS-treated rats on the ipsilateral side of the brain (Figure 1E). Contrary to our expectations, there was both gray and white matter damage. Also contrary to our expectations, there was limited brain damage in the LPS-treated HI rats (henceforth referred to as the dual hit group) (Figure 1). To further evaluate the effects of LPS on the neocortex and WM, we performed high-power imaging of ipsilateral H&E brain slices from the naïve, LPS+Sham, dual hit, and PBS+HI groups. Under 40X magnification, the PBS+HI group showed low tissue density and shrunken nuclei in the neocortex within the territory of the middle cerebral artery, which was not observed in the LPS-treated group, that looked more similar to the brains of the naïve group (Figure 1F–I). In the subcortical WM there was extensive tissue loss in the PBS+HI brains (Figure 1M). In contrast, the dual hit rats showed much less tissue damage compared to the PBS+HI group and more resembled the naïve group (Figure 1J–L). Taken altogether, LPS administration 14 h before HI surgery protected the ipsilateral neocortex and WM of the P3 rat brain from HI-induced tissue loss.
Figure 1. Pre-administering LPS before HI surgery protects neocortex and white matter from HI-associated brain damage.

(A) Outline of the experimental paradigm of LPS injection and HI injury. Single-dose 0.1mg/kg LPS was administered 14 hours before artery cauterization and 75 min, 8% oxygen condition. Perfusion was performed on postnatal day 5. (B-E) Whole-brain scanning of the H&E stained p5 rats’ 1.0mm bregma sections. Images from Naïve, LPS+Sham, PBS+ HI, and LPS+HI, respectively (ipsi=ipsilateral, contra=contralateral). (F-I) Representative 40X images of H&E stained P5 brain sections from 1.0mm rostral to bregma at the neocortex. Sections are from Naïve, LPS+Sham, PBS+ HI, and LPS+HI, respectively. Scale bar= 40μm. (J-M) Representative 40X images of H&E stained P5 brain sections from 1.0mm rostral to bregma at WM. Sections are from Naïve, LPS+Sham, PBS+ HI, and LPS+HI. Scale bar= 40μm.
3.2. LPS reduces HI-induced neocortical and WM apoptosis
To further investigate the mechanism of LPS neuroprotection from the HI-induced brain damage, sections from 1.0 mm rostral to bregma were collected from pups terminated 2 days after HI and stained using the in-situ end labeling (ISEL) method to evaluate apoptosis. ISEL detects early-stage apoptotic cells by filling in fragmented DNA with digoxigenin-labeled deoxyuridine triphosphates (dUTPs) and deoxyribonucleotide triphosphates (dNTPs), and yields fewer false positives than terminal deoxynucleotidyltransferase nick-end labeling (TUNEL) [11, 12]. ISEL+ cells were readily apparent in the dorsomedial neocortex of the PBS-treated HI rats (170 ± 51.61 cells/mm2; mean ± SEM). By contrast there were few (16.13 ± 10.29) ISEL+ cells in the dual hit group and even fewer (4.88 ± 1.59) ISEL+ cells in the naïve groups (**p<0.01 comparing PBS-HI vs dual hit group) (Figure 2A, B). In the subcortical WM, significantly higher ISEL+ cells were detected in the PBS+HI group than in the naïve and the LPS-treated groups (PBS-HI 12.75 ± 2.33 cells/mm2 vs. dual hit 5.38 ± 0.68; LPS-Sham 2.50 ± 0.42; naïve 4.63 ± 0.73 cells/mm2; mean ± SEM **p<0.01 for PBS-HI vs dual hit). There was no significant difference between the LPS+Sham, dual hit and naïve rats (Figure 2C, D). These observations indicate that LPS 14 hours before HI reduced the extent of apoptotic cell deaths in both the rat neocortex and the subcortical WM.
Figure 2. LPS reduces HI-induced neocortical and WM apoptosis.
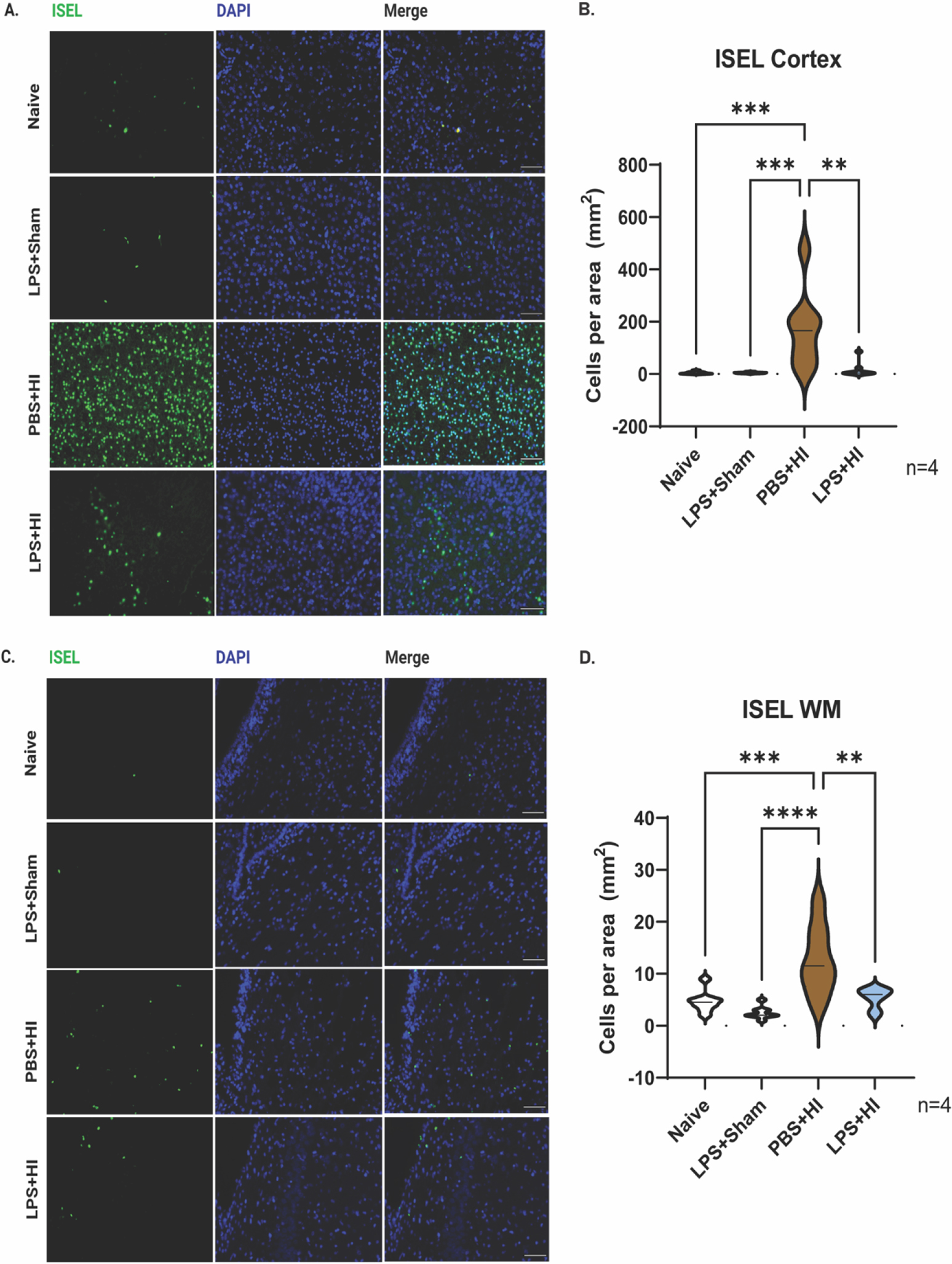
(A, C) Representative images of ISEL (green) staining of the ipsilateral neocortex and WM two days after HI. Brain sections are from Naïve, LPS+Sham, PBS+HI, LPS+HI, respectively (n=4). Scale bar= 100μm. (B, D) Violin plots showing ISEL counts per mm2 at neocortex and WM. Bold dash lines represent the median, and thin dash lines represent upper and lower quartiles. Statistical analyses were performed using one-way ANOVA with multiple-comparison tests. F (3, 28) =9.448(B) and 12.07(D), respectively.
3.3. LPS modestly suppresses microglial cell activation
We postulated that the LPS was suppressing tissue inflammation during recovery from HI based on earlier studies [13]. To test this hypothesis, sections from 1.0 mm bregma taken at 2 days after HI (P5) were stained for Iba-1 and CD68. CD68 is a transmembrane lysosomal glycoprotein expressed by tissue macrophages that indicates phagocytic activity. Iba-1 is a cytoplasmic protein considered to be a pan microglial/macrophage cell marker [14, 15]. From a pilot study, where we compared CD68 expression between controls and injured brains, we classified Iba-1+ cells as positive for CD68 if they had 3 or more punctate CD68+ dots in their cytoplasm. Using these criteria, the PBS+HI group had significantly more Iba1+/CD68+ cells in the dorsomedial neocortex compared to the naïve group (PBS-HI 10.25 ± 2.96 cells/mm2 vs naïve 0.25 ± 0.16 cells/mm2; mean ± SEM ***p<0.01 for PBS-HI vs naïve) (Figure 3A, B), consistent with previous data showing that HI increases microgliosis [16]. However, there were significantly fewer Iba1+/CD68+ cells in the dual hit group when compared to the PBS-HI (dual hit 1.13 ± 0.48 cells/mm2 vs. PBS-HI 10.25 ± 2.96 cells/mm2; mean ± SEM **p<0.01 for dual hit vs PBS-HI), indicating LPS pre-administration either reduced microglial activation or decreased the migration of macrophages into the brain. We then evaluated the subcortical WM, but no significant differences were observed between the naïve, LPS-treated, HI-PBS and dual hit groups.
Figure 3. LPS preconditioning suppresses microglial cell activation in the dual-hit rat model.
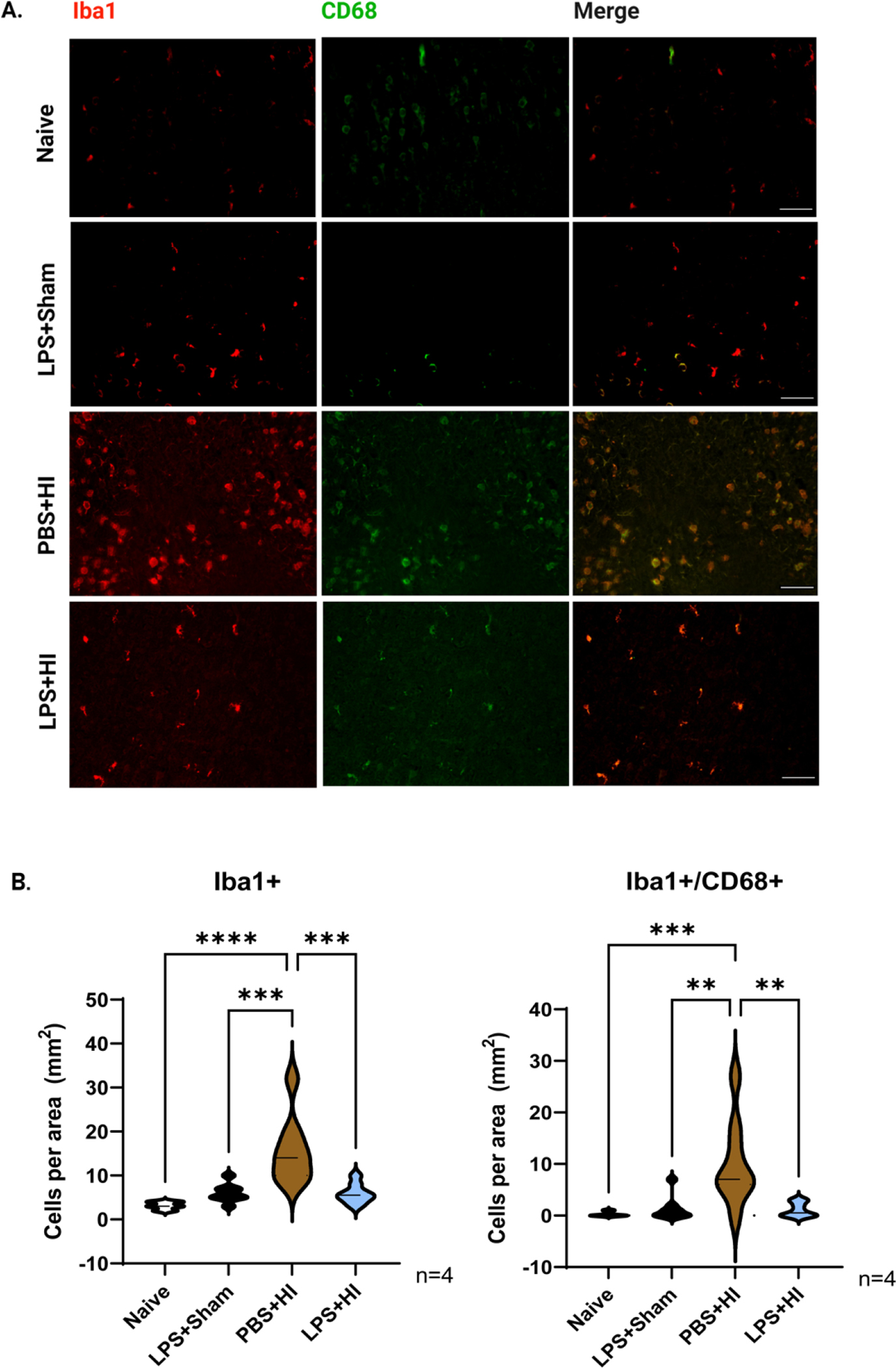
(A) Representative images of Iba-1(red)/CD68(green) staining of the neocortex 2 days after HI (P5). Brain sections are from Naïve, LPS+Sham, PBS+HI, LPS+HI, respectively (n=4). Scale bar=100 μm. (B) Violin plots showing Iba1+ and Iba1+/CD68+ counts per mm2 in the neocortex, respectively. Bold dash lines represent the median, and thin dash lines represent upper and lower quartiles. Statistical analyses were performed using one-way ANOVA with multiple-comparison tests. F (3,28) =13.83 and 9.034, respectively.
3.4. LPS preconditioning increases AKT-eNOS signaling
To further understand how the systemic inflammation induced by LPS was reducing the extent of HI brain damage, we performed qPCR analysis for Nos3, the gene encoding for endothelial nitric oxide synthase (eNOS), and we performed Western blotting to measure the phosphorylation status of eNOS (p-eNOS S1177) at 16 h after LPS administration (Figure 4A, D, E). These analyses revealed that LPS significantly increased the mRNA levels of Nos3 (vehicle 0.98 normalized units vs LPS 1.49; medians ****p<0.001 for vehicle vs LPS), as well as the protein levels of p-eNOS (vehicle 2.19 normalized units ± 0.06 vs LPS 2.39 ± 0.06; means ± SEM *p<0.05 for vehicle vs LPS). Since AKT kinase can directly phosphorylate eNOS we also evaluated both phosphorylated and total levels of AKT by Western blot (Figure 4D). Although the ratio of phosphorylated to total AKT was not different (Figure 4H), we observed an increase in both the phosphorylated (vehicle 1.33 normalized units ± 0.05 vs LPS 1.65 ± 0.07; means ± SEM **p<0.005 vehicle vs LPS) and total levels (vehicle 1.23 normalized units ± 0.04 vs LPS 1.49 ± 0.04; mean ± SEM ***p<0.001 vehicle vs LPS) of AKT kinase (Figure 4 F, G). As IGF-1 can increase the levels of p-AKT [17], we evaluated the mRNA levels of Igf1; however no significant difference was detected (Figure 4B). Finally, as the transcription factor Nrf2 has been shown to reduce HI brain injury [18], we evaluated the levels of Nrf2 mRNA; although there wasn’t a statistically significant increase in Nrf2 mRNA levels between the vehicle treated and LPS treated group, there was a trend towards higher mRNA levels in the LPS group (Figure 4C).
Figure 4. LPS upregulates the expression levels of eNOS and AKT.
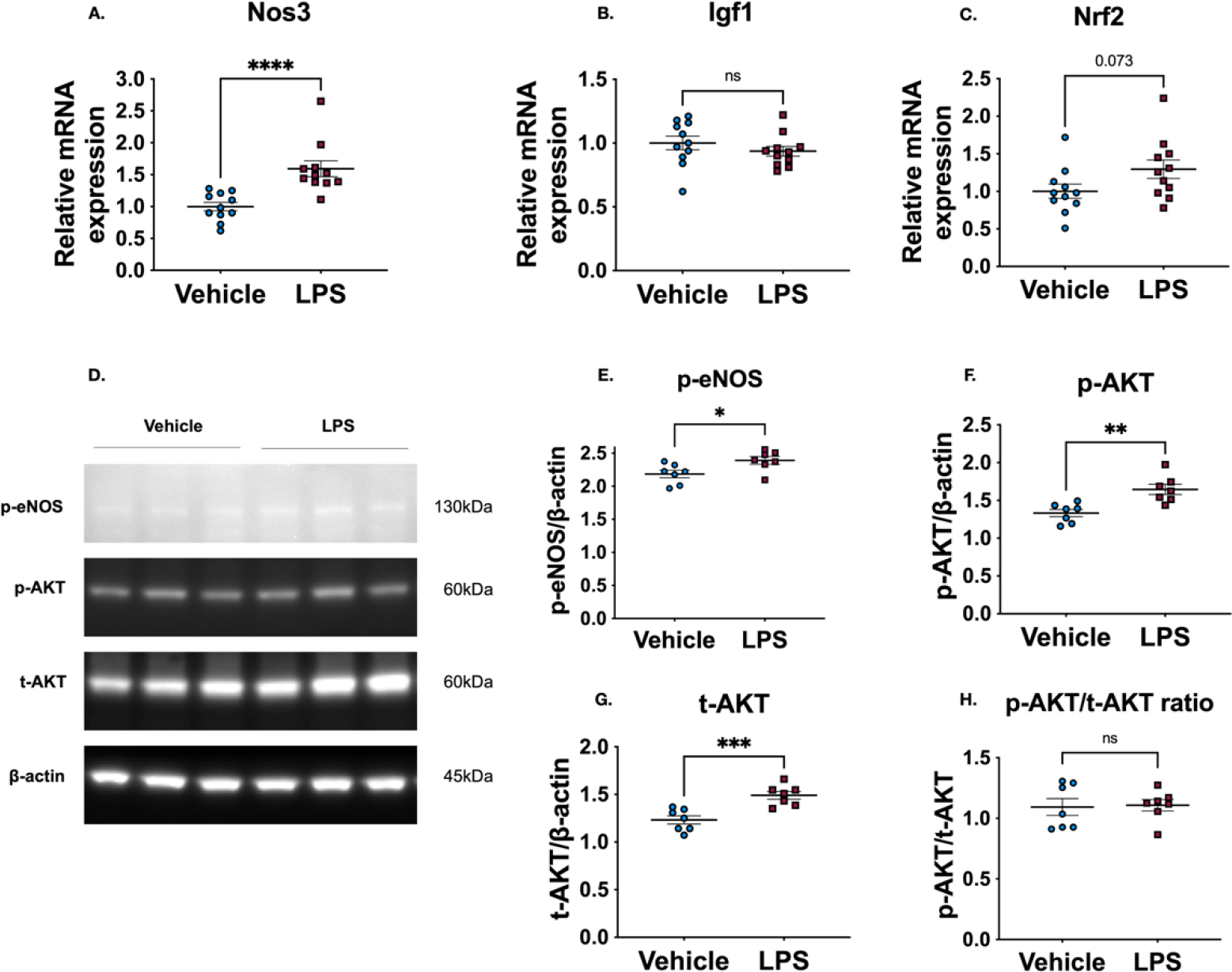
(A-C) Relative mRNA expression of Nos3, Igf1 and Nrf2 (n=11, p<0.001 for Nos3, p=0.34 for Igf1 and p=0.07 for Nrf2). (D) Representative Western blot results for p-eNOS (S1177), p-AKT (S473), t-AKT and β-actin. (E-H) Quantification of protein expression levels (n=7, p=0.03 for p-eNOS, p=0.003 for p-AKT, p<0.001 for t-AKT and p=0.87 for p-AKT/t-AKT ratio). Statistical analyses were performed using unpaired t-test for all comparisons between the vehicle and LPS groups for the different targets. The non-parametric Mann-Whitney test was only performed for the comparison of p-eNOS (S1177) target, since the LPS group did not pass the test for normal distribution.
4. Discussion
In this study, we demonstrate that LPS can serve as a preconditioning stimulus to protect the immature brain from HI-induced damage. Wistar rats on P3 are comparable, in terms of neocortical and WM development, to preterm human infants of 24–28 weeks of gestation [19]. LPS had been previously shown to exacerbate HI-induced white matter tissue loss when administered 14 h prior to HI on P9 [6]. Similarly, Wang et al., 2012 found that LPS sensitized P2 rat pups to HI [20]. By contrast, we found that LPS administered 14h before HI on P3 prevented HI-induced brain tissue loss. Our analyses of cell apoptosis using ISEL staining show that HI alone triggers widespread apoptotic cell death in both the neocortex and subcortical WM, while LPS preconditioning protects these regions. Moreover, we found that the LPS pre-administration significantly reduced microglial/macrophage activation as evaluated by quantifying the numbers of Iba1/CD68 double positive cells. These differences in our results versus those reported by Wang et al., 2009 can be attributed to several factors that include the doses of LPS, differences in immune tolerance between species and differences in developmental stage between the rats and mice used in HI models [6].
LPS can differentially affect immune tolerance between species. For example, in our study, we used a Wistar rat pup model and found that LPS acted as a pre-conditioning stimulus in contrast to studies by Wang et al., 2009 who used a C57BL/6 mouse model and found that LPS sensitized the brain to subsequent injury [6]. There are numerous differences between rats and mice that could account for their disparate responses to LPS. Phylogeographic studies have shown that the common house mouse, originated in the Himalayan mountains whereas common rats resided in the lowlands of China and India. When exposed to hypoxia, mice show increased ventilatory, metabolic, and mitochondrial responses compared to rats. By contrast, rats mounted a faster and stronger hematological response than mice with only minor ventilatory and metabolic adjustments[21]. Significant hematological differences also exist: rats have almost 3 times as many T cells in their peripheral blood compared to mice (~59% vs. ~20%). Rats also have almost 3 times as many neutrophils in their blood compared to mice (~17% vs. 5%). By contrast, mice have more B cells than rats (~47% vs. 32%). Mice also have a higher platelet count compared to rats and rat (and human) T-cells can express MHC-II, whereas mouse T-cells do not [22, 23]. These differences likely contribute to the differences in their temporal responses to LPS.
There are prior studies that showed that LPS tolerized the brain to a subsequent insult. One study investigated immune tolerance in the neonatal rat hippocampus and demonstrated that prenatal LPS exposure induced immune tolerance to subsequent postnatal LPS exposure that was accompanied by reduced microglial activation indicated by low Iba-1/CD68 expression, which is similar to our results for the neocortex [13]. Similarly a study of LPS-induced immune tolerance in the P6 rat brain showed that LPS preconditioning (0.05 mg/kg) induced eNOS in cerebral vessels, which attenuated HI-induced cerebral vessel damage, oxidative stress and apoptosis [24]. Based on that study, we hypothesized that the LPS administered to the P2 rat pups was increasing eNOS activity. Supporting that hypothesis we observed that LPS (0.1mg/kg), administered to P2 rats induced eNOS mRNA and increased levels of phosphorylated eNOS. Furthermore, we evaluated the levels of AKT, a major pro-survival kinase that can directly phosphorylate eNOS on Ser1177, which increases its enzymatic activity [25, 26]. Similarly to Lin et al., 2010 we observed higher levels of p-AKT (S473). Interestingly, we also observed a concomitant increase in total levels of AKT in the LPS group compared to the vehicle injected group 16h post LPS administration suggesting increased AKT signaling is a result of gains at both translational as well as post-translational levels. Based on a review of the literature, we surmise that increased VEGF signaling through VEGFR1 could be responsible for increasing pAKT in the neurons and the endothelial cells [24, 27]. Overall, our data suggest that LPS preconditioning increases AKT-eNOS signaling. Increased levels of AKT kinase can lead to higher eNOS enzymatic activity that, in turn, will increase nitric oxide (NO) production. As NO stimulates vasodilation, increased blood flow would lead to more efficient delivery of nutrients and oxygen that would serve as a potential neuroprotective mechanism induced by LPS to counteract the hypoxia and ischemia.
We should also note that our model differs in multiple parameters compared to that of Lin et al. 2010 as the authors administer 0.05 mg/kg to P6 Sprague-Dawley female rat pups whereas in our model both males and female Wistar rats were included for analysis (we did not detect sex differences, although our studies were not sufficiently powered to do so). In addition, the timepoint of the single 0.1 mg/kg LPS injection was administered at P2, a timepoint when both the brain and immune systems are more immature than at P6. On the other hand, it should be noted that in both rat perinatal HI models AKT-eNOS signaling seems to play a major neuroprotective role.
In addition to eNOS we evaluated whether Igf1 expression might be contributing to the increase in AKT phosphorylation, as we and others have previously shown that IGF-1 prevents white matter damage caused by hypoxia-ischemia and IGF-1 is known to activate AKT [28–32]. However, Igf1 mRNA expression was not different between the vehicle and LPS group 16 h post administration. While this does not negate a role for IGF-1, this observation does not implicate it as contributing to the observed neuroprotection. Lastly, we measured the mRNA levels of Nrf2 which also has been shown to provide neuroprotection against perinatal HI [18]. Here we observed a trend towards higher levels in the LPS group, but this increase did not reach statistical significance.
The ultimate neuropathological outcome can be affected by the timing of the inflammatory stimulus. Data from rodent studies have found that the timing of LPS administration will determine whether the injury is exacerbated or reduced in extent. For instance, Eklind et al., 2005 showed that administering LPS reduced brain injury when administered 24 h before HI in P7 rats. However, when LPS was administered either 6 h or 72 hours before HI, brain injury was increased [33]. Hickey et al., 2011 obtained neuroprotection when LPS was administered to rat pups aged P7, P9 or P14 prior to HI; but there was no neuroprotection when the LPS was administered prior to HI at P3 or P5, which differs from our results [34]. However, they used LPS from a different strain of bacteria (0111:B4) and the interval between the LPS and the HI in their study was 48 h; thus there are at least two important variables that differed between their study and ours. In a paper by Wang et al., 2012 that investigated the role of JNK signaling they found that LPS sensitized P2 rat pups to HI [20]; however, in their study they used Sprague Dawley rat pups whereas we used Wistar Rats; they also used LPS from the 0111:B4 strain of Escherichia coli. Also the interval between the LPS and the HI in their study was only 3 hours. Thus, there are at least 3 important variables to account for the differences in their results vs. ours.
Previous studies investigating the effects of HI in very immature rats (P3) showed that damage was predominantly restricted to the subcortical WM which was neuropathologically similar to the dWMI seen in very premature human infants [35, 36]. The vulnerability of the subcortical WM to HI has been explained by the vulnerability of the late oligodendrocyte (OL) progenitors to the HI insult, which are the predominant OL lineage cells present in the WM at this age [35]. The impact of HI on the gray matter in very young rat pups has not been well characterized. In 2003 McQuillen and colleagues showed that rat subplate neurons are very vulnerable to apoptosis after HI and their loss correlated with cognitive and sensory deficits, due to the deficits in connections between thalamus and the somatosensory cortex [37]. Although that study revealed the potential damage sustained by the sensory cortex, the gray matter is often considered to be spared when compared to the white matter in preterm brain injury models. Therefore, our results, which show that a unilateral HI insult delivered at P3 in the Wistar rat induces significant apoptosis and tissue damage in dorsal neopallium, challenges existing notions that the WM is the only region in rodent brain that is severely damaged by HI at this timepoint in forebrain development.
Our studies have several limitations. One limitation of this study is that the measurements were obtained only 2 days after the HI, thus it is not clear whether the LPS pre-conditioning truly protected the brain from injury or simply slowed the rate of cell death. Another limitation of this study is that we used a single lot of LPS and we used a single dose. It is conceivable that we would have obtained different results from using a more homogenous preparation of LPS, a different strain or a different dose. The experiment reported in Fig. 4 also has limitations in that we only evaluated the effects of LPS vs. saline rather than including all 4 groups as in earlier figures. Additionally, our data suggest that the LPS preconditioning is modifying cerebral blood flow, therefore, future studies should assess cerebral blood flow using this paradigm to establish whether, indeed, that is one mechanism for the neuroprotection observed.
5. Conclusion
In conclusion, LPS preconditioning when initiated 14 hours prior to HI in the P2 rat pup protects against brain damage by reducing neuroinflammation and increasing levels of phosphorylated eNOS. Clearly further work is required to fully unravel the mechanisms that will determine whether an infection will exacerbate damage to the brain or protect that brain from damage, which clearly has relevance for treating neonatal brain injuries with comorbid fetal/neonatal inflammation.
Highlights.
Hypoxia-ischemia in P3 rats damages both the neocortex and white matter
LPS pre-conditioning reduces neocortical and white matter apoptosis induced by HI on P3
LPS preconditioning suppresses microglial cell activation induced by HI on P3
LPS preconditioning activates AKT-eNOS signaling
Acknowledgements
The authors would like to thank Dana Clausen for staining some of the sections that were subsequently evaluated. This work was supported by a grant from the National Institutes of Health NS116828-01 awarded to S.W.L.
Footnotes
Declarations of interest: The authors declare that they have no relevant or material financial interests that relate to the research described in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability:
All data will be made available for inspection or use upon request to the corresponding author.
References Cited:
- [1].Stockman JA, The contribution of preterm birth to infant mortality rates in the United States, Yearbook of Pediatrics 2008 (2008) 388–390. 10.1016/s0084-3954(08)70485-8 [DOI] [PubMed] [Google Scholar]
- [2].Cannavo L, Rulli I, Falsaperla R, Corsello G, Gitto E, Ventilation, oxidative stress and risk of brain injury in preterm newborn, Ital J Pediatr 46 (2020) 100. 10.1186/s13052-020-00852-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ophelders DRMG, Gussenhoven R, Klein L, Jellema RK, Westerlaken RJJ, Hütten MC, Vermeulen J, Wassink G, Gunn AJ, Wolfs TGAM, Preterm brain injury, antenatal triggers, and therapeutics: Timing is key, Cells 9 (2020) 1871. 10.3390/cells9081871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P, The role of inflammation in perinatal brain injury, Nature reviews. Neurology 11 (2015) 192–208. 10.1038/nrneurol.2015.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].van Tilborg E, Achterberg EJM, van Kammen CM, van der Toorn A, Groenendaal F, Dijkhuizen RM, Heijnen CJ, Vanderschuren L, Benders M, Nijboer CHA, Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury, Glia 66 (2018) 78–93. 10.1002/glia.23216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C, Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner, J Immunol 183 (2009) 7471–7477. 10.4049/jimmunol.0900762 [DOI] [PubMed] [Google Scholar]
- [7].Turner RC, Naser ZJ, Lucke-Wold BP, Logsdon AF, Vangilder RL, Matsumoto RR, Huber JD, Rosen CL, Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells, Neuroimmunol Neuroinflamm 4 (2017) 6–15. 10.20517/2347-8659.2016.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Raetz CR, Whitfield C, Lipopolysaccharide endotoxins, Annu Rev Biochem 71 (2002) 635–700. 10.1146/annurev.biochem.71.110601.135414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guardia Clausi M, Levison SW, Delayed ALK5 inhibition improves functional recovery in neonatal brain injury, J Cereb Blood Flow Metab 37 (2017) 787–800. 10.1177/0271678X16638669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bain JM, Ziegler A, Yang Z, Levison SW, Sen E, TGFSS1 stimulates the over-production of white matter astrocytes from precursors of the “Brain Marrow” in a rodent model of neonatal encephalopathy, PLoS ONE 5 (2010). 10.1371/journal.pone.0009567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fehsel K, Kroncke KD, Kolb H, Kolb-Bachofen V, In situ nick-translation detects focal apoptosis in thymuses of glucocorticoid- and lipopolysaccharide-treated mice, J Histochem Cytochem 42 (1994) 613–619. 10.1177/42.5.8157933 [DOI] [PubMed] [Google Scholar]
- [12].Kim BH, Jeziorek M, Kanal HD, Contu VR, Dobrowolski R, Levison SW, Moderately Inducing Autophagy Reduces Tertiary Brain Injury after Perinatal Hypoxia-Ischemia, Cells 10 (2021). 10.3390/cells10040898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Singh G, Segura BJ, Georgieff MK, Gisslen T, Fetal inflammation induces acute immune tolerance in the neonatal rat hippocampus, J Neuroinflammation 18 (2021) 69. 10.1186/s12974-021-02119-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Simpson JE, Fernando MS, Clark L, Ince PG, Matthews F, Forster G, O’Brien JT, Barber R, Kalaria RN, Brayne C, Shaw PJ, Lewis CE, Wharton SB, Function MRCC, Ageing Neuropathology Study G, White matter lesions in an unselected cohort of the elderly: astrocytic, microglial and oligodendrocyte precursor cell responses, Neuropathol Appl Neurobiol 33 (2007) 410–419. 10.1111/j.1365-2990.2007.00828.x [DOI] [PubMed] [Google Scholar]
- [15].Walker DG, Lue LF, Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains, Alzheimers Res Ther 7 (2015) 56. 10.1186/s13195-015-0139-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin J, Niimi Y, Clausi MG, Kanal HD, Levison SW, Neuroregenerative and protective functions of Leukemia Inhibitory Factor in perinatal hypoxic-ischemic brain injury, Exp Neurol 330 (2020) 113324. 10.1016/j.expneurol.2020.113324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vincent AM, Feldman EL, Control of cell survival by IGF signaling pathways, Growth Horm IGF Res 12 (2002) 193–197 [DOI] [PubMed] [Google Scholar]
- [18].Zhang J, Tucker DongYan LD, Lu Y, Yang L, Wu C, Li Y, Zhang Q, Tert-butylhydroquinone post-treatment attenuates neonatal hypoxic-ischemic brain damage in rats, Neurochem Int 116 (2018) 1–12. 10.1016/j.neuint.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sizonenko SV, Sirimanne E, Mayall Y, Gluckman PD, Inder T, Williams C, Selective cortical alteration after hypoxic-ischemic injury in the very immature rat brain, Pediatric Research 54 (2003) 263–269. 10.1203/01.pdr.0000072517.01207.87 [DOI] [PubMed] [Google Scholar]
- [20].Wang LW, Tu YF, Huang CC, Ho CJ, JNK signaling is the shared pathway linking neuroinflammation, blood-brain barrier disruption, and oligodendroglial apoptosis in the white matter injury of the immature brain, J Neuroinflammation 9 (2012) 175. 10.1186/1742-2094-9-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Arias-Reyes C, Soliz J, Joseph V, Mice and Rats Display Different Ventilatory, Hematological, and Metabolic Features of Acclimatization to Hypoxia, Front Physiol 12 (2021) 647822. 10.3389/fphys.2021.647822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].D. W, J. W, Schalm OW, Schalm’s veterinary hematology, Wiley-Blackwell, Hoboken, NJ, 2010. [Google Scholar]
- [23].Wildner G, Are rats more human than mice?, Immunobiology 224 (2019) 172–176. 10.1016/j.imbio.2018.09.002 [DOI] [PubMed] [Google Scholar]
- [24].Lin HY, Wu CL, Huang CC, The Akt-endothelial nitric oxide synthase pathway in lipopolysaccharide preconditioning-induced hypoxic-ischemic tolerance in the neonatal rat brain, Stroke 41 (2010) 1543–1551. 10.1161/STROKEAHA.109.574004 [DOI] [PubMed] [Google Scholar]
- [25].Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC, Hypertension in mice lacking the gene for endothelial nitric oxide synthase, Nature 377 (1995) 239–242. 10.1038/377239a0 [DOI] [PubMed] [Google Scholar]
- [26].Endres M, Laufs U, Liao JK, Moskowitz MA, Targeting eNOS for stroke protection, Trends Neurosci 27 (2004) 283–289. 10.1016/j.tins.2004.03.009 [DOI] [PubMed] [Google Scholar]
- [27].Cárdenas-Rivera A, Campero-Romero AN, Heras-Romero Y, Penagos-Puig A, Rincón-Heredia R, Tovar-y-Romo LB, Early Post-stroke Activation of Vascular Endothelial Growth Factor Receptor 2 Hinders the Receptor 1-Dependent Neuroprotection Afforded by the Endogenous Ligand, Frontiers in Cellular Neuroscience 13 (2019). 10.3389/fncel.2019.00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wood TL, Loladze V, Altieri S, Gangoli N, Levison SW, Brywe KG, Mallard C, Hagberg H, Delayed IGF-1 administration rescues oligodendrocyte progenitors from glutamate-induced cell death and hypoxic-ischemic brain damage, Dev Neurosci 29 (2007) 302–310. 10.1159/000105471 [DOI] [PubMed] [Google Scholar]
- [29].Guan J, Williams C, Gunning M, Mallard C, Gluckman P, The effects of IGF-1 treatment after hypoxic-ischemic brain injury in adult rats, J Cereb Blood Flow Metab 13 (1993) 609–616 [DOI] [PubMed] [Google Scholar]
- [30].Brywe KG, Mallard C, Gustavsson M, Hedtjarn M, Leverin AL, Wang X, Blomgren K, Isgaard J, Hagberg H, IGF-I neuroprotection in the immature brain after hypoxia-ischemia, involvement of Akt and GSK3beta?, Eur J Neurosci 21 (2005) 1489–1502. 10.1111/j.1460-9568.2005.03982.x [DOI] [PubMed] [Google Scholar]
- [31].Lin S, Fan LW, Pang Y, Rhodes PG, Mitchell HJ, Cai Z, IGF-1 protects oligodendrocyte progenitor cells and improves neurological functions following cerebral hypoxia-ischemia in the neonatal rat, Brain Res 1063 (2005) 15–26 [DOI] [PubMed] [Google Scholar]
- [32].Lin S, Fan LW, Rhodes PG, Cai Z, Intranasal administration of IGF-1 attenuates hypoxic-ischemic brain injury in neonatal rats, Exp Neurol 217 (2009) 361–370.S0014–4886(09)00096-X [pii] 10.1016/j.expneurol.2009.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Eklind S, Mallard C, Arvidsson P, Hagberg H, Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain, Pediatr Res 58 (2005) 112–116. 10.1203/01.PDR.0000163513.03619.8D [DOI] [PubMed] [Google Scholar]
- [34].Hickey E, Shi H, Van Arsdell G, Askalan R, Lipopolysaccharide-induced preconditioning against ischemic injury is associated with changes in toll-like receptor 4 expression in the rat developing brain, Pediatr Res 70 (2011) 10–14. 10.1203/PDR.0b013e31821d02aa [DOI] [PubMed] [Google Scholar]
- [35].Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM, Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia, J Neurosci 22 (2002) 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sizonenko SV, Sirimanne E, Mayall Y, Gluckman PD, Inder T, Williams C, Selective cortical alteration after hypoxic-ischemic injury in the very immature rat brain, Pediatr Res 54 (2003) 263–269. 10.1203/01.PDR.0000072517.01207.87 [DOI] [PubMed] [Google Scholar]
- [37].McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM, Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia, J Neurosci 23 (2003) 3308–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data will be made available for inspection or use upon request to the corresponding author.