

Abstract
Purpose:
Several MCL-1 inhibitors (MCL-1i), including AMG-176 and AZD5991, have shown promise in preclinical studies and are being tested for the treatment of hematological malignancies. A unique feature of these agents is induction and stability of Mcl-1 protein; however, the precise mechanism is unknown. We aim to study mechanism of MCL-1i-induced Mcl-1 protein stability.
Experimental Design:
Using several B-cell leukemia and lymphoma cell lines and primary CLL lymphocytes, we evaluated molecular events associated with Mcl-1 protein stability including protein half-life, reverse-phase protein array, protein-protein interaction, phosphorylation, ubiquitination, deubiquitination, followed by molecular simulation and modeling.
Results:
Using both in vivo and in vitro analysis, we demonstrate that MCL-1i-induced Mcl-1 protein stability is predominantly associated with defective Mcl-1 ubiquitination and concurrent apoptosis induction in both cell lines and primary CLL subjects. These MCL1i also induced ERK-mediated Mcl-1Thr163 phosphorylation which partially contributed to Mcl-1 stability. Disruption of Mcl-1:Noxa interaction followed by Noxa degradation, enhanced Mcl-1 de-ubiquitination by USP9x, and Mule destabilization are the major effects of these inhibitors. However, unlike other BH3 proteins, Mule:Mcl-1 interaction was unaffected by MCL-1i. WP1130, a global deubiquitinase (DUB) inhibitor, abrogated Mcl-1 induction reaffirming a critical role of DUBs in the observed Mcl-1 protein stability. Further, in vitro ubiquitination studies of Mcl-1 showed distinct difference amongst these inhibitors.
Conclusions:
We conclude that MCL-1i blocked Mcl-1 ubiquitination via enhanced deubiquitination and dissociation of Mcl-1 from Noxa, Bak and Bax, and Mule de-stabilization. These are critical events associated with increased Mcl-1 protein stability with AMG-176 and AZD5991.
Keywords: AMG-176, AZD5991, B-cell leukemia, MCL-1 inhibitor, Mcl-1 protein, Mule E3 ligase, USP9x deubiquitinase
Introduction
The BCL-2 family proteins are divided into three major classes and all share a BCL-2 homology (BH) domain. Mcl-1 is a member of the anti-apoptotic cohort(1–3). Interactions among these proteins and their stoichiometry are central to survival and apoptosis of several types of cancer cells. The impetus to develop a small molecule pharmacological agent to neutralize the actions of Mcl-1 survival protein was two-fold. First, the successful targeting of BCL-2 by venetoclax was encouraging. Second, investigations of somatic copy number alterations identified Mcl-1 among the top genes amplified in tumors(4). Recently, several MCL-1 inhibitors (MCL-1i) have entered clinical trials including Astra Zeneca’s AZD5991 (NCT03218683)(5, 6), Amgen’s AMG-176 (NCT02675452)(7), and Novartis’s S64315 (NCT04629443)(8, 9) with proven effectiveness in pre-clinical investigations of hematological malignancies.
A common feature of treatment with these MCL-1i is an increased stability in Mcl-1 protein level (5, 7, 8, 10) as observed in breast cancer,(11) acute myelogenous leukemia,(7, 8) acute lymphoblastic leukemia, and multiple myeloma (7, 9) cell lines. However, detailed mechanistic insight into the ubiquitous observation of increased Mcl-1 protein level after treatment with MCL-1i is lacking.
Mcl-1 is a short half-life protein(12) that can be regulated at multiples levels including transcription(13), translation(14, 15), and degradation(16). Mcl-1 is degraded by the ubiquitin-proteasomal system and is under the control of ubiquitination by E3 ligases like Mule(17), Trim-17(18) or β-TRCP(19). Conversely, deubiquitinases (DUBs) like USP9x(20), KU-70(21), and USP13(22) reverse this process and contribute to Mcl-1 protein stability. Mcl-1 post-translation modifications at Thr163(23) and Ser159(24) control ubiquitination.
We hypothesized that elevation of Mcl-1 protein level after treatment with MCL-1i may involve mechanisms of Mcl-1 production, stability, and/or degradation. To test our hypothesis, we utilized hematological malignancy cell lines and primary CLL cells to determine mechanisms leading to increased Mcl-1 protein levels.
Materials and Methods
Cell lines and culture
Two different heme-malignancy cell lines including MEC1 (CLL), and Mino (mantle cell lymphoma), HEK293, USP9x WT and knockout HCT116 cell lines (RRID:CVCL_0291) and primary CLL cells were used in this study. CLL cells were obtained after patients (Supplemental Table 1) signed written informed consent for inclusion in the study, and the study was conducted in accordance with the Declaration of Helsinki. The protocol was approved by the Institutional Review Board at MD Anderson Cancer Center. Cell number and volume were determined with Coulter channelizer (Coulter Electronics). Detailed culture conditions are described in Supplemental Method.
Drugs
AMG-176 and AZD5991 were purchased from Chemietek; MG-132, Degrasyn (WP1130), Maritoclax, and trametinib were purchased from Selleckchem, Houston, TX. Agents were dissolved in DMSO (Sigma-Aldrich, Burlington, MA).
Reverse-phase protein array (RPPA)
Cells were treated with MCL-1i and lysates were prepared and printed in five serial dilutions onto slides along with the normalization and expression controls. The array included 920 samples and slides were probed with one of 386 antibodies as detailed in the Supplemental Method.
Plasmids, transfection, Lentiviral shRNA,
pCMV-Tag2A vector and HA-UB plasmids were kind gifts. pCMV-Flag-hMCL-1 (T163A) (# 25391) and pCMV-Flag-WT-hMCL-1 (# 25392) (RRID:Addgene_25392) were obtained from Addgene (Watertown, MA). Five different human Mission sh-USP9x clones, non-Target shRNA control (SHC002V), and Mission Lentiviral packaging mix (SHP001) were obtained from Sigma (Burlington, MA). Control and sh-RNA-USP9x along with Lentiviral packaging mix were individually transfected to HEK293T cells (RRID: CVCL_HA71) for virus preparation and infection as previously described(25).
Immunoprecipitation and co-Immunoprecipitation
Sub confluent HEK293 cells were transiently transfected with 3μg of plasmids using Fugene 6 transfection reagent (Promega, Madison, WI)(25). Seventy-two hours after transfection, cells were lysed in IP-lysis buffer (Pierce) supplemented with protease inhibitors cocktail (cOmplete, Protease and PhosSTOP inhibitors; Roche). Cell extracts (500μg) were immunoprecipitated with EZview anti-Flag M2 Agarose (Sigma) or EZview Red Anti-HA (Sigma) Affinity Gels overnight at 4°C and processed for immunoblot analysis. For endogenous co-IP experiments, MEC1 or Mino cells were processed as above, and complexes were captured by using Protein A/G-Agarose beads (Santa Cruz, Dallas, TX) at room temperature for 1–2 hours. The beads were washed three times with lysis buffer and immunoblotted.
Cell fractionation and immunoblot analysis
Cell fractionation and immunoblot analysis was performed as described earlier(10, 25). Detailed procedure is described in Supplemental Method. List of antibodies and their sources are provided in Supplemental Table 2.
Apoptosis Assay
MEC1, Mino cell lines and peripheral blood mononuclear cells from CLL patient samples were treated by DMSO, AMG176, or AZD5991 for 24 hours. Cells were then collected and processed for apoptosis assay by Annexin V-PI staining as described in Supplemental method.
Real-time RT-PCR
Total RNA from MEC1 and Mino cells was isolated by using RNeasy Mini Kit. RT reaction was performed by using RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fisher). Real-time RT-PCR was performed by using primers (Supplemental Table 3) and the SYBR® Green PCR Master Mix (Applied Biosystems)(25) as detailed in the Supplemental Method.
Protein purification
To purify ubiquitinated FLAG-Mcl-1, HEK293 was co-transfected with HA-ubiquitin and FLAG-Mcl-1 plasmids in 10 cm plate. Seventy-two hours post-transfection, cells were harvested, and total cell lysate was extracted using IP lysis buffer (Pierce), followed by immunoprecipitation of FLAG-Mcl-1 using EZview anti-Flag M2 Agarose (Sigma) overnight, and eluted by 3x FLAG peptide (Thermo Scientific) according to the manufacturer’s protocol. This was followed by a second immunoprecipitation using HA beads (Invitrogen) to isolate ubiquitinated FLAG-Mcl-1 as per manufacturer’s protocol. For Mule and USP9x purifications, either rabbit anti-Mule or rabbit anti-USP9x antibodies were coupled to Dynabeads™ M-280 Sheep Anti-Rabbit IgG (RRID: AB_2783009) for one hour. The beads were then washed and HEK293 protein lysates were added to the antibody bound beads and incubated by vertical rotation for two hours at 4°C. In order to maintain the activity and functionality of the purified proteins, gentle elution was done by 2M NaCl for 10 minutes.
In vitro Pulldown Assay
Full length His-tag recombinant Mcl-1 protein was allowed to bind to magnetic His-beads (Invitrogen) according to the manufacturer’s protocol. Detailed assay is described in Supplemental Method.
In vitro Ubiquitination Assay
In vitro ubiquitination of Mcl-1 protein by Mule was done using the Ubiquitinylation kit (Enzo life Sciences, Inc., Farmingdale, NY). In order to avoid extensive backgrounds produced by the biotin-conjugated ubiquitin, we replaced it with recombinant FLAG-tagged ubiquitin (R&D systems, Minneapolis). Briefly, recombinant Mcl-1 was incubated in the presence or absence of recombinant Mule with either DMSO, AMG-176, AZD5991, or Maritoclax in ubiquitination buffer containing Mg-ATP, E1 enzyme, E2 enzyme (Recombinant Human UbcH7/UBE2L3 Protein, R&D systems) according to the manufacturer’s protocol. The reactions were incubated for 4 hours at 37°C and quenched by the addition of 2x laemmli. Ubiquitination was assessed by immunoblot analysis and probed with anti-FLAG. For the in vitro ubiquitination of Mcl-1 by purified Mule from HEK293 cells, 5 μM recombinant human ubiquitin aldehyde protein (R&D systems, Minneapolis, MN) was added to inhibit deubiquitinating enzymes.
In vitro De-ubiquitination and DUB Activity Assay
Ubiquitinated HA-FLAG-Mcl-1 was purified as above and then incubated with purified USP9x from HEK293 lysate in DUB buffer at 37°C in the presence of either DMSO, AMG-176, or AZD5991 in a total reaction volume of 75μl. Reactions were allowed for 20 minutes, then quenched by the addition of 4x laemmli and subjected to immunoblot analysis and probed with anti-HA to detect ubiquitination. For detecting in vitro DUB activity, a DUB activity assay kit (Cayman Chemicals; Ann Arbor, MI) was used to assess the direct effect of AMG-176 or AZD5991 on the de-ubiquitinating enzyme USP9x. USP9x (either recombinant or purified from HEK293) or MEC1 total cell lysates pre-treated by 2μM AMG-176 or AZD5991 for 6 hours) were incubated in the presence or absence of DMSO, AMG-176, or AZD5991 in DUB buffer for 15 minutes in a 96-well plate according to manufacturer’s protocol. The fluorogenic AMC-Ub was then added to the reactions and immediately assessed for fluorescence in the microplate reader for one hour in kinetic mode. Readings were taken every minute.
Protein structural modeling
We constructed a 3D model using RaptorX (RRID:SCR_018118), which is an ab initio method (http://raptorx.uchicago.edu/). The modeled structure was combined with the known Mcl-1 crystal structure complexed with AMG-176 (PDB entry: 6OQC). To understand the effect upon inhibitor binding, we conducted molecular dynamics simulations on both AMG-176-bound and AMG-176-removed Mcl-1 structures for 50ns using GROMACS (RRID:SCR_014565) with GROMOS96 force field (http://www.gromacs.org/) as detailed in the Supplemental Method.
Statistical analysis
Statistical analysis was performed by using GraphPad Prism Software (RRID:SCR_002798). All numerical results are presented as mean ± SEM. The statistical significance of differences was analyzed using grouped analysis, ANOVA and multiple t-test (Student’s t-test).
Data availability:
The data generated in this study are available within the article and its supplementary data files. For raw data request, please contact the corresponding author at vgandhi@mdanderson.org
Results
MCL-1 inhibitors induce Mcl-1 upregulation by increasing stability of Mcl-1 protein
Reverse-phase protein array, RPPA, of MCL-1i treated MEC1 and Mino cell lines (p = 0.01) revealed selective upregulation of Mcl-1 (p<0.003) and cleaved PARP (p<0.004) protein levels; while SHIP2 protein level was downregulated (p = 0.009) (Figure 1A; Supplemental Figure 1). Interestingly, at less stringent p value (p= 0.05), AMG-176 and AZD5991 induced several non-identical proteomic changes (Supplemental Figure 1A–C). Induction in Mcl-1 protein level was confirmed by immunoblot analysis in a time and dose-dependent manner (Figure 1B) and in whole cells including mitochondria (Supplemental Figure 1D). Mcl-1 upregulation was not due to transcription (Figure 1C) but due to increased stability of Mcl-1 protein. Cycloheximide chase experiment confirmed increased half-life of the Mcl-1 protein following incubation with MCL-1i (DMSO 0.68 hr; AMG-176 2.94 hr, and AZD5991 3.50 hr, respectively) (Figure 1D), while co-incubation of cycloheximide and AMG-176 in both MEC1 and Mino cells blunted MCL-1 protein accumulation (Figure 1E). Despite higher abundance and stability of Mcl-1, MCL-1i induced apoptosis in Mino cell line (Supplemental Figure 1E) as well as peripheral blood lymphocytes from CLL-patient samples (Supplemental Figure 1F) indicating that the accumulated Mcl-1 protein did not provide any resistance to MCL-1i and lost its anti-apoptotic function. Interestingly, at 500 nM, AZD5991 was more potent than AMG-176 in inducing apoptosis in Mino cell line (p = 0.0185) and CLL patient samples (p = 0.0001).
Figure 1. MCL-1 inhibitors induce Mcl-1 upregulation by increasing the stability of Mcl-1 protein.

A. Reverse Phase Proteomic Array (RPPA) for MEC1 and Mino Cell lines. 5X106 cells were treated with DMSO, 500 nM AMG-176, or 500 nM AZD5991 for 12 hours. Proteins were extracted and analyzed by protein array as described in Materials and Methods. Protein levels were compared between drug-treated conditions and DMSO control from three biological repeat experiments. Unbiased hierarchical clustering was performed to identify proteins with significantly (*p ≤ 0.015) altered expression in MEC1 or in Mino cells (*p ≤ 0.01) after treatment with the inhibitors compared to expression in DMSO-treated cells. Mcl-1 protein level changes with MCL-1 inhibitors represented in box plot as in supplemental data (**p<0.002 in MEC1 or ****p<0.0001 in Mino cells, one-way ANOVA analysis). Protein concentrations were normalized and calculated as described in the Supplemental Methods. MEC1 is red and Mino is blue colored boxes. B. Immunoblot analysis of Mcl-1 protein levels in MEC1 and Mino cell lines treated with DMSO, AMG-176 or AZD5991. Left panel shows time-dependent changes in Mcl-1 protein levels after treatment with DMSO, AMG-176, and AZD5991 (both 1 μM). Right panel shows dose-dependent changes in Mcl-1 protein level following treatment with DMSO or AMG-176 and AZD5991 for 16 hours. C. qRT-PCR analysis of MCL-1 and cMyc mRNA expression at early (2 hours) and late (16 hours) following treatment with AMG-176 and AZD5991 in MEC1 and Mino cell lines and compared to the DMSO control. Each bar represents mean ± SEM (n=3 from separate experiments) and shown as relative to DMSO control. D. MEC1 cells were treated with DMSO, 500 nM AMG-176, or 500 nM AZD5991 for 12 hours followed by the addition of 10 μg cycloheximide (CHX). Cells were collected just before adding CHX and at 1, 2, 4, and 6 hours following CHX addition, immunoblotted, and probed for Mcl-1 (short half-life), HSP90 (long half-life), and BCL-2 (intermediate half-life) protein expression. GAPDH was probed for loading control. Mcl-1 protein expression after incubation with MCL-1 inhibitors for 12 hours was quantitated (left lower). Mcl-1 protein expression over time following CHX treatment was plotted and the rate of degradation of Mcl-1 was determined (right lower). Each bar represents mean ± SEM from n=3, separate experiments; *p<0.05; **p<0.001; ***p<0.0001 significant difference from DMSO control. E. MEC1 and Mino cells were either untreated or treated with DMSO, 500 nM AMG-176, 10 μg CHX, or combination of AMG-176 and CHX for the indicated time points and then processed for immunoblot analyses of Mcl-1, HSP90, and BCL-2. GAPDH was probed for loading control. LI-COR quantitation of Mcl-1 relative to GAPDH was calculated and expressed compared to DMSO control and indicated underneath Mcl-1 blots. AMG: AMG-176; AZD: AZD5991; CHX: Cycloheximide, CL: Cell line; TX, treatment.
MCL-1 inhibitors induce defective ubiquitination of Mcl-1 protein
The ubiquitin (UB)-proteasome system tightly regulates Mcl-1 stability and degradation(26). The increased Mcl-1 stability induced by the inhibitors prompted us to explore Mcl-1 ubiquitination. IP and reverse co-IP analysis of MEC1 cells treated with DMSO, AMG-176, or MG-132 (positive control) revealed strong inhibition of Mcl-1 ubiquitination in AMG-176-treated cells. As expected, MG-132 treatment resulted in increased recruitment and accumulation of polyubiquitinated Mcl-1 (Figure 2A). Conversely, AMG-176 treatment followed by a washout restored Mcl-1 ubiquitination. These data demonstrate that Mcl-1 protein accumulation occurs via inhibition in the Mcl-1 ubiquitination upon binding with these inhibitors. Consistently, IP and Co-IP of HEK293 transfected with WT-FLAG-MCL-1 and HA-UB confirmed disruption in HA-FLAG interactions in AMG-176-treated cells that was reversed following withdrawal of AMG-176 from the cells (Figure 2B).
Figure 2. MCL-1 inhibitors induce defective ubiquitination of Mcl-1 protein.
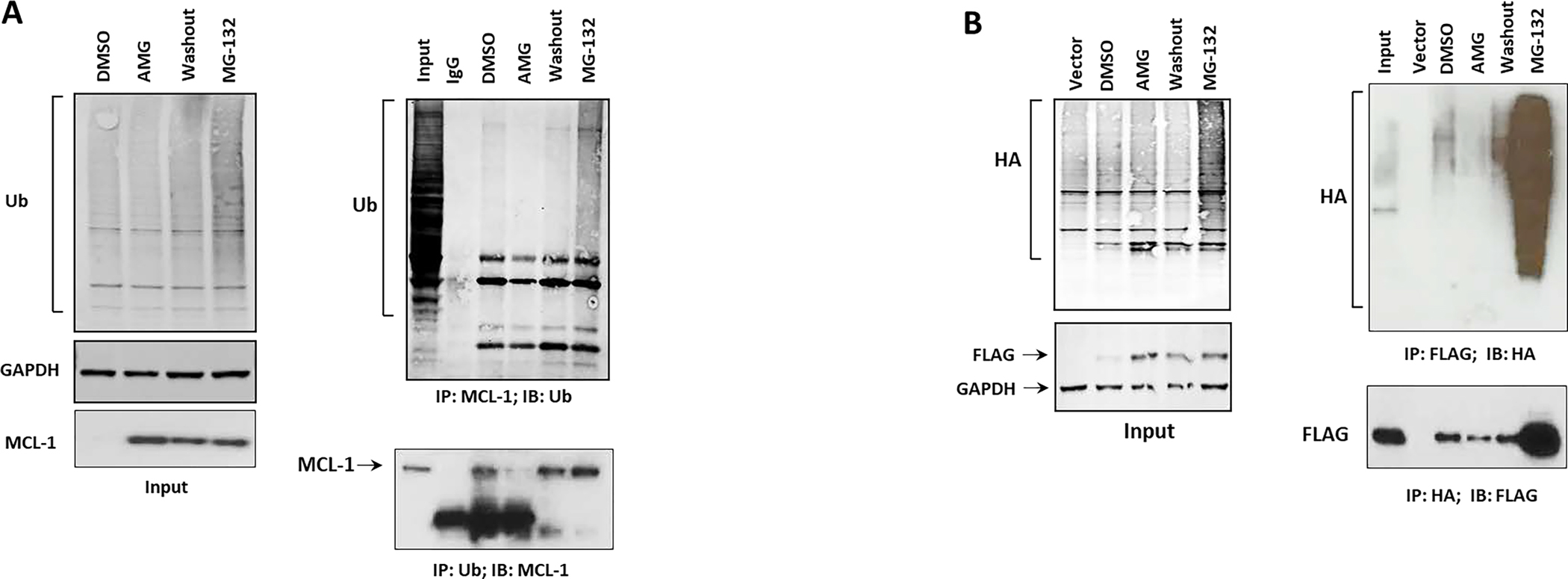
A. Endogenous reverse co-immunoprecipitation (co-IP) analysis of MEC1 cells treated with DMSO, AMG-176 (500 nM), MG-132 (1.25 μM) for 12 hours or a 4-hour washout following AMG treatment (washout). Left panel, input; right panel, equal amounts of total protein (500 μg in each treatment) were subjected to IP with mouse anti-Mcl-1 and probed with rabbit-UB antibody (upper portion) or reverse co-IP with mouse anti-UB and probed with rabbit Mcl-1 antibody (lower portion). B. HEK293 cells were transfected with FLAG-WT-MCL1 and HA-UB (3 μg each in 10-cm plate). Forty-eight hours after transfection, cells were treated with DMSO, AMG (1 μM), or MG-132 (1.5 μM) for 12 hours or a 4-hour washout following AMG-176 treatment. Cells co-transfected with an empty FLAG vector and HA-UB served as vector control. Left panel, input. Right panel, Equal amounts of total protein (500 μg in each treatment) were subjected to IP with FLAG beads (upper portion) or HA beads (lower portion) followed by immunoblot analysis with a rabbit UB antibody or rabbit FLAG respectively. IB: immunoblot; IP: immunoprecipitated; AMG: AMG-176; AZD: AZD5991.
MCL-1 inhibitors elicit ERK-mediated activation of Mcl-1Thr163 phosphorylation
Since enhanced Mcl-1Thr163 phosphorylation is associated with increased stability of Mcl-1 protein(27), we examined MCL-1i-induced post-translational modification. Treatment with MCL-1i resulted in increased total Mcl-1 protein as well as phosphorylated Mcl-1 (Mcl-1Thr163 phosphorylation) in both cell lines and primary CLL patient lymphocytes (Figure 3A–B). Moreover, a direct linear relationship was established between total Mcl-1 protein and Mcl-1Thr163 phosphorylation in either inhibitor-treated (r2=0.92 and p<0.0001 for each drug) primary CLL cells (Figure 3B). This upregulation of Mcl-1Thr163 phosphorylation may be mediated by ERK activation as we noticed an increase in ERK phosphorylation with MCL-1i (Figure 3C–E) which was downregulated upon treatment with the MEK/ERK-inhibitor, trametinib (Figure 3D–E) in cell lines as well as primary CLL lymphocytes. Trametinib partially decreased the MCL-1i-induced upregulation of both total Mcl-1 protein and Mcl-1Thr163 phosphorylation in Mino cells and CLL lymphocytes (Figure 3D–E). The use of QVD, a caspase inhibitor, did not rescue Mcl-1 downregulation in trametinib treated cells, suggesting that the downregulation of Mcl-1 with trametinib in Mino cell line and CLL patient samples is due to ERK-mediated inhibition and decrease in Mcl-1Thr163 phosphorylation rather than caspase activation and cleavage of Mcl-1 protein (Supplemental Figure 2A). However, in MEC1 cells treated with trametinib, despite loss of Mcl-1Thr163 phosphorylation, total Mcl-1 expression remained unchanged (Figure 3D and Supplemental Figure 2B). These data led us to examine the role of Mcl-1Thr163 phosphorylation on Mcl-1-protein-protein interactions rather than stability. Interestingly, overexpression of T163A-FLAG-MCL-1 in HEK293 cells showed loss of interaction with endogenous BAK, BIM, and Noxa but not BAX as compared to the WT-FLAG-MCL-1 supporting a critical role for Mcl-1Thr163 phosphorylation in modulating Mcl-1 interaction with the pro-apoptotic members (Supplemental Figure 2C). Although Mcl-1Ser159 phosphorylation also appeared to be increased in AMG-176-treated cells, it was less pronounced than the total protein of Mcl-1 (Supplemental Figure 2D). In contrast, we did not see any changes in total and GSK3βSer9 phosphorylation (Figure 3D). The use of the phospho-mutant T163A-MCL-1 did not significantly reverse the effect of MCL-1i-induced Mcl-1 upregulation in HEK293 cells (Figure 3F). Overall, we showed that Mcl-1Thr163 phosphorylation via ERK partially contributed to the observed Mcl-1 stability.
Figure 3. MCL-1 inhibitors elicit ERK-mediated Mcl-1Thr163 phosphorylation.

A. MEC1 and Mino cell lines were treated with the indicated concentrations of AMG-176 and AZD5991 for 24 hours and immunoblotted for total and Mcl-1Thr163 phosphorylation. Vinculin was probed for loading control. Data were analyzed by LI-COR quantitation to determine the amount of total and Mcl-1Thr163 phosphorylation and their ratios. Each bar represents mean ± SEM from three separate experiments (left graph: MEC1, right graph: Mino). B. Left panel immunoblot analysis of primary CLL lymphocytes showing total and Mcl-1Thr163 phosphorylation following treatment of lymphocytes with 500 nM AMG-176 or 500 nM AZD5991 for 12 hours. Vinculin was blotted for loading control. Right upper panel: Bar diagram showing quantitation of total and phospho Mcl-1Thr163 form CLL patients (n=10) immunoblots (*p<0.05; **p<0.001 significant difference from DMSO control); right lower panel: XY graph showing relationship between total and phospho-Mcl-1Thr163 following treatment with AMG (left graph) or AZD (right graph). C. Immunoblot showing induction of phospho-ERKThr202/Tyr204 in MEC1 cells treated with DMSO or 500 nM AMG-176 for 16 hours. Relative ERK or phospho-ERK was quantitated and plotted showing mean ± SEM (n=3, separate experiments); *p<0.05 significant difference from DMSO control. D. MEC1 and Mino cells were treated with DMSO, 500 nM AMG-176, 500 nM AZD5991, 10 μM trametinib (T-nib), AMG-176 plus T-nib, or AZD5991 plus T-nib for 16 hours. Immunoblot analysis of equal amount of proteins were probed with the indicated antibodies. Vinculin was probed for loading control. Because MCL-1 and GSK3β protein bands are from the same immunoblot for MEC1 cells, identical vinculin blot is shown in rows 3 and 9. Similar data obtained from three biological repeats. E. Immunoblot analysis of total and phospho-Mcl-1Thr163 and total and phospho-ERK Thr202/Tyr204 expressions from primary CLL samples treated with DMSO, 500 nM AMG-176, 500 nM AZD5991, trametinib (10 μM) alone or in combination, for 12 hours. F. Immunoblot analysis showing HEK293 cell line transiently transfected with WT- or T163A-FLAG-MCL-1. Forty-eight hours following transfection, cells were treated with DMSO, AMG-176 (500nM) or AZD5991 (500nM) for 24 hours. Equal amount of proteins was loaded and probed with indicated antibodies. FLAG expression was measured by LI-COR quantitation and represented in the bar graph (n=3) showing mean ± SEM. AMG: AMG-176; AZD: AZD5991; T-nib: Trametinib. Note. For Figures A-F, to optimally capture total and modified (phosphorylated) protein, we used one immunoblot and probed same protein band with two different species of antibodies. The membranes were imaged using Infrared Odyssey CLx machine. With different species of antibodies, we can optimally quantitate data because these are read at different wavelengths. This spectrally distinct fluorophores technique (for example, 700 nm (red) or 800 nm (green) channels) allows us to do multiplexing on the same protein band. Because immunogenicity of 2 separate antibodies are tested in the same protein blot, bands have similar appearance and features for each protein in both total and phospho-bands.
MCL-1 inhibitors induced a transient decrease in Mule and Noxa expression.
Since BH3 proteins, including the E3 ligase Mule and the pro-apoptotic Noxa, can regulate Mcl-1 ubiquitination, stability, and half-life (17, 28), we assessed the impact of MCL-1i on expression of these proteins and their interaction with Mcl-1. Interestingly, a transient decrease in Mule expression was observed one-hour following treatment with MCL-1i (Figure 4A; one-way ANOVA, p = 0.02), which increased at later time points (6 and 24-hours). Noxa expression also dramatically decreased following treatment with MCL-1i, without any changes in Trim-17 or β-TRCP expression. To further support our observation, we incubated MEC1 cell line with AZD5991 and followed up change in protein levels at shorter time intervals. Indeed, treatment by AZD5991 decreased Mule protein expression at 30 minutes followed by gradual increase to reach normal level at 6 hours, while Noxa expression decreased in a time and dose dependent manner (Supplemental Figure 3A). This transient decrease in Mule expression is thought to be due to decreased stability of Mule protein (Supplemental Figure 3B) mediated by a decreased Mule:USP9x interaction (Supplemental Figure 3C). Interestingly, our IP data showed that Mule:Mcl-1 interaction was not disrupted (Figure 4B), despite both binding to the same BH3 pocket of Mcl-1 protein. In contrast, pro-apoptotic proteins including Noxa, BAK, and BAX are disrupted in either MEC1 (endogenous; Figure 4C, left panel) or FLAG-MCL-1 and HA-UB co-transfected HEK293 cells (Figure 4C, right panel). In vitro pull-down assay of recombinant GST-tagged Mule (catalytic domain) or Mule IP lysate from HEK293 cells by His-tagged recombinant full length Mcl-1 confirmed that Mule:Mcl-1 interaction was maintained in the presence of inhibitors (Figure 4D). Interestingly, Mule was found to dimerize under non-denaturing condition (Supplemental Figure 3D). We performed an in vitro ubiquitination assay to assess Mule’s role to ubiquitinate Mcl-1 in the presence of MCL-1i. AMG-176 was found to enhance Mcl-1 ubiquitination in similar magnitude as maritoclax, while AZD5991 failed to ubiquitinate Mcl-1 significantly compared to DMSO (Figure 4E). Similar observations were seen with the full-length Mule IP lysate in an in vitro Mcl-1 ubiquitination study (Supplemental Figure 3E), so much so, that even in the absence of the recombinant Mule catalytic domain, E2 conjugating enzyme can ubiquitinate Mcl-1 protein (Figure 4E, lane 5). This observation indicates that MCL-1i can directly induce enhanced (AMG-176) or decreased (AZD5991) Mcl-1 ubiquitination independent of Mule. However, Mule as a catalyst can accelerate the ubiquitination process. No significant change in Bim or PUMA expression was noticed (Figure 4F). The transient decrease in Mule expression, loss of Noxa:Mcl-1 interaction, and decreased Noxa protein may contribute to more Mcl-1 protein accumulation induced by MCL-1i.
Figure 4. MCL-1 inhibitors induced a transient decrease in Mule and Noxa expression.
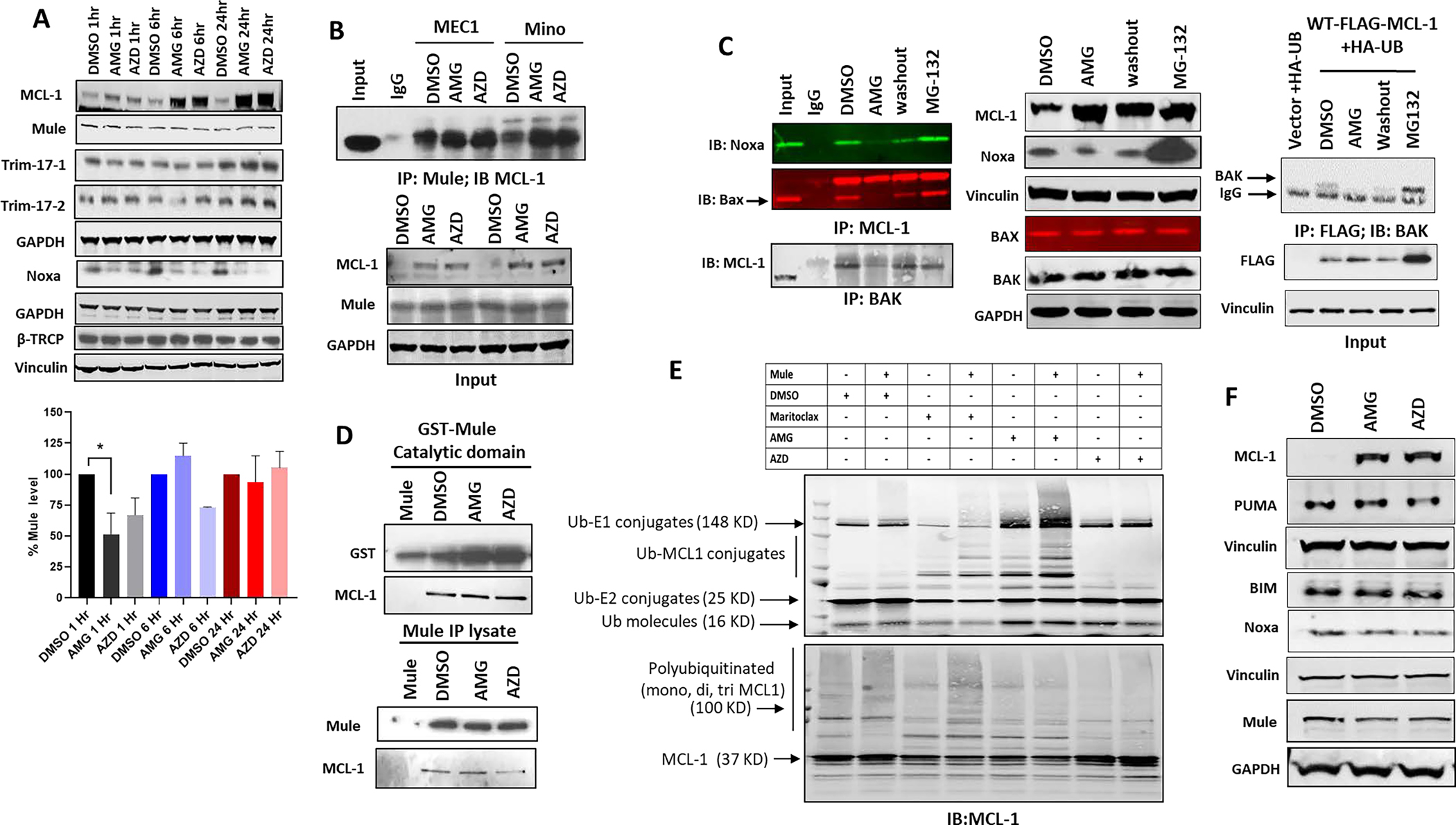
A. Upper panel: Immunoblot for MEC1 cell line treated with DMSO, AMG-176 (1μM) or AZD5991 (1μM) for the indicated time points. Blot was probed with different E3 ligases (Mule, Trim-17–1, 17–2, β-TRCP) and Noxa. Lower panel: bar graph showing relative Mule expression (*p<0.05 significant difference from DMSO control). B. Upper panel: Immunoblot for MEC1 and Mino cell lines were treated with DMSO. AMG-176 (1μM) or AZD5991(1μM) for 16 hours. Equal amount of protein (500μg) was IP with rabbit Mule followed by IB of mouse Mcl-1 antibody. Lower panel, showing input. GAPDH was used for loading control. C. Left panel: Immunoblot for MEC1 cell line treated with DMSO, AMG-176 (500nM), AMG-176 (500nM) followed by wash out for four hours, or MG-132(1.5 μM) for 16 hours. Equal amount of proteins (500 μg) were immunoprecipitated by either mouse Mcl-1 antibody and probed with Noxa and Bax antibodies or immunoprecipitated with mouse BAK antibody and probed with Mcl-1 antibody respectively. Middle panel: showing input. Vinculin and GAPDH was used for loading controls. Right panel: HEK293 cells were co-transfected with HA-Ub and FLAG-MCL-1 for 48 hours followed by treatment by either DMSO, AMG-176, or AMG-176 washout (upper right panel) for 4 hours or MG-132 (1.5 μM) for 16 hours. Equal amount of proteins (500 μg) were immunoprecipitated by FLAG beads and probed with BAX. Bottom right panel shows input for FLAG. Vinculin was used for loading control. D. Immunoblot showing in vitro pull-down assay of His-tag recombinant Mcl-1 with either GST-tagged recombinant Mule (upper panel) or endogenous full-length Mule immunoprecipitated lysate from untreated HEK293 (lower panel) as described in methods section. Reaction was made for one hour in the presence of DMSO, AMG-176 (10mM) or AZD5991 (10mM) in a total volume of 1ml. A control sample without Mcl-1 protein was added. Samples were immunoblotted for Mule and Mcl-1. E. Immunoblot assay following in vitro ubiquitination assay of His-tag recombinant Mcl-1 protein in the presence of absence of Mule. Samples were untreated or treated with DMSO, AMG-176 (10mM), AZD5991 (10mM) or maritoclax (10mM) and incubated for two hours in ubiquitination buffer containing FLAG-tagged ubiquitin as described in method section. Samples were immunoblotted and probed with either anti-FLAG antibody to detect ubiquitination (upper panel) or rabbit Mcl-1 (lower panel). F. Immunoblot analysis for MEC1 cell line treated with either DMSO, AMG-176 (500nM), or AZD5991 (500nM) for 24 hours and probed with different BH3 proteins. Vinculin and GAPDH were probed for loading control. AMG: AMG-176; AZD: AZD5991.
MCL-1 inhibitors target deubiquitinases for enhanced Mcl-1 de-ubiquitination
Since both DUBs and multiple E3 ligases, including Mule(20–22), modulate Mcl-1 stability, we sought to explore the role of DUBs in Mcl-1 stability induced by MCL-1i. MCL-1i treatment did not significantly change protein levels of DUBs (USP9x, USP13 or KU-70, one-way ANOVA, p =0.35) (Figure 5A). Co-IP experiment in MEC1 cells treated with MCL-1i showed increased USP9x interaction with Mcl-1 compared to DMSO control. However, this might be due to increased amount of Mcl-1 protein in the MCL-1i-treated samples (relative ratio of USP9x:Mcl-1 is 0.9 with AMG-176 and AZD5991 and 1 with DMSO) (Figure 5B). In order to test the DUB functional activity, we used in vitro de-ubiquitination assay and demonstrated that MCL-1i enhanced the de-ubiquitination of Mcl-1 by USP9x as compared to DMSO (Figure 5C). However, the enhanced DUB activity is less likely to be a result of direct effect of MCL-1i on USP9x as indicated by normal DUB assay activity in the presence of MCL-1i as compared to DMSO (Figure 5D, Supplemental Figure 4 A–C). Interestingly, AMG-176 showed an intrinsic fluorescence property as compared to AZD5991. Even in the absence of recombinant USP9x, AMG-176 showed a very high relative fluorescence (Supplemental Figure 4D), that decreased with lower doses of AMG-176 as compared to AZD5991 (Supplemental Figure 4E).
Figure 5. MCL-1 inhibitors target deubiquitinases for enhanced Mcl-1 de-ubiquitination.
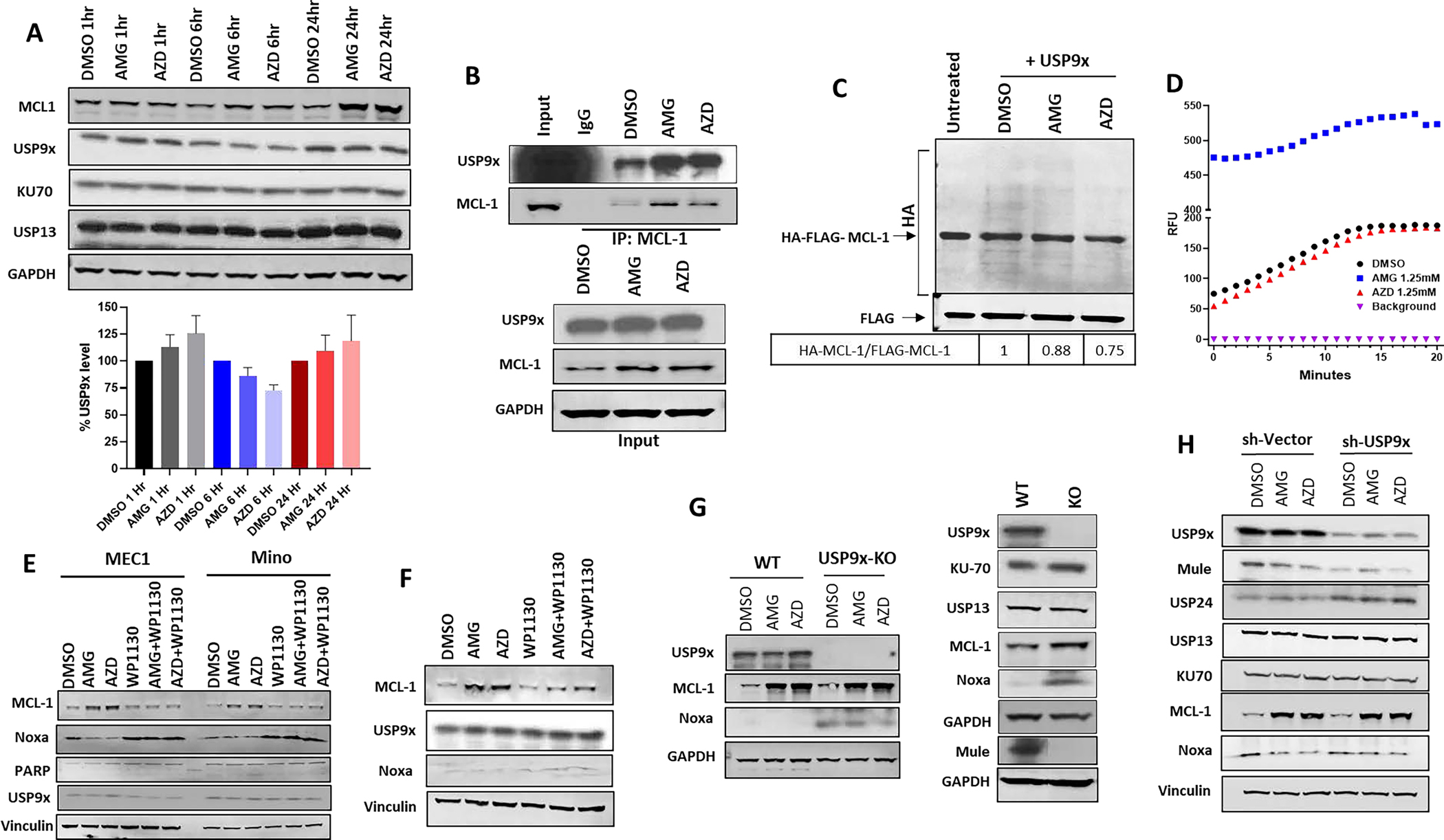
A. Immunoblot analysis for MEC1 cells treated with either DMSO, AMG-176 (1μM) or AZD5991 (1μM) for the indicated time points (upper panel). Bar graph showing relative USP9x expression (n=3; lower panel). B. Upper panel showing immunoblot analysis of MEC1 cell line treated with either DMSO, AMG-176 (1μM), or AZD5991 (1μM) for 16 hours. Equal amount of proteins (500μg) were immunoprecipitated with mouse Mcl-1 antibody and immunoblotted with rabbit USP9x and Mcl-1. Lower panel showing input control. GAPDH was used for loading control. C. Immunoblot analysis showing in vitro de-ubiquitination assay of purified full-length HA-Ub-FLAG-Mcl-1 protein by purified USP9x in the presence of either DMSO, AMG-176 (10mM), or AZD5991 (10mM) as described in method section followed by probing with anti-HA to detect FLAG-Mcl-1 ubiquitination. Relative ratio of AMG and AZD-induced Mcl-1 ubiquitination compared to DMSO control is shown underneath. D. Line graph showing the Relative Fluorescence Unit (RFU) of recombinant USP9x after incubation with either DMSO, AMG (1.25mM) or AZD (1.25mM) in DUB buffer containing the fluorogenic Ubiquitin-AMC substrate in 96 well plate as described in method section. E. Immunoblot analysis of MEC1 and Mino cell lines treated with DMSO, AMG-176 (1μM), AZD5991(1μM), WP1130 (5μM), either alone or in combination with AMG-176 or AZD5991 for six hours. All samples were also pre-treated by the pan-caspase inhibitor QVD (20μM) to inhibit apoptosis. F. Immunoblot analysis from primary CLL patient sample either treated with DMSO, AMG-176 (500nM), AZD5991 (500nM), WP1130 (10μM), either alone or in combination with AMG-176 or AZD5991 for six hours. All lanes are pre-treated by the pan-caspase inhibitor, QVD, (20μM), to inhibit apoptosis. G. Left panel; Immunoblot analysis of WT and USP9x knockout HCT-116 cell line treated with DMSO, AMG-176 (1μM) or AZD5991 (1μM) for 16 hours and probed with the indicated antibodies. Right panel; Immunoblot analysis of untreated WT and USP9X knockout HCT-116 cell line and probed with the indicated antibodies. H. Immunoblot of MEC1-shRNA-USP9x knockdown treated by DMSO, AMG-176 (1μM) or AZD5991 (1μM) for 16 hours and probed with the indicated antibodies. All lanes are pre-treated by the pan-caspase inhibitor QVD (20μM) to inhibit apoptosis. AMG: AMG-176; AZD: AZD5991.
To further confirm the role for DUBs in the observed Mcl-1 protein stability, we tested the effect of the DUB inhibitor-WP1130, which abrogated Mcl-1 induction in response to MCL-1i in both MEC1 and Mino cell lines (Figure 5E) as well as in CLL lymphocytes, and upregulated Noxa level (Figure 5F). WP1130 has been tested before to inhibit DUB activity in a dose dependent manner (1–5μM); at a dose of 5 μM, WP1130 inhibited 80% of DUB activity(29). This data demonstrates that inhibition of DUBs can completely reverse the effect of MCL-1i on Noxa and Mcl-1 protein. However, USP9x knockout in HCT116 (Figure 5G) or USP9x knockdown in MEC1 cells (Figure 5H) did not abrogate Mcl-1 protein stability. This may be due to the overexpression of other DUBs (e.g. USP24) which may retain overall DUB activity(30). Indeed, a compensatory increase in KU-70 (HCT116-USP9x-KO) and USP24(MEC1-shRNA-USP9x) were observed while strong upregulation of Noxa in HCT116-USP9x-KO was seen similar to WP1130-treated cells. In contrast, Mule expression was dramatically decreased by loss of USP9x, reflecting an important role for USP9x in regulating Mule (Figure 5G, H).
Conformation change upon inhibitor binding altered post-translation modification, interaction and stability in the Mcl-1 protein
Primary process by which MCL-1i elicit their effect is through binding to Mcl-1 protein which results in conformation change. Our in-silico modeling suggests that residues 1–172 in the Mcl-1 protein might have largely disordered structures with high structural flexibility. This may be why, to date, no crystal structures have been obtained for these residues, which is in agreement with a previous report(5). However, our modeling also revealed that residues 150–172-fold back and form close interactions with the inhibitor binding site in Mcl-1. Molecular dynamics simulation showed that binding of AMG-176 or AZD5991 to Mcl-1 protein significantly affects the conformation of these 23 residues. In particular, the inhibitor binding pushes the residues away from one of the α-helices and increases the solvent-accessible surface area of Thr163 from 22.01 Å2 to 64.79 Å2 and Ser159 from 35.63 Å2 to 42.76 Å2 (Figure 6A). These findings indicate that the two residues, especially Thr163 (>42 Å2 difference), may become more accessible to other partnering proteins, such as protein kinases, and may lead to increased phosphorylation and stabilization of the Mcl-1 protein (Figure 3). AMG-176 and AZD5991 bind to the same BH3 pocket such as the BH3 domains of BAX, BAK, (Figure 6B, C) and Mule (Figure 6D). So, the binding of MCL-1i can disrupt the binding of other BH3 proteins. Indeed, BAX-BAK interaction was disrupted but not Mule (Figure 4B, C). Moreover, the interaction of USP9x, which binds to the N-terminus of Mcl-1, can be enhanced by these N-terminus conformation changes where several residues in Mcl-1, including Ser155, Ser159 and Thr163 (cyan sticks), are spaced out and become more exposed (see SASA data), and thus prone to phosphorylation. Once this occurs, the increased negative charges (phosphate groups) will enhance their binding to USP9x (orange) by interacting with several positively charged residues including Arg1936, Arg1940, Arg1941, and Lys1943 on USP9x (Figure 6E). Based on our data, we provide a schematic summary for the mechanisms of MCL-1i induced stability of Mcl-1 protein (Figure 6F).
Figure 6. Conformation change upon inhibitor binding altered post-translation modification, interaction and stability in the Mcl-1 protein.
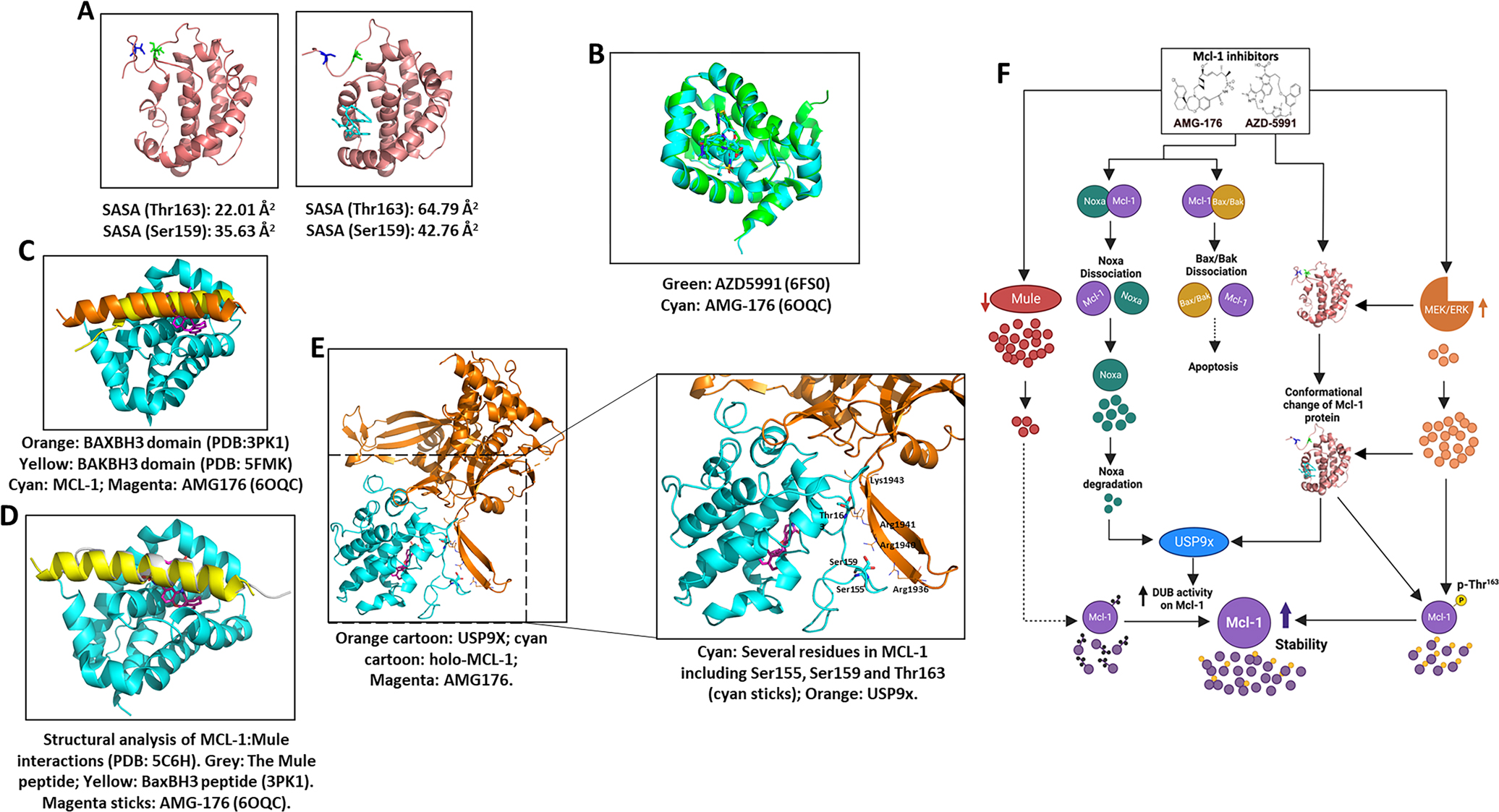
A. Mcl-1 apo structure after 10-ns molecular dynamics simulation. Left panel, binding efficacy (solvent-accessible surface area; SASA) without an MCL-1 inhibitor; right panel, complex of AMG-176 and Mcl-1 protein. Pink ribbon, Mcl-1; Blue, Ser159 residue; Green, Thr163; Cyan, AMG-176. B. Binding site of AMG-176 and AZD5991, Cyan: AMG176-Mcl-1 complex structure (PDB: 6OQC); Green: AZD5991 (PDB: 6FS0). C. Binding site for AMG-176 and BAX-BAK BH3 domains: Orange: BAXBH3 domain (PDB:3PK1); Yellow: BAKBH3 domain (PDB: 5FMK); Cyan: Mcl1; Magenta: AMG-176 (6OQC). D. Structural analysis of MCL1-Mule interactions (PDB: 5C6H). The Mule peptide (grey) binds to MCL1 (cyan) in the same region as BaxBH3, also partially overlapped with inhibitor binding (AMG-176). E. Left panel, MD simulations showed that the inhibitor binding can cause large conformational change of Mcl-1 protein and make the N-terminus more extended and less packed. Orange cartoon: USP9x; cyan cartoon: holo-MCL1; magenta: AMG-176. Right panel, Zoom-in view of MCL1-USP9x interactions. Several residues in Mcl-1 (cyan) including Ser155, Ser159 and Thr163 (cyan sticks) are spaced out and more exposed (see SASA data), thus prone to phosphorylation. Once phosphorylation occurs, the increased negative charges (phosphate groups) will enhance their binding to USP9x (orange) by interacting with several positively charged residues including Arg1936, Arg1940, Arg1941, and Lys1943 on USP9x. F. Schema summary showing mechanisms of MCL-1i induced stability of Mcl-1 protein. Anti- and pro-apoptotic proteins (Mcl-1, Bax, Bak, Noxa) are indicated by circles. Ubiquitinase and deubiquitinase proteins are oval shaped. Dissociation of Mcl-1 from BAX/BAK may be a trigger for apoptosis induction. Noxa disruption and degradation (e.g. AMG and AZD), along with MCL-1i induced conformation that does not favor ubiquitination and enhances de-ubiquitination by USP9x (e.g. AZD) can stabilize MCL-1. ERK phosphorylates Thr-163 on Mcl-1 which is facilitated by conformation change. Phosphorylated Mcl-1 is more stable. Mule adds ubiquitins on Mcl-1 protein however increased USP9x DUB activity removes these ubiquitins, increasing Mcl-1 protein stability. SASA: Solvent-Accessible Surface Area.
Discussion
MCL-1i-induced upregulation of Mcl-1 protein was shown to be due to increased stability, without any change in transcription, and it correlated with target engagement of MCL-1i to Mcl-1 protein, which can serve as a biomarker of target engagement(5). While disruption/inhibition of Mule binding to Mcl-1 by the MCL-1i for targeting Mcl-1 proteasomal degradation could be an attractive explanation for the upregulation of Mcl-1 protein, data supporting this hypothesis are inconclusive and lack mechanistic experiments by MCL-1i(17). Consistent with the data of Mcl-1 upregulation by MCL-1i, we showed that AMG-176 and AZD5991 upregulated Mcl-1 protein in B-cell lines and primary CLL lymphocytes due to increased Mcl-1 stability rather than increased transcription. However, this increased Mcl-1 stability failed to offer any resistance to the action of MCL-1i as evidenced by loss of BAK:Mcl-1 interaction and concomitant induction of apoptosis.
This data indicates that disruption of Mcl-1 protein interaction with proapoptotic molecules is enough to induce apoptosis regardless of Mcl-1 protein accumulation. However, this Mcl-1 protein accumulation serves as a biomarker for target engagement, as shown previously using FRET studies by Tron et al, 2018, where Mcl-1 protein level increases with MCL-1i targeting Mcl-1 and is reversed with washout of Mcl-1i as shown in Figure 2. Sensitivity to MCL-1i varies among cell lines and CLL patient samples. Current mechanistic investigations further confirm our prior report that there is no association of apoptosis with Mcl-1 protein induction(10). Due to the presence of caspase cleavage sites, Mcl-1 protein undergoes cleavage and disintegration once apoptosis initiates. We also showed decreased Mcl-1 ubiquitination by AMG-176 that was reversible upon AMG-176 washout, suggesting MCL-1i-induced defective Mcl-1 ubiquitination as a possible mechanism for this stability.
Treatment with MCL-1i induced an ERK-mediated increase in Mcl-1Thr163 phosphorylation in B-cell lines and CLL lymphocytes that may be enhanced by increasing SASA to T163 and S159 due to Mcl-1 conformation change (Figure 6A). Although the use of trametinib, a MEK-inhibitor, abolished Mcl-1Thr163 phosphorylation, the change in total Mcl-1 level was variable among cell lines and patient samples, suggesting a partial and cell-type specific effect. Further, the use of the phospho-mutant T163A-MCL-1 did not significantly reverse the effect of MCL-1i-induced Mcl-1 upregulation. Conversely, no significant changes were observed in Mcl-1ser159 or GSK3β expression. Collectively, Mcl-1Thr163 phosphorylation only partially associates with MCL-1i-induced Mcl-1 upregulation.
BH3 only proteins were shown to regulate Mule-dependent ubiquitination of Mcl-1 where Bim opposes Mule:Mcl-1 interaction leading to its stability(31). In contrast, Noxa promotes Mule-dependent Mcl-1 ubiquitination by inhibiting USP9x:Mcl-1 interaction leading to Mcl-1 degradation (32). MCL-1i are also BH3 mimetic molecules(5, 7) and hence may regulate Mcl-1 protein level. With this respect, Mule expression transiently decreased following treatment with MCL-1i and compensated by gradual increase at later time points. Further, this transient loss in Mule expression was associated with decreased USP9x:Mule interaction affecting Mule stability. In addition, MCL-1i induced loss of Noxa level following disruption of Mcl-1:Noxa interaction, triggering Noxa degradation. The Noxa downregulation is may be due to decreased stability following its disruption. The disruption of Noxa interaction and its decreased expression may, in part, explain the decreased Mule:USP9x interaction where Noxa over-expression can increase Mule:USP9x and decrease Mcl-1:USP9x interaction(32).
Despite the loss of interaction of proapoptotic BH3 proteins (e.g. BAX, BAK and Noxa) to Mcl-1 by MCL-1i, Mule:Mcl-1 interaction wasn’t disrupted by MCL-1i in both endogenous co-IP as well as in an in vitro pull-down assay. One possible mechanism for the observed lack of disruption of Mcl-1:Mule interaction in the presence of MCL-1i could be the access and availability of additional site(s) of interaction outside the BH3 binding groove of Mcl-1 where Mule could also bind to the N-terminal 30 amino acids of Mcl-1(33). This in turn can allow unrestricted Mule:Mcl-1 binding even when a BH3-only protein interacts with Mcl-1. This is supported by the evidence that other BH3 mimetics like Noxa and maritoclax can bind to the BH3 groove of Mcl-1 and, at the same time, potentiate Mule interaction(32, 34, 35). However, this fails to explain why Bim inhibited Mule ubiquitination to Mcl-1 in in vitro studies(31). Hence, we characterize here two categories of BH3-only proteins that regulate Mule interaction to Mcl-1. BH3-only proteins, such as Bim and PUMA, that bind non-specifically to Mcl-1 tend to block Mule interaction to Mcl-1(31). On the other hand, BH3-only proteins that bind specifically to Mcl-1, such as Noxa, do not interfere with Mule interaction to Mcl-1. Specific MCL-1i such as AMG-176 and AZD5991 are predicted to bind to Mcl-1 in a similar manner as Noxa; although Noxa does not impair Mule interaction to Mcl-1.
In order to verify if Mule binding is still functional, we performed in vitro ubiquitination studies of Mcl-1 protein in the presence of MCL-1i with or without Mule. Interestingly, AMG-176 was shown to enhance Mcl-1 ubiquitination in a similar way to martioclax, an MCL-1i that is known to ubiquitinate and degrade Mcl-1 protein. Ubiquitination occurred even in the absence of Mule, suggesting a direct effect of AMG-176 on Mcl-1 ubiquitination, independent of Mule. Indeed, the addition of Mule catalyzed this process. On the other hand, the magnitude of Mcl-1 ubiquitination in the presence of AZD5991 is far less than AMG-176. These data suggest that a possible conformation change induced by AMG-176 and maritoclax result in Mcl-1 ubiquitination. Conversely, AZD5991 does not induce similar conformation change in the Mcl-1 protein and hence had little effect on Mcl-1 ubiquitination compared to AMG-176. The reason for this discrepancy between AMG-176 and AZD5991 is not clear. However, it may be due to binding differences between AMG-176 and AZD5991 to the BH3 groove of Mcl-1. AZD5991 has an extra binding site to the BH3 groove of Mcl-1 where the 17-chloride atom of AZD5991 was shown to bind to the backbone carbonyl alanine-227 whereas with AMG-176 the chlorine atom is buried in the induced fit hydrophobic pocket (5, 36). Also different BH3 molecules can induce different Mcl-1 Q221R222N223 motif conformation either towards a non-helical (e.g. Bim) conformation that does not favor ubiquitination or a helical (e.g. maritoclax, Noxa, Mule) one that favors ubiquitination (35, 37). However, further studies are needed to explain this discrepancy. Elucidating differences between MCL-1i might help guide the development of potent Mcl-1i since AZD5991 is shown to be more potent than AMG-176 (Supplemental Figure 1E, F). Another interesting point, if AMG-176 can enhance Mcl-1 ubiquitination in in vitro ubiquitination assays like maritoclax, then why does AMG-176 stabilize Mcl-1 while maritoclax degrades Mcl-1 in treated cells? One possible explanation may be the difference in Noxa expression where maritoclax does not affect Noxa expression significantly(34) whereas we showed that AMG-176 downregulated Noxa expression. Normally, Noxa regulates USP9x interaction to Mcl-1.
The observed AMG-176-induced ubiquitination of Mcl-1 in in vitro ubiquitination studies triggered us to explore the role of DUBS in the observed stability, since it can reverse the ubiquitination process. Following treatment with MCL-1i, our IP data showed similar increase for either USP9x or Mcl-1. However, in vitro DUB assay revealed enhanced USP9x activity following treatment with MCL-1i compared to DMSO that is more prominent with AZD5991. While the observed in vitro high DUB activity is not related to the direct effect of MCL-1i on USP9x, we predict that MCL-1i-induced change in Mcl-1 conformation may expose N-terminus offering enhanced USP9x activity. Interestingly, we also report here for the first time the intrinsic fluorescence properties of AMG-176 which can be clinically useful in tracking AMG-176 pharmacokinetics.
Pharmacological inhibition of DUBs by WP1130 completely abrogated Mcl-1 induction following treatment with MCL-1i; however, genetic knockout of USP9x was ineffective in our study. Failure of USP9x knockout to control Mcl-1 stability in HCT116 has been previously reported(38). Later, Peterson et.al,(30) showed that the HCT116 USP9x-knockout retained its DUB activity. They attributed it to the possible compensatory increase in other DUBs (e.g., USP24) that may compensate for USP9x deficiency and the need for complete depletion of USP9x since small level of the protein can take over the function. Besides, it is generally more effective to inhibit multiple DUBs(30, 39). This may explain why WP1130, but not USP9x knockout, abrogated the MCL-1i induced stability of Mcl-1 protein. Here we also showed KU-70 upregulation in HCT116-USP9x knockout. Similar observation was noted in MEC1-shRNA-USP9x with compensatory increase in USP24. Further, we reported that the loss of USP9x downregulated Mule expression significantly in either HCT116-USP9x-KO or MEC1-shRNA-USP9x. This may stabilize MCL-1 and explain why sometimes USP9x knockdown does not affect Mcl-1 stability. It is interesting to see a dramatic decrease in Mule compared to no significant change in Mcl-1 expression with USP9x downregulation. This highlights a dual function for USP9x where, on one hand, it stabilizes Mule, possibly enhancing Mcl-1 degradation, and, on the other hand, it de-ubiquitinates Mcl-1. The switch between the two functions can possibly be regulated by Noxa. Pharmacological or genetic loss of USP9x results in an increase in Noxa. While this was not fully evaluated, a possibility is that WP1130-mediated ER stress may induce ATF4, a transcription factor for Noxa (40).
The AZD5991 clinical trial (NCT03218683) has been on hold recently after reporting a case with asymptomatic elevation in cardiac parameters (https://www.ashclinicalnews.org/online-exclusives/fda-places-clinical-hold-trials-azd5991). In concert, another MCL-1i, S63845, impaired contractility and myofibril assembly in human induced pluripotent stem cells cardiomyocytes(41). Our data showed that MCL-1i decreased Mule and Noxa stability with a potential role in MCL-1i-induced cardiotoxicity, although further investigations are required. Mule was suggested to protect the heart against oxidative stress; its cardiac knockdown in mice lead to cardiac hypertrophy and left ventricular dysfunction(42). Also, Noxa was shown to regulate autophagy, and its downregulation mediated phenylephrine-induced cardiac hypertrophy(43–46). Collectively these indirect mechanisms and direct targeting of Mcl-1 may be responsible for drug-induced cardiotoxicity.
In conclusion, MCL-1i are not only important in clinic, they serve as powerful tools to explore Mcl-1 protein regulation by BH3 mimetics. MCL-1i enhanced Mcl-1 de-ubiquitination through Noxa disruption, enhanced de-ubiquitination and Mule destabilization. They bind to Mcl-1 specifically, like Noxa, and hence they do not impair the BH3 only protein E3 ligase Mule interaction to Mcl-1, unlike other BH3 proteins (e.g. Bim, PUMA). Although MCL-1i are similar in their mechanism of action, striking differences were elaborated that can help guide development of more potent MCL-1i. Molecular changes of Noxa downregulation and Mule de-stabilization following MCL-1i treatment may be potential inducers of the reported cardiotoxicity induced by MCL-1i, although further investigations are required.
Supplementary Material
Translational Relevance.
Bcl-2 family antiapoptotic proteins provide survival advantage to many tumors. The founding member, Bcl-2, has been effectively targeted by venetoclax. This success provided impetus to design inhibitors for Mcl-1, another member of Bcl-2 antiapoptotic family. MCL-1 inhibitors (MCL-1i) including AMG-176 and AZD5991, are currently in clinic for hematological malignancies. Preclinical studies suggest that binding of these inhibitors augment Mcl-1 protein accumulation; however precise mechanism is unknown. Apart from transcription and translation, several posttranslational processes contribute to Mcl-1 stability, accumulation, and degradation. Therefore, a detailed understanding of molecular changes associated with Mcl-1 protein stability is imperative for the optimal use of these agents in the clinic. Here we show that MCL-1i-induced disruption of Mcl-1 protein ubiquitination via engaging enhanced deubiquitination and Mule destabilization play a major role in Mcl-1 protein stability and establishes a rationale for designing next generation of more effective MCL-1i for the treatment of hematological malignancies.
Acknowledgments
This work was supported in part by a CLL Global Research Foundation Alliance grant and MD Anderson Cancer Center’s CLL Moon Shot™ program. Zhi Tan receives research funding (RR220039) from Cancer Prevention and Research Institute of Texas (CPRIT). S. Tantawy received a scholarship from the Administration of Cultural Affairs and Missions, Egypt, to work as a visiting student at MD Anderson. Authors are thankful to Dr. Rong Chen, Dr Betty Lamothe and Dr. H. Amin, The University of Texas MD Anderson Cancer Center for providing, pCMV-Tag2A vector, HA-UB plasmids and Mino cell lines respectively. Authors are thankful to Dr. Fred Bunz, John Hopkins University for providing WT and USP9x knockout HCT116 cell lines. We thank Stephanie Deming of Scientific Publications, Research Medical Library, MD Anderson Cancer Center, for editing the manuscript and Danielle Walsh, Scientific Project Director, CLL Moon Shot, for reviewing final revised manuscript.
Support:
This work was supported in part by an Alliance grant from the CLL Global Research Foundation (to V.G.); by MD Anderson’s Chronic Lymphocytic Leukemia Moon Shot program (to V.G. and W.G.W.); and by the NIH/NCI through MD Anderson’s Cancer Center Support Grant, CA016672. Zhi Tan receives research funding (RR220039) from Cancer Prevention and Research Institute of Texas (CPRIT).
Footnotes
Conflict-of-interest disclosure
The authors do declare no potential conflicts of interest.
References
- 1.Davids MS, Deng J, Wiestner A, Lannutti BJ, Wang L, Wu CJ, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood, The Journal of the American Society of Hematology. 2012;120(17):3501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K, et al. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. The American journal of pathology. 1995;146(6):1309. [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell KJ, Dhayade S, Ferrari N, Sims AH, Johnson E, Mason SM, et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell death & disease. 2018;9(2):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nature communications. 2018;9(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch R, Christie AL, Crombie JL, Palmer AC, Plana D, Shigemori K, et al. Biomarker-driven strategy for MCL1 inhibition in T-cell lymphomas. Blood, The Journal of the American Society of Hematology. 2019;133(6):566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer discovery. 2018;8(12):1582–97. [DOI] [PubMed] [Google Scholar]
- 8.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–82. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, He S, Look AT. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia. 2019;33(1):262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi X, Sarkar A, Kismali G, Aslan B, Ayres M, Iles LR, et al. AMG-176, an Mcl-1 antagonist, shows preclinical efficacy in chronic lymphocytic leukemia. Clinical Cancer Research. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leverson J, Zhang H, Chen J, Tahir S, Phillips D, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell death & disease. 2015;6(1):e1590–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BCL-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. Journal of cellular physiology. 1996;166(3):523–36. [DOI] [PubMed] [Google Scholar]
- 13.Jourdan M, De Vos J, Mechti N, Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death & Differentiation. 2000;7(12):1244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritsch RM, Schneider G, Saur D, Scheibel M, Schmid RM. Translational repression of MCL-1 couples stress-induced eIF2α phosphorylation to mitochondrial apoptosis initiation. Journal of Biological Chemistry. 2007;282(31):22551–62. [DOI] [PubMed] [Google Scholar]
- 15.Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin C-J, et al. mTORC1 promotes survival through translational control of Mcl-1. Proceedings of the National Academy of Sciences. 2008;105(31):10853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells. 2014;3(2):418–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–95. [DOI] [PubMed] [Google Scholar]
- 18.Magiera M, Mora S, Mojsa B, Robbins I, Lassot I, Desagher S. Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death & Differentiation. 2013;20(2):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Q, He X, Hsu J-M, Xia W, Chen C-T, Li L-Y, et al. Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Molecular and cellular biology. 2007;27(11):4006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103–7. [DOI] [PubMed] [Google Scholar]
- 21.Wang B, Xie M, Li R, Owonikoko T, Ramalingam S, Khuri F, et al. Role of Ku70 in deubiquitination of Mcl-1 and suppression of apoptosis. Cell Death & Differentiation. 2014;21(7):1160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nature communications. 2018;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23(31):5301–15. [DOI] [PubMed] [Google Scholar]
- 24.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Molecular cell. 2006;21(6):749–60. [DOI] [PubMed] [Google Scholar]
- 25.Sarkar A, Stellrecht CM, Vangapandu HV, Ayres M, Kaipparettu BA, Park JH, et al. Ataxia telangiectasia mutated interacts with Parkin and induces mitophagy independent of kinase activity. Evidence from mantle cell lymphoma. Haematologica. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neutzner A, Li S, Xu S, Karbowski M, editors. The ubiquitin/proteasome system-dependent control of mitochondrial steps in apoptosis. Seminars in cell & developmental biology; 2012: Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong J, Wang P, Tan S, Chen D, Nikolovska-Coleska Z, Zou F, et al. Mcl-1 degradation is required for targeted therapeutics to eradicate colon cancer cells. Cancer research. 2017;77(9):2512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakajima W, Hicks MA, Tanaka N, Krystal GW, Harada H. Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer. Cell death & disease. 2014;5(2):e1052–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer research. 2010;70(22):9265–76. [DOI] [PubMed] [Google Scholar]
- 30.Peterson LF, Sun H, Liu Y, Potu H, Kandarpa M, Ermann M, et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood, The Journal of the American Society of Hematology. 2015;125(23):3588–97. [DOI] [PubMed] [Google Scholar]
- 31.Warr MR, Acoca S, Liu Z, Germain M, Watson M, Blanchette M, et al. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS letters. 2005;579(25):5603–8. [DOI] [PubMed] [Google Scholar]
- 32.Gomez-Bougie P, Ménoret E, Juin P, Dousset C, Pellat-Deceunynck C, Amiot M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochemical and biophysical research communications. 2011;413(3):460–4. [DOI] [PubMed] [Google Scholar]
- 33.Warr MR, Mills JR, Nguyen M, Lemaire-Ewing S, Baardsnes J, Sun KL, et al. Mitochondrion-dependent N-terminal processing of outer membrane Mcl-1 protein removes an essential Mule/Lasu1 protein-binding site. Journal of Biological Chemistry. 2011;286(28):25098–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doi K, Li R, Sung S-S, Wu H, Liu Y, Manieri W, et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. Journal of Biological Chemistry. 2012;287(13):10224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song T, Wang Z, Ji F, Feng Y, Fan Y, Chai G, et al. Deactivation of Mcl-1 by Dual-Function Small-Molecule Inhibitors Targeting the Bcl-2 Homology 3 Domain and Facilitating Mcl-1 Ubiquitination. Angewandte Chemie International Edition. 2016;55(46):14250–6. [DOI] [PubMed] [Google Scholar]
- 36.Hird AW, Tron AE. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacology & therapeutics. 2019;198:59–67. [DOI] [PubMed] [Google Scholar]
- 37.Miles JA, Hobor F, Trinh CH, Taylor J, Tiede C, Rowell PR, et al. Selective Affimers Recognise the BCL-2 Family Proteins BCL-xL and MCL-1 through Noncanonical Structural Motifs. ChemBioChem. 2021;22(1):232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris DR, Mims A, Bunz F. Genetic disruption of USP9X sensitizes colorectal cancer cells to 5-fluorouracil. Cancer biology & therapy. 2012;13(13):1319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’arcy P, Brnjic S, Olofsson MH, Fryknäs M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nature medicine. 2011;17(12):1636–40. [DOI] [PubMed] [Google Scholar]
- 40.Bianchetti E, Bates S, Carroll SL, Siegelin MD, Roth KA. Usp9X regulates cell death in malignant peripheral nerve sheath tumors. Scientific reports. 2018;8(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen ML, Taneja N, Neininger AC, Wang L, Robertson GL, Riffle SN, et al. MCL-1 inhibition by selective BH3 mimetics disrupts mitochondrial dynamics causing loss of viability and functionality of human cardiomyocytes. IScience. 2020;23(4):101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dadson K, Hauck L, Hao Z, Grothe D, Rao V, Mak TW, et al. The E3 ligase Mule protects the heart against oxidative stress and mitochondrial dysfunction through Myc-dependent inactivation of Pgc-1α and Pink1. Scientific reports. 2017;7(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu J, Hu W, Song Z-P, Chen Y-G, Zhang D-D, Wang C-Q. Rapamycin inhibits cardiac hypertrophy by promoting autophagy via the MEK/ERK/Beclin-1 pathway. Frontiers in physiology. 2016;7:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson ÅB, Mentzer RM, et al. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic research in cardiology. 2009;104(2):157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boengler K, Schulz R, Heusch G. Loss of cardioprotection with ageing. Cardiovascular research. 2009;83(2):247–61. [DOI] [PubMed] [Google Scholar]
- 46.Xu M, Wan C-x, Huang S-h, Wang H-b, Fan D, Wu H-M, et al. Oridonin protects against cardiac hypertrophy by promoting P21-related autophagy. Cell death & disease. 2019;10(6):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. For raw data request, please contact the corresponding author at vgandhi@mdanderson.org