

Abstract
Objective
To further clarify genotype:phenotype correlations associated with variants in KCNC1 encoding the voltage‐gated potassium (K+) channel subunit Kv3.1 and which are an emerging cause of a spectrum of neurological disease including intellectual disability, isolated myoclonus, progressive myoclonus epilepsy, and developmental and epileptic encephalopathy.
Methods
We describe the clinical and genetic characteristics of a series of three patients with de novo heterozygous missense variants in KCNC1 associated with nonspecific developmental delay/intellectual disability and central hypotonia without epilepsy or ataxia. All three variants lead to amino acids alterations with mild predicted differences in physicochemical properties yet are localized to the S6 pore region of the Kv3.1 protein between the selectivity filter and PXP motif important for K+ channel gating. We performed whole‐cell voltage clamp electrophysiological recording of wild‐type versus variants in a heterologous mammalian expression system.
Results
We demonstrate a prominent leftward (hyperpolarized) shift in the voltage dependence of activation and slowed deactivation of all variants in the clinically defined series.
Interpretation
Electrophysiological recordings are consistent with a gain of K+ channel function that is predicted to exert a loss of function on the excitability of Kv3‐expressing high frequency‐ firing neurons based on the unique electrophysiological properties of Kv3 channels. These results define a clinical‐genetic syndrome within the spectrum of KCNC1‐related neurological disorders.
Introduction
Potassium (K+) channels are critical determinants of neuronal excitability via setting the resting membrane potential and mediating repolarization of the action potential. 1 Variants in many K+ channel genes have been associated with epilepsy. 2 K+ channels exhibit broad molecular and genetic diversity and are organized into families and subfamilies based on DNA sequence similarities, with the voltage‐gated K+ channels (Kv) representing the largest family. 3 Previously identified missense variants in the Kv3 subfamily member gene KCNC1 encoding the K+ channel subunit Kv3.1 act via loss of function (LoF) at the level of the ion channel and are associated with a spectrum of neurological dysfunction including isolated nonprogressive myoclonus, nonspecific developmental delay/intellectual disability (ID), progressive myoclonus epilepsy type 7 (EPM or PME7), and developmental and epileptic encephalopathy (DEE), including a case of epilepsy of infancy with migrating focal seizures. 4 , 5 , 6 , 7 , 8 , 9 However, genotype–phenotype correlation remains unclear. Kv3.1 is one of four members of the Kv3 subfamily of voltage‐gated K+ channels (Kv3.1–3.4), which contribute to high‐frequency firing in specific subsets of neurons throughout the nervous system based on unique biophysical properties, including a depolarized voltage dependence of activation, fast activation kinetics, and rapid deactivation, relative to other Kv channels. 10 , 11
Here, we describe a category of KCNC1 neurological disease characterized by nonspecific developmental delay/ID and gross motor impairment without epilepsy, associated with KCNC1 variants that exhibit a unique and previously unreported biophysical abnormality of gain of function (GoF) at the ion channel level. Whole‐cell voltage clamp electrophysiological recordings of K+ channels formed by disease‐associated variants in a heterologous mammalian cell line revealed a left (hyperpolarized) shift in the voltage‐dependence of activation and slowed deactivation. These effects are predicted to exert a loss of function effect on Kv3‐expressing fast‐spiking neurons in the brain.
Methods
Study subjects, genetic testing, and variant interpretation
Patients were enrolled in a study approved by the Institutional Review Board of The Children's Hospital of Philadelphia (#15‐12226). Informed consent for participation was provided by each patient's parents as all patients were minors.
Patient 1 was a female born at term without issue. An electroencephalogram performed at age 2 months to evaluate episodes of paroxysmal arm jerking was without electrographic correlate but showed occasional sharply contoured waveforms at C3 and Cz during sleep. Neurological examination revealed apparent sensory nystagmus and concern for cortical visual impairment. An MRI of the Brain performed at age 5 months to evaluate nystagmus was normal. Ophthalmologic examination demonstrated mildly pale optic nerves but normal anterior and posterior chambers; an electroretinogram and an ocular coherence tomography were normal. A repeat EEG at age 6 months was normal. By age 1 year, the patient could roll over, prop sit, and sit without support, but exhibited nystagmus, axial hypotonia, and weakness with limited antigravity movement. At age 18 months, the patient remained non‐ambulatory, but could roll supine‐to‐prone and prone‐to‐supine and maintain a seated position for 2–3 min; she could grasp and bring objects to the midline. She smiled responsively and giggled, and expressive language included nonspecific consonant sounds but no words. She did not track or follow with concern for cortical visual impairment. There was no history of seizure. Trio whole genome sequencing identified a de novo heterozygous variant in KCNC1 c.1300G>C (p.Val434Leu), which is not present in the Exome Aggregation Consortium (ExAC) or genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org) and has a corresponding CADD score of 26.8. 12 This variant is localized to the S6 pore of the protein and converts the small hydrophobic amino acid valine to the slightly larger leucine, with a Grantham score of 32.
Patient 2 was a male born at term without issue. Central hypotonia was noted at age 3 months. He walked independently at age 23 months. At first evaluation at age 3 years, gait remained unsteady and he was unable to run or jump but could ascend or descend stairs while holding a railing. He had an extensive vocabulary and could speak in full sentences, but speech was difficult to understand. There was no nystagmus, tremor or myoclonus, or seizure. An MRI of the Brain was normal; EEG has not been performed. Trio whole exome sequencing identified a de novo heterozygous variant in KCNC1 c.1294G>A (p.Val432Met), which is not present in the Exome Aggregation Consortium (ExAC) or genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org) and has a corresponding CADD score of 27.6. This variant is localized to the S6 pore of the protein and converts the small hydrophobic amino acid valine to the slightly larger sulfur containing methionine associated with a Grantham score of 21.
Patient 3 was a female born at 32 and 4/7 weeks gestation without complications. Early mild gross motor developmental delay was initially attributed to prematurity; she walked at age 20 months corrected age. The patient was enrolled in speech therapy at age 2 years due to speech/language delay. There was no history of nystagmus, tremor, ataxia, or prior seizure. Neurological examination performed most recently at age 5 years and revealed mild central hypotonia. Trio whole exome sequencing to evaluate the cause of developmental delay identified a de novo heterozygous variant in KCNC1 c.1290G>A (p.Met430Ile), which is not present in the Exome Aggregation Consortium (ExAC) or genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org) and has a corresponding CADD score of 26.9. This variant is localized to S6 pore converts the hydrophobic sulfur containing methionine to the smaller isoleucine associated with a Grantham score of 10.
All identified variants were interpreted according to the American College of Medical Genetics and Genomics standards and guidelines for the interpretation of sequence variants.
Plasmid preparation, cell culture and transfection
A cDNA plasmid encoding human KCNC1 (reference sequence NM_001112741.2) and identified variants were synthesized and subcloned into a pCAG plasmid. HEK‐293T cells (ATCC, CRL‐3216) were grown at 37°C and 5% co 2 DMEM supplemented with 10% heat‐inactivated fetal calf serum and 1% penicillin–streptomycin in 35 mm dishes. Cells were transfected with 0.1 μg of pCAG.EGFP and 0.2 μg of either wild‐type (WT) or variant hKCNC1 cDNA using PolyFect transfection reagent (QIAGEN; Germanton, MD) according to the manufacturer's instructions. Twenty‐four hours after transfection, cells were trypsinized and seeded at low density and single GFP‐positive cells were identified for patch‐clamp experiments.
Electrophysiology
Whole‐cell patch clamp biophysical experiments were performed at room temperature using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) in an extracellular Tyrode's solution consisting of the following: 150 mM NaCl, 2 mM KCl, 1.5 mM CaCl2, 2 mM MgCl2, 10 mM HEPES and 10 mM glucose; pH was adjusted to 7.4 with NaOH. Intracellular solution contained, in mM: 125 KCl, 25 KOH; 1 CaCl2, 2 MgCl2, 4 Na2‐ATP, 10 EGTA, 10 HEPES, with pH adjusted to 7.2 with KOH and osmolarity to 305 mOsm/L with sucrose.
Patch pipettes were fashioned from thin‐walled borosilicate glass (Harvard apparatus, Holliston, MA) and fire‐polished (Zeitz) to a resistance of 1.7–2.5 MΩ in the whole cell configuration. Voltage errors were reduced via series resistance compensation. Currents were filtered at 2 kHz by a low‐pass Bessel filter and digitized at 30 kHz. Data were acquired with pClamp 11 and analyzed with Clampfit (Axon Instruments, San Jose, CA). Transient potassium currents were measured by performing 100 ms step depolarizations to between −85 and +55 mV in increments of 5 mV from a holding potential of −120 mV, and the current–voltage relation was constructed; this was followed by a 100 ms pulse to −40 mV to facilitate measurement of tail current. Activation conductance was normalized, plotted against voltage, and fit with a Boltzmann function to determine V 1/2 of activation. Kinetics of deactivation was calculated via a single exponential fit of the decay of the tail current.
All recordings and data analysis (below) were performed blind to experimental group.
Data analysis
Data for electrophysiological parameters was obtained from at least n = 10 cells from multiple transfections. Data were analyzed using custom Matlab scripts, Clampfit 11 (Molecular Devices), and Sigma Plot 11 (Systat Software, Inc., San Jose, CA). Results are presented as the mean ± standard error of the mean (SEM) and statistical significance was established using the p value calculated from a one‐way analysis of variance (ANOVA), with significance established at p < 0.05.
Results
A single‐center series of patients with KCNC1 variants and nonspecific developmental delay without epilepsy
We identified three patients with nonspecific global developmental delay without epilepsy found to have de novo heterozygous variants in KCNC1 via genome‐wide sequencing analysis that localized to a five amino acid extent at the C‐terminal end of the S6 transmembrane segment of the Kv3.1 protein (see Patients, Materials, and Methods). All three patients exhibited mild–moderate speech/language and gross motor developmental delay and central hypotonia with onset in the first year of life. Two of the three patients were ambulatory at time of last follow up. One of three patients had nystagmus.
Electrophysiological properties of identified KCNC1 variants
To investigate the mechanistic basis of the association between the clinical features described above and the seemingly minor missense variants in the region of amino acid residues 430–434 of the Kv3.1 protein, we expressed wild‐type (WT) and disease‐associated KCNC1 variants in a heterologous cells system (HEK‐293T cells) for voltage‐clamp electrophysiological recording.
All variants were recorded as outward non‐inactivating delayed rectifier currents via depolarizing steps from −120 mV (Fig. 1A). We found no difference in peak current density (measured at +40 mV; normalized to cell capacitance, as pA/pF) (Fig. 1A; Table 2). Peak current (at +40 mV) is: for Kv3.1‐WT, 1564 ± 211 pA/pF (n = 11); for Kv3.1‐p.Met430Ile, 1143 ± 196 pA/pF (n = 9; p < 0.001 vs. Kv3.1‐WT via one‐way ANOVA with post‐hoc correction for multiple comparisons using Dunn's multiple comparison test); for Kv3.1‐p.Val432Met, 1736 ± 257 pA/pF (n = 11; p < 0.001 vs. Kv3.1‐WT); and for Kv3.1‐p.Val434Leu, 1578 ± 145 pA/pF (n = 11; p < 0.001 vs. Kv3.1‐WT). There was a large leftward (hyperpolarized) shift in the voltage dependence of activation (Fig. 1B). The voltage at half‐maximal activation (V 1/2) was: for KCNC1‐WT, +20.2 ± 0.6 mV (n = 11); KCNC1‐p.Met430Ile, 3.2 ± 1.9 mV (n = 6; p < 0.001 vs. WT via one‐way ANOVA with post‐hoc correction using Dunn's multiple comparison test); KCNC1‐p.Val432Met, −0.8 ± 1.7 mV (n = 13; p < 0.001); and KCNC1‐p.Val434Leu, −4.1 ± 3.3 mV (n = 11; p < 0.001).
Figure 1.
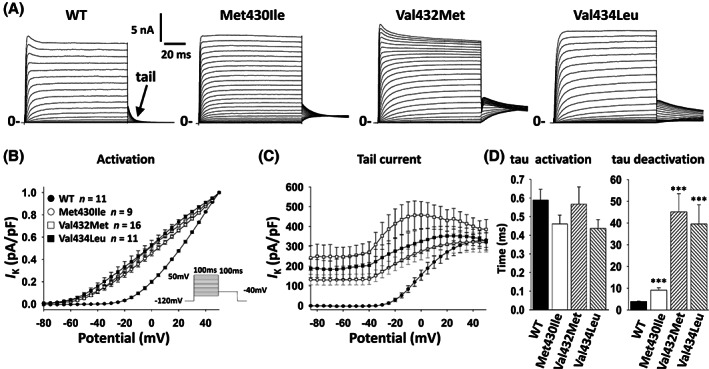
Biophysical properties of novel KCNC1 gain of function variants. Wild‐type (WT) or gain of function variants were expressed in HEK‐293T cells and outward delayed‐rectifier potassium currents were recorded via whole‐cell voltage clamp. (A) Representative examples of recording from of Kv3.1‐WT, Met430Ile, Val432Met and Val434Leu. Arrow in WT indicates the tail current upon return from the test potential to −40 mV. (B) Voltage‐dependence of activation presented as current density normalized to peak I K for each variant. (C) Voltage dependence and peak current density of tail currents. (D) Time constant of activation (left) and deactivation (right) at +40 mV.
Table 2.
Biophysical properties of wild‐type and variant KCNC1.
| Group | n | Peak current density (+40 mV) | Tail current (−40 mV) | Voltage dependence of activation (V 1/2) | Slope factor | Time constant of activation | Time constant of deactivation | Resting membrane potential (V m ) |
|---|---|---|---|---|---|---|---|---|
| pA/pF | mV | k | t (ms) | t (ms) | mV | |||
| WT | 11 | 1564 ± 211 | ‐2 ± 2 | 20.2 ± 0.6 | 0.034 ± 0.001 | 0.59 ± 0.06 | 3.9 ± 0.2 | −38.8 ± 2.4 |
| Met430Ile | 9 | 1143 ± 196 | 134 ± 28*** | 3.2 ± 1.9*** | 0.021 ± 0.001*** | 0.46 ± 0.05 | 9.1 ± 1.1*** | −85.7 ± 3.5*** |
| Val432Met | 16 | 1736 ± 257 | 270 ± 53*** | −0.8 ± 1.7*** | 0.023 ± 0.001*** | 0.57 ± 0.09 | 45.2 ± 8.4*** | −75.6 ± 2.3*** |
| Val434Leu | 11 | 1578 ± 145 | 204 ± 59*** | −4.1 ± 3.3 | 0.02 ± 0.002*** | 0.44 ± 0.05 | 39.6 ± 8.8*** | −83.4 ± 2.6*** |
| WT + Met430Ile | 11 | 811 ± 69* | 125 ± 23*** | 0.3 ± 1.6*** | 0.021 ± 0.001*** | 0.43 ± 0.05 | 9.1 ± 0.6*** | −81.0 ± 2.7*** |
| WT + Val432Met | 6 | 1321 ± 346 | 126 ± 20*** | 5.8 ± 1.1*** | 0.021 ± 0.001*** | 0.40 ± 0.07 | 17.1 ± 3.2*** | −63.2 ± 2.5*** |
| WT + Val434Leu | 7 | 1794 ± 319 | 233 ± 42*** | −0.4 ± 1.9*** | 0.022 ± 0.001*** | 0.38 ± 0.06 | 23.1 ± 7.7*** | −86.0 ± 2.0*** |
*p < 0.05; **p < 0.01; ***p < 0.001; vs. WT via one‐way ANOVA with post‐hoc correction via Dunn's test.
Rapid deactivation kinetics is hypothesized to be a critical biophysical feature whereby Kv3.1 subunits facilitate high‐frequency firing. Deactivation was assessed via a single exponential fit to the decay of the tail current upon return to −40 mV (Fig. 1A,C,D). The time constant (τ) of deactivation was: for KCNC1‐WT, 3.9 ± 0.2 ms (n = 11); KCNC1‐p.Met430Ile, 9.1 ± 1.1 ms (n = 9; p = 0.001 vs. WT via one‐way ANOVA with post‐hoc correction using Dunn's multiple comparison test); KCNC1‐p.Val432Met, 45.2 ± 8.4 ms (n = 4; p = 0.001); and KCNC1‐p.Val434Leu, 28.9 ± 19 ms (n = 4; p = 0.001).
It was also noted that HEK‐293T cells expressing variant KCNC1 exhibited a hyperpolarized resting membrane potential of: for KCNC1‐WT, −38.8 ± 2.4 mV (n = 5); KCNC1‐p.Met430Ile, −85.7 ± 3.5 mV (n = 3; p < 0.001 vs. WT via one‐way ANOVA with post‐hoc correction using Dunn's multiple comparison test); KCNC1‐p.Val432Met, −75.6 ± 2.3 mV (n = 6; p < 0.001); and KCNC1‐p.Val434Leu, −83.4 ± 2.6 mV (n = 9; p < 0.001) (Table 2).
As Kv3 channels are tetrameric assemblies of Kv3 subunits and patients with KCNC1‐related disorders will harbor one WT and one variant copy, we tested the effect of the identified variants on the properties of heteromeric channels composed of Kv3.1‐WT and variant subunits via co‐transfection at a 1:1 ratio. This experiment largely recapitulated the results obtained with variants alone (Fig. 2; Table 2).
Figure 2.
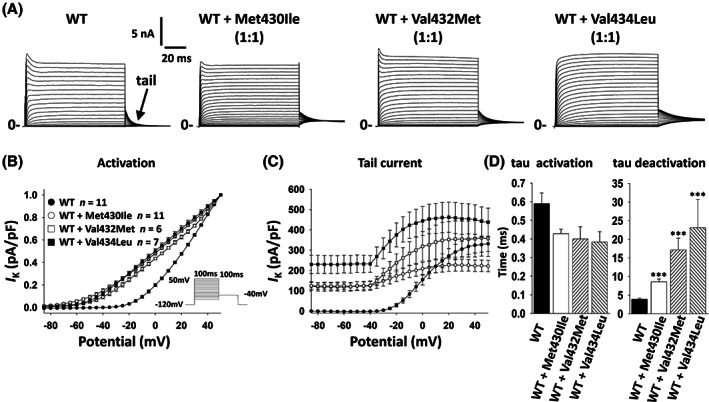
Biophysical properties of KCNC1‐WT co‐expressed with KCNC1 variants. WT and variants were co‐transfected in a 1:1 ratio and expressed in HEK‐293T cells, and outward delayed‐rectifier potassium currents were recorded via whole‐cell voltage clamp. (A) Representative examples of recording from of Kv3.1‐WT, WT + Met430Ile, WT + Val432Met and WT + Val434Leu. (B) Voltage‐dependence of activation presented as current density normalized to peak I K for each group. (C) Voltage dependence and peak current density of tail currents. (D) Time constant of activation (left) and deactivation (right) at +40 mV. Note that each WT:variant combination exhibits similar results to variant alone in Figure 1.
Discussion
A single‐center series of KCNC1 variants with nonspecific developmental delay without epilepsy
Here, we report detailed clinical and genetic data on three patients with nonspecific global developmental delay/intellectual disability and central hypotonia without epilepsy found to have de novo variants in KCNC1. Whole‐cell voltage clamp analysis of K+ channels formed by wild‐type versus variant Kv3.1 subunits in a heterologous systems revealed a clear GoF at the level of the ion channel for all three variants, including a left/hyperpolarized shift in the voltage dependence of activation and slowed deactivation kinetics, which will lead to increased K+ conductance due to activation at more hyperpolarized voltages and slowing of current decay following channel activation, respectively. Co‐expression of each of the three variants with WT Kv3.1 essentially reproduced the results obtained with the variants alone, suggesting a dominant effect of the variant on heteromeric Kv3 channels. This work establishes a new clinical‐genetic‐electrophysiological phenotype within the KCNC1‐related neurological disorders. This clinical entity can be distinguished from other KCNC1‐related neurological disorders, including that due to the recurrent variant KCNC1‐p.Arg320His (exhibiting moderate LoF, with decreased conductance and dominant‐negative effect of the variant on tetrameric Kv3.1‐containing channels) and associated with EPM7, and KCNC1‐Ala421Val (exhibiting severe LoF) associated with DEE.
All three variants described here are located within five amino acid residues of one another in the Kv3.1 protein (between residues 430 and 434) and represent amino acid changes associated with only mild alterations in physicochemical properties. Interestingly, there are zero nonsynonymous gnomAD variants localized to S6 of Kv3.1, further supporting the deleterious effect of even minor physicochemical changes in this region. These three residues are located between the selectivity filter and the conserved PXP motif in S6 (PVP in Kv3 subunits); the PXP motif which forms a “kink” that has been shown to be important for gating of K+ channels 13 and the identified variants could decrease the stability of the closed state and favor channel opening. Recent publication of the cryo‐EM structure of human Kv3.1 provides unprecedented insight into the mechanism of gating of Kv3 channels. 14 The Met430 residue has been shown to contribute to ion conduction and regulate Kv3.1 pore stability as mutation of this residue to a lysine induces channel flicker, 15 while the cryo‐EM structure reveals that the Met430 residue in S6 forms a intersubunit sulfo‐aromatic interaction with the F345 residue in S5 of adjacent subunits. Why the variant KCNC1‐p.Ala421Val located slightly more proximal – and which also converts between two small amino acids with hydrophobic side chains – instead produces a profound LoF (and the more severe DEE) remains unclear.
A limitation of the present study is the small number of patients in the series and the relative young age of the patients. While none of the patients has a history of seizures/epilepsy, only one underwent EEG (Table 1) and hence we cannot definitely rule out the presence of EEG abnormalities in sleep or wake. Future studies may provide additional information as to the full clinical spectrum associated with KCNC1 GoF variants, such as whether ataxia and/or epilepsy appear with age.
Table 1.
Clinical features of patients in the study.
| Patient | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Variant | c.1300G>C (p.Val434Leu) | c.1294G>A (p.Val432Met) | c.1290G>A (p.Met430Ile) |
| Inheritance | De novo | De novo | De novo |
| Age at symptom onset (current age) | 2 months (18 months) | 3 months (3 years) | 2 years (5 years) |
| First sign/symptom | Nystagmus | Hypotonia | Gross motor delay (walked at age 20 mo) |
| Developmental delay | Moderate–severe | Mild–moderate | Mild |
| Central hypotonia | Yes | Yes | Yes |
| Seizures/epilepsy | No | No | No |
| Ataxia | No | Mild gait ataxia | No |
| Myoclonus | None | None | None |
| Progression | None | None | None |
| Other | Nystagmus | None | None |
| EEG | C3/Cz sharps during sleep | Not performed | Not performed |
| MRI brain | Normal | Normal | Not performed |
Proposed mechanisms of KCNC1‐related neurological disorders
Kv3.1 is selectively expressed in fast‐spiking neurons throughout the brain including parvalbumin‐positive GABAergic interneurons (PV‐INs) of neocortex, hippocampus, and basal ganglia nuclei, and is also prominently expressed in the reticular thalamic nucleus as well as granule cells of the cerebellum and in the deep cerebellar nuclei. 16 , 17 , 18 Kv3.1 expression overlaps with that of Kv3.2; Kv3 channels in PV‐INs are likely heteromultimers of Kv3.1 and Kv3.2 subunits, which are electrophysiologically near‐identical and possess biophysical properties unique among ion channels that facilitate high‐frequency firing, including a depolarized voltage dependence of activation relative to other members of the Kv family and rapid kinetics of activation and deactivation with lack of inactivation. 10 , 11 Prior work has demonstrated LoF of varying degrees of severity at the level of the ion channel with a dominant‐negative effect mediated by the tetramerization of voltage‐gated K+ channels. Hence, while exhibiting GoF at the ion channel level, our prediction is that changes observed for these Kv3.1 variants act via LoF at the level of neurons, as suggested previously for the EPM7‐associated KCNC1‐p.Arg320His variant. 7
The prominent left‐shift in voltage dependence of activation observed here is predicted to lead to channel recruitment during the rising phase of the action potential and counteract inward sodium current, leading to impaired high‐frequency firing; slowed deactivation kinetics will render it such that K+ current does not completely deactivate between successive action potentials in a high‐frequency train, which will lead to accumulation of K+ conductance and a progressive increase in the inter‐spike interval, leading to impaired high‐frequency firing. Such effects would be predicted to impair fast‐spiking of PV‐INs and lead to decreased GABAergic inhibition in cerebral cortex circuits. This in turn may underlie developmental delay/ID and risk of epilepsy in this clinical‐genetic series, while dysfunction of Kv3.1‐expressing neurons in cerebellum may underlie hypotonia and gross motor developmental delay. Mice lacking Kcnc1 (Kcnc1−/− mice) exhibit motor dysfunction but not epilepsy 19 while deletion of KCNC1 in human is associated with ID without seizures/epilepsy. 9 Hence, the GoF effect observed here may ultimately exert a more mild effect on the excitability of Kv3.1‐expressing neurons than dominant negative‐acting LoF variants associated with more severe clinical entities along the spectrum of KCNC1‐related neurological disorders (such as EPM7 and DEE). Future work in experimental neuronal systems will be required to further address this issue.
Conclusion
In summary, we have established a syndrome of developmental delay/intellectual disability associated with central hypotonia without myoclonus or early‐onset epilepsy due to de novo heterozygous GoF variants in KCNC1. The mechanism whereby variants in KCNC1 lead to disease at the level of neurons and circuits remains to be established, as does the underlying basis of the clinical spectrum observed for KCNC1‐related neurological disorders.
Author Contributions
EMG contributed to the conception and design of the study; JC, NG, GC, and EMG contributed to acquisition and analysis of the data; JC and EMG contributed to preparing the figures; and EMG wrote the manuscript.
Conflict of Interest
None declared.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) R01 NS122887 to EMG and U54 NS108874. We thank the patients enrolled in the study and their families for participation in research, as well as the cooperation of the clinicians involved in the care of these patients.
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS122887 and U54 NS108874.
References
- 1. Jan LY, Jan YN. Voltage‐gated potassium channels and the diversity of electrical signalling. J Physiol. 2012;590(11):2591‐2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cooper EC. Potassium channels (including KCNQ) and epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado‐Escueta AV, eds. Jasper's Basic Mechanisms of the Epilepsies. 4th ed. National Center for Biotechnology Information; 2012. Accessed October 22, 2021. http://www.ncbi.nlm.nih.gov/books/NBK98164/ [PubMed] [Google Scholar]
- 3. Gutman GA, Chandy KG, Adelman JP, et al. International Union of Pharmacology. XLI. Compendium of voltage‐gated ion channels: potassium channels. Pharmacol Rev. 2003;55(4):583‐586. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Ali SR, Nabbout R, Barcia G, Kaczmarek LK. A KCNC1 mutation in epilepsy of infancy with focal migrating seizures produces functional channels that fail to be regulated by PKC phosphorylation. J Neurophysiol. 2021;126(2):532‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cameron JM, Maljevic S, Nair U, et al. Encephalopathies with KCNC1 variants: genotype–phenotype‐functional correlations. Ann Clin Transl Neurol. 2019;6(7):1263‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park J, Koko M, Hedrich UBS, et al. KCNC1‐related disorders: new de novo variants expand the phenotypic spectrum. Ann Clin Transl Neurol. 2019;6(7):1319‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015;47(1):39‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oliver KL, Franceschetti S, Milligan CJ, et al. Myoclonus epilepsy and ataxia due to KCNC1 mutation: analysis of 20 cases and K+ channel properties. Ann Neurol. 2017;81(5):677‐689. [DOI] [PubMed] [Google Scholar]
- 9. Poirier K, Viot G, Lombardi L, Jauny C, Billuart P, Bienvenu T. Loss of function of KCNC1 is associated with intellectual disability without seizures. Eur J Hum Genet. 2017;25(5):560‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaczmarek LK, Zhang Y. Kv3 channels: enablers of rapid firing, neurotransmitter release, and neuronal endurance. Physiol Rev. 2017;97(4):1431‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rudy B, McBain CJ. Kv3 channels: voltage‐gated K+ channels designed for high‐frequency repetitive firing. Trends Neurosci. 2001;24(9):517‐526. [DOI] [PubMed] [Google Scholar]
- 12. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Webster SM, Del Camino D, Dekker JP, Yellen G. Intracellular gate opening in shaker K+ channels defined by high‐affinity metal bridges. Nature. 2004;428(6985):864‐868. [DOI] [PubMed] [Google Scholar]
- 14. Chi G, Liang Q, Sridhar A, et al. Cryo‐EM structure of the human Kv3.1 channel reveals gating control by the cytoplasmic T1 domain. Nat Commun. 2022;13(1):4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aiyar J, Nguyen AN, Chandy KG, Grissmer S. The P‐region and S6 of Kv3.1 contribute to the formation of the ion conduction pathway. Biophys J. 1994;67(6):2261‐2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chow A, Erisir A, Farb C, et al. K(+) channel expression distinguishes subpopulations of parvalbumin‐ and somatostatin‐containing neocortical interneurons. J Neurosci. 1999;19(21):9332‐9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weiser M, Vega‐Saenz de Miera E, Kentros C, et al. Differential expression of Shaw‐related K+ channels in the rat central nervous system. J Neurosci. 1994;14(3 Pt 1):949‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiser M, Bueno E, Sekirnjak C, et al. The potassium channel subunit KV3.1b is localized to somatic and axonal membranes of specific populations of CNS neurons. J Neurosci. 1995;15(6):4298‐4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho CS, Grange RW, Joho RH. Pleiotropic effects of a disrupted K+ channel gene: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci U S A. 1997;94(4):1533‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]