

Abstract
Objective
To compare serum levels of the astrocyte biomarker glial fibrillary acidic protein (GFAP) in patients with amyotrophic lateral sclerosis (ALS) and neurologically healthy controls and to analyze the relations between serum GFAP (sGFAP) and phenotype in ALS.
Methods
We studied 114 ALS patients and 38 controls. sGFAP was quantified with single molecule array (Simoa) technology.
Results
In both ALS patients and controls, sGFAP moderately correlated with age. ALS patients had higher sGFAP levels compared to controls, but this yielded a weak discriminative performance (AUC = 0.6198). In ALS, sGFAP was not associated with most of the motor phenotypic features, including site of onset, functional status, disease progression rate, disease stage, and indices of upper (UMN) and lower motor neuron (LMN) impairment. However, sGFAP negatively correlated with cognitive scores regarding ALS‐nonspecific functions, particularly memory (r = −0.2082) and tended to be higher in ALS patients with eye movement abnormalities (p = 0.0628). sGFAP also correlated with polysomnographic indices of oxygen desaturation (ODI; r = 0.2639) and apnea‐hypopnea (AHI; r = 0.2858). In a multivariate analysis, sGFAP was negatively associated with survival (HR = 1.005). Relevantly, we found a negative correlation between sGFAP and estimated glomerular filtration rate (eGFR; r = −0.3500).
Interpretation
Our work provides neurochemical evidence of astrocyte involvement in ALS pathophysiology and particularly in the development of extra‐motor manifestations (namely, cognitive – memory – impairment) and respiratory dysfunction. The negative correlation between sGFAP and eGFR has practical relevance and should not be disregarded in future investigations.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder selectively affecting upper motor neurons (UMNs) of the cerebral cortex and lower motor neurons (LMNs) of the brainstem and spinal cord and causing progressive paralysis of voluntary muscles. There is no effective cure and death occurs after a median time of 3–5 years from onset, usually due to respiratory failure. 1 While the majority of ALS cases are sporadic (sporadic ALS, sALS), up to 10–15% show a family history (familial ALS, fALS), in most instances in the presence of a dominantly inherited mutation in one of >30 genes, the four main ones being C9orf72, SOD1, TARDBP, and FUS. 2 Notably, almost 50% of ALS cases display cognitive or behavioral alterations of frontal type, with up to 15% fulfilling the criteria for a diagnosis of behavioral‐variant frontotemporal dementia (bvFTD). Indeed, the two entities also share genetic and neuropathological aspects, thus belonging to the same clinicopathological spectrum, i.e. the so‐called TDP‐43 proteinopathies. 3
Although the diagnosis of ALS is mainly clinical, it may be supported by neurochemical biomarkers. These latter also have the potential additional functions of helping predict disease course, and, in the setting of therapeutic trials, of stratifying patients, verifying target engagement, and measuring treatment effect. 4 To date, the only established neurochemical biomarkers for ALS – except for specific genetic forms – are neurofilaments, whose levels in the cerebrospinal fluid (CSF) are increased as a consequence of their release from degenerating motor neuron (MN) axons. Importantly, thanks to recently developed highly sensitive technologies –mainly the single molecule array (Simoa) platform–, neurofilaments can now be quantified also in peripheral blood, where they are present at much lower concentrations. 5 However, there is an urgent need for further neurochemical biomarkers reflecting other pathophysiological mechanisms in ALS, as they would allow advancements in the development of targeted treatments. One such mechanism is astrocyte activation. Astrocytes are the most numerous glial cells of the central nervous system (CNS) and have several supporting functions related to fluid and ion homeostasis, energy metabolism, synapse activity, and blood brain barrier (BBB) integrity. 6 In ALS, astrocytosis occurs in both the cerebral cortex and the spinal cord, 7 , 8 and astrocytes are involved in the non‐cell‐autonomous process leading to MN degeneration, as exemplified by the toxicity exerted by astrocytes derived from ALS patients on healthy MNs in vitro. 9 A specific neurochemical biomarker of astrocyte activation – and/or damage – is the 432‐amino‐acid‐long intermediate filament glial fibrillary acidic protein (GFAP), the most abundant cytoskeletal protein of this cell type. 10 Similarly to neurofilaments, current highly sensitive technologies enable to measure GFAP also in peripheral blood, 11 where, for instance, it may support diagnosis and prognosis of traumatic brain injury (TBI) 12 and shows promise as an early biomarker for Alzheimer's disease (AD), being increased in amyloid‐positive individuals also before the onset of cognitive impairment. 13 The study of GFAP as an ALS biomarker is still limited. An early finding of slightly increased CSF GFAP levels compared to neurological controls 14 was not confirmed by a later investigation mainly focusing on genetic cases. 15 However, a recent study measured serum GFAP with Simoa in a large ALS cohort and found higher levels in ALS patients with cognitive and/or behavioral impairment or full‐blown frontotemporal dementia (FTD) compared to those with purely motor disease; moreover, serum GFAP weakly correlated with an UMN score and disease progression rate (DPR) and, in univariate analysis, was negatively associated with survival. 16
In our work, we measured serum levels of GFAP with Simoa in a large, deeply phenotyped cohort of ALS patients and in neurologically healthy controls with the aims of: (1) testing whether serum GFAP is increased in ALS; (2) investigating possible associations between the biomarker and disease features.
Methods
ALS patients and controls
We investigated N = 114 patients with ALS who were diagnosed at the Department of Neurology of IRCCS Istituto Auxologico Italiano, Milan, between 2015 and 2022 and whose serum samples were collected in our biobank. We also included N = 38 neurologically healthy controls whose sera had been sampled in 2020–2021. The diagnosis of ALS was made according to the revised El Escorial criteria. 17 Blood sampling occurred at the same time of clinical evaluation, which in the large majority of cases coincided with the diagnosis. In addition to basic demographic and clinical data (sex; age at evaluation; presence or absence of ALS family history, i.e. fALS vs. sALS; age at onset; site of onset, i.e. bulbar vs. spinal), patients were subdivided according to ALS motor phenotype into the following categories: classic, bulbar, respiratory, UMN‐predominant (UMN‐p), primary lateral sclerosis (PLS), flail arm syndrome, flail leg syndrome, and progressive muscular atrophy (PMA). 18 In order to enable further comparisons, the abovementioned individual motor phenotypes were also collected in the three main groups UMN/LMN (classic, bulbar, and respiratory), UMN (UMN‐p and PLS), and LMN (flail arm/leg and PMA). Quantification of the revised version of the ALS Functional Rating Scale (ALSFRS‐R) 19 allowed calculation of the DPR, according to the following formula: (48 – ALSFRS‐R score at clinical evaluation)/months elapsed between symptom onset and evaluation. 20 A composite score of muscle strength was calculated as the sum of the points of the following muscle groups/actions according to the Medical Research Council (MRC) scale, with a total range from 0 to 60: shoulder abduction, elbow flexion, wrist extension, thigh flexion, leg extension, and foot dorsiflexion. 21 In addition to the Penn UMN score, 22 a LMN score was quantified as proposed by Devine et al. 23 with a modification consisting in adding one point each for LMN signs in the bulbar and thoracic regions. 24 Clinical measures of UMN and LMN dysfunction were complemented by parameters derived from neurophysiological investigations –needle electromyography (EMG) and transcranial magnetic stimulation (TMS)–, namely semiquantitative indices of active and chronic limb denervation, central motor conduction time and cortical silent period. Disease stage was defined according to both King's 25 and Milano‐Torino Staging (MiToS) 26 systems. Eye movement abnormalities were investigated and categorized as previously described. 27
Patients also underwent neuropsychological evaluation. The cognitive and behavioral status was assessed in all patients with the Edinburgh Cognitive and Behavioral ALS Screen (ECAS). 28 In addition to scores for the five cognitive domains of executive functions, verbal fluency, language, memory, and visuospatial functions, composite ALS‐specific (i.e. executive + verbal fluency + language) and ALS‐nonspecific (i.e. memory + visuospatial) sub‐scores were computed. The ECAS enabled classification of ALS patients into the following 4 categories according to the criteria of Strong et al.: purely motor ALS, ALS with cognitive impairment (ALSci), ALS with behavioral impairment (ALSbi), and ALS with cognitive and behavioral impairment (ALScbi). 29 For the diagnosis of bvFTD, the criteria of Rascovsky et al. were used. 30 Global cognition was also evaluated with the Montreal Cognitive Assessment (MoCA), 31 while disturbances of frontal type were investigated with the Frontal Assessment Battery (FAB). 32 Behavioral impairment was additionally assessed through the Frontal Behavioral Inventory (FBI), consisting of two parts exploring negative (FBI‐A) and positive (FBI‐B) behaviors, respectively. 33 Furthermore, psychological aspects were assessed with the following scales: Beck Depression Inventory (BDI), including two subscales investigating cognitive‐affective and somatic symptoms, respectively 34 ; and State–Trait Anxiety Inventory (STAI‐Y), comprising two scores referring to state (STAI‐Y1) and trait (STAI‐Y2) anxiety, respectively. 35
Sub‐cohorts of patients underwent the following respiratory investigations: pulmonary function testing, providing forced vital capacity (FVC), expressed as percentage of the normal value predicted for the relative sex, height, and age; arterial blood gas (ABG) analysis, providing partial arterial pressures of oxygen (Pa o 2) and of carbon dioxide (Pa co 2) and blood bicarbonate () levels; and nocturnal polysomnography, allowing calculation of average peripheral oxygen saturation (Spo 2), oxygen desaturation index (ODI; the number of desaturation events per hour, defined as SpO2 drops of >3% of the baseline), and apnea‐hypopnea index (AHI; number of apnea or hypopnea events per hour). 36
All patients underwent measurement of routine laboratory blood parameters, among which we selected serum creatinine and creatine kinase (CK) levels. From serum creatinine, the estimated glomerular filtration rate (eGFR) was calculated according to the CKD‐EPI equation. 37 Finally, all fALS and a sub‐cohort of sALS patients were genotyped for the four main ALS‐associated genes, i.e. C9orf72, SOD1, TARDBP, and FUS.
Measurement of serum GFAP
Samples were handled and biobanked according to international recommendations. 38 Blood was sampled in the morning in fasting conditions, subsequently kept at room temperature for 15–30 min to allow clot formation, then refrigerated at 4°C, and finally centrifuged (2000 × g, 10 min) and stored at −80°C in 0.5‐ or 1‐mL aliquots in polypropylene vials within 4 h from sampling. sGFAP was measured with a commercial kit (catalog number, 102336) on the Simoa SR‐X instrument (Quanterix, Lexington, MA, USA). Samples were measured in duplicates, with coefficients of variation (CVs) <20%. All samples had sGFAP levels above the limit of detection (LOD) and lower limit of quantification (LLOQ), which were 0.26 and 1.37 pg/mL, respectively.
Statistical analysis
Distributions of categorical variables among groups of individuals were analyzed with chi‐square test. Differences in continuous variables between two and more groups were evaluated with Mann–Whitney U test and Kruskal‐Wallis test, respectively, with the latter followed by Dunn's post‐hoc comparisons in case of statistically significant differences. The diagnostic performance of sGFAP in discriminating between ALS patients and controls was analyzed by a receiver operating characteristic (ROC) curve, and a cut‐off was chosen as the value maximizing the Youden index (sensitivity + specificity – 1). Correlations between continuous variables were assessed by means of Spearman's rank correlation. In order to analyze the contribution of a continuous variable of interest to a continuous dependent variable correcting for other covariates, partial rank correlation was employed. Survival was assessed by Kaplan–Meier curves and the difference between curves was evaluated with the log‐rank test. Cox proportional hazards model was used to analyze the effect of multiple variables on survival. As to survival, tracheostomy was considered equivalent to death. Patients who were still alive at last follow‐up information available were censored. For all tests, the level of statistical significance was set at p < 0.05. Statistical analyses were performed with Prism, version 9 (GraphPad, La Jolla, CA, USA) and, in the case of partial rank correlation, with SPSS, version 26 (IBM Corp., Armonk, NY, USA).
Results
sGFAP levels in ALS patients and controls
ALS patients (N = 114; males, N = 71; females, N = 43) and neurologically healthy controls (N = 38; males, N = 24; females, N = 14) did not differ for sex distribution (p = 0.9229) or median age at evaluation (ALS, 66 years; controls, 62.5 years; p = 0.5720). Among controls, males and females did not significantly differ for age at evaluation (p = 0.1583) or sGFAP (p = 0.0995); however, sGFAP moderately correlated with age (r = 0.5318; 95% confidence interval –CI–, 0.2464 to 0.7324; p = 0.0006). Among ALS patients, sGFAP correlated with age at evaluation as well (r = 0.5590; 95% CI, 0.4135 to 0.6767; p < 0.0001; Fig. 1). At the same time, sGFAP was higher in ALS female patients compared to males (median, 131.5 vs. 104.1 pg/mL; p = 0.0289). This seemed to be at least partially independent from the fact that female patients had an older median age compared to male ones (68 vs. 65 years, without statistical significance, p = 0.1148). In ALS patients, sGFAP levels were significantly higher compared to controls (median, 117.0 vs. 92.1 pg/mL; p = 0.0269; Fig. 2A). This resulted, however, in a weak diagnostic performance, with an area under the ROC curve (AUC) of 0.6198 (95% CI, 0.5199 to 0.7198; p = 0.0272) for the discrimination between ALS patients and controls, corresponding to a sensitivity of 61.40% (95% CI, 52.23% to 69.83%) and a specificity of 63.16% (95% CI, 47.28% to 76.62%) at a cut‐off of 103.3 pg/mL (Fig. 2B).
Figure 1.
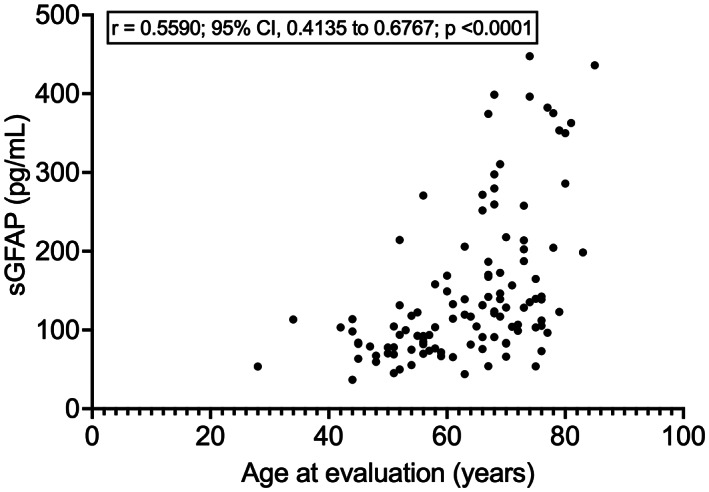
Correlation between sGFAP and age at evaluation in ALS patients. ALS, amyotrophic lateral sclerosis; CI, confidence interval; sGFAP, serum glial fibrillary acidic protein.
Figure 2.
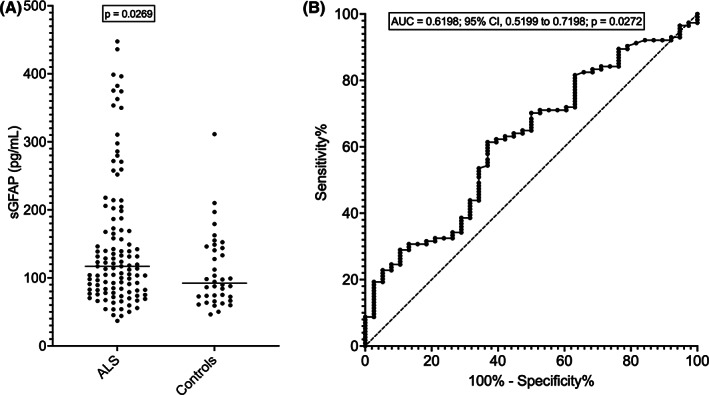
sGFAP levels in ALS and controls. (A) sGFAP levels in patients with ALS and neurologically healthy controls. The horizontal bars represent median values. (B) ROC curve of sGFAP for the discrimination between ALS patients and controls. ALS, amyotrophic lateral sclerosis; AUC, area under the ROC curve; ROC, receiver operating characteristics; sGFAP, serum glial fibrillary acidic protein.
Relation between sGFAP levels and phenotype in ALS
Table 1 reports phenotypic features of ALS patients. Disease onset was bulbar in 27 (23.7%) cases and spinal in 87 (76.3%). Median ages at onset and at evaluation were 65 and 66 years, respectively. One ALS patient also had bvFTD (ALS‐FTD). As expected, given the observed correlations between sGFAP and age at evaluation and between age at evaluation and age at onset (r = 0.9935; 95% CI, 0.9904 to 0.9955; p < 0.0001), sGFAP also correlated with age at onset (r = 0.5641; 95% CI, 0.4197 to 0.6807; p < 0.0001). On the contrary, sGFAP did not differ between patients with fALS versus sALS (p = 0.8843), nor between patients with bulbar versus spinal onset (p = 0.4606). Similarly, no significant differences were observed among ALS motor phenotypes, both when analyzed separately (p = 0.8564) and when grouped into the three large UMN/LMN, UMN, and LMN categories (p = 0.3373). We found no significant correlations between sGFAP and ALSFRS‐R score, disease duration at evaluation, or DPR (p > 0.05 in all cases). In agreement with this, patients in the different disease stages according to King's and MiToS systems did not have significantly different sGFAP levels (p = 0.1071 and p = 0.0766, respectively). We observed no significant correlations between sGFAP and Penn UMN score, composite MRC score (N = 106), or LMN score (p > 0.05 for all tests). Accordingly, sGFAP levels did not correlate with EMG‐ and TMS‐derived parameters (p > 0.05 in all cases). On the contrary, median sGFAP was higher in patients with clinically evident eye movement abnormalities (square wave jerks, conjugate gaze palsy, slow pursuit dysfunction, saccade alterations, or oculomotor apraxia; N = 7; median sGFAP, 164.9 pg/mL) compared to those without (N = 107; median sGFAP, 113.7 pg/mL), although the difference did not reach statistical significance (p = 0.0628; Fig. 3).
Table 1.
Demographic and clinical features of ALS patients and results of instrumental and laboratory investigations.
| Sex | |
| Males | 71 (62.3%) |
| Females | 43 (37.7%) |
| Age at evaluation (years) | 66 (56–72) |
| Age at onset (years) | 65 (54–71) |
| Family history of ALS | |
| fALS | 23 (20.2%) |
| sALS | 91 (79.8%) |
| Site of onset | |
| Bulbar | 27 (23.7%) |
| Spinal | 87 (76.3%) |
| Motor phenotype | |
| Classic | 62 (54.4%) |
| Bulbar | 24 (21.1%) |
| Respiratory | 4 (3.5%) |
| UMN‐p | 9 (7.9%) |
| PLS | 5 (4.4%) |
| Flail arm | 4 (3.5%) |
| Flail leg | 2 (1.8%) |
| PMA | 4 (3.5%) |
| Disease duration at evaluation (months) | 12 (7–20) |
| ALSFRS‐R (N = 101) | 41 (36–44) |
| DPR (N = 101) | 0.611 (0.273–1.032) |
| Penn UMN score | 9 (3–15) |
| LMN score | 5 (3–6) |
| Composite MRC score (N = 106) | 54 (48–58) |
| King's staging system | |
| Stage 1 | 9 (7.9%) |
| Stage 2 | 28 (24.6%) |
| Stage 3 | 74 (64.9%) |
| Stage 4 | 3 (2.6%) |
| Stage 5 | 0 (0%) |
| MiToS system | |
| Stage 0 | 86 (75.4%) |
| Stage 1 | 25 (21.9%) |
| Stage 2 | 3 (2.6%) |
| Stage 3 | 0 (0%) |
| Stage 4 | 0 (0%) |
| Stage 5 | 0 (0%) |
| Eye movement abnormalities | |
| Absent | 107 (93.9%) |
| Present | 7 (6.1%) |
| ECAS scores | |
| Executive | 37 (30–40) |
| Verbal fluency | 18 (14–20) |
| Language | 25 (22–27) |
| Memory | 15 (12–18) |
| Visuospatial | 12 (11–12) |
| ALS‐specific | 79 (66–86) |
| ALS‐nonspecific | 27 (23–30) |
| Total | 105 (91–113) |
| Cognitive‐behavioral classification according to ECAS | |
| ALS | 50 (43.9%) |
| ALSci | 29 (25.4%) |
| ALSbi | 21 (18.4%) |
| ALScbi | 14 (12.3%) |
| MoCA score (N = 92) | 24 (22–26.5) |
| FAB score (N = 99) | 16.3 (15.0–17.9) |
| FBI scores (N = 88) | |
| A | 1 (0–3) |
| B | 0 (0–1) |
| Total | 1 (0–3) |
| BDI scores (N = 101) | |
| Cognitive‐affective | 5 (2–7) |
| Somatic | 8 (5–10) |
| Total | 13 (8–17) |
| STAI scores (N = 104) | |
| Y1 | 52 (46.5–58.5) |
| Y2 | 49.5 (43–58) |
| FVC (pulmonary function testing; N = 35) | 81 (60–100.5) |
| ABG parameters (N = 66) | |
| pa o 2 (mmHg) | 80.5 (69.0–89.0) |
| pa co 2 (mmHg) | 42.0 (38.0–44.0) |
| (mmol/L) | 27.9 (26.2–29.9) |
| Polysomnographic parameters (N = 70) | |
| Average Spo 2 | 93.4% (92.1–94.9%) |
| ODI | 6.1 (1.8–11.2) |
| AHI | 5.4 (1.8–10.6) |
| Serum GFAP (pg/mL) | 117.0 (82.4–172.0) |
| eGFR (mL/min) | 94.3 (82.8–102.7) |
| Serum CK (U/L) | 181.5 (120–259) |
| Patients with gene mutations | |
| C9orf72 | 6 (N = 77) |
| SOD1 | 1 (N = 23) |
| TARDBP | 5 (N = 25) |
| FUS | 0 (N = 23) |
For continuous variables, median and interquartile range are reported, if not otherwise specified. When not all patients were evaluated for a given parameter, the number of evaluated patients is provided. For gene mutations, the number of tested patients (N) is indicated in brackets after the number of patients with mutations in the relative gene.
Abbreviations: ABG, arterial blood gas; AHI, apnea‐hypopnea index; ALS, amyotrophic lateral sclerosis; ALSbi, ALS with behavioral impairment; ALScbi, ALS with cognitive and behavioral impairment; ALSci, ALS with cognitive impairment; ALSFRS‐R, amyotrophic lateral sclerosis functional rating scale, revised; BDI, Beck Depression Inventory; CK; creatine kinase; DPR, disease progression rate; ECAS, Edinburgh Cognitive and Behavioral ALS Screen; eGFR, estimated glomerular filtration rate; FAB, Frontal Assessment Battery; fALS, familial amyotrophic lateral sclerosis; FVC, forced vital capacity; IQR, interquartile range; LMN, lower motor neuron; MiToS, Milano‐Torino Staging; MoCA, Montreal Cognitive Assessment; MRC, Medical Research Council; ODI, oxygen desaturation index; PLS, primary lateral sclerosis; PMA, progressive muscular atrophy; sALS, sporadic amyotrophic lateral sclerosis; Spo 2, peripheral oxygen saturation; STAI, State–Trait Anxiety Inventory; UMN, upper motor neuron; UMN‐p, upper‐motor‐neuron‐predominant.
Figure 3.
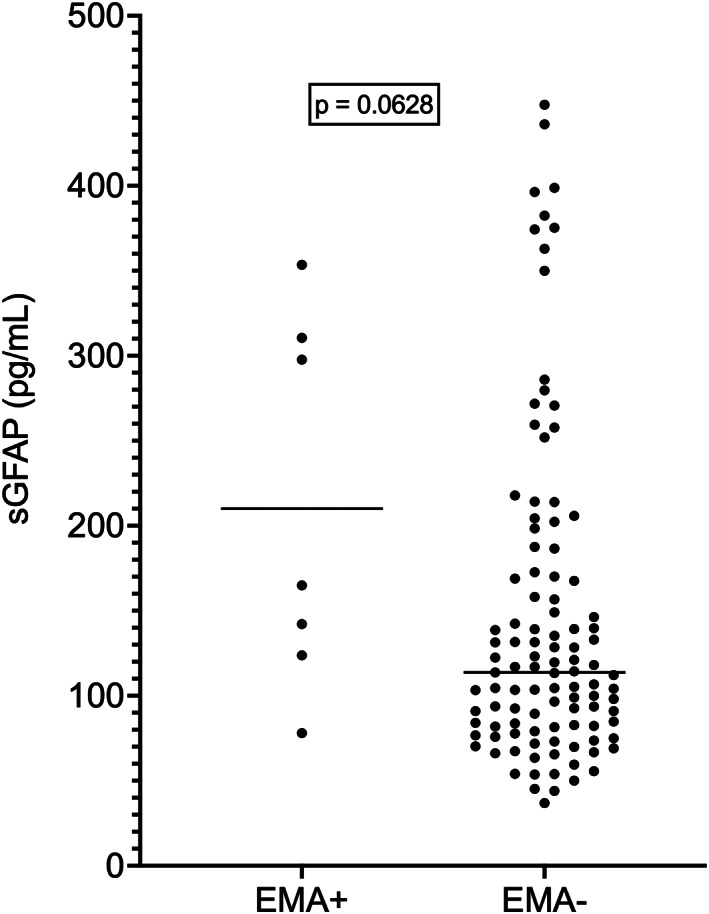
sGFAP levels in ALS patients with and without eye movement abnormalities. Horizontal bars represent median values. ALS, amyotrophic lateral sclerosis; EMA+, patients with eye movement abnormalities; EMA−, patients without eye movement abnormalities. sGFAP, serum glial fibrillary acidic protein.
We next examined the relations between sGFAP levels and cognitive‐behavioral phenotype. sGFAP did not differ among the 4 classes of ALS, ALSci, ALSbi, and ALScbi (p = 0.3917), between patients with versus without cognitive impairment – i.e. (ALSci + ALScbi) versus (ALS + ALSbi); p = 0.2244 –, or among patients with versus without behavioral impairment – i.e. (ALSbi + ALScbi) versus (ALS + ALSci); p = 0.2118 –. Whereas sGFAP did not correlate with the ALS‐specific ECAS composite cognitive score nor with the three single scores of executive functions, verbal fluency, and language (p > 0.05 in all cases), we observed a negative correlation between sGFAP and the ALS‐nonspecific ECAS composite cognitive score (r = −0.2091; 95% CI, −0.3832 to −0.02071; p = 0.0256), but this was not confirmed when controlling for age (p = 0.526). Similarly, we found a trend towards a negative correlation between sGFAP and total ECAS cognitive score (r = −0.1784; 95% CI, −0.3557 to 0.01115; p = 0.0575), with a correspondingly higher median sGFAP level in patients having low (i.e. abnormal) scores (138.7 pg/mL; N = 35) compared to those with normal scores (106.7 pg/mL; N = 79; p = 0.0226), but the trend was not confirmed after controlling for age (p = 0.430). As regards single ALS‐nonspecific functions, while sGFAP did not correlate with visuospatial score (p = 0.4792), we found a significant negative correlation between sGFAP and memory score (r = −0.2082; 95% CI, −0.3823 to −0.01972; p = 0.0262; Fig. 4), which, however, was not retained after controlling for age (p = 0.342). sGFAP had a weak negative correlation with MoCA score (r = −0.2723; 95% CI, −0.4568 to −0.06540; p = 0.0086; N = 92), which, however, was not confirmed after controlling for age (p = 0.422). There were no significant correlations between sGFAP and scores on FAB (N = 99), FBI (FBI‐A, FBI‐B, or total scores; N = 88), BDI (cognitive‐affective, somatic, or total scores; N = 101), or STAI (Y1 or Y2 scores; N = 104; p > 0.05 in all cases).
Figure 4.
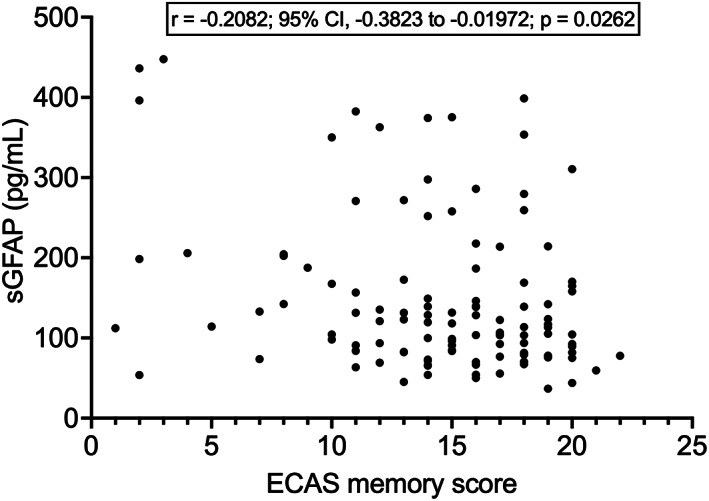
Correlation between sGFAP levels and ECAS memory score. ECAS, Edinburgh Cognitive and Behavioral ALS Screen; sGFAP, serum glial fibrillary acidic protein.
As regards respiratory function, we observed no significant correlations between sGFAP and FVC on pulmonary function testing (N = 35; p = 0.1474) or ABG analysis parameters (Pa o 2, Pa co 2, ; N = 66; p > 0.05 in all cases). However, sGFAP correlated with both ODI and AHI on nocturnal polysomnography (ODI: r = 0.2639; 95% CI, 0.02378 to 0.4753; p = 0.0273; AHI: r = 0.2858; 95% CI, 0.04744 to 0.4934; p = 0.0165; for both parameters, N = 70; Fig. 5), in the absence of a significant correlation between those indices and age (p > 0.05 in both cases). On the contrary, the observed weak negative correlation between sGFAP and average Spo 2 (r = −0.2215; 95% CI, −0.4396 to 0.02133; N = 70) did not reach statistical significance (p = 0.0654).
Figure 5.
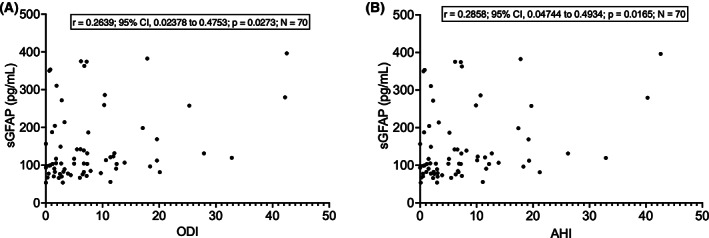
Correlations between sGFAP levels and polysomnographic parameters. (A) Correlation between sGFAP and ODI. (B) Correlation between sGFAP and AHI. AHI, apnea‐hypopnea index; ODI, oxygen desaturation index; sGFAP, serum glial fibrillary acidic protein.
As for laboratory parameters, sGFAP had a moderate negative correlation with eGFR (r = −0.3500; 95% CI, −0.5057 to −0.1722; p = 0.0001; Fig. 6). sGFAP also had a weak negative correlation with serum CK (r = −0.1870; 95% CI, −0.3633 to 0.002349; p = 0.0464), which, however, was not confirmed after controlling for eGFR (p = 0.248). Given the correlation between sGFAP and eGFR, we tested whether the correlation between sGFAP and age at evaluation was still confirmed after taking eGFR into account: in partial rank correlation, sGFAP retained a moderate correlation with age at evaluation also when controlling for eGFR (r = 0.468; p < 0.001). sGFAP did not differ between patients with versus without mutations in C9orf72 and TARDBP genes (C9orf72: N = 6 vs. N = 71; p = 0.4058; TARDBP: N = 5 vs. N = 20; p = 0.2667); the analysis could not be conducted for SOD1 because there was only one mutated patient out of 23 tested, nor for FUS, for which no mutations were found among 24 patients tested.
Figure 6.
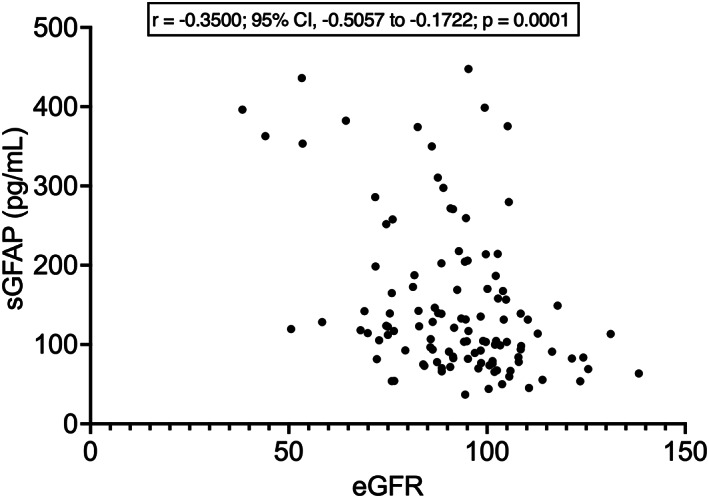
Correlation between sGFAP levels and estimated glomerular filtration rate. eGFR, estimated glomerular filtration rate (calculated according to the formula of the CKD‐EPI equation); sGFAP, serum glial fibrillary acidic protein.
Finally, we examined the relation between sGFAP and survival. 38 (33.3%) of the 114 patients were deceased at the time of last information available, whereas one had a tracheostomy. The remaining 75 patients (65.8%) were censored. Log‐rank test performed on Kaplan–Meier curves indicated no significant difference between survival of patients with sGFAP levels below versus above the median (p = 0.0914). Nevertheless, a Cox proportional hazards model considering sGFAP, age at onset, site of onset (bulbar vs. spinal), DPR, and presence/absence of cognitive impairment (as determined by the ECAS) as covariates demonstrated that sGFAP was an independent, albeit weak, predictor of survival (hazard ratio – HR – 1.005; 95% CI, 1.000 to 1.009; p = 0.0256), with DPR being the best predictor (HR, 7.568; 95% CI, 3.943 to 15.24; p < 0.0001), whereas the other three parameters were not independently associated with survival.
Discussion
We quantified serum levels of the astrocyte biomarker GFAP in a cohort of deeply phenotyped patients with ALS, comparing them with those measured in neurologically healthy controls and analyzing the relations between this biomarker and disease features. In both ALS patients and controls, sGFAP correlated with age. Patients had significantly higher sGFAP levels than controls, but this resulted in an only weak diagnostic performance in the discrimination between the two groups. Among ALS patients, sGFAP was higher in females – in agreement with recent observations on plasma GFAP in large healthy or mixed cohorts 13 , 39 – but was not associated with most of the main phenotypic parameters considered, including site of onset, functional status, disease progression rate, disease stage, and indices of UMN or LMN impairment. On the contrary, sGFAP negatively correlated with cognitive scores regarding ALS‐nonspecific functions, particularly memory – although statistical significance was not retained after correction for age – and tended to be higher in ALS patients with eye movement abnormalities. Moreover, sGFAP correlated with polysomnographic indices of nocturnal respiratory dysfunction. In a multivariate analysis, the biomarker also showed an independent, albeit weak, negative association with survival. Finally, we found a negative correlation between sGFAP and eGFR.
Our results are in line with an early investigation reporting increased CSF levels of GFAP in ALS patients compared to neurological disease controls. 14 More important is, however, the comparison with the recent elegant work of Falzone and colleagues, which, similarly to our study, measured serum GFAP and used the Simoa technology. 16 At variance with our results, in their study sGFAP levels did not significantly differ between ALS patients and healthy controls. However, the positive correlation between sGFAP and age was similar to that observed in our cohort. Other results are partially consistent between our two investigations, with the previous study reporting increased sGFAP levels in ALS patients with cognitive and/or behavioral alterations and an association between the biomarker and survival only in a univariate analysis. 16 On the contrary, only that study found significant correlations of sGFAP with DPR and an UMN score.
Our data have to be interpreted from a pathophysiological standpoint. It is well‐known that astrocytosis occurs in ALS in sites of MN degeneration. 8 An early study on cultures of human fetal motor cortex tissue indicated proliferation of GFAP‐expressing cells upon exposure to CSF from ALS patients. 40 Moreover, diseased astrocytes play a role in promoting MN death. 41 Importantly, astrocytes themselves have been demonstrated to degenerate in the proximity of dying MNs in a transgenic ALS mouse model expressing mutant SOD1. 42 Therefore, although GFAP increases in CSF or blood in a wide range of inflammatory as well as degenerative neurological diseases are generally interpreted as reflecting astrocyte activation, 11 which is known to up‐regulate the expression of this structural protein, 43 we cannot exclude that, at least in ALS, this neurochemical finding also reflects degeneration of this glial cell population. Our findings suggest that astrocyte involvement in ALS does not characterize specific motor phenotypes and is not associated with specific aspects of the motor disease, including UMN or LMN impairment, disease stage – neither in a chronological sense, as reflected by disease duration at evaluation, nor in an anatomic/functional one, as indexed by the King's and MiToS systems–, and DPR, which mirrors the biological aggressiveness of the disease. In this sense, considering the significant difference with respect to healthy controls, sGFAP increase seems to be an underlying pathophysiological phenomenon accompanying ALS independently from the anatomical and clinical expressions of the motor disease. However, our data, in remarkable agreement with those of Falzone et al., 16 also suggest that a peculiar contribution to sGFAP increase in ALS could be provided by disease manifestations other than the classical motor ones, most importantly cognitive impairment. Interestingly, in addition to the findings of the previous work, 16 we demonstrate that sGFAP elevation is associated with ALS‐nonspecific aspects of neuropsychological dysfunction, especially memory deficit. This observation is particularly interesting when considering that sGFAP increases in the blood characterize AD to a greater extent than other neurodegenerative diseases, as opposed to CSF levels whose elevations are less AD‐specific, 13 with the difference between the two biological fluids hypothetically reflecting the location of AD astrocytes at the anatomical interface between CNS and peripheral blood (neurovascular unit and BBB). 39 According to our observations, astrocytes might play an additional, and possibly even more prominent, role in ALS‐related neurodegeneration occurring in brain areas not belonging to the motor system. Since this investigation did not include measurement of CSF or plasma AD biomarkers, our data do not allow to argue whether this is an ALS‐specific, although non‐classical, phenomenon, or rather reflects subclinical, possibly age‐related, AD pathology, as might be hypothesized from the loss of the statistical significance of our findings after correction for age. Furthermore, albeit regarding a small number of patients and without reaching the threshold of statistical significance, our data suggest an additional link between sGFAP increase and non‐classical phenotypic manifestations of ALS, namely eye movement abnormalities. Notably, these latter have been recently shown by our group to be in turn associated with cognitive impairment itself. 27 Similarly to the neuropsychological aspect, also in this case it may be hypothesized that astrocytes are involved in neurodegeneration occurring in brain areas other than the main targets of ALS pathology, namely the frontal and supplementary eye fields of the cerebral cortex. Finally, the correlation with polysomnographic indices suggests a possible specific connection between astrocyte pathology and degeneration of MNs controlling respiratory muscles. While this might reflect a prominent susceptibility of respiratory MNs to toxic factors released by astrocytes or a particular dependence of these MNs on astroglial homeostatic support, it cannot be excluded, on the other hand, that sGFAP increase represents a pathophysiological response to hypoxia induced by (nocturnal) respiratory impairment, as suggested by experimental reports of increased GFAP expression in rat brain following hypoxic–ischemic injury. 44 Anyway, it is possible that the observed correlation of sGFAP with nocturnal respiratory dysfunction is involved in the weak negative association with survival found in the multivariate analysis, where, on the contrary, cognitive impairment was not demonstrated to be an independent predictor.
As regards the potential clinical implications of our study, at notable variance with Falzone et al., 16 we provide evidence of increased sGFAP levels in ALS patients compared to neurologically healthy controls. However, the overlap of values between these two groups is large, not enabling a clinically meaningful discrimination, as demonstrated by the weak diagnostic capability shown by the ROC curve. This contrasts with the good performance of neurofilaments documented by several investigations and indicates a low potential of sGFAP as for the neurochemical diagnosis of ALS in clinical practice. It should also be mentioned that our investigation did not include ALS‐mimic conditions, which represent the most suitable control group in studies of ALS diagnostic biomarkers. On the other hand, we cannot exclude a future role of sGFAP measurement in ALS as an aid to cognitive and respiratory prognostication – as part of multi‐parameter predictive algorithms–, and, more importantly, as a biomarker of astrocyte activation/pathology for patient stratification, demonstration of target engagement and monitoring of treatment effects in therapeutic trials targeting neuroinflammation and astrocyte‐dependent disease mechanisms. Regarding the potential clinical employment of sGFAP measurement, our finding of a negative correlation with eGFR – possibly reflecting reduced clearance of the molecule from the blood with decreasing renal function – cannot be overemphasized, in contrast with the insufficient consideration which, in our opinion, this theme receives in most investigations on blood biomarkers. The influence from renal function is a typical drawback of such innovative, easy‐to‐measure and versatile peripheral neurochemical markers: given the high prevalence of kidney impairment in the population and the age‐dependent nature of the most common neurodegenerative diseases, the impact of renal function on sGFAP and other blood biomarkers requires further large‐scale investigations, especially regarding the practical relevance in a potential context of routine clinical use in elderly patients with multiple medical comorbidities.
We acknowledge that our study has some limitations: (1) We did not investigate other neurochemical biomarkers, such as neurofilament light chain (NFL) and especially AD biomarkers, which could have been informative regarding the association with cognitive deficits; (2) For some phenotypic parameters, not all patients of the cohort had available data; (3) Our phenotyping did not include neuroimaging features; (4) sGFAP was not measured longitudinally; (5) We did not investigate a validation cohort. At the same time, we believe that one of the main strengths of our work, which is among the first ones investigating sGFAP as a candidate biomarker in ALS, is represented by the detailed phenotyping of our cohort, including the neuropsychological profile.
In conclusion, we have demonstrated that GFAP levels are increased in the serum of ALS patients and suggest that this increase might also partly reflect an astrocyte contribution to neurodegeneration occurring in areas of the CNS involved in cognition, oculomotion and respiratory function. Future investigations in the field are needed, including analysis of multiple neurochemical biomarkers, comparison between GFAP levels in CSF, serum and plasma, longitudinal measurements, correlation with neuroradiological indices of UMN impairment, evaluation of ALS‐mimic diseases, and parallel neurochemical‐neuropathological assessments aiming at elucidating the role of GFAP as a biomarker of astrocyte activation and/or degeneration in ALS.
Author contributions
Study conception and design, F.V.; Data acquisition/analysis, F.V., I.M., A.M., E.C., S.T., F.S., A.D., F.G., A.M., R.B., S.P., S.M., L.M., C.M., B.P., A.R., V.S., N.T.; Statistical analysis, F.V., N.T.; Drafting of the manuscript, F.V.; Critical revision of the manuscript, A.R., V.S., N.T.
Conflict of Interest
The authors report no conflict of interest.
Acknowledgments
The authors are thankful to patients, patients' families, and healthcare professionals involved in patient care. The authors also thank Mrs. P. Nelli for her role in collecting survival data of patients.
Funding InformationFunding was provided by the Italian Ministry of Health (Ricerca Corrente to IRCCS Istituto Auxologico Italiano).
Funding Statement
This work was funded by Italian Ministry of Health.
References
- 1. van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084‐2098. doi: 10.1016/S0140-6736(17)31287-4 [DOI] [PubMed] [Google Scholar]
- 2. Goutman SA, Hardiman O, Al‐Chalabi A, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21(5):465‐479. doi: 10.1016/S1474-4422(21)00414-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burrell JR, Halliday GM, Kril JJ, et al. The frontotemporal dementia‐motor neuron disease continuum. Lancet. 2016;388(10047):919‐931. doi: 10.1016/S0140-6736(16)00737-6 [DOI] [PubMed] [Google Scholar]
- 4. Verde F, Silani V, Otto M. Neurochemical biomarkers in amyotrophic lateral sclerosis. Curr Opin Neurol. 2019;32(5):747‐757. doi: 10.1097/WCO.0000000000000744 [DOI] [PubMed] [Google Scholar]
- 5. Verde F, Otto M, Silani V. Neurofilament light chain as biomarker for amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. 2021;15:679199. doi: 10.3389/fnins.2021.679199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7‐35. doi: 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nagy D, Kato T, Kushner PD. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J Neurosci Res. 1994;38(3):336‐347. doi: 10.1002/jnr.490380312 [DOI] [PubMed] [Google Scholar]
- 8. Schiffer D, Cordera S, Cavalla P, Migheli A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci. 1996;139:27‐33. doi: 10.1016/0022-510x(96)00073-1 [DOI] [PubMed] [Google Scholar]
- 9. Haidet‐Phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824‐828. doi: 10.1038/nbt.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. 2011;93(3):421‐443. doi: 10.1016/j.pneurobio.2011.01.005 [DOI] [PubMed] [Google Scholar]
- 11. Abdelhak A, Foschi M, Abu‐Rumeileh S, et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat Rev Neurol. 2022;18(3):158‐172. doi: 10.1038/s41582-021-00616-3 [DOI] [PubMed] [Google Scholar]
- 12. Korley FK, Jain S, Sun X, et al. Prognostic value of day‐of‐injury plasma GFAP and UCH‐L1 concentrations for predicting functional recovery after traumatic brain injury in patients from the US TRACK‐TBI cohort: an observational cohort study. Lancet Neurol. 2022;21(9):803‐813. doi: 10.1016/S1474-4422(22)00256-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐beta but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505‐3516. doi: 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benninger F, Glat MJ, Offen D, Steiner I. Glial fibrillary acidic protein as a marker of astrocytic activation in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J Clin Neurosci. 2016;26:75‐78. doi: 10.1016/j.jocn.2015.10.008 [DOI] [PubMed] [Google Scholar]
- 15. Oeckl P, Weydt P, Steinacker P, et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90(1):4‐10. doi: 10.1136/jnnp-2018-318868 [DOI] [PubMed] [Google Scholar]
- 16. Falzone YM, Domi T, Mandelli A, et al. Integrated evaluation of a panel of neurochemical biomarkers to optimize diagnosis and prognosis in amyotrophic lateral sclerosis. Eur J Neurol. 2022;29(7):1930‐1939. doi: 10.1111/ene.15321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases . El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293‐299. doi: 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 18. Chio A, Calvo A, Moglia C, Mazzini L, Mora G, PARALS Study Group . Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2011;82(7):740‐746. doi: 10.1136/jnnp.2010.235952 [DOI] [PubMed] [Google Scholar]
- 19. Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169(1–2):13‐21. doi: 10.1016/s0022-510x(99)00210-5 [DOI] [PubMed] [Google Scholar]
- 20. Kimura F, Fujimura C, Ishida S, et al. Progression rate of ALSFRS‐R at time of diagnosis predicts survival time in ALS. Neurology. 2006;66(2):265‐267. doi: 10.1212/01.wnl.0000194316.91908.8a [DOI] [PubMed] [Google Scholar]
- 21. Kleyweg RP, van der Meche FG, Schmitz PI. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain‐Barre syndrome. Muscle Nerve. 1991;14(11):1103‐1109. doi: 10.1002/mus.880141111 [DOI] [PubMed] [Google Scholar]
- 22. Quinn C, Edmundson C, Dahodwala N, Elman L. Reliable and efficient scale to assess upper motor neuron disease burden in amyotrophic lateral sclerosis. Muscle Nerve. 2020;61(4):508‐511. doi: 10.1002/mus.26764 [DOI] [PubMed] [Google Scholar]
- 23. Devine MS, Ballard E, O'Rourke P, Kiernan MC, McCombe PA, Henderson RD. Targeted assessment of lower motor neuron burden is associated with survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(3–4):184‐190. doi: 10.3109/21678421.2015.1125502 [DOI] [PubMed] [Google Scholar]
- 24. Maranzano A, Poletti B, Solca F, et al. Upper motor neuron dysfunction is associated with the presence of behavioural impairment in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2022;29(5):1402‐1409. doi: 10.1111/ene.15243 [DOI] [PubMed] [Google Scholar]
- 25. Roche JC, Rojas‐Garcia R, Scott KM, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135(Pt 3):847‐852. doi: 10.1093/brain/awr351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chio A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86(1):38‐44. doi: 10.1136/jnnp-2013-306589 [DOI] [PubMed] [Google Scholar]
- 27. Poletti B, Solca F, Carelli L, et al. Association of clinically evident eye movement abnormalities with motor and cognitive features in patients with motor neuron disorders. Neurology. 2021;97(18):e1835‐e1846. doi: 10.1212/WNL.0000000000012774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poletti B, Solca F, Carelli L, et al. The validation of the Italian Edinburgh cognitive and behavioural ALS screen (ECAS). Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(7–8):489‐498. doi: 10.1080/21678421.2016.1183679 [DOI] [PubMed] [Google Scholar]
- 29. Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS‐FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):153‐174. doi: 10.1080/21678421.2016.1267768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456‐2477. doi: 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Conti S, Bonazzi S, Laiacona M, Masina M, Coralli MV. Montreal cognitive assessment (MoCA) – Italian version: regression based norms and equivalent scores. Neurol Sci. 2015;36(2):209‐214. doi: 10.1007/s10072-014-1921-3 [DOI] [PubMed] [Google Scholar]
- 32. Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal assessment battery at bedside. Neurology. 2000;55(11):1621‐1626. doi: 10.1212/wnl.55.11.1621 [DOI] [PubMed] [Google Scholar]
- 33. Alberici A, Geroldi C, Cotelli M, et al. The frontal behavioural inventory (Italian version) differentiates frontotemporal lobar degeneration variants from Alzheimer's disease. Neurol Sci. 2007;28(2):80‐86. doi: 10.1007/s10072-007-0791-3 [DOI] [PubMed] [Google Scholar]
- 34. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561‐571. doi: 10.1001/archpsyc.1961.01710120031004 [DOI] [PubMed] [Google Scholar]
- 35. Siciliano M, Trojano L, Trojsi F, Monsurro MR, Tedeschi G, Santangelo G. Assessing anxiety and its correlates in amyotrophic lateral sclerosis: the state‐trait anxiety inventory. Muscle Nerve. 2019;60(1):47‐55. doi: 10.1002/mus.26475 [DOI] [PubMed] [Google Scholar]
- 36. Engel M, Glatz C, Helmle C, Young P, Drager B, Boentert M. Respiratory parameters on diagnostic sleep studies predict survival in patients with amyotrophic lateral sclerosis. J Neurol. 2021;268(11):4321‐4331. doi: 10.1007/s00415-021-10563-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604‐612. doi: 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Otto M, Bowser R, Turner M, et al. Roadmap and standard operating procedures for biobanking and discovery of neurochemical markers in ALS. Amyotroph Lateral Scler. 2012;13(1):1‐10. doi: 10.3109/17482968.2011.627589 [DOI] [PubMed] [Google Scholar]
- 39. Benedet AL, Mila‐Aloma M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471‐1483. doi: 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Silani V, Pizzuti A, Redaelli LM, et al. ALS cerebrospinal fluid enhances human foetal astroglial cell proliferation in vitro. Adv Exp Med Biol. 1987;209:79‐81. doi: 10.1007/978-1-4684-5302-7_13 [DOI] [PubMed] [Google Scholar]
- 41. Tong J, Huang C, Bi F, et al. Expression of ALS‐linked TDP‐43 mutant in astrocytes causes non‐cell‐autonomous motor neuron death in rats. EMBO J. 2013;32(13):1917‐1926. doi: 10.1038/emboj.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rossi D, Brambilla L, Valori CF, et al. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008;15(11):1691‐1700. doi: 10.1038/cdd.2008.99 [DOI] [PubMed] [Google Scholar]
- 43. Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26(18):4930‐4939. doi: 10.1523/JNEUROSCI.5480-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burtrum D, Silverstein FS. Hypoxic‐ischemic brain injury stimulates glial fibrillary acidic protein mRNA and protein expression in neonatal rats. Exp Neurol. 1994;126(1):112‐118. doi: 10.1006/exnr.1994.1047 [DOI] [PubMed] [Google Scholar]