

Abstract
Background and Objectives
Anti-CD20 monoclonal antibody (mAb) B-cell depletion is a remarkably successful multiple sclerosis (MS) treatment. Chimeric antigen receptor (CAR)-T cells, which target antigens in a non-major histocompatibility complex (MHC)–restricted manner, can penetrate tissues more thoroughly than mAbs. However, a previous study indicated that anti-CD19 CAR-T cells can paradoxically exacerbate experimental autoimmune encephalomyelitis (EAE) disease. We tested anti-CD19 CAR-T cells in a B-cell–dependent EAE model that is responsive to anti-CD20 B-cell depletion similar to the clinical benefit of anti-CD20 mAb treatment in MS.
Methods
Anti-CD19 CAR-T cells or control cells that overexpressed green fluorescent protein were transferred into C57BL/6 mice pretreated with cyclophosphamide (Cy). Mice were immunized with recombinant human (rh) myelin oligodendrocyte protein (MOG), which causes EAE in a B-cell–dependent manner. Mice were evaluated for B-cell depletion, clinical and histologic signs of EAE, and immune modulation.
Results
Clinical scores and lymphocyte infiltration were reduced in mice treated with either anti-CD19 CAR-T cells with Cy or control cells with Cy, but not with Cy alone. B-cell depletion was observed in peripheral lymphoid tissue and in the CNS of mice treated with anti-CD19 CAR-T cells with Cy pretreatment. Th1 or Th17 populations did not differ in anti-CD19 CAR-T cell, control cell-treated animals, or Cy alone.
Discussion
In contrast to previous data showing that anti-CD19 CAR-T cell treatment exacerbated EAE, we observed that anti-CD19 CAR-T cells ameliorated EAE. In addition, anti-CD19 CAR-T cells thoroughly depleted B cells in peripheral tissues and in the CNS. However, the clinical benefit occurred independently of antigen specificity or B-cell depletion.
Multiple sclerosis (MS) is a CNS inflammatory autoimmune demyelinating disease that affects approximately 1 million patients in the United States.1 A large percentage of patients with MS accumulate significant neurologic disability, including the weakness, vision loss, and cognitive decline. The dramatic success of systemic anti-CD20 B-cell–depleting monoclonal antibodies (mAbs) in treating relapsing remitting MS revealed that B cells play an important role in MS immune pathophysiology.2-5 However, the effect of B-cell–targeted therapy on preventing long-term disability in progressive forms of MS is modest,4,5 possibly because progressive MS reflects a neurodegenerative process. It is also possible that current treatments do not effectively deplete CNS resident B cells6-8 associated with meningeal and subpial inflammation (e.g., ectopic lymphoid follicles) in progressive MS.8-14 Thus, treatments that better deplete B cells in both the periphery and the CNS may usher in a new era of MS therapies.
Chimeric antigen receptor (CAR)-T cells are autologous T cells engineered to express a non-major histocompatibility complex (MHC) target antigen-specific receptor with an intracellular CD3 domain fused to costimulatory domains such as CD28 or 4-1BB, bypassing the need for an antigen-presenting cell.15-17 CAR-T cells targeted against CD19+ B cells are revolutionizing the treatment of B-cell leukemias, achieving >80% remission rates.16 Furthermore, case reports document successful treatment of CNS leukemia and lymphoma with anti-CD19 CAR-T cells, highlighting their capability to cross the blood-brain barrier.18 CAR-T cell therapy has been well tolerated and rarely associated with irreversible CNS adverse events.15-18 Thus, anti-CD19 CAR-T cells represent a highly potent tool for depleting CD19+ B cells in the periphery and the CNS, making them a viable treatment strategy for testing in patients with MS, as well as in other autoimmune diseases.19
A recent study explored treatment using anti-CD19 CAR-T cells in a model of experimental autoimmune encephalomyelitis (EAE) in mice that contain T cells and B cells which both target the CNS autoantigen myelin oligodendrocyte glycoprotein (MOG).20 In that model, mice develop spontaneous EAE in association with meningeal B-cell aggregates. They found that anti-CD19 CAR-T cell treatment reduced the size of meningeal B-cell follicles but exacerbated clinical disease. This is similar to what was seen when anti-CD20 B-cell depletion caused paradoxical worsening in the same spontaneous EAE or in EAE induced by immunization with MOG peptide (p) 35–55.21,22 Here, we evaluated anti-CD19 CAR-T cells in EAE induced by immunization with the extracellular domain of recombinant human (rh) MOG protein, a model that is B cell-dependent.21-23 In this model, anti-CD20 mAb treatment emulates the beneficial treatment response in patients with MS.2-6 We sought to understand if CAR-T cell–induced B-cell depletion may also show benefit in this EAE model and thus may be a potential option for patients with MS.
Methods
Mice
Wild type (WT) female C57BL/6 (B6), and muMT female mice (6–8 weeks of age) were purchased from Jackson Laboratory. Mice were housed in a 1:1 light:dark animal facility at the Sandler Neurosciences Center of University of California, San Francisco (UCSF).
Standard Protocol Approvals and Registrations
All animal experiments were conducted in accordance with UCSF's Institutional Animal Care and Use Committee. Standard protocol approvals and registrations were obtained (AN188984-02B).
EAE Induction
rhMOG was produced and purified according to previously published protocols.23 Mouse MOG p35-55 (MEVGWYRSPFSRVVHLYRNGK) was purchased from Genemed Synthesis (San Antonio, TX). MOG p35-55 induces a B-cell–independent EAE with direct MHC peptide presentation resulting in activation of pathogenic MOG-specific T cells. By contrast, EAE induced by the rhMOG, which is the extracellular domain of human MOG (rhMOG 1–117), is B cell-dependent possibly reflecting a need for B cell processing of MOG protein for antigen presentation.24 B6 mice were immunized with 100 µg of rhMOG or 100 µg of MOG p35-55 emulsified in M. tuberculosis H37Ra (Difco Laboratories, Franklin Lakes, NJ) and incomplete Freund adjuvant (BD) subcutaneously (sc). Afterward, 200 ng of B. pertussis toxin (List Biological, Campbell, CA) was injected by the intraperitoneal (IP) route on the day of induction and 2 days later. All mice were monitored daily for signs of clinical EAE for 28 days. Individual mice were rated on a clinical scoring system: 0 = no clinical disease, 1 = loss of tail tone only, 2 = mild monoparesis or paraparesis, 3 = severe paraparesis, 4 = paraplegia and/or quadriparesis, and 5 = moribund or death.
Primary Mouse T-Cell Isolation and Culture
Primary CD8+ T cells were isolated from B6 mice by negative selection (STEMCELL Technologies, Vancouver, Canada). After isolation, T cells were activated with CD3/CD28 Dynabeads (Thermo Fisher Scientific 11453d, Waltham, MA) and cultured in media consisting of RPMI-1640 + 2mM Glutamax (Thermo Fisher Scientific 61870036), 10% FBS serum (UCSF Tissue Culture Facility), 20mM HEPES (Thermo Fisher Scientific 15-630-106), 1X Pen/Strep (Thermo Fisher Scientific 15140122), 1mM Sodium Pyruvate (Thermo Fisher Scientific 11360070), and 0.05mM 2-betamercaptoethanol (Thermo Fisher Scientific 21985023) supplemented with 100 units/mL IL-2 (NCI BRB Preclinical Repository) for all experiments.
Retroviral Transduction of Mouse T Cells
We obtained an anti-CD19 CAR plasmid (AddGene 1D3-28Z. 1–3 mut, based on the 1D3 antibody that recognizes the mouse CD19).25 This plasmid was originally cloned into the mouse stem cell virus–based splice-gag vector (MSGV) retroviral backbone with the following components in-frame from the 5′ to the 3′ ends: the signal sequence of the light chain of the 1D3 antibody, the 1D3 antibody light chain variable region, a (GGGGS)3 linker, the 1D3 antibody heavy chain variable region, the murine CD28 portion (amino acids IEFMY to the 3′ terminus) in which a dileucine motif was changed to diglycine to enhance CAR expression, and the cytoplasmic portion of the murine CD3-ζ (amino acids RAKFS to the 3′ terminus) in which the first and third ITAMs were inactivated to reduce CAR-T cell apoptosis. For our study, we modified the AddGene 1D3-28Z. 1–3 mut to include a green fluorescent protein (GFP) under its own promoter (PGK) downstream of the CAR to be able to track the CAR-expressing cells. The control GFP vector consisted of the GFP sequence cloned into MSGV retroviral backbone. The following timeline was used to transduce the mouse T cells. On day 1, retroviral plasmids (anti-CD19 CAR and control GFP) were transfected into Platinum (Plat-E) retroviral packaging cells (Cell Biolabs RV-101, San Diego, CA). On day 2, primary CD8 T cells were isolated and activated with CD3/CD28 Dynabeads (Thermo Fisher Scientific 11453d). CD3/CD28 Dynabeads are beads covered with anti-CD3 and anti-CD28 antibodies that allow direct activation and expansion of T cell without the need of feeder cells (antigen-presenting cells) or antigen. On day 3, the viral supernatant was harvested, mixed with primary mouse T cells, and spinduced on RetroNectin coated plates (Takara T100B, Kusatsu, Shiga, Japan) for 2 hours, at 2,000g, 32°C to increase the transduction efficiency. Viral supernatant was then removed, and cells were resuspended in growth media and transferred to a new plate. On day 5, beads were removed, and cells were sorted and expanded in growth media for another 5 days before being used either for in vitro analysis or in vivo. Untransduced activated CD8+ T cells were isolated and suspended in growth media with activation beads similar duration to CAR-T cells. For in vitro analyses, engineered K562 cells (ATCC) were engineered to express murine CD19 and grown in DMEM, 10% FBS. For killing assays, the CAR-T cells and the K562 cells were cocultured for 24–48 hours, and K562 survival was assessed by flow cytometry. For in vivo injections, 2 million CAR-T cells or control cells were injected IV through tail vein 24 hours after treatment with cyclophosphamide (Cy) 100 mg/kg IP, similar to human protocols.15-27,26–28
Anti-CD20 Monoclonal Antibody
Anti-mouse-CD20 mAb (clone 5D2) was generously provided by Genentech, Inc. Mice were treated with 200 µg IP anti-CD20 mAb weekly for 3 weeks
Flow Cytometry Analysis
5 µL of peripheral blood was obtained from tail snipping into 200 µL heparin weekly. Samples were incubated with red blood cell (RBC) lysis buffer twice and then stained with CD45 (FITC, BL 103108), CD11b (PerCP-Cy7, Invitrogen 45-0112-82, Waltham, MA), CD20 (PE, BL 150410), CD19 (PE Cy7, BL 115520), CD8 (brilliant violet, BL 100738), and CD4 (APC, TONBO 20–00420U100). Flow cytometry was run the same day. The mice were sacrificed at two end points of day 17 and day 28. Mice were treated with 1 mL of Avertin after which the spleen and inguinal lymph nodes were dissected. The femur was removed for bone marrow (BM) harvesting. After perfusion with 10mL PBS, CNS mononuclear cells were isolated as described previously.22 The spleen, lymph nodes, and BM were also incubated with RBC lysis before wash and resuspension. For staining of Tregs, the aqua dead cell stain kit (Molecular Probes, Eugene, OR), antibodies to CD45 (FITC), CD11b (PerCp-Cy7), CD4 (brilliant violet, BL 100438), CD25 (APC, eBioscience 17-0251-82), and FoxP3 (PE, eBioscience 12-5773-82) were used. Proinflammatory T-cell polarization was evaluated by in vitro stimulation with 50 ng/mL PMA (Sigma-Aldrich, St. Louis MO) and 1 µg/mL ionomycin (Sigma-Aldrich) in the presence of 1 µg/mL BD GolgiStop (BD Bioscience, Franklin Lakes, NJ) for 4 hours. Intracellular cytokine staining for T-cell cytokines was performed using the cytofix/cytoperm Plus Fixation/Permeabilization kit (BD), aqua dead cell stain kit (Molecular Probes) and antibodies to CD45 (FITC), CD11b (PerCp-Cy7), CD4 (BV), IFN-g (APC, Invitrogen 17-7311-82), and IL-17 (PE, Invitrogen 12-7177-81). Lymphocytes were examined on a Fortessa flow cytometer using FACSDiva software (BD).
Thymidine Proliferation
Mice were immunized with 100 µg of rhMOG (1–117) or MOG p35-55 sc in CFA containing 400 μg M tuberculosis H37Ra (Difco Laboratories). Lymph nodes were harvested on days 10–12 and cultured at 2 × 105/well with the respective peptide used for immunization for 72 hours. The incorporation of 3H-thymidine was measured in triplicate wells.
Histology
Brain, spinal cord, and optic nerve tissue samples were fixed in 10% neutral-buffered formalin, paraffin-embedded, sectioned, and stained with hematoxylin and eosin before examination by light microscopy. Meningeal and parenchymal inflammatory lesions and areas of demyelination were counted in a blinded manner as previously described.22,29
Phenotyping of Transfer Cells
Cells were pelleted and flash frozen. RNA was extracted using the Zymo Research Quick-DNA/RNA Miniprep Plus kit (D7003T). RNA was then prepared for bulk RNA sequencing using the NEB Ultra II RNA Library Prep kit (E7770S/L), and paired end reads of 75 base pair length were obtained on an Illumina MiSeq according to the manufacturer's protocol. These data were analyzed using GRCm39 vM30 for the reference genome, STAR-2.7.10a for the alignments, R v4.2.1 for the heatmap generation, and DESeq2 (a R package) for the differential gene expression analysis.
Statistics
For all statistical analyses, multiple t tests were completed on GraphPad Prism 9 software.
Data Availability
Data not provided in the article because of space limitations, can be shared at the request of other investigators.
Results
rhMOG-Induced EAE Is B-Cell Dependent
Wild-type (WT), but not B-cell–deficient (muMT) mice developed EAE after immunization with rhMOG, whereas WT and muMT mice were both susceptible to EAE induced by MOG p35-55 (eFigure 1, A and B, links.lww.com/NXI/A790).21-24,30 As expected, anti-CD20 mAb B-cell depletion protected mice from rhMOG-induced EAE (eFigure 1A).21-24
Anti-CD19 CAR-T Cell Characterization
The retroviral transduction efficiency of CD8+ T cells with the anti-CD19 CAR was approximately 50% (eFigure 2A, links.lww.com/NXI/A790). In vitro cytotoxicity assays showed that anti-CD19 CAR-T cells lysed 60% of CD19+ K562 target cells (eFigure 2B). Cytotoxicity was not observed in control cells. There were no differences in the differentially expressed genes (DEGs) across anti-CD19 CAR-T cells and the control cells. When comparing anti-CD19 CAR-T cells and control cells against untransduced activated CD8+ T cells, there was no difference in genes commonly associated with cell differentiation or activation. Among the DEGs, there was an increased expression of Lilrb4, an immunosuppressive gene, within the anti-CD19 CAR-T cell and control cells31,32 (eFigure 2C). Similarly, there was an increase in expression of Flt31, a cell proliferation and survival gene.33 Additional data are listed in eTable 1 (links.lww.com/NXI/A791).
Anti-CD19 CAR-T Cells Ameliorate EAE
One day before anti-CD19 CAR-T cell infusion, recipient mice were pretreated with Cy, similar to the standard preconditioning regimen for administration of CAR-T cells in humans.15-19,26-28 Mice were immunized with rhMOG 2 weeks after anti-CD19 CAR-T transfer (Figure 1A). There was a significant reduction in clinical EAE scores in recipients of anti-CD19 CAR-T cells from days 15–26 and in recipients of control cells from days 16–19 (Figure 1B and Table 1) when compared with WT mice, from scores averaging around 1 (limp tail) compared with WT scores of 3 (monoparesis). The clinical benefit of anti-CD19 CAR-T cells was similar to anti-CD20–mediated mAb (eFigure 1A, links.lww.com/NXI/A790). Cy treatment delayed EAE onset (around day 15), coinciding with lymphocyte reconstitution (Figure 2A). However, the magnitude of the effect was less than what was seen in the anti-CD19 CAR-T cell treatment group from days 21–28. There were no differences in EAE scores between WT mice and mice that received anti-CD19 CAR-T cells or control cells that were not pretreated with Cy or mice treated with untransduced activated CD8+ T cells alone (eFigure 3A). The histology scores similarly showed that the anti-CD19 CAR-T cell and control cell groups both had markedly reduced perivascular mononuclear inflammatory cell foci compared with the WT and Cy alone groups, with the anti-CD19 CAR-T cell group having a complete absence of these foci (Figure 1, C and D).
Figure 1. EAE Disease on CAR-T Cell Therapy.

(A) Experimental timeline. (B) Average EAE clinical score after immunization with rhMOG. Multiple t tests revealed p < 0.05 from days 15–26 between WT vs cyclophosphamide (Cy) + anti-CD19 (aCD19) CAR-T cells and from days 21–28 from between Cy alone vs Cy + aCD19 CAR-T cells. Shown is average ± SD. (C) Cerebellar histology 28 days after EAE induction. WT and cyclophosphamide treated mice show typical perivascular mononuclear inflammatory cell foci in the white matter (indicated by arrows), whereas the Cy treated with aCD19 CAR-T cells or control cells are normal. Bar = 100 µm and applies to all. (D) Representative total numbers of meningeal and parenchymal foci within spinal cord and brain per sacrificed mouse shown, 2 mice per group. aCD20 as anti-CD20, Cy as cyclophosphamide, and aCD19 CAR as anti-CD19 CAR-T cell. CAR = chimeric antigen receptor; Cy = cyclophosphamide; EAE = experimental autoimmune encephalomyelitis; MOG = myelin oligodendrocyte protein; WT = wild type.
Table 1.
The p Values Between Treatment Groups per Days Post Immunization
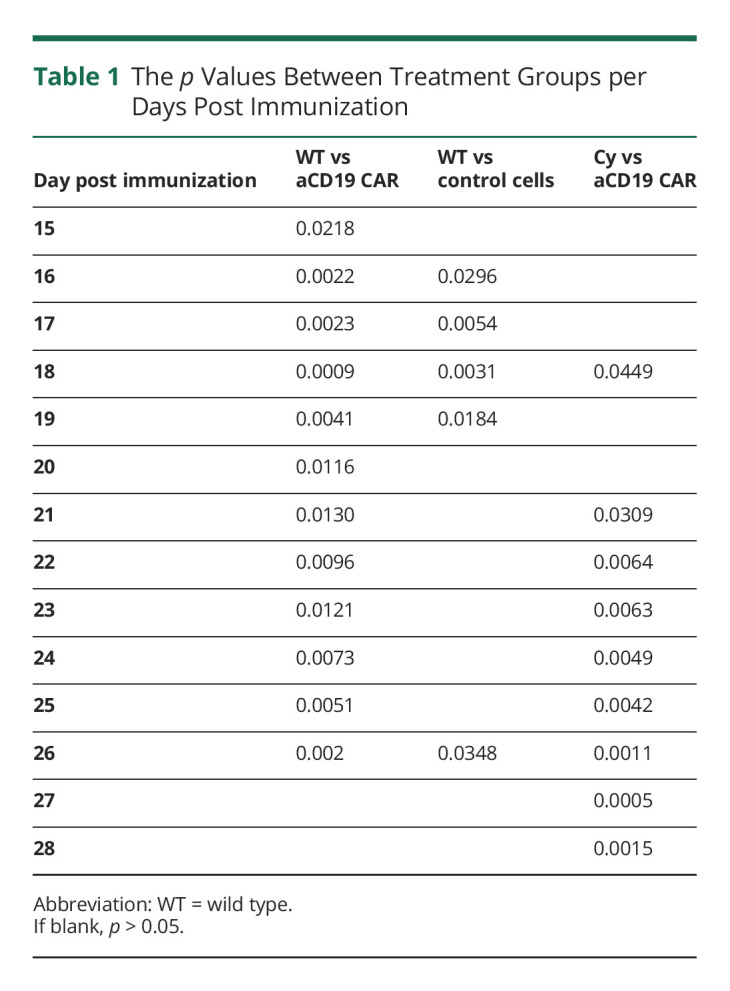
Figure 2. B Cell Depletion Based on Treatment.

(A) Frequency of CD45+CD11b-CD19+ cells, average of 5 mice peripheral blood from 5 µL of tail vein blood as ratio against WT. (B) Frequency of CD45+CD11b-CD19+ cells, average of 2 mouse per disease cohort per compartment which included spleen, inguinal lymph node, and femoral bone marrow, and 1 mouse per CNS (brain and spinal cord) on day 17 postimmunization. Shown is average ± SD. (C) Frequency of CD45+CD11b-CD19+ cells, average of 2 mouse per disease cohort per compartment which included spleen, inguinal lymph node, and femoral bone marrow, and CNS (brain and spinal cord) on day 28 postimmunization. Paired t test, p value <0.05 considered significant (*), if p < 0.009 (**). WT = wild type.
Although immune effector cell-associated neurotoxicity syndrome is inherently limited in mice, the mice did not show evidence of increased systemic toxicity, such as increased weight loss, coat changes (i.e., piloerection), or grooming changes associated with toxicity in mice.26,27,34
Anti-CD19 CAR-T Cell Pharmacokinetics/Dynamics
Anti-CD19 CAR-T cells were tracked using flow cytometry. Approximately 2% of CD8 cells in 5 µL of peripheral blood were CAR-T cells for the first 2 weeks after IV injection and then the cells were not detected within the blood or within other compartments (spleen, lymph nodes, BM, and CNS [brain and spinal cord]) (data not shown).
Anti-CD19 CAR-T Cells Efficiently and Durably Deplete B Cells In Vivo
The anti-CD19 CAR-T cell treated group showed complete depletion of CD19+-positive cells in peripheral blood within the first week that persisted >25 weeks (Figure 2A). Given this persistence, there were no concerns regarding loss of therapeutic effect due to an intrinsic anti-GFP immune response, which can be present in B6 mice35 despite lack of CAR-T cell detection. Mice treated with Cy alone exhibited transient declines in CD19+ and CD3+ cells that nadir in week 1 but returned to WT baseline 4–6 weeks later. Mice treated with the anti-CD20 mAb showed complete loss of peripheral CD19+ cells by week 1 and reconstitution beginning at 6–8 weeks, similar to previously published data.22
The frequency of CD19+ (and CD20+) cells in various tissue compartments were analyzed by flow cytometry on day 17. CD19+ cells were depleted all tissues in anti-CD19 CAR-T cell therapy and anti-CD20 mAb (Figure 2B) but were near WT levels in mice treated with Cy alone and control cell groups. Persistent CD19+ B-cell depletion was confirmed on day 28 for anti-CD19 CAR-T cell and anti-CD20 mAB (Figure 2C, eFigure 3B, links.lww.com/NXI/A790). B-cell depletion (both CD19+ and CD20+ B cells) was more extensive in the anti-CD19 CAR-T cell group, in particular in the CNS. Cy alone or control cell groups had restored levels of CD19+ cells, comparable with WT.
Anti-CD19 CAR-T Cells Do Not Have an Effect on Endogenous T Cells
In mice treated with Cy alone, there were increased frequencies of Th1 and Th17 cells in the CNS on day 17 (Figure 3A) compared with other treatment groups corresponding with increasing clinical EAE scores. There were no differences in the frequencies of Th1 and Th17 cells among all treatment groups by day 28 (Figure 3B). There was no statistically significant difference in the frequency of regulatory T cells (Treg) (Figure 3C). This was similar to T-cell treatment groups without Cy pretreatment (eFigure 3, C–D, links.lww.com/NXI/A790). There was no difference in MOG-specific T-cell proliferation among groups (eFigure 4).
Figure 3. Endogenous T Cell Response Based on Treatment.

(A) Frequency of CD45+CD4+IL17+ (Th17) or CD45+CD4+IFNg+ (Th1) cells of 1 mouse per disease cohort from periphery (spleen) or CNS (brain and spinal cord) on day 17 postimmunization. (B) Frequency of CD45+CD4+IL17+ (Th17) or CD45+CD4+IFNg+ (Th1) cells, average of 3 mice per disease cohort from periphery (spleen) or CNS (brain and spinal cord) on day 28 postimmunization (C) Frequency of CD4+CD25+FOXp3+ cells average of 2 mice per disease cohort per compartment on day 28 postimmunization. Shown is average ± SD.
Discussion
Although successes have been achieved in the treatment of relapsing MS, advances in progressive MS have been modest.4,5 Our understanding suggests that progressive MS may in part result from ongoing CNS damage associated with meningeal ectopic lymphoid follicles. The ability of CAR-T cells to penetrate the CNS more readily may offer a therapeutic benefit over existing mAb therapies that only deplete B cells in the periphery.6,7,18 In this study, we found that anti-CD19 CAR-T cells thoroughly depleted B cells in the periphery and within the CNS. The depth of CNS B-cell depletion with anti-CD19 CAR-T cells was sustained beyond that observed by anti-CD20 mAb treatment. Furthermore, as anti-CD20 mAb treatment of B-cell–dependent EAE emulates the clinical benefit in patients with MS, here we observed clinical improvement with anti-CD19 CAR-T cell treatment.
A recent study evaluated the efficacy of anti-CD19 CAR-T cells in spontaneous EAE,20 a model that previously showed lack of disease protection with anti-CD20 mAb therapy.36 In this study, although treatment with anti- CD19 CAR-T cells enhanced clearance of meningeal B-cell aggregates, the mice paradoxically had more severe EAE, an observation that we confirmed (data not shown). The authors postulated that the clinical worsening could reflect depletion of CD19+ regulatory IL-10-producing B (B10) cells. Of interest, a previous study demonstrated that constitutive CD19 overexpression on B cells protected mice from EAE and was associated with elevated levels of B10 cells, whereas CD19 deficiency exacerbated EAE.37 However there was another study showing treatment with humanized anti-CD19 mAb ameliorated disease.38 These differences reflect the diverse autoimmune mechanisms that drive tissue pathology in various EAE models and emphasize the vital importance of employing a variety of models when considering the potential of any therapeutic strategy to translate to the human disease.26,27
Although our B-cell–dependent EAE model induced by immunization with the extracellular domain of rhMOG highlighted the efficacy of anti-CD19 CAR-T cell therapy, we were surprised to find that mice treated with control cells also derived clinical benefit despite the fact that their B-cell populations returned to WT levels after the Cy pretreatment. Thus, B-cell depletion did not clearly account for the observed clinical benefit. In clinical care, Cy is used as a standard pretreatment for CAR-T cell therapy and is required for CAR-T cell–induced B-cell depletion in the mice. Although Cy alone has been used to treat EAE, the results have been inconsistent.39,40 In our study, Cy treatment alone only delayed the onset of clinical disease. Thus, Cy does not account for the benefit observed in either of our improved EAE groups. Furthermore, anti-CD19 CAR-T cells without pretreatment or Cy with untransduced activated cells were not found to be beneficial, suggesting a protective effect from the combination of transduced activated cells with Cy pretreatment.
Studies have demonstrated that Foxp3+ Treg cells can protect against EAE, whereas Treg deficiency exacerbates disease.41-43 We considered the possibility that increased Treg cell populations in the peripheral blood and CNS in the adoptively transferred treatment mice might have induced an immune regulatory environment that ameliorated EAE but did not find statistically significant differences in the Treg populations between treatment groups. Furthermore, their transcriptional profiles were not significantly different. Lilrb4 expression, however, was noted to be elevated in both anti-CD19 CAR-T cells and control cells when compared with untransduced activated cells. Lilrb4 has been identified to play a role in tumor microenvironment immune suppression, and its blockade results in increased T-cell effector activity.31,32 One could postulate that increased Lilrb4 expression promotes immunomodulation that results in less severe disease but occurs at a threshold undetectable by our assays of the endogenous response. We also considered if the overrepresentation of nonspecific CD8+ T cells affected the type of T cells that repopulated after Cy pretreatment.44,45 Many studies have investigated bystander CD4+ and CD8+ T cell activity that seem to promote an immune response.46-49 However, in this case, the bystander cells for both anti-CD19 CAR-T (∼50% of the injected cells) and control cells were rested for 2 weeks before EAE immunization. It is possible that earlier IL-2 secretion could have promoted Treg differentiation from effector cells.43,48 Unfortunately, we were unable to collect sufficient donor T cells in recipient mice to evaluate their phenotype after in vivo treatment.
This study has limitations. A comprehensive characterization of the adaptive immune system, including B regulatory cells, was not performed but will be important in future work. A more complete understanding of changes in cytokine profiles might also help clarify the mechanisms responsible for differential treatment effects. Future work should consider if an anti-CD20 CAR-T cell treatment strategy might preserve the CD19+ B10 cell compartment and provide more targeted B-cell depletion.38,50 Finally, we studied CAR-T cells in EAE prevention. It will be important to evaluate their use in established clinical disease as we consider translation to MS therapy.
This study demonstrates a benefit of anti-CD19 CAR-T cells in the treatment of a B-cell–dependent model of EAE without any identified systemic toxicity. Although anti-CD20 mAb therapy depleted B cells in the CNS and the periphery in this mouse model, it is known that anti-CD20 mAbs do not penetrate the CNS well in humans,6-8 whereas anti-CD19 CAR-T cells do.18 It is quite encouraging that profound and durable B-cell depletion was achieved by the anti-CD19 CAR-T cell regimen not only as expected in the periphery but also the CNS, reinforcing the concept that anti-CD19 CAR-T cells hold promise for patients with particular autoimmune diseases. While modeling disease is an important first step for evaluating the safety and efficacy of novel therapeutics, the fact that anti-CD19 CAR-T cells are already approved by multiple regulatory agencies and that they recently showed promise for treating another human autoimmune disease, systemic lupus erythematosus,19 argues for consideration of this treatment modality in patients with MS.
Acknowledgment
The authors thank Genentech, Inc for supplying them with murine anti-CD20 mAb. The authors would also like to thank Peggy Ho, PhD and Lawrence Steinman, MD of Stanford University for helpful conversations.
Glossary
- BM
bone marrow
- CAR
chimeric antigen receptor
- Cy
cyclophosphamide
- DEG
differentially expressed gene
- EAE
experimental autoimmune encephalomyelitis
- GFP
green fluorescent protein
- IP
intraperitoneal
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- MOG
myelin oligodendrocyte protein
- MSVG
mouse stem cell virus–based splice-gag vector
- UCSF
University of California, San Francisco
Appendix. Authors

Study Funding
This project was supported by funding from the UCSF Weill Institute for Neurosciences (S.G., M.S., S.A.S., C.S., W.L., M.R.W., S.S.Z.), the National MS Society (NMSS) fellowship (S.G., M.S.) and Alpha Stem Cell-California Institute for Regenerative Medicine fellowship (S.G.), the Westridge Foundation (M.R.W.), and the Valhalla Foundation. S.S.Z. and R.A.S. are supported by RO1 AI31624-01A1. SSZ is supported by NMSS RG1701-26628 and the Maisin Foundation. G.F.W. is supported by R21AI171994 (NIAID), R01AI165771 (NIAID), and RG-1808-32345 (NMSS). S.L.H. is supported by R35NS11164 (NINDS).
Disclosure
S. Gupta reports no disclosures relevant to the manuscript. M. Simic reports no disclosures relevant to the manuscript. S.A. Sagan reports no disclosures relevant to the manuscript. C. Shepherd reports no disclosures relevant to the manuscript. J. Duecker reports no disclosures relevant to the manuscript. R.A. Sobel reports no disclosures relevant to the manuscript. R Dandekar reports no disclosures relevant to the manuscript. G.F. Wu reports consulting for Sage research group, Progentec, Sangamo, Novartis, Roche, Speaking for EMD Serono, Editorial Board for Neurology: Neuroimmunology & Neuroinflammation, Journal of Neuroimmunology, research support from EMD Serono, Biogen, Roche. W. Wu reports no disclosures relevant to the manuscript. J.E. Pak reports no disclosures relevant to the manuscript. S.L. Hauser reports being a part of the Board of Directors for Neurona, Scientific Advisory Board for Annexon, Alector, and Accure; one-time consulting for BD, Moderna, and NGM Bio; and has received travel reimbursement and writing assistance from F. Hoffmann-La Roche Ltd and Novartis for CD20-related meetings and presentations. W. Lim reports equity in Gilead Sciences and Intellia Therapeutics, is an adviser for Allogene Therapeutics, and has filed patents related to this work. M.R. Wilson reports unrelated research grants from Roche/Genentech and Novartis and speaking fees from Takeda, WebMD, Novartis, and Genentech outside the submitted work. S.S. Zamvil reports being Deputy Editor of Neurology: Neuroimmunology & Neuroinflammation and is an Associate Editor for Frontiers in Immunology and Frontiers in Neurology, he serves on the Advisory Committee for the American Congress on Treatment and Research in MS (ACTRIMS) and is a standing member of the research grant review committee for the National MS Society (NMSS), he has served on the Editorial Board of the Journal of Clinical Investigation, The Journal of Immunology, and The Journal of Neurologic Sciences, and has been a charter member of the grant review committee for the NIH (NIH) Clinical Neuroimmunology and Brain Tumors (CNBT), he has served, or serves, as a consultant and received honoraria from Alexion, Biogen-Idec, EMD-Serono, Genzyme, Novartis, Roche/Genentech, and Teva Pharmaceuticals, Inc., and has served on Data Safety Monitoring Boards for Lilly, BioMS, Teva and Opexa Therapeutics. Go to Neurology.org/NN for full disclosures.
References
- 1.Wallin MT, Culpepper WJ, Campbell JD, et al. The prevalence of MS in the United States: a population-based estimate using health claims data [published correction appears in Neurology. 2019,93(15):688]. Neurology. 2019,92(10):e1029-e1040. doi: 10.1212/WNL.0000000000007035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017,376(3):221-234. doi: 10.1056/NEJMoa1601277 [DOI] [PubMed] [Google Scholar]
- 3.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011,378(9805):1779-1787. doi: 10.1016/S0140-6736(11)61649-8 [DOI] [PubMed] [Google Scholar]
- 4.Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009,66(4):460-471. doi: 10.1002/ana.21867 [DOI] [PubMed] [Google Scholar]
- 5.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017,376(3):209-220. doi: 10.1056/NEJMoa1606468 [DOI] [PubMed] [Google Scholar]
- 6.Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol. 2005,62(2):258-264. doi: 10.1001/archneur.62.2.258 [DOI] [PubMed] [Google Scholar]
- 7.Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006,180(1-2):63-70. doi: 10.1016/j.jneuroim.2006.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mainero C, Louapre C. Meningeal inflammation in multiple sclerosis: the key to the origin of cortical lesions? [published correction appears in Neurology. 2015 Sep 15,85(11):1010]. Neurology. 2015,85(1):12-13. doi: 10.1212/WNL.0000000000001586 [DOI] [PubMed] [Google Scholar]
- 9.Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011,134(Pt 9):2755-2771. doi: 10.1093/brain/awr182 [DOI] [PubMed] [Google Scholar]
- 10.Choi SR, Howell OW, Carassiti D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. 2012,135(Pt 10):2925-2937. doi: 10.1093/brain/aws189 [DOI] [PubMed] [Google Scholar]
- 11.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007,130(Pt 4):1089-1104. doi: 10.1093/brain/awm038 [DOI] [PubMed] [Google Scholar]
- 12.Lehmann-Horn K, Wang SZ, Sagan SA, Zamvil SS, von Büdingen HC. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight. 2016,1(20):e87234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters A, Pitcher LA, Sullivan JM, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011,35(6):986-996. doi: 10.1016/j.immuni.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004,14(2):164-174. doi: 10.1111/j.1750-3639.2004.tb00049.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu D, Jin G, Chai D, et al. The development of CAR design for tumor CAR-T cell therapy. Oncotarget. 2018,9(17):13991-14004. doi: 10.18632/oncotarget.24179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018,379(1):64-73. doi: 10.1056/NEJMra1706169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017,168(4):724-740. doi: 10.1016/j.cell.2017.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abramson JS, McGree B, Noyes S, et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N Engl J Med. 2017,377(8):783-784. doi: 10.1056/NEJMc1704610 [DOI] [PubMed] [Google Scholar]
- 19.Mackensen A, Müller F, Mougiakakos D, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022,28(10):2124-2132. doi: 10.1038/s41591-022-02017-5 [DOI] [PubMed] [Google Scholar]
- 20.Mitsdoerffer M, Di Liberto G, Dötsch S, et al. Formation and immunomodulatory function of meningeal B cell aggregates in progressive CNS autoimmunity [published correction appears in Brain. 2021 Jul 14]. Brain. 2021,144(6):1697-1710. doi: 10.1093/brain/awab093 [DOI] [PubMed] [Google Scholar]
- 21.Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010,68(3):369-383. doi: 10.1002/ana.22081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Häusler D, Häusser-Kinzel S, Feldmann L, et al. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc Natl Acad Sci USA. 2018,115(39):9773-9778. doi: 10.1073/pnas.1810470115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devaux B, Enderlin F, Wallner B, Smilek DE. Induction of EAE in mice with recombinant human MOG, and treatment of EAE with a MOG peptide. J Neuroimmunol. 1997,75(1-2):169-173. doi: 10.1016/s0165-5728(97)00019-2 [DOI] [PubMed] [Google Scholar]
- 24.Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci USA. 2005,102(39):13992-13997. doi: 10.1073/pnas.0504979102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010,116(19):3875-3886. doi: 10.1182/blood-2010-01-265041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegler EL, Wang P. Preclinical models in chimeric antigen receptor-engineered T-cell therapy. Hum Gene Ther. 2018,29(5):534-546. doi: 10.1089/hum.2017.243 [DOI] [PubMed] [Google Scholar]
- 27.Stromnes IM, Schmitt TM, Chapuis AG, Hingorani SR, Greenberg PD. Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol Rev. 2014,257(1):145-164. doi: 10.1111/imr.12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheadle EJ, Gornall H, Baldan V, Hanson V, Hawkins RE, Gilham DE. CAR T cells: driving the road from the laboratory to the clinic. Immunol Rev. 2014,257(1):91-106. doi: 10.1111/imr.12126 [DOI] [PubMed] [Google Scholar]
- 29.Lehmann-Horn K, Sagan SA, Bernard CC, Sobel RA, Zamvil SS. B-cell very late antigen-4 deficiency reduces leukocyte recruitment and susceptibility to central nervous system autoimmunity. Ann Neurol. 2015,77(5):902-908. doi: 10.1002/ana.24387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013,210(13):2921-2937. doi: 10.1084/jem.20130699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma N, Atolagbe OT, Ge Z, Allison JP. LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med. 2021,218(7):e20201811. doi: 10.1084/jem.20201811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng M, Gui X, Kim J, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. 2018,562(7728):605-609. doi: 10.1038/s41586-018-0615-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019,33(2):299-312. doi: 10.1038/s41375-018-0357-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018,24(6):731-738. doi: 10.1038/s41591-018-0041-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stripecke R, Carmen Villacres M, Skelton D, Satake N, Halene S, Kohn D. Immune response to green fluorescent protein: implications for gene therapy. Gene Ther. 1999,6(7):1305-1312. doi: 10.1038/sj.gt.3300951 [DOI] [PubMed] [Google Scholar]
- 36.Brand RM, Friedrich V, Diddens J, et al. Anti-CD20 depletes meningeal B cells but does not halt the formation of meningeal ectopic lymphoid tissue. Neurol Neuroimmunol Neuroinflamm. 2021,8(4):e1012. doi: 10.1212/NXI.0000000000001012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late- phase immunopathogenesis. J Immunol. 2010,185(4):2240-2252. doi: 10.4049/jimmunol.1001307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen D, Blazek M, Ireland S, et al. Single dose of glycoengineered anti-CD19 antibody (MEDI551) disrupts experimental autoimmune encephalomyelitis by inhibiting pathogenic adaptive immune responses in the bone marrow and spinal cord while preserving peripheral regulatory mechanisms. J Immunol. 2014,193(10):4823-4832. doi: 10.4049/jimmunol.1401478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.CALNE DB, LEIBOWITZ S. Suppression of experimental allergic encephalomyelitis by cytotoxic drugs. Nature. 1963,197:1309-1310. doi: 10.1038/1971309b0 [DOI] [PubMed] [Google Scholar]
- 40.Mangano K, Nicoletti A, Patti F, et al. Variable effects of cyclophosphamide in rodent models of experimental allergic encephalomyelitis. Clin Exp Immunol. 2010,159(2):159-168. doi: 10.1111/j.1365-2249.2009.04050.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber MS, Prod'homme T, Youssef S, et al. Type II monocytes modulate T cell–mediated central nervous system autoimmune disease. Nat Med. 2007,13(8):935-943. doi: 10.1038/nm1620 [DOI] [PubMed] [Google Scholar]
- 42.Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol Commun. 2014,2:163. doi: 10.1186/s40478-014-0163-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YC, Zhang AH, Yoon J, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun. 2018,92:77-86. doi: 10.1016/j.jaut.2018.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rambaldi B, Kim HT, Reynolds C, et al. Impaired T- and NK-cell reconstitution after haploidentical HCT with posttransplant cyclophosphamide. Blood Adv. 2021,5(2):352-364. doi: 10.1182/bloodadvances.2020003005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J, Kim MS, Kim EY, et al. Mycophenolate mofetil promotes down-regulation of expanded B cells and production of TNF-alpha in an experimental murine model of colitis. Cytokine. 2008,44(1):49-56. doi: 10.1016/j.cyto.2008.06.006 [DOI] [PubMed] [Google Scholar]
- 46.Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol. 2016,16(2):102-111. doi: 10.1038/nri.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim TS, Shin EC. The activation of bystander CD8+ T cells and their roles in viral infection. Exp Mol Med. 2019,51(12):1-9. doi: 10.1038/s12276-019-0316-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramanathan S, Gagnon J, Ilangumaran S. Antigen-nonspecific activation of CD8+ T lymphocytes by cytokines: relevance to immunity, autoimmunity, and cancer. Arch Immunol Ther Exp (Warsz) 2008,56(5):311-323. doi: 10.1007/s00005-008-0033-2 [DOI] [PubMed] [Google Scholar]
- 49.Lee HG, Lee JU, Kim DH, Lim S, Kang I, Choi JM. Pathogenic function of bystander-activated memory- like CD4+ T cells in autoimmune encephalomyelitis. Nat Commun. 2019,10(1):709. doi: 10.1038/s41467-019-08482-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parker KR, Migliorini D, Perkey E, et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell. 2020,183(1):126.e17-142.e17. doi: 10.1016/j.cell.2020.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not provided in the article because of space limitations, can be shared at the request of other investigators.