

Abstract
To deeply understand late onset Alzheimer’s disease (LOAD), it may be necessary to change the concept that it is a disease exclusively driven by aging processes. The onset of LOAD could be associated with a previous peripheral stress at the level of the gut (changes in the gut microbiota), obesity (metabolic stress), and infections, among other systemic/environmental stressors. The onset of LOAD, then, may result from the generation of mild peripheral inflammatory processes involving cytokine production associated with peripheral stressors that in a second step enter the brain and spread out the process causing a neuroinflammatory brain disease. This hypothesis could explain the potential efficacy of Sodium Oligomannate (GV-971), a mixture of acidic linear oligosaccharides that have shown to remodel gut microbiota and slow down LOAD. However, regardless of the origin of the disease, the end goal of LOAD-related preventative or disease modifying therapies is to preserve dendritic spines and synaptic plasticity that underlay and support healthy cognition. Here we discuss how systemic/environmental stressors impact pathways associated with the regulation of spine morphogenesis and synaptic maintenance, including insulin receptor and the brain derived neurotrophic factor signaling. Spine structure remodeling is a plausible mechanism to maintain synapses and provide cognitive resilience in LOAD patients. Importantly, we also propose a combination of drugs targeting such stressors that may be able to modify the course of LOAD by acting on preventing dendritic spines and synapsis loss.
Keywords: BDNF, dendritic spines, late onset Alzheimer’s disease, neuroinflammation, obesity, type 2 diabetes mellitus
INTRODUCTION
In recent years, investments in public health and research have allowed for the increase of the life expectancy of the human population in developed countries due to new discoveries in biomedicine, especially in high mortality diseases such as cancer and coronary heart diseases. However, one of the most important consequences associated to the increased life expectancy in the world population is the appearance of age-related diseases such as Alzheimer’s disease (AD), the most common form of dementia that affects 40 million people over the world and which is expected to triplicate the cases by 2050 [1]. This pathology not only creates a substantial personal burden within the community, but also leads to an unsustainable increase in the cost of public and private care [1, 2].
AD is hallmarked by a gradual decline in memory and other cognitive domains in both forms of the pathology, familial AD and idiopathic AD, which normally correlates with late onset AD (LOAD). Although the first diagnosis was performed more than 100 years ago, the etiology of LOAD remains unclear [2, 3]; however, several theories have been proposed, among which age is considered as the clear primary risk factor. A recent new hypothesis supports that it may not be just the inherent aging process but rather an accumulation of stressors through the lifetime that are responsible for the development of LOAD [4]. Such recently identified factors include alterations in gut microbiota, and subsequent inflammatory processes, dependent or independent of systemic changes, which leads to posterior appearance of neurodegeneration [5–10]. Moreover, its origin would not be at the point of the first clinical symptoms but rather 15–20 years earlier and slowly progressing to the full-fledged form of the pathology [11]. Specifically, brain symptoms would evolve from cognitive anomalies, to subjective cognitive decline, to mild cognitive impairment (MCI), and, finally, dementia [11].
At the molecular level, the classical hypothesis of the disease asserts that symptoms are a downstream consequence of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangle (NFT) formation, the latter caused by the hyperphosphorylation of tau [1–3, 12, 13]. Although both elements are of significant relevance, Aβ-linked alterations do not correlate at all with the actual symptoms of the pathology and only show a very significant role in the neuropathology in 3% of all AD patients (familial AD). In fact, the role of Aβ in the pathology has become controversial since, on the one hand, designed drugs focus on eliminating the Aβ plaques have failed in clinical trials and, on the other hand, these plaques have been detected in people without LOAD or, as recently highlighted, without familial AD [12–16]. Because of these inconsistencies, the hypothesis is now shifting toward a multifactorial origin, affecting multiple systems and structures which favor neuronal degeneration causing a vicious circle [17–20]. These alterations include a demise of neuronal function, synaptic dysfunction, stress derived of mitochondrial and endoplasmic reticulum alterations, and neuroinflammation, among others [20]. In fact, new studies suggest that neuronal synapse loss and alterations are more closely correlated with cognitive impairment evidencing that the maintenance of synaptic integrity and functions are critical for the neural circuits and behavioral functions, which raise new questions that contribute to design new therapeutic strategies aiming to delay and prevent LOAD [20–24].
In this review, we will summarize and discuss evidence on how some elements that play a crucial role in synaptic loss and subsequent neuronal death and cognitive deficits. Moreover, discussion about using these elements as targets for novel pharmacological therapies has also been undertaken.
RISK FACTORS CONTRIBUTING TO LOAD
It is well known that in the development of familial AD, genetic factors generating a dramatic amount of Aβ play a key role in disease development [1–3]. Despite the importance of genetics in AD development, it is becoming equally clear that these factors are not the only or even main responsible factors of the cognitive loss observed in LOAD [31–34]. The exponential increase of LOAD patients after the age of 65 have suggested the involvement of other age-related factors. Some of these factors stem from cumulative impact of infections, or peripheral diseases such as high blood pressure or metabolic stress syndromes such as obesity and type 2 diabetes (T2DM). These, in turn, are thought to increase inflammatory processes that underlay glial activation and cytokine release leading to brain insulin resistance appearance associated to insulin receptor (IR) inhibition, in consequence, brain glucose metabolism and mitochondrial activity are altered, and the blood-brain barrier loses its integrity, among others [35–47].
Apolipoprotein E and LOAD
The only genetic correlation to LOAD so far is the presence of apolipoprotein E (APOE) ε4 allele, which is responsible for 95% of AD cases [1–3, 15]. In fact, recent work published about a carrier of two copies of the APOE3 Christchurch (R136S) mutation, who presented with unusually high brain Aβ and limited tau/NFTs and neurodegenerative measurements, suggests that APOE not only is implicated in pathogenesis but may also become an important therapeutic target for treatment and prevention of LOAD [15, 25–30]. Moreover, APOE ε4 allele is considered one of the main risk factors in LOAD, due to its relation to alterations in energy metabolism and mitochondrial activity, in addition to being involved in Aβ clearance and aggregation [28, 30]. Of note, Ji and colleagues demonstrated that aged mice and humans expressing APOE ε4 gene showed a reduction in dendritic spine density in the dentate gyrus, thus supporting a contributing role of APOE ε4 on cognitive deficits in LOAD [33].
Type 2 diabetes and LOAD
Focusing on T2DM as a LOAD risk factor, it is well known that this metabolic pathology is associated with the presence of brain insulin resistance. In line with this concept, several authors have reported the importance of brain IRs in the cognitive process of LOAD [37, 40, 41]. It has been demonstrated that the reduction or inhibition of these receptors and consequent change in downstream intracellular pathways favors the increase of Aβ levels and tau phosphorylation. Likewise, Grillo and colleagues reported that the intrahippocampal administration of a lentiviral vector expressing an IR antisense sequence (LV-IRAS) induces synaptic transmission deficit associated with alterations in long-term potentiation (LTP) in which alterations in the expression and phosphorylation of glutamate receptor subunits are observed, suggesting that all this in whole will be responsible for the impairment in hippocampal learning [42]. At the molecular level, insulin activates PI3K/Akt/mTOR signaling pathway, which in turn promotes Ras-related C3 botulinum toxin substrate 1 (Rac1) dependent dendritic spine formation [44]. Therefore, at the brain level, IR signaling activation is involved in the regulation of dendritic spine formation and excitatory synapse development. For this reason, either drugs that stimulate the IR at the brain level and specifically at the hippocampus or also antidiabetic drugs can improve the cognitive process by increasing synaptic connections [44, 45].
Inflammatory response and LOAD
It has been widely demonstrated that inflammatory processes are clearly involved in the development of LOAD, due to multiple risk factors cited above are involved in trigger this response [51–56]. For example, in some patients, multiple association of risk factors (aging+obesity+T2DM+cumulative infections) lead to systemic inflammation which contributes synergistically to accelerate LOAD pathology. In addition, vascular factors (atherosclerosis hypertension, etc.), independently from initiating inflammatory process, can generate a cerebral blood flow decrease and/or damage in blood-brain barrier vasculature which can also results in LOAD progression. In line with this, Merlini and colleagues demonstrated that fibrinogen, a vascular protein, is key in initiating a microglial activation that can trigger a spine and dendrite loss associated with a cognitive impairment in preclinical mice model of AD [57], reinforcing the link between vascular pathology, inflammation, and synaptic loss in this pathology. In this complex process, the authors hypothesize that fibrinogen can yield its impact independently of Aβ through increased oxidative stress derived from low vascular tone and cooperatively with by the linkage of these two proteins to produce brain fibrin deposition. This deposition can then deal to microglial activation, spine loss, and cognitive impairment.
Additional risk factors involved in LOAD
Other factors involve specific cellular events that become progressively less efficient or more vulnerable in the aging process, namely increases oxidative stress or decreases in endogenous antioxidants, downregulation of neurotrophic factors, and alterations in the neurotransmitter levels such as glutamate or acetylcholine [1–4]. Moreover, with the age, neural circuits also become progressively more vulnerable to injury, especially glutamatergic pyramidal neurons of the hippocampus and prefrontal cortex; areas involved in memory processes whose alterations correlate with cognitive impairment and dendritic spines disturbances [48–50].
The multifactorial and synergic nature of insults that drive LOAD makes targeting such factors separately, extremely difficult and probably therapeutically inefficient. However, all risk factors associated with LOAD merge into a common path involving synaptic loss. Thus, making this end-result pharmacologically targetable and specially therapeutically relevant since spine/synaptic density is key in retaining cognitive function (cognitive resilience). In fact, it is possible to distinguish individuals with or without clinical dementia in accordance with the state of their synaptic structures [59, 60]. Therefore, even though the precise mechanisms of how synaptic remodeling contributes to cognitive resilience and protects individuals with LOAD remains unclear, therapeutically targeting such common end-result (synaptic loss) may be a more promising strategy [59, 60]. Certainly, the exploration and targeting of mechanisms implicated in dendritic spines is, thus, described maintenance to avoid the transition from preclinical LOAD to symptomatic LOAD is worth considering and is thus described in the following section.
CONTRIBUTION OF SYNAPTIC SPINE LOSS TO LOAD
Dendritic spines are highly dynamic neural structures that can change in size and shape in response to activity of neurotransmitters in the central nervous system [59–62]. According to their morphology, dendritic spines have been mainly classified in several types: thin, stubby, and mushroom [59–65]. The morphology of dendritic spines influences excitatory neurotransmission and synaptic plasticity. More specifically, it has been suggested that thin spines actively participate in the learning process [63]. Mushroom spines, since they are more stable and can last a long time, would act in storage of long-term information [63, 64]. However, the role of stubby spines is still unclear, although it has been proposed that they regulate neuronal excitability.
Spines behave as points for the establishment of synapses and help transmit electrical signals to the soma of the neuron by increasing the synaptic contact area in the dendrite. Moreover, it has been showed that dendritic spines are the primary site of structural plasticity in the adult brain and its alterations correlate with cognitive impairment [63–68]. Therefore, it is important to understand the precise molecular factors involved in spine deterioration in LOAD to develop therapeutic strategies that target cognitive loss in this process [69, 70].
In accordance with the functional role of dendritic spines, a relevant study evaluated their structure related to the maintenance of cognitive function in AD patients. In this study, patients were divided into three groups: cognitively normal subjects without AD, cognitively normal control subjects showing moderate to severe AD pathology at autopsy and AD cases with severe pathology [60]. The results demonstrated that the maintenance of the dendritic spines plasticity was a key mechanism of cognitive resilience, protecting elderly people with AD physiopathology from developing dementia. Likewise, studies in non-human primates indicate that synaptic loss are responsible of cognitive aging decline in aged monkeys in area 7a in cortex correlating loss of glutamatergic synapses with disturbances in working memory [71, 72]. Moreover, Dumitriu and colleagues reported a prominent selective loss of thin spines related to synaptic changes in layer III of 46 area in prefrontal cortex of rhesus monkeys, which was associated to age-related memory impairment compared to young subjects [71–73]. For this reason, due to the prevalence of cognitive impairment related to aging in the elderly population, the concept of maintaining synaptic health in aging has been proposed [74]. Moreover, in a preclinical study by Pereira and colleagues, they treated rats with riluzole, a blocker of TTX-sensitive sodium channels [74]. They aimed to ameliorate age-related cognitive decline by improving glutamatergic circuits and retention of hippocampal-dependent memory performance. Their results showed an increase of thin dendritic spine clustering in the hippocampus and prefrontal cortex, suggesting to its potential as a new therapeutic approach for age-associated cognitive impairment. In the study, they also observed increased brain-derived neurotrophic factor (BDNF) expression whose involvement in synaptic structures will be tackled in the next section.
Significant data in AD models and humans are in accordance with earlier works that demonstrated the correlation of synapse loss with cognitive dysfunction that somehow overshadowed the importance of Aβ plaques and NFTs in the development of LOAD [48, 49]. Moreover, Scheff and colleagues showed significant synapse loss and spine alterations in the CA1 area of the hippocampus in early stages of LOAD [23, 70]. Furthermore, numerous literature points toward the involvement of Aβ deposition as a mediator of synaptic loss in LOAD [75–77]. Specifically, Aβ overexpression correlates with the severity of the disease, showing neurotoxic properties that trigger AD pathophysiology such as LTP inhibition, dendritic spine loss, and learning and spatial memory impairments in preclinical models [72–74]. Likewise, Aβ has an extracellular localization which not only is spread through the brain, but also throughout the peripheral tissues which allows it to bind to several types of receptors such as the brain IR, receptor for advanced glycation end products (RAGE), and cellular prion protein (PrPC), among others, that can induce synaptic and neuronal dysfunction and death [13]. For all these reasons, its potential participation in the initiation of the cognitive loss progression should not be ruled out [13, 16, 75, 77]. However, whether Aβ plays a crucial role in synaptic loss in LOAD remains unclear. In fact, clinical studies using positron emission tomography (PET) scans have established that subgroups of patients with cognitive impairment may have little or no accumulation of Aβ, while elderly people with normal cognition may have extensive Aβ accumulation. This questions the direct role of Aβ as triggering factor for LOAD and may provide a potential explanation for the overwhelming failure of therapies solely targeting Aβ [78].
Taken together, as discussed in the previous section and based on the evidence above, the importance of dendritic spines and thus, preventing, maintaining or being able to recover their functionality, represents an inherent protective target that delays the onset of dementia. This hypothesis introduces a new concept focusing on a “catch-all” disease endpoint centered on life quality in adults (“good synaptic health”). Therefore, the molecular factors involved in these processes and the development of therapeutic targets constitute an urgent unmet medical need that needs to be addressed.
AMELIORATING LATE ONSET ALZHEIMER’S DISEASE: POSSIBLE SPINE/SYNAPTIC TARGETS
The synaptic loss hypothesis associated with LOAD cognitive loss was widely studied by Terry’s work in the 1990s and incorporated the importance of glial activation associated with another key component, the loss of trophic factors [48, 49, 67, 68]. Also important, while the pathophysiology of AD was believed to be centralized in the brain, over time, a diverse array of research studies has been able to observe that there are many peripheral elements [75, 76]. This is especially true for factors involving inflammatory processes.
More recently, the role of peripheral factors that contribute to inflammatory processes and link peripheral disease to LOAD are also increasingly gaining attention. Two of the main factors include metabolic syndrome and dysregulations of gut microbiota [6, 20]. In an interesting review by Herrup, from the two-hypothesis postulating that multiple independent foci contribute to LOAD [79, 80], the researcher suggested that LOAD progresses in a well-defined three phase progression system [4]. The first stage would be “the trigger”, an alteration or group of alterations of diverse nature that would initiate the downwards process. These would include different forms of metabolic or environmental stresses like an infection, insulin resistance, or simply day-to-day habits. The second step would be “the convergence point”. That is, where all such paths would focalize in the appearance of chronic inflammatory processes, which would, in turn, damage multiple tissues including the brain. Alterations would include but not be restricted to microglial activation, appearance of oxidative stress, cytokine release, and synapse loss. Finally, in the third step, and considering the assault suffered in the previous stages, cells would suffer a biological change that would predispose them to degenerate through apoptotic mechanisms (Fig. 1). This final stage would coincide with the appearance of the first visual clinical symptoms. Thus, considering the hypothesis by Herrup, it seems that the second stage, would provide a good target and, modifying the development of inflammatory responses may be an effective approach to prevent and ameliorate neurodegenerative states [4, 81–83].
Fig. 1.
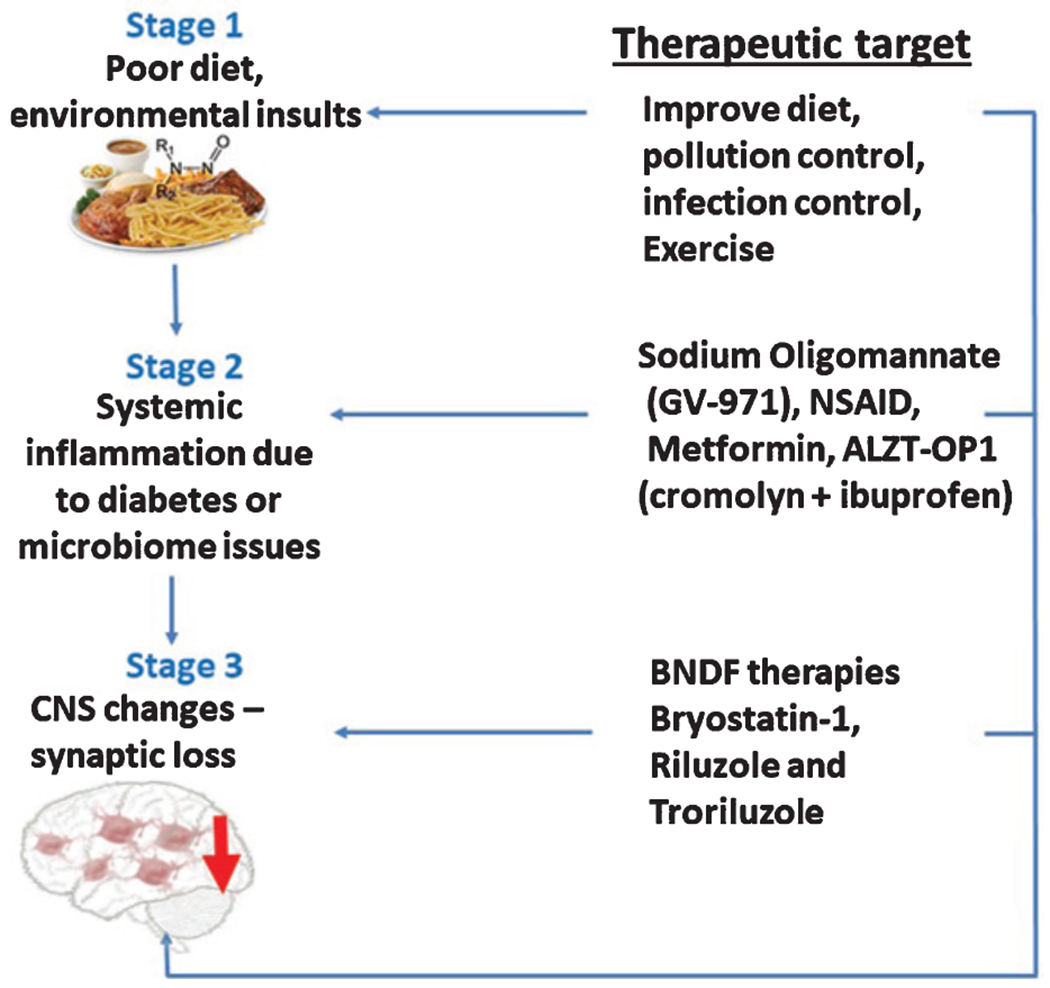
Effects of unhealthy diet, infections, sedentary lifestyle, and other neurotoxic factors, as potential first responsible for triggering cognitive loss in Alzheimer’s disease. In a second step, systemic inflammation will amplify the initial stress signal that will finally favor the process of cognitive loss. Likewise, it can be observed non-pharmacological and therapeutic strategies, proposed to stop this process of memory loss.
Targeting inflammatory responses in the central nervous system (CNS) and periphery
Inflammation is a common denominator to several of these LOAD risk factors. To this end, neuroinflammation, depending upon the phenotypic polarization, ‘M1’ phenotype and ‘M2’ phenotype, displays a central and dual role in LOAD pathology: while initially beneficial for clearance of extracellular aggregates of Aβ, the sustained pro-inflammatory profile can be extremely deleterious to neuronal function. The dysregulation of the microglial M1/M2 ratio causes deleterious alterations in synaptic structure and function. Specifically, M1 microglia generates a harmful effect through the enhanced response of inflammatory cytokine release leading to neuronal damage [52, 53, 81–85]. In addition, IL-1β is the main immune player in LTP regulation in the hippocampus [9]. By contrast, M2 microglia displays a neuroprotective effect releasing neuroprotective cytokines such as IL-10, transforming growth factor-β (TGF-β), and insulin like growth factor 1 (IGF-1). Thus, it could be hypothesized that, the modulation of proinflammatory processes such as obesity related to T2DM together with other such as gut microbiota could be a suitable strategy for LOAD treatment, since both generate a swift on stimuli signal involved in the activation of brain M1 [83–85].
The peripheral mechanisms involved in the association between obesity and cognitive impairment highlights the role of neuroimmune pathways. In particular, adipokines (cytokines synthesized in the adipose tissue) are increased in obesity patients and actively favor or alter the evolution of a low-grade inflammatory response that increase the risk of multiple systems damage, including the SNC [20, 45–47, 85]. In this process, adipocyte hypertrophy triggers the recruitment of innate immunity cells, macrophages, which are important contributors to the overall systemic inflammatory process where proinflammatory cytokines release happen [20, 85–87]. These proteins reach the circulatory system, cross the blood-brain barrier, and trigger the neuroinflammatory process, which activate resident microglia to M1 in brain areas linked to dementia and cognitive loss such as the hypothalamus and hippocampus. This process involves the activation of several molecular pathways including c-Jun N-terminal kinase (JNK), which in the brain plays a prominent role in the regulation of brain insulin resistance, which apart from inducing an inflammatory response, is thought to generate a positive feedback loop [86, 87]. Taken together, it could be hypothesized that the brain resident microglia must be working in concert with peripheral immune cells to modulate synaptic function (Fig. 2).
Fig. 2.
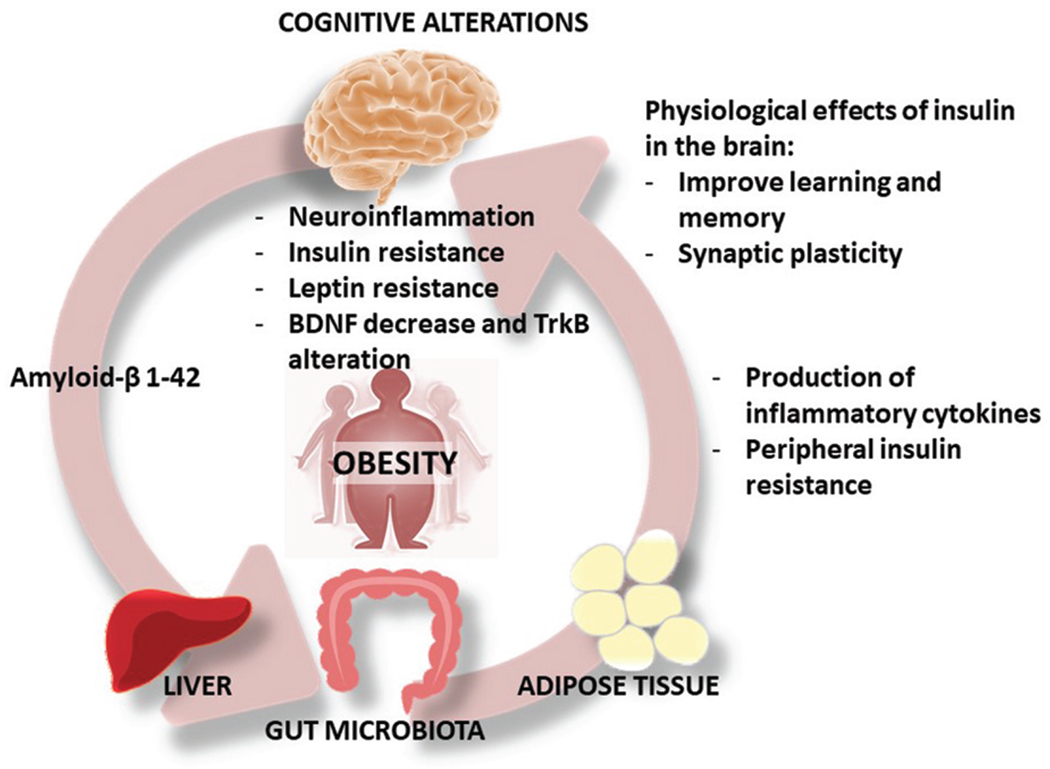
Alzheimer’s disease should be viewed as a disease of the whole body. The production of amyloid in the brain can promote the development of T2DM, since Aβ can bind directly to insulin receptor. Likewise, obesity associated with T2DM, gut microbiota composition and adipose tissue alteration develop insulin resistance-related diseases. All these processes favor a peripheral and central inflammatory process that induces resistance to insulin, leptin and alteration of the BDNF/TrkB receptor. The final stage is the process of memory loss due to the alteration of the dendritic spines and alterations of the synaptic connections.
Likewise, another key component in this inflammatory response is the nucleotide-binding oligomerization domain (NOD) - leucine-rich repeat-(LRR)- and pyrin domain-containing protein 3 (NLRP3) inflammasome. NLRP3 inflammasome is composed of three key components: Nod-like receptor protein NLRP3 (also called cyopyrin or NALP3), apoptosis-associated speck-like protein (ASC), and cysteine protease precursor procaspase-1 (consist of a C-terminal caspase recruitment domain [CARD] and a caspase domain). In addition, varieties of different stimulators have been confirmed to induce NLRP3-dependent caspase-1 activation, including for example bacterial infection, tau, and Aβ. Thus, NLRP3 would be responsible for detecting a cell stress signal, causing or triggering a defense process through innate immunity that recruits ASC and procaspase-1, resulting in the activation of caspase-1 and the processing of cytoplasmic targets, including the pro-inflammatory cytokines. Thus, NLRP3 inflammasome may be a potential link between peripheral diseases that act as stressors (T2DM, gut microbiota) and LOAD.
Moreover, T2DM is associated with mitochondrial dysfunction and consequent oxidative stress and also with NLRP3 inflammasome activation. Zhou and colleagues reported that mitochondrial dysfunction is a key modulator of oxidative stress, which is essential for the activation of both the NLRP3 inflammasome and apoptosome and, at the same time, is involved in the modulation of apoptotic process [88]. Therefore, NLRP3 inflammasome is related to a variety of metabolic and inflammatory diseases, including gout, diabetes, atherosclerosis, and neurodegenerative diseases [89–93]. Concerning to AD, it has been reported that Aβ might stimulate NLRP3 activation, which is mainly localized in glial cells, through the binding to microglial surface receptors CD36 and TLR4 and thereby increasing the release of proinflammatory cytokines, and consequently amplifying the neuroinflammatory process and aggravating the neuropathological LOAD [91]. Furthermore, the neuropathological inflammatory circuit involving mitochondrial alterations associated with the NLRP3 inflammasome in T2DM causes an endothelial dysfunction that contributes to blood-brain barrier dysfunction [90].
Recently, and supporting the above claims, a new drug, Sodium Oligomannate (GV-971), has been conditionally approved for the treatment of LOAD exclusively in China [94, 95]. To this end, Wang and coworkers reported that an alteration of the intestinal microbiota in preclinical mice models of AD increased the peripheral infiltration of immune system cells such as peripheral type 1 T-helper (Th1) cell to allow their local crosstalk with the M1 microglia and, in turn, triggers the microglia differentiation towards a M1 pro-inflammatory state [96]. Likewise, these authors also reported a significant alteration in some amino acids in blood and feces of their AD mouse model, primarily related to the phenylalanine pathway such as phenylalanine and isoleucine (Phe/Ile). Both amino acids play a significant role promoting the differentiation and proliferation of peripheral inflammatory Th1 cells.
Taken together, these studies suggest a potential peripheral LOAD initiation where gut dysbiosis contributes to Phe/Ile elevation, which leads to the proliferation/differentiation and brain infiltration of Th1 cells and this neuroinflammation promoting synapses loss and support the notion that peripheral stimuli may contribute to neuronal damage and memory impairment independent of Aβ accumulation.
The next step is to understand the possible mechanisms involved in how the activation of the microglia causes the loss of the synapse. In this regard, Hong and colleagues reported in an interesting manuscript, the implication of the microglia as potential cellular mediator of synapse loss. In particular, this process in AD is orchestrated by soluble Aβ oligomers and it is mediated through CR3 complement-dependent pathway and microglia that engulfs synapses which position microglia as potential cellular mediators of synapse loss independently or before brain plaques formation [84]. Thus, this research study suggest that microglia is a potential early therapeutic target in LOAD involving synaptic dysfunction associated with cognitive decline.
The Rho family GTPases, including Ras homologous member A (RhoA) and Rac1, have been found to play an important role in synaptic morphology of dendritic spines and synaptic plasticity [62–64, 96]. It has been reported that RhoA activation leads to spine pruning, whereas Rac1 activation is involved in spine formation, growth, and stabilization [63]. In line with this, previous studies have been demonstrated that some non-steroidal anti-inflammatory drugs (NSAID), which are widely used to treat mild to moderate pain and inflammation, among them ibuprofen, inhibits RhoA in neurons suggesting that this effect on Rho GTPases activity might provide a new strategy for reversing synapse loss and dysfunction, and improving memory and cognitive ability in AD [97]. In fact, Cummings and colleagues reported a recent study about ALZT-OP1 (cromolyn+ibuprofen) demonstrating that an association of two compounds, a mast cell stabilizer (cromolyn) and an anti-inflammatory (ibuprofen), is currently in phase III for AD treatment [98, 99]. Cromolyn could affect the route of Aβ and ibuprofen constitutes a multiple target drug, acting on the process of Aβ production (by α-secretase activation), microglia inhibition, activation of PPAR γ, and on Rho family GTPases. Therefore, the inhibition of Rho family GTPases by ibuprofen and the synaptic improvement could contribute to cognitive resilience in LOAD treatment.
Targeting trophic factors (BDNF) in the CNS
BDNF is the most abundant neurotrophin in the CNS, widely distributed in different areas of the brain such as the hypothalamus, amygdala, hippocampus, and neocortex [100]. It is a non-glycosylated polypeptide that is synthesized as a prepropeptide of 247 aminoacids and is later processed until its mature form of 119 aminoacids. Its physiological activity is modulated through the Tropomyosin receptor kinase B (TrkB) receptor. After binding, the receptor dimerizes and activates extracellularly regulated protein kinase (ERK) pathway [100–102]. Importantly, BDNF has a role in neuronal survival processes, axonal and dendritic growth and branching, synaptic structure and plasticity, neurotransmission, just like in the induction and maintenance of LTP or, the regulation of dendritic spine number and morphology [102]. By contrast, its genetic deletion in rats leads to impairment of hippocampal LTP and hippocampal-dependent memory function loss [103–106]. Therefore, this signaling pathway is of key relevance in the formation of synaptic plasticity and memory and, could be a possible target for LOAD treatment [105].
Targeting the increase of brain BDNF levels
Not pharmacological interventions such as aerobic exercise are known to stimulate BDNF brain production which is associated to modulation and preservation of hippocampal volume [100, 101], providing a therapeutic avenue for LOAD [106]. Likewise, it has been demonstrated that exercise in humans is involved in the neurotransmitter regulation of glutamate release from excitatory synapses, activating glutamatergic α-amino-3-hydroxy-5-methyl-4-iso-xazole propionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors which increase the efficiency of synaptic transmission [107–109]. In preclinical studies, Li and colleagues reported that exercise induced an increase in Rac1 or cell division control protein 42 (Cdc42) activity (two molecules associated with synaptic plasticity improvement) in the hippocampus of aged animals [110]. Furthermore, this research study demonstrated the role of exercise training on the modulatory regulation of Rho-GTPase in rodents. Thus, the potential association of physical exercise together with the administration of BDNF analogue drugs may constitute a good strategy to improve cognitive resilience in LOAD [109–112].
When trying to increase the levels of BDNF pharmacologically aimed to retard the underlying process of AD, researchers encountered the problem that like most trophic factors, BDNF could not cross the blood-brain barrier [100–105]. Thus, new methods had to be developed in order to stimulate its production in an indirect manner.
In this area, the transcription factor CREB plays a critical role in cognition and its activation facilitates the transcription of proteins for the synaptic process, mainly of BDNF.
In this context, Caccamo and colleagues not only were able to increase BDNF levels in the hippocampus of rodent model of AD, but also enhanced NMDA activity, using viral delivery of the CREB-binding protein. This pathway stimulates transcription and translation of genes involved in the formation of memory through cyclic adenosine monophosphate (cAPM) (CREB) phosphorylation (via cAMP/PKA pathway), thus, creating a positive feedback process [113].
Additionally, Blurton-Jones and co-workers reported that neuronal stem cells transplantation improved learning and memory deficits in 18-month-old 3xTg-AD mice by increasing BDNF levels [114]. In support of these findings, Nagahara and colleagues demonstrated that the administration of a BDNF lentivirus improved learning and memory deficits in several animal models of AD and aged rats [115, 116].
An additional strategy for increasing the brain BDNF levels is the regulation of (cAMP) through the modulation of the enzyme phosphodiesterase-4 (PDE-4) [117]. Inhibition of PDE-4 by specific drugs enhances cAMP levels activates Protein kinase A (PKA), which results in an increase in phosphorylation of pCREB, increasing the expression of BDNF which, facilitates neuronal plasticity and LTP in hippocampus [117–119]. One example of PDE-4 inhibitor is Roflumilast, a highly selective second-generation drug, currently used for the treatment of chronic obstructive pulmonary disease [118–120]. It has been reported that at the preclinical level in APP/PS1 mice, chronic roflumilast treatment improves cognitive impairment through the upregulation of the cAMP/CREB/BDNF pathway [120].
Likewise, de Pins and co-workers delivered engineered astrocytes with a BDNF gene under the control of the GFAP promoter in 5xFAD mice [121]. These animals showed a recovery in BDNF levels in the hippocampus and related areas when compared to normal 5xFAD mice and cognitive improvements. Additional assays evidenced a restoration of dendritic spine density and morphology, and a recovery of the levels of several other presynaptic and postsynaptic markers like synaptophysin and PSD-95. Interestingly, these animals showed no changes in Aβ load or the rate of hippocampal neurogenesis.
Supporting this idea, Fingolimod, a drug which has been undergone several preclinical models of AD and Huntington’s disease, has shown to be associated with synaptic plasticity improved and amelioration of dendritic spine loss, effects mediated through the increase of BDNF mRNA levels and BDNF protein release by the activation of Sphingosine-1-phosphate receptors in the brain [123].
Delving deeper into possible molecular mechanisms, Hedrick and colleagues suggest a complex model of facilitation of neuron plasticity where BDNF plays a key role [123]. In this postsynaptic process, Rho GTPase proteins Rac1 together with RhoA and Cdc42 are involved in the regulation of dendritic spines actin cytoskeleton and, therefore, its structural and functional plasticity [124]. More specifically, it has been demonstrated that Ca2 + /calmodulin-dependent kinase 2 (CaMKII), which is known to be essential for LTP induction, is activated by postsynaptic NMDA receptors. In this process, Rho GTPase proteins serve as a molecular link between CaMKII and the expression of LTP [123–125]. Furthermore, BDNF regulates the activity of Rac1 and Cdc42 which depend of BDNF source in the cell and autocrine activation of its receptor, TrkB. This could explain the synaptic accumulation of BDNF observed in vesicles into presynaptic terminals and dendrites. In this way, when glutamate is bound to its receptors, generates a presynaptic depolarization that triggers the Ca2+-dependent release of BDNF, which binds to the TrkB tyrosine kinase receptor to activate Rac1. Rac1 acts through a cascade of serine / threonine-protein kinase PAK1-LIM domain kinase 1 (LIMK1) to phosphorylate and inhibit cofilin and block its separation from actin [126–127].
Besides, BDNF/TrkB receptor also regulates dendritic spines through the synthesis of several proteins including Arc, Homer 2, and LIMK1 which promote actin polymerization and so enlargement of spine heads that determine its morphology [102–106].
Targeting BDNF receptor: TrkB agonists
Another potential approach to protect and restore synaptic connections may be to target the BDNF receptor itself (Table 1). By stimulating TrkB receptors through the administration of drug agonists, it may be possible to increase its intracellular signaling. Specifically, these compounds potentiate neurite/spine/dendrite growth, synapse formation, neuronal maturation, and synaptic plasticity via the PLCγ/PI3K/MAPK signaling cascade [128, 129].
Table 1.
Therapies using TrkB agonists
| Active compound | In vitro studies | In vivo studies | References |
|---|---|---|---|
| 7,8-dihydroxyflavone (7,8-DHF) | Crosses the blood-brain barrier Activates TrkB to modulate cognitive function | 7, 8-DHF in a mice model of AD, induced an increase in the density of the dendritic spines. | [130–134] |
| Selectively improved spine density in AD mice and did not affect control mice | |||
| LM22A-4 | Inhibits neuronal death in in vitro neurodegenerative disease models | Favors the amelioration of dysfunctional motor activity, administered intranasally in rodents after traumatic brain injury. | [128] |
| Chronic peripheral treatment in a mice model of Rett syndrome improved motor and cognitive function by restoring deficits in synaptic plasticity in the hippocampus | |||
| AS86 (TrkB agonist antibody) | Activates BDNF-TrkB brain signaling pathways | Improves dendritic spine growth and facilitates synaptic transmission and plasticity in preclinical models | [135] |
| Peptide TAT-BDNF | Intraperitoneal administration improved memory in APPswe mice | [136] | |
| TDP6 (BNDF mimetic peptide) | TDP6 induces TrkB phosphorylation in vitro. | [137] | |
| TDP6 promotes oligodendrocyte myelination in vitro. | |||
| The pro-myelinating effect of TDP6 is TrkB-dependent. | |||
| Sitagliptin | Administrated by oral gavage protects the cognition function of the Alzheimer’s disease mice through activating glucagon-like peptide-1 and BDNF-TrkB signaling pathways. | [138] |
The 7,8-dihydroxyflavone (7,8-DHF) compound was reported to cross the blood-brain barrier and activate TrkB in the brain to modulate cognitive function [130–134]. Castello and colleagues reported that a 7, 8-DHF treatment, in a preclinical mice model of AD, induced an increase in the density of the dendritic spines [133]. Specifically, this compound selectively improved spine density in AD mice and did not affect control mice, suggesting that TrkB receptor signaling activation interacts with other injury-initiated signals to facilitate remodeling of the spine [132, 134].
Another similar compound, LM22A-4, inhibits neuronal death in in vitro neurodegenerative disease models and favors the amelioration of dysfunctional motor activity, when administered intranasally in rodents after traumatic brain injury. In addition, chronic peripheral treatment of LM22A-4 in a mice model of Rett syndrome disease improved motor and cognitive function by restoring deficits in synaptic plasticity in the hippocampus [128].
Moreover, AS86 is a new TrkB agonist antibody that activates the BDNF-TrkB signaling pathways in the brain. Wang and colleagues demonstrated that AS86 improved dendritic spine growth and facilitates synaptic transmission and plasticity in preclinical studies. Thus, this drug has been proposed as a new therapeutic strategy for neurodegenerative diseases including LOAD due to its capacity to repair synapsis [135].
Wu and colleagues reported in an APPswe/PS1dE9 mice preclinical study that the intraperitoneal administration of the TAT-BDNF fusion peptide improved memory. The improvement in the cognitive process is mediated through the activation of the TrkB receptor-signaling pathway, such as Erk1/2, PI3K/Akt, upregulation of CREB, as well as PSD95, a postsynaptic protein [136].
TDP6 is a peptide mimetic, which induces TrkB phosphorylation and activation [137] (see Table 1). Sitagliptin, a dipeptidyl peptidase 4 inhibitor used as an anti-diabetic drug, in preclinical models of AD improved the cognition function through the activation of BDNF-TrkB signaling pathway [138].
Targeting protein kinase C isozyme epsilon (PKCε)
Another strategy to increase brain BDNF levels involves the administration of molecules that activate protein kinase C isozyme epsilon (PKCε) [139–141]. PKCε constitutes a very attractive target because it is directly involved in the regulation of the BDNF signaling pathway in the brain, playing an essential role in the maintenance of synaptic functions and structures and in the cognitive process [139]. To this end, Sen and colleagues reported that PKCε reduction is associated with increased Aβ in AD brain patients and preclinical transgenic AD mice models [141]. Moreover, they demonstrated that PKCε inhibition modulates the activity of γ-secretase (activate) and α-secretase (inhibits) involved in AβPP processing. It has been reported that PKCε activators 8-[2-(2-pentylcyclopropylmethyl)-cyclopropyl]-octanoic acid (DCP-LA), a synthesized linoleic acid derivative specific activator of PKCε and bryostatin-1 which activates PKCε and also PKCα, prevented synaptic loss, amyloid plaque deposition, and memory deficits in mice models of AD through and significant increasing expression of BDNF in the hippocampal CA1 area [139–141]. In addition, PKCε activation by both compounds could improve synapsis and cognitive resilience through the decrease of levels of soluble Aβ [139]. The first Bryostatin-1 clinical trial was promising because the drug was safe, showing favorable pharmacokinetics [142].
Although Bryostatin is a novel powerful activator of PKCε, it has also shown benefits for synaptic loss and maturation in animal models of AD, as well as other neurological disorders. However, in humans it does not seem to be effective. The clinical study (NCT03560245) randomized, placebo-controlled, phase II multicenter trial was to assess the safety, efficacy, and tolerability of intravenous Bryostatin-1 20 μg in 108 participants with AD who were not taking memantine. Participants were randomly assigned 1:1 to Bryostatin-1 or placebo for 12 weeks for a total of 7 doses. Participants were between 55 and 85 years old and for at least 30 days before the start of study drug treatment, they should not have taken memantine. At week 13, authors evaluated the study’s primary endpoint, the change in total score of the Severe Impairment Battery scale score. They found no statistically significant differences between treatment and placebo groups. However, although the treatment of Bryostatin-1 in monotherapy in patients with AD has failed, its potential use in the future in a multiple combinatory therapy should not be ruled out.
Rho-associated coiled-coil containing protein kinases (ROCK) inhibitors as strategy to induce dendritic spine resilience
The protection of dendritic spines against the degeneration caused by LOAD is critical to modify the disease course and as a preventive therapeutic strategy, since synapse or dendritic spine loss correlates more strongly with cognitive impairment than Aβ or NFT hallmarks. In LOAD, it was reported that Aβ would be responsible for causing the dendritic cytoskeleton damage by activating the RhoA guanosine triphosphatase (GTPase) and its downstream effector the Rho-associated protein kinase (ROCK) [143]. Two isoforms have been characterized, ROCK1 and ROCK2. ROCKs are involved in the regulation of actin-myosin–mediated cytoskeleton contractility, and increased activity of ROCKs have harmful damages on the morphology of dendritic spines contributing to cognitive loss in LOAD [144]. Moreover, Rush and colleagues demonstrated that Aβ induced synaptic loss, which is associated with cognitive loss in LOAD which was prevented using Fasudil, a ROCK inhibitor, in neuronal cell cultures [143]. Thus, several ROCK inhibitors have been evaluated in preclinical AD models and neuronal cell cultures such as Fasudil and Y-27632 FSD-C10, due to constitute a potential strategy for treatment of LOAD trough the increase of cognitive resilience [144, 145]. On the other hand, Ripasudil, another ROCK inhibitor, is being evaluated for glaucoma treatment [146].
COMPLEMENTARY DRUG STRATEGIES
Besides cognitive problems, AD runs in parallel with many other alterations. Patients suffer from metabolic dysregulations, vascular affectations, sleep disorders, etc., that contribute significantly to dendritic spine loss. Thus, it is possible that future disease-modifying treatments will require to be effective in multiple fronts or be composed of different molecules that will modulate more than one system at a time with the aim to retard the underlying process of LOAD [98, 99, 147, 148].
Likewise, the use of antidiabetic drugs for the T2DM treatment (such as metformin) and antihypertensive (losartan) and anticholesterolemic drugs (statins) are examples of drugs that are currently under investigation in phase III [98, 99]. These treatments may be effective to prevent the development of pathology and thus, it could become a complementary element for future therapeutic [147–149]. In fact, preclinical studies based in this preventive strategy have shown beneficial effects in rodents. Interestingly, preclinical data demonstrated that statins inhibit the RhoA/LIMK/cofilin pathway, which, as discussed above, is involved in the formation of synapses and cognitive resilience [149].
Likewise, BHV4157 (troriluzole) is a third-generation prodrug (pharmaceutical formulation of riluzole) which is currently in phase III (NCT03605667) as disease modified molecule. Interestingly, it is suggested that the beneficial effects of this drug are mediated through the modulation of synaptic levels of excitatory neurotransmitter glutamate and, which improves synaptic functioning.
Additionally, anti-inflammatory drugs could be necessary in these drug cocktails in early stages, even though NSAIDs therapy alone in human trials has, thus far, failed. However, some anti-inflammatory drugs, among them, ibuprofen, apart from acting on the microglia decreasing its activation, constitute a multi-drug target since regulates the formation of amyloid, acting on Aβ secretion and more importantly, blocking RhoA-activation [92]. NSAIDs can also inhibit the nuclear translocation of NF-κB, which is essential for transcription of NLRP3 and pro-IL-1β.
A possible explanation for the failure of drugs in LOAD may be that, so far, most clinical trials have begun too late when cognitive symptoms are already present. Thus, success of these therapies may be maximized when initiated 15–20 years before the apparition of the first symptoms of LOAD, when the microglia is still in M1 phase and synapses loss is not yet present.
In addition, in this combinatory drug strategy it could be necessary to add the classical drugs in the market such as memantine (glutamate antagonist) and acetylcholinesterase inhibitors, which increase the brain levels of acetylcholine and improve the cognitive process.
Lastly, the administration of a drug targeting brain insulin signaling pathway could be key to improve spines and synapses, attenuating peripheral and central Aβ levels, modulating BDNF/TrkB signaling [150, 151].
CONCLUSIONS
The maintenance of the integrity of the synaptic functions is fundamental for the suitable functioning of the neural circuits and therefore of the cognitive functions. In this line, synapse loss is highly correlated with the progression of LOAD. In addition, another important point is that LOAD has various etiological origins that give rise to various co-morbidities; thus, a potential therapeutic success may depend on combination of therapies that should star 15–20 years before the appearance of disease symptoms (cognitive loss). The hypothesis that LOAD begins with an initiating injury, such as a metabolic stress, has both theoretical and practical relevance. Likewise, as it has been previously stated, the initial injury (gut, adipose tissue, infections, and others) could generate a peripheral inflammatory process, realizing cytokines, which are able to propagate throughout peripheral and central tissues. Therefore, the disease could have a peripheral origin and for this reason, it must be considered as a global disease of the whole organism, not only exclusively of the brain. It should also be taken into account that this peripheral inflammatory process can favor an activation of protein tyrosine phosphatase PTP1B that can inhibit at the hippocampal level the insulin, TkrB and leptin receptors, which would possibly also alter dendritic spines and cognitive loss [87]. Moreover, alterations in hypothalamus also have been associated with inflammatory processes since it has been widely demonstrated that it regulates glucose and insulin levels [150, 151]. Therefore, preventive anti-inflammatory drugs targeted to both peripheral and central tissues could be an effective approach for LOAD.
Finally, the main objective of therapeutic strategies is to improve the life quality of patients avoiding the cognitive decline; therefore, the maintenance of dendritic spines and synapses improving cognitive resilience are key in the design of new therapeutic strategies [60, 152]. Thus, all these studies point out the importance of the administration of combined drug therapies to which we can also associate strategies such as physical exercise that increases BDNF levels, due to the complexity of the pathologic mechanism involved LOAD [152–155].
ACKNOWLEDGMENTS
This study was partly supported by funds from the Spanish Ministerio de Economía y Competitividad (SAF2017-84283-R to AC), the Generalitat de Catalunya (2014SGR-525 to AC). CIBER de Enfermedades Neurodegenerativas (CIBERNED) (Grant CB06/05/2004 to AC; Instituto de Salud Carlos III).
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1106r1).
REFERENCES
- [1].2020 Alzheimer’s disease facts and figures. Alzheimers Dement, doi: 10.1002/alz.12068 [DOI] [PubMed] [Google Scholar]
- [2].Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2016) Alzheimer’s disease. Lancet 388, 505–517. [DOI] [PubMed] [Google Scholar]
- [3].Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL (2015) Alzheimer’s disease. Nat Rev Dis Primers 1, 15056. [DOI] [PubMed] [Google Scholar]
- [4].Herrup K (2010) Reimagining Alzheimer’s disease–an age-based hypothesis. J Neurosci 30, 16755–16762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li B, He Y, Ma J, Huang P, Du J, Cao L, Wang Y, Xiao Q, Tang H, Chen S (2019) Mild cognitive impairment has similar alterations as Alzheimer’s disease in gut microbiota. Alzheimers Dement 15, 1357–1366. [DOI] [PubMed] [Google Scholar]
- [6].Noble JM, Scarmeas N, Celenti RS (2014) Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One 9, e114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fülöp T, Itzhaki RF, Balin BJ, Miklossy J, Barron AE (2018) Role of microbes in the development of Alzheimer’s disease: state of the art - An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front Genet 9, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Itzhaki RF, Golde TE, Heneka MT, Readhead B (2020) Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol 16, 193–197. [DOI] [PubMed] [Google Scholar]
- [9].Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25, 181–213. [DOI] [PubMed] [Google Scholar]
- [10].Wang M, Gamo NJ, Yang Y, Jin LE, Wang XJ, Laubach M, Mazer JA, Lee D, Arnsten AF (2011) Neuronal basis of age-related working memory decline. Nature 476, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McDade E, Bateman RJ (2017) Stop Alzheimer’s before it starts. Nature 547, 153–155. [DOI] [PubMed] [Google Scholar]
- [12].Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol Med 8, 95–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Viola KL, Klein WL (2015) Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129, 183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim J, Yoon H, Basak J, Kim J (2014) Apolipoprotein E in synaptic plasticity and Alzheimer’s disease: potential cellular and molecular mechanisms. Mol Cells 37, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT (2019) Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med 25, 1680–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Viola KL, Velasco PT, Klein WL (2008) Why Alzheimer’s is a disease of memory: the attack on synapses by A beta oligomers (ADDLs). J Nutr Health Aging 12, 51S–57S. [DOI] [PubMed] [Google Scholar]
- [17].Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espírito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG (2013) TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab 18, 831–843. [DOI] [PubMed] [Google Scholar]
- [18].Wilcox KC, Lacor PN, Pitt J, Klein WL (2011) Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol Neurobiol 31, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ferreira ST, Klein WL (2011) The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem 96, 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].De Felice FG, Ferreira ST (2014) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63, 2262–2272. [DOI] [PubMed] [Google Scholar]
- [21].Singh A, Jones OD, Mockett BG, Ohline SM, Abraham WC (2019) Tumor necrosis factor-α-mediated metaplastic inhibition of ltp is constitutively engaged in an Alzheimer’s disease model. J Neurosci 39, 9083–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Scheff SW, Price DA (2006) Alzheimer’s disease-related alterations in synaptic density: Neocortex and hippocampus. J Alzheimers Dis 9 (3 Suppl), 101–115. [DOI] [PubMed] [Google Scholar]
- [23].Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27, 1372–1384. [DOI] [PubMed] [Google Scholar]
- [24].Coleman P, Federoff H, Kurlan R (2004) A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63, 1155–1162. [DOI] [PubMed] [Google Scholar]
- [25].Martínez-Martínez AB, Torres-Perez E, Devanney N, Del Moral R, Johnson LA, Arbones-Mainar JM (2020) Beyond the CNS: The many peripheral roles of APOE. Neurobiol Dis 138, 104809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS (2009) ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci 29, 15317–15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gérard HC,Wildt KL, Whittum-Hudson JA, Lai Z, Ager J, Hudson AP (2005) The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb Pathog 39, 19–26. [DOI] [PubMed] [Google Scholar]
- [28].Wu L, Zhang X, Zhao L (2018) Human ApoE isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J Neurosci 38, 6665–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N, Krasemann S, Lichtenthaler SF, Müller SA, Colombo A, Monasor LS, Tahirovic S, Herms J, Willem M, Pettkus N, Butovsky O, Bartenstein P, Edbauer D, Rominger A, Ertürk A, Grathwohl SA, Neher JJ, Holtzman DM, Meyer-Luehmann M, Haass C (2019) Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 22, 191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jones NS, Watson KQ, Rebeck GW (2019) Metabolic disturbances of a high-fat diet are dependent on APOE genotype and sex. eNeuro 6, ENEURO.0267-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G (2019) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neuro 15, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shinohara M, Sato N (2019) The roles of apolipoprotein E, lipids, and glucose in the pathogenesis of Alzheimer’s disease. Adv Exp Med Biol 1128, 85–101. [DOI] [PubMed] [Google Scholar]
- [33].Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T (2003) Apolipoprotein E isoform- specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience 122, 305–315. [DOI] [PubMed] [Google Scholar]
- [34].Arendt T, Schindler C, Brückner MK, Eschrich K, Bigl V, Zedlick D, Marcova L (1997) Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J Neurosci 17, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kellar D, Craft S (2020) Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol 19, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Plucińska K, Dekeryte R, Koss D, Shearer K, Mody N, Whitfield PD, Doherty MK, Mingarelli M, Welch A, Riedel G, Delibegovic M, Platt B (2016) Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia 59, 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hoyer S (1998) Risk factors for Alzheimer’s disease during aging. Impacts of glucose/energy metabolism. J Neural Transm Suppl 54, 187–194. [DOI] [PubMed] [Google Scholar]
- [38].Morris G, Berk M, Maes M, Puri BK (2019) Could Alzheimer’s disease originate in the periphery and if so how so? Mol Neurobiol 56, 406–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Long JM, Holtzman DM (2019) Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Salkovic-Petrisic M, Hoyer S (2007) Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl 72, 217–233. [DOI] [PubMed] [Google Scholar]
- [41].Hoyer S, Lee SK, Löffler T, Schliebs R (2000) Inhibition of the neuronal insulin receptor. An in vivo model for sporadic Alzheimer disease? Ann N Y Acad Sci 920, 256–258. [DOI] [PubMed] [Google Scholar]
- [42].Grillo CA, Piroli GG, Lawrence RC, Wrighten SA, Green AJ, Wilson SP, Sakai RR, Kelly SJ, Wilson MA, Mott DD, Reagan LP (2015) Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes 64, 3927–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].De la Monte SM, Wands JR (2008) Alzheimer’s disease is type 3 diabetes–evidence reviewed. J Diabetes Sci Technol 2, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM (2018) Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].De Felice FG (2013) Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J Clin Invest 123, 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Melo HM, Seixas da Silva GDS, Sant’Ana MR, Teixeira CVL, Clarke JR, MiyaCoreixas VS, de Melo BC, Fortuna JTS, Forny-Germano L, Ledo JH, Oliveira MS, Figueiredo CP, Pardossi-Piquard R, Checler F, Delgado-García JM, Gruart A, Velloso LA, Balthazar MLF, Cintra DE, Ferreira ST, De Felice FG (2020) Palmitate is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro-inflammatory TNF-α. Cell Rep 30, 2180–2194. [DOI] [PubMed] [Google Scholar]
- [47].Forny-Germano L, De Felice FG, Vieira MNDN (2019) The role of leptin and adiponectin in obesity-associated cognitive decline and Alzheimer’s disease. Front Neurosci 12, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Terry RD (1996) The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 55, 1023–1025. [PubMed] [Google Scholar]
- [49].Terry RD (1994) Neuropathological changes in Alzheimer disease. Prog Brain Res 101, 383–390. [DOI] [PubMed] [Google Scholar]
- [50].Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA (2001) Copernicus revisited: amyloid beta in Alzheimer’s disease. Neurobiol Aging 22, 131–146. [DOI] [PubMed] [Google Scholar]
- [51].McGeer PL, Rogers J, McGeer EG (2016) Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimers Dis 54, 853–857. [DOI] [PubMed] [Google Scholar]
- [52].McGeer PL, Schulzer M, Edith GM (1996) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease. Neurology 47, 425–432. [DOI] [PubMed] [Google Scholar]
- [53].Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 12, 719–732. [DOI] [PubMed] [Google Scholar]
- [54].Morris JK, Honea RA, Vidani ED, Swerdlow RH, Burns JM (2014) Is Alzheimer’s disease a systemic disease? Biochim Biophys Acta 1842, 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Costello DA, Keenan K, McManus RM, Falvey A, Lynch MA (2016) The age-related neuroinflammatory environment promotes macrophage activation, which negatively impacts synaptic function. Neurobiol Aging 43, 140–148. [DOI] [PubMed] [Google Scholar]
- [56].Lynch MA (2015) Neuroinflammatory changes negatively impact on LTP: a focus on IL-1β. Brain Res 1621, 197–204. [DOI] [PubMed] [Google Scholar]
- [57].Merlini M, Rafalski VA, Rios Coronado PE, Gill TM, Ellisman M, Muthukumar G, Subramanian KS, Ryu JK, Syme CA, Davalos D, Seeley WW, Mucke L, Nelson RB, Akassoglou K (2019) Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron 101, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wood H (2019) Fibrinogen links vascular pathology to cognitive decline. Nat Rev Neurol 15, 187. [DOI] [PubMed] [Google Scholar]
- [59].Walker CK, Herskowitz JH (2020) Dendritic spines: mediators of cognitive resilience in aging and Alzheimer’s disease. Neuroscientist, doi: doi: 10.1177/1073858420945964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Boros BD, Greathouse KM, Gentry EG, Curtis KA, Birchall EL, Gearing M, Herskowitz JH (2017) Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann Neurol 82, 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K (2005) Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. [DOI] [PubMed] [Google Scholar]
- [62].Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10, 647–658. [DOI] [PubMed] [Google Scholar]
- [63].Chidambaram SB, Rathipriya AG, Bolla SR, Bhat A, Ray B, Mahalakshmi AM, Manivasagam T, Thenmozhi AJ, Essa MM, Guillemin GJ, Chandra R, Sakharkar MK (2019) Dendritic spines: Revisiting the physiological role. Prog Neuropsychopharmacol Biol Psychiatry 92, 161–193. [DOI] [PubMed] [Google Scholar]
- [64].Knott G, Holtmaat A (2008) Dendritic spine plasticity–current understanding from in vivo studies. Brain Res Rev 58, 282–289. [DOI] [PubMed] [Google Scholar]
- [65].Scheff SW, Price D, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27, 1372–1384. [DOI] [PubMed] [Google Scholar]
- [66].Bourne J, Harris KM (2007) Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol 17, 381–386. [DOI] [PubMed] [Google Scholar]
- [67].Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580. [DOI] [PubMed] [Google Scholar]
- [68].Masliah E, Miller A, Terry RD (1993) The synaptic organization of the neocortex in Alzheimer’s disease. Med Hypotheses 41, 334–340. [DOI] [PubMed] [Google Scholar]
- [69].Neuman KM, Molina-Campos E, Musial TF, Price AL, Oh KJ, Wolke ML, Buss EW, Scheff SW, Mufson EJ, Nicholson DA (2015) Evidence for Alzheimer’s disease-linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons. Brain Struct Funct 220, 3143–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Scheff SW, Neltner JH, Nelson PT (2014) Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem Pharmacol 88, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, Rapp PR, Morrison JH (2010) Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci 30, 7507–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Motley SE, Grossman YS, Janssen WGM, Baxter MG, Rapp PR, Dumitriu D, Morrison JH (2018) Selective loss of thin spines in area 7a of the primate intraparietal sulcus predicts age-related working memory impairment. J Neurosci 38, 10467–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Morrison JH, Baxter MG (2014) Synaptic health. JAMA Psychiatry 71, 835–837. [DOI] [PubMed] [Google Scholar]
- [74].Pereira AC, Lambert HK, Grossman YS, Dumitriu D, Waldman R, Jannetty SK, Calakos K, Janssen WG, McEwen BS, Morrison JH (2014) Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc Natl Acad Sci U S A 111, 18733–18738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cline EN, Bicca MA, Viola KL, Klein WL (2018) The amyloid-β oligomer hypothesis: beginning of the third decade. J Alzheimers Dis 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Clarke JR, Lyra E Silva NM, Figueiredo CP, Frozza RL, Ledo JH, Beckman D, Katashima CK, Razolli D, Carvalho BM, Frazão R, Silveira MA, Ribeiro FC, Bomfim TR, Neves FS, Klein WL, Medeiros R, LaFerla FM, Carvalheira JB, Saad MJ, Munoz DP, Velloso LA, Ferreira ST, De Felice FG (2015) Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol Med 7, 190–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ding Y, Zhao J, Zhang X, Wang S, Viola KL, Chow FE, Zhang Y, Lippa C, Klein WL, Gong Y (2019) Amyloid beta oligomers target to extracellular and intracellular neuronal synaptic proteins in Alzheimer’s disease. Front Neurol 10, 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ (2016) PET imaging of tau deposition in the aging human brain. Neuron 89, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zhu X, Raina AK, Perry G, Smith MA (2004) Alzheimer’s disease: the two-hit hypothesis. Lancet Neurol 4, 219–226. [DOI] [PubMed] [Google Scholar]
- [80].Zhu X, Lee HG, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta 1772, 494–502. [DOI] [PubMed] [Google Scholar]
- [81].Rajendran L, Paolicelli RC (2018) Microglia-mediated synapse loss in Alzheimer’s disease. J Neurosci 38, 2911–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yao K, Zu HB (2020) Microglial polarization: novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 28, 95–110. [DOI] [PubMed] [Google Scholar]
- [84].Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Clarke JR, Ribeiro FC, Frozza RL, De Felice FG, Lourenco MV (2018) Metabolic dysfunction in Alzheimer’s disease: from basic neurobiology to clinical approaches. J Alzheimers Dis 64, S405–S426. [DOI] [PubMed] [Google Scholar]
- [86].Vieira MNN, Lima-Filho RAS, De Felice FG (2018) Connecting Alzheimer’s disease to diabetes: Underlying mechanisms and potential therapeutic targets. Neuropharmacology 136, 160–171. [DOI] [PubMed] [Google Scholar]
- [87].De Felice FG, Lourenco MV, Ferreira ST (2014) How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10(1 Suppl), S26–32. [DOI] [PubMed] [Google Scholar]
- [88].Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]
- [89].Zhang Y, Zhao Y, Zhang J, Yang G (2020) Mechanisms of NLRP3 inflammasome activation: its role in the treatment of Alzheimer’s disease. Neurochem Res 45, 2560–2572. [DOI] [PubMed] [Google Scholar]
- [90].Söderbom G, Zeng BY (2020) The NLRP3 inflammasome as a bridge between neuro-inflammation in metabolic and neurodegenerative diseases. Int Rev Neurobiol 154, 345–391. [DOI] [PubMed] [Google Scholar]
- [91].Liu Y, Dai Y, Li Q, Chen C, Chen H, Song Y, Hua F, Zhang Z (2020) Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci Lett 736, 135279. [DOI] [PubMed] [Google Scholar]
- [92].Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R (2020) Focus on the role of NLRP3 inflammasome in diseases. Int J Mol Sci 21, 4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pellegrini C, Antonioli L, Calderone V, Colucci R, Fornai M, Blandizzi C (2020) Microbiota-gut-brain axis in health and disease: Is NLRP3 inflammasome at the crossroads of microbiota-gut-brain communications? Prog Neurobiol 191, 101806. [DOI] [PubMed] [Google Scholar]
- [94].Syed YY (2020) Sodium oligomannate: first approval. Drugs 80, 441–444. [DOI] [PubMed] [Google Scholar]
- [95].Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, Xie Z, Chu X, Yang J, Wang H, Chang S, Gong Y, Ruan L, Zhang G, Yan S, Lian W, Du C, Yang D, Zhang Q, Lin F, Liu J, Zhang H, Ge C, Xiao S, Ding J, Geng M (2019) Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res 29, 787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Arrazola Sastre A, Luque Montoro M, Gálvez-Martín P, Lacerda HM, Lucia AM, Llavero F, Zugaza JL (2020) Small GTPases of the Ras and Rho families switch on/off signaling pathways in neurodegenerative diseases. Int J Mol Sci 21, E6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Fu Q, Hue J, Li S (2007) Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J Neurosci 11, 4154–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K (2020) Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y) 6, e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Cummings JL, Tong G, Ballard C (2019) Treatment combinations for Alzheimer’s disease: current and future pharmacotherapy options. J Alzheimers Dis 67, 779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Caffino L, Mottarlini F, Fumagalli F (2020) Born to protect: leveraging BDNF against cognitive deficit in Alzheimer’s disease. CNS Drugs 34, 281–297 [DOI] [PubMed] [Google Scholar]
- [101].Zhao H, Alam A, San CY, Eguchi S, Chen Q, Lian Q, Ma D (2017) Molecular mechanisms of brain-derived neurotrophic factor in neuro-protection: Recent developments. Brain Res 1665, 1–21. [DOI] [PubMed] [Google Scholar]
- [102].Kim BY, Lee SH, Graham PL, Angelucci F, Lucia A, Pareja-Galeano H, Leyhe T, Turana Y, Lee IR, Yoon JH, Shin JI (2017) Peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease and mild cognitive impairment: a comprehensive systematic review and meta-analysis. Mol Neurobiol 54, 7297–7311. [DOI] [PubMed] [Google Scholar]
- [103].Chen JJ, Wang T, An CD, Jiang CY, Zhao J, Li S (2016) Brain-derived neurotrophic factor: a mediator of inflammation-associated neurogenesis in Alzheimer’s disease. Rev Neurosci 27, 793–811. [DOI] [PubMed] [Google Scholar]
- [104].Tanila H (2017) The role of BDNF in Alzheimer’s disease. Neurobiol Dis 97, 114–118. [DOI] [PubMed] [Google Scholar]
- [105].Song JH, Yu JT, Tan L (2015) Brain-derived neurotrophic factor in Alzheimer’s disease: risk, mechanisms, and therapy. Mol Neurobiol 52, 1477–1493. [DOI] [PubMed] [Google Scholar]
- [106].Lu B, Nagappan G, Lu Y (2014) BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol 220, 223–250. [DOI] [PubMed] [Google Scholar]
- [107].Wang R, Holsinger RMD (2018) Exercise-induced brain-derived neurotrophic factor expression: Therapeutic implications for Alzheimer’s dementia. Ageing Res Rev 48, 109–121. [DOI] [PubMed] [Google Scholar]
- [108].Lourenco MV, Frozza RL, de Freitas GB, Zhang H, Kincheski GC, Ribeiro FC, Gonçalves RA, Clarke JR, Beckman D, Staniszewski A, Berman H, Guerra LA, Forny-Germano L, Meier S, Wilcock DM, de Souza JM, Alves-Leon S, Prado VF, Prado MAM, Abisambra JF, Tovar-Moll F, Mattos P, Arancio O, Ferreira ST, De Felice FG (2019) Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat Med 25, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Müller P, Duderstadt Y, Lessmann V, Müller NG (2020) Lactate and BDNF: key mediators of exercise induced neuroplasticity? J Clin Med 9, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Li Y, Zhao L, Gu B, Cai J, Lv Y, Yu L (2017) Aerobic exercise regulates Rho/cofilin pathways to rescue synaptic loss in aged rats. PLoS One 12, e0171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Marosi K, Kim SW, Moehl K, Scheibye-Knudsen M, Cheng A, Cutler R, Camandola S, Mattson MP (2016) 3-Hydroxybutyrate regulate energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem 139, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hongpaisan J, Xu C, Sen A, Nelson TJ, Alkon DL (2013) PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol Dis 55, 44–62. [DOI] [PubMed] [Google Scholar]
- [113].Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S (2010) CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 107, 22687–22692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM (2009) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 106, 13594–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Nagahara AH, Wilson BR, Ivasyk I, Kovacs I, Rawalji S, Bringas JR, Pivirotto PJ, Sebastian WS, Samaranch L, Bankiewicz KS, Tuszynski MH (2018) MR-guided delivery of AAV2-BDNF into the entorhinal cortex of non-human primates. Gene Ther 25, 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bhat A, Ray B, Mahalakshmi AM, Tuladhar S, Nandakumar DN, Srinivasan M, Essa MM, Chidambaram SB, Guillemin GJ, Sakharkar MK (2020) Phosphodiesterase-4 enzyme as a therapeutic target in neurological disorders. Pharmacol Res 160, 105078. [DOI] [PubMed] [Google Scholar]
- [118].Sugin LJS, Murugesan A, Bindu M, Sunil KN (2020) Roflumilast: A potential drug for the treatment of cognitive impairment? Neurosci Lett 736, 135281. [DOI] [PubMed] [Google Scholar]
- [119].Feng H, Wang C, He W, Wu X, Li S, Zeng Z, Wei M, He B (2019) Roflumilast ameliorates cognitive impairment in APP/PS1 mice via cAMP/CREB/BDNF signaling and anti-neuroinflammatory effects. Metab Brain Dis 34, 583–559. [DOI] [PubMed] [Google Scholar]
- [120].Wang H, Zhang FF, Xu Y, Fu HR, Dan Wan X, Wang L, Chen W, Xu XY, Gao YF, Zhang JG, Zhang HT (2020) The phosphodiesterase-4 inhibitor roflumilast, a potential treatment for the comorbidity of memory loss and depression in Alzheimer’s disease: a preclinical study in APP/PS1 transgenic mice. Int J Neuropsychopharmacol, doi: 10.1093/ijnp/pyaa048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].de Pins B, Cifuentes-Díaz C, Farah AT, López-Molina L, Montalban E, Sancho-Balsells A, López A, Ginás S, Delgado-García JM, Alberch J, Gruart A, Girault JA, Giralt A (2019) Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of Alzheimer disease. J Neurosci 39, 2441–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Patnaik A, Spiombi E, Frasca A, Landsberger N, Zagrebelsky M, Korte M (2020) Fingolimod modulates dendritic architecture in a BDNF-dependent manner. Int J Mol Sci 21, 3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, Yasuda R (2016) Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, McNamara JO (2016) Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hedrick NG, Yasuda R (2017) Regulation of Rho GTPase proteins during spine structural plasticity for the control of local dendritic plasticity. Curr Opin Neurobiol 45, 193–201. [DOI] [PubMed] [Google Scholar]
- [126].Yoshii A, Constantine-Paton M (2010) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70, 304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Yoshii A, Constantine-Paton M (2007) BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci 10, 702–711. [DOI] [PubMed] [Google Scholar]
- [128].Li W, Bellot-Saez A, Phillips ML, Yang T, Longo FM, Pozzo-Miller L (2017) A small-molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis Model Mech 10, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K (2010) A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A 107, 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Zeng Y, Lv F, Li L, Yu H, Dong M, Fu Q (2012) 7,8-dihydroxyflavone rescues spatial memory and synaptic plasticity in cognitively impaired aged rats. J Neurochem 122, 800–811. [DOI] [PubMed] [Google Scholar]
- [131].Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K (2014) 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Gao L, Tian M, Zhao HY, Xu QQ, Huang YM, Si QC, Tian Q, Wu QM, Hu XM, Sun LB, McClintock SM, Zeng Y (2016) TrkB activation by 7, 8-dihydroxyflavone increases synapse AMPA subunits and ameliorates spatial memory deficits in a mouse model of Alzheimer’s disease. J Neurochem 136, 620–636. [DOI] [PubMed] [Google Scholar]
- [133].Castello NA, Nguyen MH, Tran JD, Cheng D, Green KN, LaFerla FM (2014) 7,8-Dihydroxyflavone, a small molecule TrkB agonist, improves spatial memory and increases thin spine density in a mouse model of Alzheimer disease-like neuronal loss. PLoS One 9, e91453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wang X, Romine JL, Gao X, Chen J (2017) Aging impairs dendrite morphogenesis of newborn neurons and is rescued by 7, 8-dihydroxyflavone. Aging Cell 16, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wang S, Yao H, Xu Y, Hao R, Zhang W, Liu H, Huang Y, Guo W, Lu B (2020) Therapeutic potential of a TrkB agonistic antibody for Alzheimer’s disease. Theranostics 10, 6854–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Wu Y, Luo X, Liu X, Liu D, Wang X, Guo Z, Wang JZ (2015) Intraperitoneal administration of a novel TATBDNF peptide ameliorates cognitive impairments via modulating multiple pathways in two Alzheimer’s rodent models. Sci Rep 5, 15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Wong AW, Giuffrida L, Wood R, Peckham H, Gonsalvez D, Murray SS, Xiao J (2014) TDP6, a brain-derived neurotrophic factor-based trkB peptide mimetic, promotes oligodendrocyte myelination. Mol Cell Neurosci 63, 132–140. [DOI] [PubMed] [Google Scholar]
- [138].Dong Q, Teng SW, Wang Y, Qin F, Li Y, Ai LL, Yu H (2019) Sitagliptin protects the cognition function of the Alzheimer’s disease mice through activating glucagonlike peptide-1 and BDNF-TrkB signalings. Neurosci Lett 696, 184–190. [DOI] [PubMed] [Google Scholar]
- [139].Hongpaisan J, Sun MK, Alkon D (2011) PKCε Activation prevents synaptic loss, Aε elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J Neurosci 31, 630–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Sun MK, Alkon DL (2019) Neuro-regeneration therapeutic for Alzheimer’s dementia: perspectives on neurotrophic activity. Trends Pharmacol Sci 40, 655–668. [DOI] [PubMed] [Google Scholar]
- [141].Sen A, Nelson TJ, Alkon DL, Hongpaisan J (2018) Loss in pkc epsilon causes downregulation of MnSOD and BDNF expression in neurons of Alzheimer’s disease hippocampus. J Alzheimers Dis 63, 1173–1189. [DOI] [PubMed] [Google Scholar]
- [142].Nelson TJ, Sun MK, Lim C, Sen A, Khan T, Chirila FV, Alkon DL (2017) Bryostatin effects on cognitive function and PKCε in Alzheimer’s disease phase IIa and expanded access trials. J Alzheimers Dis 58, 521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Rush T, Martinez-Hernandez J, Dollmeyer M, Frandemiche ML, Borel E, Boisseau S, Jacquier-Sarlin M, Buisson A (2018) Synaptotoxicity in Alzheimer’s disease involved a dysregulation of actin cytoskeleton dynamics through cofilin 1 phosphorylation. J Neurosci 38, 10349–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Swanger SA, Mattheyses AL, Gentry EG, Herskowitz JH (2016) ROCK1 and ROCK2 inhibition alters dendritic spine morphology in hippocampal neurons. Cell Logist 19, e1133266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Yu JZ, Li YH, Liu CY, Wang Q, Gu QF, Wang HQ, Zhang GX, Xiao BG, Ma CG (2017) Multitarget therapeutic effect of fasudil in APP/PS1 transgenic mice. CNS Neurol Disord Drug Targets 16, 199–209. [DOI] [PubMed] [Google Scholar]
- [146].Testa V, Ferro Desideri L, Della Giustina P, Traverso CE, Iester M (2020) An update on ripasudil for the treatment of glaucoma and ocular hypertension. Drugs Today (Barc) 56, 599–608 [DOI] [PubMed] [Google Scholar]
- [147].Bauzon J, Lee G, Cummings J (2020) Repurposed agents in the Alzheimer’s disease drug development pipeline. Alzheimers Res Ther 12, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Yarchoan M, Arnold SE (2014) Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 63, 2253–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kim ML, Sung KR, Kwon J, Shin JA (2020) Statins suppress TGF-β2-mediated MMP-2 and MMP-9 expression and activation through RhoA/ROCK inhibition in astrocytes of the human optic nerve head. Invest Ophthalmol Vis Sci 61, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ganguli M, Beer JC, Zmuda JM, Ryan CM, Sullivan KJ, Chang CH, Rao RH (2020) Aging, diabetes, obesity, and cognitive decline: a population-based study. J Am Geriatr Soc 68, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Kivipelto M, Mangialasche F, Ngandu T (2018) Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol 14, 653–666. [DOI] [PubMed] [Google Scholar]
- [152].Grutzendler J, Kasthuri N, Gan WB (2002) Long-term dendritic spine stability in the adult cortex. Nature 420, 812–816. [DOI] [PubMed] [Google Scholar]
- [153].Sabbagh MN (2020) Alzheimer’s disease drug development pipeline. J Prev Alzheimers Dis 7, 66–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Tolar M, Abushakra S, Sabbagh M. Huang LK, Chao SP, Hu CJ (2020) Clinical trials of new drugs for Alzheimer disease. J Biomed Sci 27, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Sun MK, Nelson TJ, Alkon DL (2015) Towards universal therapeutics for memory disorders. Trends Pharmacol Sci 36, 384–394. [DOI] [PubMed] [Google Scholar]