

Abstract
Aggregated α-synuclein, a major constituent of Lewy bodies plays a crucial role in the pathogenesis of α-synucleinopathies (SPs) such as Parkinson’s disease (PD). PD is affected by the innate and adaptive arms of the immune system, and recently both active and passive immunotherapies targeted against α-synuclein are being trialed as potential novel treatment strategies. Specifically, dendritic cell-based vaccines have shown to be an effective treatment for SPs in animal models. Here, we report on the development of adoptive cellular therapy (ACT) for SP and demonstrate that adoptive transfer of pre-activated T-cells generated from immunized mice can improve survival and behavior, reduce brain microstructural impairment via magnetic resonance imaging (MRI), and decrease α-synuclein pathology burden in a peripherally induced preclinical SP model (M83) when administered prior to disease onset. This study provides preclinical evidence for ACT as a potential immunotherapy for LBD, PD and other related SPs, and future work will provide necessary understanding of the mechanisms of its action.
Keywords: Parkinson’s disease, gut−brain axis, adoptive cellular therapy, alpha synuclein, diffusion MRI, gait analysis
Introduction
Synucleinopathies (SPs) including Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), dementia with Lewy bodies (DLB), and multiple systems atrophy (MSA) are neurodegenerative diseases characterized by accumulation of alpha-synuclein (α-syn) in the brain. Current treatment options provide symptomatic relief but do not modify underlying pathology, representing an area of unmet medical need.1 Deposition of aggregated α-syn protein into Lewy bodies is a central neuropathological feature of SPs including PD.2−4 The pivotal discovery that certain familial forms of PD are caused by mutations in the α-syn gene (SNCA1) suggests its importance in pathophysiology.5 The physiological role of α-syn is not fully understood, yet it is involved in immune cell activation in the enteric nervous system,6,7 and its presynaptic localization suggests its role in the release of neurotransmitters and in regulating exo- and endocytosis.8,9 α-syn is predominantly an intracellular protein, yet studies have demonstrated that extracellular α-syn also plays a role in disease and is involved in neutralizing toxic proteins released from cells, preventing their propagation to neighboring cells.10 Intercellular transfer of prion-like aggregates comprising amyloid-type proteins can contribute to the pathological spread of misfolded proteins in neurodegenerative disease.11 These “infectious” aggregates can propagate from one cell to another and initiate aggregation once in the recipient cell.11 Evidence for cell-to-cell spread of α-syn in PD came from post-mortem studies of patients who had undergone embryonic neuronal transplantation.12,13 In vivo experiments by Desplats et al. showed that α-syn was transferred from rodent hosts to transplanted embryonic neurons in wild-type recipient mice.14 Given that α-syn aggregates play a central role in the processes leading to neurodegeneration, the reduction of α-syn deposition and oligomerization could have disease modifying effects.
The identification of natural antibodies in PD patients against α-syn15−17 indicates the role of the immune system in clearing pathogenic α-syn. Yet very few immunotherapies have been developed for proteinopathies to date, with autoimmunity presenting a huge challenge.18−21 Active in clinical trials are UB-312 and ABBV-0805. UB-312 (Vaxxinity) is a synthetic peptide vaccine, which elicits an enhanced B-cell response, avoiding harmful pro-inflammatory T-cell responses.22,23 ABBV-0805 (AbbVie) is a humanized monoclonal antibody whose efficacy was demonstrated in the murine intracerebral α-syn preformed fibril (PFF) injection model, where a significant reduction in the burden of pathology was noted at 16 weeks post PFF injection.24 Also ongoing are trials of PRX002, BIIB054, MEDI1341, PD03A, and PD01A/PD03A, all showing good safety profiles in patients with PD.21 Recent results from two Phase 2 trials (SPARK25 and PASADENA26) of monoclonal antibodies showed no efficacy with respect to imaging measures when compared to control group even though there was a suggestion that low-dose prasinezumab improved the MDS-UPDRS part III score. These negative results reinforce the need for further research into different modalities of immunotherapies since a targeted approach in genetic forms of the disorder might be crucial to their success.
There is a critical need for non-invasive biomarkers for SPs that can characterize structural and functional properties of the brain and the effects of therapeutic interventions. Diffusion MRI (dMRI) has been widely used in neurological studies for in vivo structural imaging. Fractional anisotropy (FA) is the primary diffusion metric used to characterize white matter tracts27,28 where it quantifies axonal density and myelination.29 FA is useful in characterizing gray matter areas30−32 where it is related to the number of dopaminergic cells in the SN,31 with studies showing reduced FA in PD.30,33
A subset of PD patients has T-cell responses to specific α-syn epitopes, suggesting that PD has some autoimmune features34−36 and the transfer of T-cells engineered to recognize and target particular forms of α-syn could become a way to reduce pathology burden.37 Alterations in the adaptive immune system likely promotes PD and immunotherapies targeting dysregulated T-cells could be beneficial. Here, we report on the development of adoptive cellular therapy (ACT) and investigate its efficacy in ameliorating disease progression in the peripheral injection model of α-synucleinopathy using in vivo neuroimaging.
Results and Discussion
Development of an Antigen-Specific T-Cell-Based Therapy
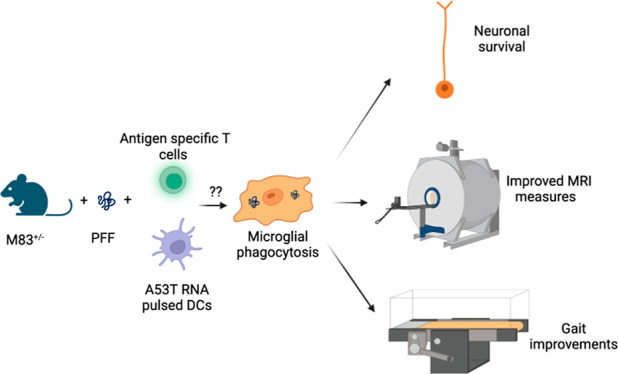
An overview of the study is shown in Figure 1.
Figure 1.

Schematic representation of immunization and testing scheme. Three groups of M83+/– Tg mice were examined in this study: mice injected with PFFs (disease group; PFF), PFFs and ACT (therapy group; PFF + ACT), and PBS (healthy control group; PBS). A gait assessment (Digigait) and MRI scans were performed prior to intramuscular (IM) seeding and at the end of the 12 week study period. Immunohistochemistry (IHC) was performed at 12 weeks post-seeding.
ACT is a personalized therapy, which involves in vitro isolation and expansion of antigen-specific T-cells to provide a “boost” to the host’s immunity.53 Here, dendritic cells were pulsed with α-syn (A53T) RNA to generate DC vaccine, and A53T α-syn-specific T-cells (CD4+, CD8+) were generated ex vivo for adoptive transfer into M83 mice expressing this α-syn mutation. To promote engraftment and persistence of T-cells, A53T α-syn-specific DC vaccine followed the adoptive T-cell transfer. To determine in vitro efficacy and ensure sufficiently potent T-cell responses were generated by this method, we conducted experiments where the α-syn-specific T-cells were co-cultured overnight with either A53T α-syn expressing target cells (DC2.4 A53T) or GFP expressing target cells (DC2.4 GFP). The supernatant was collected and analyzed for cytokine interferon gamma (IFNγ) (Figure 2). IFNγ was significantly increased when A53T α-syn-specific T-cells were cultured against A53T α-syn expressing target cells. This demonstrates the feasibility of generating α-syn-specific T-cells and that this technology is capable of generating strong immune responses against the antigen target. It has recently been postulated that one of the mechanisms for the reduction in phosphorylated α-syn pathology in mice that received adoptive transfer of T-cells could be via activation of resting microglia to become active via the presence of T-cells releasing IFNγ and enhancing phagocytosis of PFFs.37
Figure 2.

T-cells secrete IFNγ in vitro upon exposure to cognate antigen. Bone marrow-derived dendritic cells (DC)s were electroporated with A53T α-syn RNA (DC2.4A53T) and used to activate and expand A53T α-syn-specific T-cells in vitro (T-cells + DC2.4A53T). Upon recognition of cognate antigen, A53T α-syn-specific T-cells secrete IFNγ, present in low amounts when A53T α-syn-specific T-cells were co-cultured against cells that did not express A53T α-syn, (DC2.4 GFP). p value = 0.0003. One-way ANOVA statistical significance was determined at p value < 0.05 (*** = p < 0.005). p-values were corrected for multiple comparisons using the false discovery rate (FDR) method at p < 0.05.46
Feasibility of Delivering Adoptive T-Cells in an Animal Model of α-Synucleinopathy and Survival Effect Following Adoptive T-Cell Transfer
The A53T familial mutation in α-syn results in early onset PD (∼45 years of age).54,55 Homozygous (M83+/+) M83 mice expressing human A53T α-syn display a dramatic and robust phenotype, which results in their death coinciding with α-syn aggregation and subsequent neuronal dysfunction (at around 8–10 months), hence making this an excellent transgenic (Tg) model of α-synucleinopathies.56 Furthermore, these α-syn inclusions recapitulate the typical characteristics of human inclusions, with significant α-syn aggregation in neuronal cell bodies as well as processes. These pathologies can also be observed in hemizygous M83+/– mice but after 20 months of age.56 In an effort to rapidly and synchronously induce a motor phenotype and α-syn pathology, PFFs injected in the periphery (gastrocnemius muscle) result in prion-type seeding followed by neuroinvasion and spreading into CNS inducing severe motor dysfunction (paralysis).57 This animal model of α-synucleinopathy is a valuable tool as disease onset in heterozygous M83+/– mice can be predictable, shortened considerably, and synchronized. Efficient and rapid induction of α-syn pathology in the CNS induced by peripheral injections of PFFs to induce rapid, lethal motor phenotype (50–120 d) has been described by the Giasson group.57,58 Mice injected with PFFs in this manner developed α-syn inclusion pathology seen in aged (>8 mo old), homozygous M83 mice. We employed peripheral injections in the heterozygous M83 mouse model as published57,58 to test the feasibility and efficacy of the ACT platform. Mice that were treated adoptively survived significantly longer (141 ± 9 days post-seeding, P < 0.05) compared to PFF-injected mice (Figure 3). PFF-seeded mice had a survival time (paralysis) of 117 ± 6 days post-seeding, and all mice in the PBS (healthy control) group survived past 170 days post-injection.42 In our hands, we observed no significant differences in survival between males and females.
Figure 3.
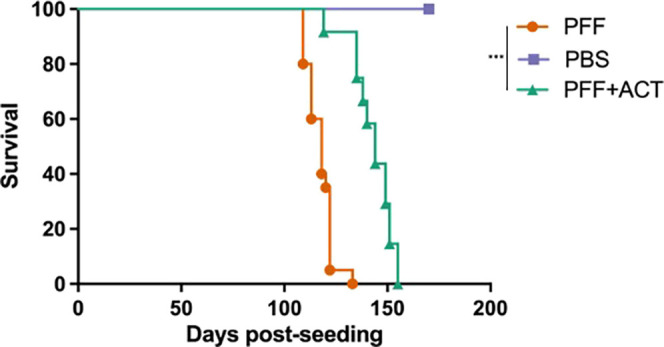
ACT treatment prolongs survival in a mouse model of synucleinopathy. Overall survival was plotted to compare time to terminal state between the three groups of mice. ACT treatment shows increased time to terminal state (paralysis) for M83+/– Tg PFF seeded mice, compared to M83+/– Tg PFF only mice. Neither group survived as long as M83+/– Tg PFF unseeded (PBS only) mice.42 Mice were monitored for morbidity endpoints as approved by UF IACUC, and survival data were collected on these mice (PFF: n = 20, PFF + ACT: n = 12, and PBS: n = 20). Survival analysis was performed using log-rank (Mantel Cox) test. Statistical significance was determined at p value < 0.05 (*** = p < 0.005).
Moderate Improvement in Gait Performance Post ACT
There is very little in the literature describing quantitative measures of ambulatory gait in M83 mice with PFFs. Paumier et al. describe gait behavior in homozygous M83 mice that develop gait abnormalities with age and demonstrate a significant decrease in hind limb stride length and frequency at 12 months compared to wild-type littermates.59 Pre-seeding, animals in all groups showed no significant differences in gait metrics. At 12 weeks post-seeding, we observed no significant differences between swing, stance, or paw area variability in any of the groups. We noted that stride length was significantly different between ACT-treated and untreated male mice (did not reach significance in female mice), whereas there was no significant difference between PBS and PFF + ACT mice at this time-point (Figure 4). Stride length is defined as the spatial length that a paw traverses through a given stride and should normally be equal among the four paws of a given mouse. It has been noted that ethanol exposure results in significantly altered stride length,60 as does arthritis.61 There have been several studies to date in PD patients aimed at detecting gait alterations using quantitative technology-based assessments62,63 and gait impairment in PD evolves throughout the course of the disease and is detectable in early, moderate, as well as advanced disease states.64 Abnormalities in gait pattern in patients with PD have been characterized by a reduction in gait velocity and stride length.65 In our previous study,42 we assessed differences in locomotion using the rotorod at 12 weeks post-PFF seeding. Digigait is a more sensitive mode of detection of gait abnormality than the rotorod; however, the Digigait data from our study suggest that there were not many differences in gait in the ACT v PFF mice before and at 12 weeks post-PFF seeding. This is in line with our previous study where we were unable to discern significant differences in locomotion using the rotorod at 12 weeks. One of the likely explanations could be that severe motor functions in our mouse model probably do not manifest until after 12 weeks post-PFF seeding. It will be valuable in future studies to assess locomotion in PFF-seeded animals at another timepoint after 12 weeks prior to onset of severe paresis.
Figure 4.
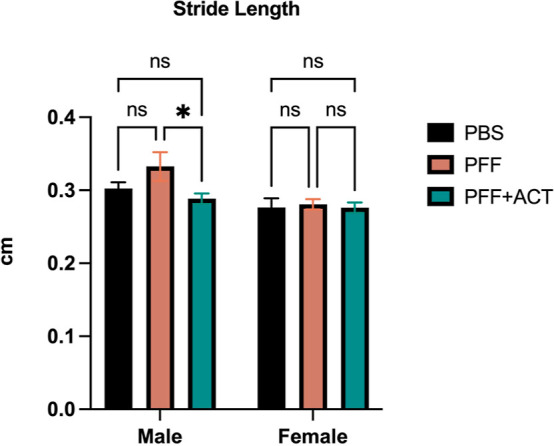
Digigait assessment. Digigait testing provides quantitative metrics of gait to assess changes in strength, balance, and coordination. Animals in all groups were tested at baseline, and 12 weeks post PFF seeding. Data were analyzed using 2-way ANOVA. All p-values were corrected for multiple comparisons using the FDR method at p < 0.05.46 Graphs show that stride length is significantly altered between PFF and PFF + ACT male mice only at 12 weeks post seeding. Gait measures were collected on mice at pre-seeding and 12 weeks post-seeding (PFF: n = 20, PFF + ACT: n = 12, and PBS: n = 20).
ACT Reduces Pathology Burden
Data from IHC using antibody 81A, specific for Ser129 phosphorylated α-syn showed a decrease in pathology burden in the PFF + ACT-treated group compared to the PFF-injected group in the pons, motor cortex, and medulla regions (Figure 5A). IHC using 2H6 (an antibody developed to target an epitope in residues 2–21) and 94-3A10 (an antibody developed to target an epitope in residues 130–140) showed decreased pathology burden in the PFF + ACT-treated group compared to the PFF untreated group (Figure 5B,C). The α-syn inclusion pathology in untreated PFF mice was robust in the brainstem and midbrain structures, aligning with previously published work.57 The PFF + ACT-treated mice showed a significant reduction in pathology compared to untreated PFF mice.
Figure 5.

Assessment of α-syn pathology. Immunohistochemistry using (A) 81A, (B) 2H6, and (C) 94-3A10 antibodies in the pons, motor cortex, and midbrain of PBS (n = 3), PFFs (n = 3), and PFF + ACT (n = 3) mice at 12 weeks post-seeding. For each marker studied, the percentage area of immunoreactivity over two consecutive sections was performed using Image J software. 20x images were first converted to 8 bit grayscale and a threshold was set for each section using the triangle setting, following which images were converted to a black background. The fraction of each area (pons, thalamus, motor cortex) that was positive was recorded and expressed as a percentage. All values are expressed as ± SEM. Differences in means between the groups were analyzed using a Student’s t-test (GraphPad Prism software, v9). Regions with significant between-group differences are designated with asterisk(s). All p-values were corrected for the false discovery rate (* = pfdr<0.05, ** = pfdr<0.01, *** = pfdr<0.005). Student’s t-test between groups shows a reduced burden of synuclein pathology in the ACT treatment group when compared to the PFF-only group.
We next characterized immune alterations using glial fibrillary acidic protein (GFAP) and Iba1 markers. We detected elevated astrogliosis at 12 weeks post-seeding in the caudal regions of the brain of the untreated PFF group compared to the PBS group, aligning with previously published work.66 The PFF + ACT-treated group showed a significantly reduced amount of astrogliosis when compared to the untreated PFF group (Figure 6A). We next explored microglial activation using Iba1. Microglial activity mirrored that of astrocytosis, with elevated activation at 12 weeks in the PFF group compared to the PBS group. The burden of microglia was reduced in the ACT + PFF group compared to the PFF group in the cortex and pons regions (Figure 6B). However, to fully comprehend whether microglia play a role in the reduction of the observed α-syn pathology, future work will require morphometric assessments of Iba1 reactive microglia.
Figure 6.

Assessment of astrogliosis. Immunohistochemistry using (A) GFAP and (B) Iba1 antibodies in the pons, motor cortex, and midbrain of PBS (n = 3), PFF (n = 3), and PFF + ACT (n = 3) mice at 12 weeks post-seeding. All values are expressed as ± SEM. Differences in means between the groups were analyzed using a Student’s t-test (GraphPad Prism software, v9). Regions with significant between-group differences are designated with asterisk(s). All p-values were corrected for the false discovery rate (* = pfdr<0.05, ** = pfdr<0.01, *** = pfdr<0.005). Student’s t-test between groups shows a reduced burden of astrogliosis in the ACT treatment group when compared to the PFF-only group.
ACT Induces Microstructural Brain Changes
Using the seeding model of α-synucleinopathy, we have previously examined progressive microstructural degeneration and functional activation of brain circuits subsequent to the advancing of α-syn pathology along the neuroaxis using MRI tools.42 In this study, dMRI was employed to investigate the temporal progression of microstructural changes in the peripheral injection α-syn mouse model. Results from this previous study revealed that there are transient microstructural and functional activity brain changes at 4 weeks post-seeding of PFFs. Furthermore, at 12 weeks post-seeding, we observed microstructural deficits in the pons and thalamus that were consistent with a previous work.66 Here, mice seeded with PFFs and mice that were seeded with PFFs and treated with ACT were imaged pre-seeding, and at 12 weeks post-seeding. FA was calculated from diffusion images within relevant regions of interest that spanned the large majority of the brain (Figure 7A). Pre-seeding, no significant differences between the PFF + ACT, PFF, and PBS groups were found (Figure 7B). At 12 weeks post-seeding, ACT-treated mice had higher FA compared to untreated PFF-injected mice in the pons and thalamus (Figure 7C). FA is a robust measure of brain tissue microstructure and lower FA has been reported in humans with PD,30,67,68 LBD,69−71 and MSA72,73 compared to healthy controls.
Figure 7.

Diffusion Tensor Imaging. FA was quantified in nine regions of interest throughout the brain in mice from three experimental groups: PFF + ACT (treatment group; n = 12), PFF (disease group; n = 20), and PBS (healthy control group; n = 20). PFF and PBS data were reproduced from a previous publication42 for comparison to the PFF + ACT group. Independent samples t-tests were used to compare between the three groups. Regions with significant between-group differences are designated with an asterisk. All p-values were corrected for the false discovery rate (* = pfdr<0.05). (A) Regions of interest: cerebellum (CER; orange), vermis (VER; yellow), posterior medulla (pMED; light green), and anterior medulla (aMED; dark green), pons (PON; pink), midbrain (MID; light blue), thalamus (THA; dark blue), somatosensory cortex (SS; purple), and striatum (STR; red) (top). (B) Pre-seeding, there were no significant differences between the three groups. (C) 12 weeks post-seeding, the PFF + ACT and PBS mice had higher FA compared to untreated PFF mice in the pons and thalamus.
A major driver of pathogenesis in LBD, PD, and related α-synucleinopathies are fibrillar α-syn aggregates, thought to propagate throughout the brain in a prion-like fashion.74−76 Hence, it is crucial to develop therapies that specifically target pathogenic α-syn and block its intracellular propagation. To address this, we established an adoptive T-cell-based platform that in a preclinical setting reduces the accumulation of pathological α-syn aggregates, improves survival, restores brain microstructure, and thus holds promise as a potential therapeutic strategy against SPs.
Several studies have investigated the effects of immunotherapies, including active vaccination strategies and passive administration of antibodies in rodents.81−85 Results from these preclinical studies clearly demonstrate the potential for immunotherapies to reduce α-syn pathological burden in transgenic models of α-synucleinopathies.
Recently, it has been reported that α-syn fibril-specific antibodies can reduce α-syn pathology.77 However, antibodies are typically poor at membrane penetration and do not function optimally in a reducing cytosolic environment.78 Therefore, they are typically capable of only targeting extracellular α-syn fibrils,79−81 limiting their impact on intracellular α-syn aggregates. Recently, results of two Phase II trials of monoclonal antibodies for the treatment of PD were reported in the New England Journal of Medicine.25,26 The investigations using Prasinezumab were reported by Pagano et al. [PASADENA]26 and using Cinpanemab were reported by Lang et al. (SPARK).25 They were both double-blind trials and used the change in the sum of MDS-UPDRS86 scores on parts I, II, and III at 52 weeks as the primary endpoint. Subsequently, each study entered a blinded phase, where all participants received active therapy. Secondary endpoints were motor and nonmotor MDS-UPDRS subscale scores and neuroimaging (DaT-SPECT). Both trials failed to show benefit with respect to either primary or secondary endpoints, yet were relatively safe, with no concerns of immunogenicity. Antibodies are notorious for poor penetrance through the blood–brain barrier (BBB) as has been shown extensively in the field of brain tumors such as glioblastoma. The recent Phase 2 trials25,26 that were unable to clarify binding of the antibodies to the α-syn targets are only further proof of this point and demonstrate the need for developing novel immunotherapeutic modalities for targeting α-syn that could still be beneficial if delivered in prodromal stages or genetic forms of PD. We believe our work provides a proof-of-concept that adoptive transfer of antigen-specific T-cells exploits a highly effective natural mechanism for induction and maintenance of peripheral tolerance, that can effectively reduce pathological α-syn propagation.
Several years ago, an antigen-loaded DC-based vaccine was tested against a preclinical model of AD.87 This approach resulted in a long-lasting antibody response without any significant inflammation. A similar approach was tested in a preclinical model of PD targeting α-syn. Results from the study suggest that dendritic cells sensitized with α-syn are capable of triggering antibody formation.88 However, a targeted T-cell-based approach has not yet been sufficiently explored in transgenic models of α-synucleinopathies. Based on their ability to induce both antibodies as well as T-cell responses, nucleic acids encoding antigen targets can be utilized as immunotherapeutic platforms for transfection of antigen presenting cells (APCs).89 These immune responses are lower in DNA vaccines likely due to a variety of reasons including reduced levels of DNA-sensing machinery.89 RNA vaccines are, however, a promising alternative as they only need cytoplasm for entry, cannot integrate into the genome, and are relatively simple to manufacture.90 Our study describes the development of an mRNA encoded vaccine antigen, amplified to generate copious amounts of A53T specific antigen, and delivered to our preclinical model after ex vivo priming of autologous dendritic cells. The role of extracellular α-syn in our model is thought to be prion-like; however, at this time we are unable to ascertain if the effect of ACT is via reduction of the spread of α-syn or if it functions by improving the degradation of intracellular α-syn aggregates. Previous immune-based studies have used a DC approach in the M83 homozygous model, wherein DCs sensitized to antigenic α-syn have been shown to be effective in generating specific antibody responses, with amelioration of locomotion indicating the feasibility and development of such adaptive cell-mediated immunotherapies for use in this model.88 Furthermore, it has been shown that reconstituting T-cell populations in immunocompromised mice reduces α-syn pathology burden resulting from striatal PFF injections, suggesting a key role that T-cells might play in modulating the accumulation of phosphorylated α-syn.37 Nevertheless, we are not aware of other T-cell-based therapies using M83 PFF models and hence direct data comparison is difficult. A protective vaccination strategy is thought to prevent or slow down pathology via inducing tolerance. We posit that in order to achieve this goal, it may be necessary to modulate the peripheral immune system such that it can recruit a long-lasting T-cell response and avoid neuroinflammation. It is possible that ACT reduces α-syn pathology via microglial activation91,92 and T-cells can stimulate microglia by the release of IFNγ (Figure 2), which are required for MHC II upregulation in microglia but is difficult to conclusively tell since microglial activation was ameliorated in the pons and not in the midbrain. This could be due to a variety of factors including the single time point of immunohistochemical assessment and the fact that microglia have distinct region-dependent molecular and morphological profiles; hence, further work is required to comprehend morphological changes in microglia over time post ACT treatment.
Our study also employed dMRI as an in vivo marker of disease progression in this model. The major findings provide evidence for brain microstructural changes with the disease and amelioration post ACT. Decreased FA has also been observed in animal models of PD.31,93 We have previously reported reduced FA in the pons and thalamus at 12 weeks post PFF seeding, which coincides with astrocytosis and microgliosis, which play key roles in neuroinflammation.94 Specifically, in a previously published study by our group, longitudinally comparing 20 PFF injected mice to 20 PBS-injected mice, we showed that the disease group had reduced FA compared to healthy controls at 4 weeks and 12 weeks post-seeding despite no observable deficits in rotarod performance.42 Furthermore, the regions in which we found reduced FA in PFF-injected mice compared to PBS controls at 12 weeks post-seeding (pons and thalamus) were the same regions in which we found reduced FA in PFF-injected mice compared to PFF + ACT-treated mice. Together, these findings suggest that the ACT treatment had a neuroprotective effect on the PFF-seeded mice, such that neurodegeneration is reduced in the ACT-treated mice compared to untreated PFF mice. The reduced neurodegeneration in the ACT-treated mice at 12 weeks post PFF seeding provides in vivo evidence that ACT delays pathological disease progression by showing that it delays the neurodegeneration that results from pathological disease progression. Although only a 12 and 14% increase in FA was observed, in the pons and thalamus, respectively, the difference was statistically significant and is indicative of a larger cumulative change that may be observable at later time points. Our data suggest that adoptive transfer of preactivated T-cells is a possible therapeutic modality for SPs, reduces brain microstructural impairment (dMRI), and prevents α-syn aggregates in the brain of M83 mice peripherally injected with PFFs.
In summary, we have demonstrated the feasibility of ACT to reduce pathology burden, preserve brain tissue microstructure, and improve overall survival in a preclinical model of a genetic form of PD. As a research tool, targeted T-cell therapy would enable the interrogation of peripheral immune functions in disease progression. ACT could be utilized to study further downstream effects such as metabolomics and microbiota and their effects on the alleviation of survival, behavioral, and cognitive deficits. Further investigation and development of ACT related approaches could inform on treatment approaches in LBD, PD, and related SPs.
Materials and Methods
Animals
All animals were maintained on a 12/12 h light/dark cycle with food and water ad libitum. Animals were acquired and cared for in accordance with the National Institutes of Health Guide for the Care and Use of Experimental Animals. All procedures were approved by the University of Florida Institutional Animal Care and Use Committee (UF IACUC). At around 8 weeks of age, α-syn Tg Ala53Thr hemizygous (M83+/–) mice received bilateral intramuscular (biceps femoris) injections of 10 μg of α-syn fibrils or PFFs (disease group) or 5 μL of sterile PBS (healthy control group). The treatment group received PFFs intramuscularly and ACT via tail vein injection. M83+/– Tg mice were maintained on a C57Bl/C3H background strain. Both groups contained equal numbers of males and females.
Experimental Design
Three groups of M83+/– Tg mice were examined in this study: mice intramuscularly injected with PFFs (disease group; PFF), mice injected with PFFs and ACT (therapy group; PFF + ACT), and genotype-matched mice intramuscularly injected with PBS (healthy control group; PBS). Figure 1 shows the experimental timeline. Gait measures and MRI scans were collected on mice at pre-seeding and 12 weeks post-seeding (PFF: n = 20, PFF + ACT: n = 12, and PBS: n = 20). The mice were monitored for morbidity endpoints as approved by UF IACUC, and survival data were collected on these mice. Mice for immunohistochemical analyses were sacrificed at 12 weeks post-seeding and the brains were collected (PBS: n = 3, PFF: n = 3, and PFF + ACT: n = 3).
MRI Equipment
Experiments were conducted on an 11.1 Tesla Magnex Scientific horizontal bore magnet (Agilent; BFG-240/120-S6 gradient system with 120 mm inner gradient bore size; maximum gradient strength 1000 mT/m and rise time of 200 μs) interfaced to a Bruker Avance III HD console and controlled by Paravision 6.01 software (Brucker BioSpin). All imaging was conducted at the AMRIS Facility at the McKnight Brain Institute, University of Florida. Images were collected using an in-house 2.5 × 3.5 cm quadrature surface transmit/receive coil affixed to the top of the skull and tuned to 470.7 MHz (H1 resonance) for B1 excitation and signal detection.
MRI Data Acquisition
Mice were anesthetized for the entire duration of the experiment. Isoflurane was delivered using compressed medical-grade air (70% N2/30% O2) using a Surgivet vaporizer, which was connected to a charcoal trap. Mice were induced at 3% isoflurane for 1–2 min in an enclosed induction chamber. Anesthesia was reduced to 2% for animal setup and then maintained between 1.0 and 1.5% for the duration of MRI acquisition.38,39 Mice were placed in the prone position on a custom-made 3D-printed plastic mouse bed. The mouse bed was equipped with a bite bar that immobilized the head and delivered anesthesia during the time of scanning. Respiratory vitals were monitored using a respiration pad. Body temperature was constantly monitored and maintained at 36–37 °C using a recirculating water heating system (SA Instruments).40
Diffusion MRI Scan Parameters
dMRI with echo-planar imaging distortion correction (dMRI-EPI) scans were acquired with the following parameters: repetition time (TR) = 4000 ms; echo time (TE) = 19.17 ms; slices = 17; coronal orientation; thickness = 0.7 mm; gap = 0 mm; FOV = 15 × 11 mm; data acquisition matrix = 128 × 96 in-plane; 2 b0 images; b-values = 600, 2000 s/mm2; and directions = 52 total (6 at b-value 600; 46 at b-value 2000). The time between gradient pulses: 8 ms; diffusion gradient duration: 3 ms.
MRI Data Analysis
The dMRI data analyses were performed using custom-designed UNIX shell scripts. Linear and nonlinear registration between images were performed using the Advanced Normalization Tools (ANTs) package.41 The dMRI pre-processing was performed using previously described methods and a rodent-modified diffusion analysis pipeline.42−44 FMRIB Software Library (FSL) was used to correct for distortions due to eddy currents and head motion using affine transformations. Gradient directions were rotated in response to eddy current corrections and non-brain tissue was removed. FA maps were calculated from pre-processed images.45 To standardize data, FA images from each mouse were nonlinearly registered to a template FA image created from a prior work.42 FA maps were used as they provide good contrast between white matter, gray matter, and CSF. The diffusion scans had a high in-plane resolution (117 × 115 μm) with a relatively large slice thickness (700 μm). Thus, to prevent interslice warping, each slice was independently and nonlinearly registered to the matching template image slice. We used regions of interest (ROIs) in template-space that were created in the prior work.42 The cerebellum, vermis, medulla oblongata, pons, midbrain, thalamus, cortex, and striatum were examined in this study. ROIs were applied to the FA maps and then compared between groups. Independent sample t-tests were used to compare between the PFF + ACT, PFF, and PBS groups. All p-values were corrected for multiple comparisons using the FDR method at p < 0.05.46
DigiGait Data Acquisition
Ambulation of the mice was characterized and quantified using the DigiGait Imaging System (Mouse Specifics, Inc). DigiGait employs Ventral Plane Imaging (VPI) Technology, a high-speed, 147 frames-per-second video camera mounted inside a stainless steel treadmill chassis below a transparent treadmill belt to capture ventral images of the subject to continuously image the underside of walking animals to generate “digital paw prints” and dynamic gait signals. This generates a temporal record of paw placement relative to the treadmill belt. Images obtained are automatically digitized by DigiGait Video Imaging Acquisition software (Mouse Specifics, Inc) and imported for analyses. The software defines the area of each paw and vectors associated with each paw generate a set of periodic waveforms used to describe the kinetics of the paws and can identify parts of the paw in contact with the treadmill belt in the stance phase of stride and track the foot through the swing phase of the stride. After a brief period of habituation, the mice were placed on the treadmill and accelerated to 20 cm/s for analysis. The total time of recording was approximately one min. Quantitative metrics of gait assess changes in strength, balance, and coordination, to compare ambulation in treated versus untreated animal groups. Animals in all groups were tested at pre-seeding and 12 weeks post PFF seeding. Data were analyzed using 2-way ANOVA. P-values were corrected for multiple comparisons using the FDR method at p < 0.05.46 Graphs and statistical analysis were generated using Prism 9.
ACT
ACT vaccines were generated using the DC platform as previously described.47 In brief, α-syn RNA was in vitro transcribed. Autologous DCs were extracted following standard sterile procedures from bone marrow cells, electroporated with mutant α-syn RNA, and re-plated. Matured α-syn RNA–DC were collected and stimulated with murine T-cells. Two million T-cells per well in 24-well plates were co-cultured with appropriate numbers of α-syn RNA–DC in RPMI-1640 medium supplemented with cytokines. Flow cytometry was performed using FACSCalibur and FACSCanto (BD Biosciences). Cell viability was determined by a combination of forward angle light scatter and 7-AAD staining of dead cells. Cell surface expression of CD3, CD4, CD8, CD25, CD27, CD28, CD44, CD70, CD62L, and CD45RO were measured using fluorescent-conjugated antibodies (BD). IFNγ, IL-2, and FOXP3 levels were determined by intracellular staining. All FACS data were analyzed using FlowJo 8.1.1 software (FlowJo, Ashland, OR, USA). After 10 days of ex vivo expansion, M83 mice were injected with 0.5–1 × 106 α-syn RNA–DC intradermally (ID) and infused with 1 × 106 T-cells via tail vein injection. Mice in the treatment group were vaccinated 1 week after receiving intramuscular PFF injections. On days 7 and 14 after ACT, the mice were boosted twice with 0.5–1 × 106 α-syn RNA–DC ID.
Immunohistochemistry
To evaluate the neurodegenerative consequences of α-syn inclusion pathology formation and the effect of ACT treatment, at 12 weeks post PFF seeding the mice were anesthetized with an overdose of isoflurane, perfused with phosphate-buffered saline (PBS), followed by 70% ethanol/150 mM NaCl or PBS-buffered formalin.48 The brains were surgically removed, and tissue was fixed for 24 h in the respective fixative. Samples were then dehydrated through a stepwise series of graded ethanol solutions to xylene at room temperature, infiltrated with paraffin at 60 °C as previously described,49 and sectioned serially at 5 μm. Immunocytochemistry was conducted as previously described50,51 with anti-pSer129 α-syn antibody (81A),48,51 2H6, 94 3A10,52 GFAP (Abcam, USA), and Iba1 (Millipore, USA). Images were captured and processed on the Keyence System (BZ-X800E series). For each marker studied, the percentage area of immunoreactivity over two consecutive sections was performed using Image J software. PBS group N = 3, PFF group N = 3, PFF + ACT group N = 3.20× images were first converted to 8 bit grayscale, and a threshold was set for each section using the triangle setting, following which images were converted to a black background. The fraction of each area (pons, thalamus, cortex) that was positive was recorded and expressed as a percentage. All values are expressed as ± SEM. Differences in means between the groups were analyzed using a Student’s t-test (GraphPad Prism software, v9). All p-values were corrected for multiple comparisons using the FDR method at p < 0.05.46
Ethics Statement
All experimental protocols were approved by institutional guidelines from the University of Florida Institutional Animal Care and Use Committee.
Glossary
Abbreviations
- α-syn
alpha synuclein
- IFNγ
interferon gamma
- IHC
immunohistochemistry
- APC
antigen presenting cells
- ACT
adoptive cellular therapy
- DC
dendritic dells
- PD
Parkinson’s disease
- SP
synucleinopathy
- PDD
Parkinson’s disease dementia
- DLB
dementia with Lewy bodies
- MSA
multiple systems atrophy
- AD
Alzheimer’s disease
- IM
intramuscular
- PFF
preformed fibrils
- MRI
magnetic resonance imaging
- dMRI
diffusion magnetic resonance imaging
- DTI
diffusion tensor imaging
- FA
fractional anisotropy
Author Contributions
W.C.: Design and execution of MRI experiments, statistical analysis, and manuscript preparation. A.G.: Execution of project related to gait analyses and immunohistochemistry, data collection and analyses, and manuscript preparation. J.H.: Execution of project related to gait analyses and immunohistochemistry, data collection and analyses, and manuscript preparation. A.B.: Execution of project related to gait data collection and analyses. S.R.: Execution of project related to gait data collection and analyses. M.S.O.: Review and critique of manuscript. C.F.: Assistance with ACT experiments and review and critique of manuscript. B.G.: Review and critique of manuscript. D.V.: Design of MRI experiments, statistical analysis, and review and critique of manuscript. V.V.-M.: Project conception, organization, and execution, design and execution of ACT experiments, immunohistochemistry, data analysis, and manuscript preparation.
This study was supported by funds to VVM from NINDS (1R56NS112401-01), the Mangurian Foundation, UF Moonshot funds, UF Foundation funds, and by funds to DV from NIH (T32082168).
The authors declare no competing financial interest.
This paper was published ASAP on December 26, 2022. Additional text corrections were made on pages one and seven of the article, and the corrected version was reposted on December 28, 2022.
References
- Hutter-Saunders J. A.; Mosley R. L.; Gendelman H. E. Pathways towards an effective immunotherapy for Parkinson’s disease. Expert Rev Neurother 2011, 11, 1703–1715. 10.1586/ern.11.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J.; Sharma M.; Tsetsenis T.; Buchman V.; Etherton M. R.; Südhof T. C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G.; Schmidt M. L.; Lee V. M.; Trojanowski J. Q.; Jakes R.; Goedert M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Chadchankar H.; Ihalainen J.; Tanila H.; Yavich L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. 2011, 1382, 37–44. 10.1016/j.brainres.2011.01.064. [DOI] [PubMed] [Google Scholar]
- Houlden H.; Singleton A. B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol 2012, 124, 325–338. 10.1007/s00401-012-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie V.; Brundin P. Alpha-Synuclein to the Rescue: Immune Cell Recruitment by Alpha-Synuclein during Gastrointestinal Infection. J Innate Immun 2017, 9, 437–440. 10.1159/000479653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Yang D.; Li X. Q.; et al. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep 2022, 38, 110090. 10.1016/j.celrep.2021.110090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Wang B.; Li X.; Fu C.; Wang C.; Kang X. α-Synuclein: A Multifunctional Player in Exocytosis, Endocytosis, and Vesicle Recycling. Front Neurosci 2019, 13, 28. 10.3389/fnins.2019.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D.; Edwards R. H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. 10.1111/jnc.14810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E.; Elenis D.; Papasilekas T.; et al. Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PloS one 2011, 6, e22225 10.1371/journal.pone.0022225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M.; Zurzolo C. The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 2013, 452, 1–17. 10.1042/bj20121898. [DOI] [PubMed] [Google Scholar]
- Olanow C. W.; Prusiner S. B. Is Parkinson’s disease a prion disorder?. Proc Natl Acad Sci U S A 2009, 106, 12571–12572. 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P.; Barker R. A.; Parmar M. Neural grafting in Parkinson’s disease Problems and possibilities. Prog. Brain Res. 2010, 184, 265–294. 10.1016/s0079-6123(10)84014-2. [DOI] [PubMed] [Google Scholar]
- Desplats P.; Lee H. J.; Bae E. J.; et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13010–13015. 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Koudstaal W.; Fletcher L.; et al. Naturally occurring antibodies isolated from PD patients inhibit synuclein seeding in vitro and recognize Lewy pathology. Acta Neuropathol 2019, 137, 825–836. 10.1007/s00401-019-01974-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel S.; Gold M.; Deuschle C.; et al. Naturally occurring alpha-synuclein autoantibodies in Parkinson’s disease: sources of (error) variance in biomarker assays. PloS one 2014, 9, e114566 10.1371/journal.pone.0114566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood H. Naturally occurring antibodies target Parkinson disease pathology. Nat Rev Neurol 2019, 15, 186–187. 10.1038/s41582-019-0167-3. [DOI] [PubMed] [Google Scholar]
- Ha D.; Stone D. K.; Mosley R. L.; Gendelman H. E. Immunization strategies for Parkinson’s disease. Parkinsonism Relat Disord 2012, 18, S218–S221. 10.1016/s1353-8020(11)70067-0. [DOI] [PubMed] [Google Scholar]
- Schneeberger A.; Mandler M.; Mattner F.; Schmidt W. AFFITOME(R) technology in neurodegenerative diseases: the doubling advantage. Hum Vaccin 2010, 6, 948–952. 10.4161/hv.6.11.13217. [DOI] [PubMed] [Google Scholar]
- Nimmo J. T.; Kelly L.; Verma A.; Carare R. O.; Nicoll J. A. R.; Dodart J. C. Amyloid-beta and alpha-Synuclein Immunotherapy: From Experimental Studies to Clinical Trials. Front Neurosci 2021, 15, 733857. 10.3389/fnins.2021.733857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.; Kim H. J.; Jeon B. Immunotherapy Targeting Neurodegenerative Proteinopathies: alpha-Synucleinopathies and Tauopathies. J. Mov. Disord. 2020, 13, 11–19. 10.14802/jmd.19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Y.; Finstad C. L.; Walfield A. M.; et al. Site-specific UBITh amyloid-beta vaccine for immunotherapy of Alzheimer’s disease. Vaccine 2007, 25, 3041–3052. 10.1016/j.vaccine.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Wang C. Y.; Wang P. N.; Chiu M. J.; et al. UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (N Y) 2017, 3, 262–272. 10.1016/j.trci.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström E.; Eriksson F.; Sigvardson J.; et al. ABBV-0805, a novel antibody selective for soluble aggregated alpha-synuclein, prolongs lifespan and prevents buildup of alpha-synuclein pathology in mouse models of Parkinson’s disease. Neurobiol. Dis. 2021, 161, 105543. 10.1016/j.nbd.2021.105543. [DOI] [PubMed] [Google Scholar]
- Lang A. E.; Siderowf A. D.; Macklin E. A.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 408–420. 10.1056/nejmoa2203395. [DOI] [PubMed] [Google Scholar]
- Pagano G.; Taylor K. I.; Anzures-Cabrera J.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. 10.1056/nejmoa2202867. [DOI] [PubMed] [Google Scholar]
- Kraus M. F.; Susmaras T.; Caughlin B. P.; Walker C. J.; Sweeney J. A.; Little D. M. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain 2007, 130, 2508–2519. 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- Mori S.; Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron 2006, 51, 527–539. 10.1016/j.neuron.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Song S. K.; Sun S. W.; Ramsbottom M. J.; Chang C.; Russell J.; Cross A. H. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage 2002, 17, 1429–1436. 10.1006/nimg.2002.1267. [DOI] [PubMed] [Google Scholar]
- Vaillancourt D. E.; Spraker M. B.; Prodoehl J.; et al. High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology 2009, 72, 1378–1384. 10.1212/01.wnl.0000340982.01727.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boska M. D.; Hasan K. M.; Kibuule D.; et al. Quantitative diffusion tensor imaging detects dopaminergic neuronal degeneration in a murine model of Parkinson’s disease. Neurobiology of disease 2007, 26, 590–596. 10.1016/j.nbd.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A.; Adalsteinsson E.; Rohlfing T.; Sullivan E. V. Diffusion tensor imaging of deep gray matter brain structures: effects of age and iron concentration. Neurobiology of aging 2010, 31, 482–493. 10.1016/j.neurobiolaging.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan W.; Kang G. A.; Glass G. A.; et al. Regional alterations of brain microstructure in Parkinson’s disease using diffusion tensor imaging. Mov Disord 2012, 27, 90–97. 10.1002/mds.23917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garretti F.; Agalliu D.; Lindestam Arlehamn C. S.; Sette A.; Sulzer D. Autoimmunity in Parkinson’s Disease: The Role of alpha-Synuclein-Specific T Cells. Front Immunol 2019, 10, 303. 10.3389/fimmu.2019.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindestam Arlehamn C. S.; Dhanwani R.; Pham J.; et al. alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. 10.1038/s41467-020-15626-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D.; Alcalay R. N.; Garretti F.; et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. 10.1038/nature22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S.; Tyson T.; Rey N. L.; et al. T Cells Limit Accumulation of Aggregate Pathology Following Intrastriatal Injection of alpha-Synuclein Fibrils. J. Parkinson’s Dis. 2021, 11, 585–603. 10.3233/jpd-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron J. F.; Kroeger D.; Chever O.; Amzica F. Cortical inhibition during burst suppression induced with isoflurane anesthesia. J. Hist. Neurosci. 2009, 29, 9850–9860. 10.1523/jneurosci.5176-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Zhu X. H.; Zhang Y.; Chen W. The change of functional connectivity specificity in rats under various anesthesia levels and its neural origin. Brain topography 2013, 26, 363–377. 10.1007/s10548-012-0267-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann H. M.; Hentschel J.; Marek J.; et al. Normothermic Mouse Functional MRI of Acute Focal Thermostimulation for Probing Nociception. Scientific reports 2016, 6, 17230. 10.1038/srep17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants B. B.; Epstein C. L.; Grossman M.; Gee J. C. Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal 2008, 12, 26–41. 10.1016/j.media.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W. T.; DeSimone J. C.; Riffe C. J.; et al. α-Synuclein Induces Progressive Changes in Brain Microstructure and Sensory-Evoked Brain Function That Precedes Locomotor Decline. J. Neurosci. 2020, 40, 6649–6659. 10.1523/jneurosci.0189-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofori E.; Pasternak O.; Planetta P. J.; et al. Longitudinal changes in free-water within the substantia nigra of Parkinson’s disease. Brain 2015, 138, 2322–2331. 10.1093/brain/awv136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSimone J. C.; Febo M.; Shukla P.; et al. In vivo imaging reveals impaired connectivity across cortical and subcortical networks in a mouse model of DYT1 dystonia. Neurobiol. Dis. 2016, 95, 35–45. 10.1016/j.nbd.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basser P. J.; Mattiello J.; LeBihan D. MR diffusion tensor spectroscopy and imaging. Biophys. J. 1994, 66, 259–267. 10.1016/s0006-3495(94)80775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 1995, 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Flores C.; Pham C.; Snyder D.; et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 2015, 4, e994374 10.4161/2162402x.2014.994374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman E. A.; Duda J. E.; Giasson B. I. Characterization of antibodies that selectively detect alpha-synuclein in pathological inclusions. Acta Neuropathol 2008, 116, 37–46. 10.1007/s00401-008-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski J. Q.; Schuck T.; Schmidt M. L.; Lee V. M. Distribution of tau proteins in the normal human central and peripheral nervous system. J. Histochem. Cytochem. 1989, 37, 209–215. 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- Duda J. E.; Giasson B. I.; Gur T. L.; et al. Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol 2000, 59, 830–841. 10.1093/jnen/59.9.830. [DOI] [PubMed] [Google Scholar]
- Duda J. E.; Giasson B. I.; Mabon M. E.; Lee V. M.; Trojanowski J. Q. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 2002, 52, 205–210. 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Dhillon J. S.; Riffe C.; Moore B. D.; et al. A novel panel of alpha-synuclein antibodies reveal distinctive staining profiles in synucleinopathies. PloS one 2017, 12, e0184731 10.1371/journal.pone.0184731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González H.; Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation 2014, 11, 201. 10.1186/s12974-014-0201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbe L. I.; Lazzarini G.; Duvoisin G.; et al. Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol 1996, 40, 767–775. 10.1002/ana.410400513. [DOI] [PubMed] [Google Scholar]
- Spira P. J.; Sharpe D. M.; Halliday G.; Cavanagh J.; Nicholson G. A. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neuro 2001, 49, 313–319. 10.1002/ana.67. [DOI] [PubMed] [Google Scholar]
- Giasson B. I.; Duda J. E.; Quinn S. M.; Zhang B.; Trojanowski J. Q.; Lee V. M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Sacino A. N.; Brooks M.; Thomas M. A.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10732–10737. 10.1073/pnas.1321785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers J. I.; Brooks M. M.; Rutherford N. J.; et al. Robust Central Nervous System Pathology in Transgenic Mice following Peripheral Injection of alpha-Synuclein Fibrils. J Viroly 2017, 91. 10.1128/JVI.02095-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumier K. L.; Sukoff Rizzo S. J.; Berger Z.; et al. Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PloS one 2013, 8, e70274 10.1371/journal.pone.0070274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley E. P.; Barron S. The behavioral and neuroanatomical effects of prenatal alcohol exposure in animals. Ann N Y Acad Sci 1989, 562, 173–177. 10.1111/j.1749-6632.1989.tb21015.x. [DOI] [PubMed] [Google Scholar]
- Williams J. M.; Zurawski J.; Mikecz K.; Glant T. T. Functional assessment of joint use in experimental inflammatory murine arthritis. J. Orthop. Res. 1993, 11, 172–180. 10.1002/jor.1100110204. [DOI] [PubMed] [Google Scholar]
- Mirelman A.; Bonato P.; Camicioli R.; et al. Gait impairments in Parkinson’s disease. Lancet Neurol 2019, 18, 697–708. 10.1016/s1474-4422(19)30044-4. [DOI] [PubMed] [Google Scholar]
- Pistacchi M.; Gioulis M.; Sanson F.; et al. Gait analysis and clinical correlations in early Parkinson’s disease. Funct Neurol 2017, 32, 28–34. 10.11138/fneur/2017.32.1.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. R.; Lee Y. Y.; Cheng S. J.; Lin P. Y.; Wang R. Y. Relationships between gait and dynamic balance in early Parkinson’s disease. Gait Posture 2008, 27, 611–615. 10.1016/j.gaitpost.2007.08.003. [DOI] [PubMed] [Google Scholar]
- O’Sullivan J. D.; Said C. M.; Dillon L. C.; Hoffman M.; Hughes A. J. Gait analysis in patients with Parkinson’s disease and motor fluctuations: influence of levodopa and comparison with other measures of motor function. Mov Disord 1998, 13, 900–906. 10.1002/mds.870130607. [DOI] [PubMed] [Google Scholar]
- Sorrentino Z. A.; Xia Y.; Funk C.; et al. Motor neuron loss and neuroinflammation in a model of alpha-synuclein-induced neurodegeneration. Neurobiol. Dis. 2018, 120, 98–106. 10.1016/j.nbd.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péran P.; Cherubini A.; Assogna F.; et al. Magnetic resonance imaging markers of Parkinson’s disease nigrostriatal signature. Brain 2010, 133, 3423–3433. 10.1093/brain/awq212. [DOI] [PubMed] [Google Scholar]
- Du G.; Lewis M. M.; Styner M.; et al. Combined R2* and diffusion tensor imaging changes in the substantia nigra in Parkinson’s disease. Mov Disord 2011, 26, 1627–1632. 10.1002/mds.23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota M.; Obata T.; Akine Y.; et al. Age-related degeneration of corpus callosum measured with diffusion tensor imaging. Neuroimage 2006, 31, 1445–1452. 10.1016/j.neuroimage.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Watson R.; Blamire A. M.; Colloby S. J.; et al. Characterizing dementia with Lewy bodies by means of diffusion tensor imaging. Neurology 2012, 79, 906–914. 10.1212/wnl.0b013e318266fc51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K.; Avula R.; Senjem M. L.; et al. Dementia with Lewy bodies and Alzheimer disease: neurodegenerative patterns characterized by DTI. Neurology 2010, 74, 1814–1821. 10.1212/wnl.0b013e3181e0f7cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga K.; Yamada K.; Yoshikawa K.; Mizuno T.; Nishimura T.; Nakagawa M. Local tissue anisotropy decreases in cerebellopetal fibers and pyramidal tract in multiple system atrophy. J Neurol 2005, 252, 589–596. 10.1007/s00415-005-0708-0. [DOI] [PubMed] [Google Scholar]
- Wang P. S.; Wu H. M.; Lin C. P.; Soong B. W. Use of diffusion tensor imaging to identify similarities and differences between cerebellar and Parkinsonism forms of multiple system atrophy. Neuroradiology 2011, 53, 471–481. 10.1007/s00234-010-0757-7. [DOI] [PubMed] [Google Scholar]
- Li J. Y.; Englund E.; Holton J. L.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Li W.; Englund E.; Widner H.; et al. Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 6544–6549. 10.1073/pnas.1605245113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower J. H.; Chu Y.; Hauser R. A.; Freeman T. B.; Olanow C. W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Henderson M. X.; Covell D. J.; Chung C. H.; et al. Characterization of novel conformation-selective alpha-synuclein antibodies as potential immunotherapeutic agents for Parkinson’s disease. Neurobiol. Dis 2020, 136, 104712. 10.1016/j.nbd.2019.104712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slastnikova T. A.; Ulasov A. V.; Rosenkranz A. A.; Sobolev A. S. Targeted Intracellular Delivery of Antibodies: The State of the Art. Front Pharmacol 2018, 9, 1208. 10.3389/fphar.2018.01208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman B. T.; Phelps C. H.; Beach T. G.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012, 8, 1–13. 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae E. J.; Lee H. J.; Rockenstein E.; et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J. Hist. Neurosci. 2012, 32, 13454–13469. 10.1523/jneurosci.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D.; Valera E.; Spencer B.; et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. 10.1523/jneurosci.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield D. J.; Irving L.; Calo L.; et al. Preclinical development of a high affinity alpha-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular alpha-synuclein and attenuate alpha-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. 10.1016/j.nbd.2019.104582. [DOI] [PubMed] [Google Scholar]
- Weihofen A.; Liu Y.; Arndt J. W.; et al. Development of an aggregate-selective, human-derived alpha-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis 2019, 124, 276–288. 10.1016/j.nbd.2018.10.016. [DOI] [PubMed] [Google Scholar]
- Masliah E.; Rockenstein E.; Adame A.; et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Sanchez-Guajardo V.; Annibali A.; Jensen P. H.; Romero-Ramos M. alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol 2013, 72, 624–645. 10.1097/nen.0b013e31829768d2. [DOI] [PubMed] [Google Scholar]
- Goetz C. G.; Tilley B. C.; Shaftman S. R.; et al. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008, 23, 2129–2170. 10.1002/mds.22340. [DOI] [PubMed] [Google Scholar]
- Cao C.; Lin X.; Zhang C.; et al. Mutant amyloid-beta-sensitized dendritic cells as Alzheimer’s disease vaccine. J. Neuroimmunol 2008, 200, 1–10. 10.1016/j.jneuroim.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Ugen K. E.; Lin X.; Bai G.; et al. Evaluation of an alpha synuclein sensitized dendritic cell based vaccine in a transgenic mouse model of Parkinson disease. Hum Vaccin Immunother 2015, 11, 922–930. 10.1080/21645515.2015.1012033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer J. B.; Mason P. W.; Geall A.; Mandl C. W. RNA-based vaccines. Vaccine 2012, 30, 4414–4418. 10.1016/j.vaccine.2012.04.060. [DOI] [PubMed] [Google Scholar]
- Kreiter S.; Diken M.; Selmi A.; Türeci O.; Sahin U. Tumor vaccination using messenger RNA: prospects of a future therapy. Curr. Opin. Immunol. 2011, 23, 399–406. 10.1016/j.coi.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Schetters S. T. T.; Gomez-Nicola D.; Garcia-Vallejo J. J.; Van Kooyk Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front Immunol 2017, 8, 1905. 10.3389/fimmu.2017.01905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindestam Arlehamn C. S.; Garretti F.; Sulzer D.; Sette A. Roles for the adaptive immune system in Parkinson’s and Alzheimer’s diseases. Curr. Opin. Immunol. 2019, 59, 115–120. 10.1016/j.coi.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Camp N.; Blockx I.; Verhoye M.; et al. Diffusion tensor imaging in a rat model of Parkinson’s disease after lesioning of the nigrostriatal tract. NMR Biomed 2009, 22, 697–706. 10.1002/nbm.1381. [DOI] [PubMed] [Google Scholar]
- McGeer P. L.; McGeer E. G. Glial reactions in Parkinson’s disease. Mov Disord 2008, 23, 474–483. 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]