

Abstract
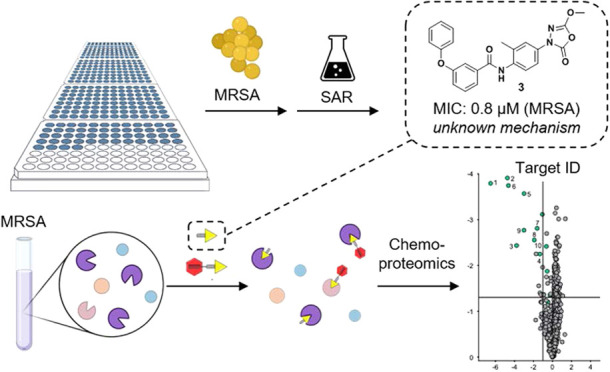
Phenotypic screening is a powerful approach to identify novel antibiotics, but elucidation of the targets responsible for the antimicrobial activity is often challenging in the case of compounds with a polypharmacological mode of action. Here, we show that activity-based protein profiling maps the target interaction landscape of a series of 1,3,4-oxadiazole-3-ones identified in a phenotypic screen to have high antibacterial potency against multidrug-resistant Staphylococcus aureus. In situ competitive and comparative chemical proteomics with a tailor-made activity-based probe, in combination with transposon and resistance studies, revealed several cysteine and serine hydrolases as relevant targets. Our data showcase oxadiazolones as a novel antibacterial chemotype with a polypharmacological mode of action, in which FabH, FphC, and AdhE play a central role.
Introduction
The emergence of multidrug-resistant bacteria combined with a dearth of new antibiotics poses a serious threat to global health.1−3 Among Gram-positive pathogens, methicillin-resistant Staphylococcus aureus (MRSA) remains the most worrisome. Recent data indicate that in 2019, drug-resistant staphylococcal infections, due predominantly to MRSA, were associated with a staggering 750,000 deaths worldwide.4 New antibiotics with unprecedented modes of action (MoAs) are urgently required to counteract antimicrobial drug resistance.
While target-based screens are commonly applied to identify small molecule hits in traditional drug discovery projects, such strategies are less successful in antibiotic discovery.5 Phenotypic screening has instead emerged as a promising approach to identify antibiotics with novel MoAs.6−8 However, for hits found by phenotypic screening, target elucidation often presents a challenge. Recently, chemical proteomics has emerged as a powerful chemical biology technique to map target interaction landscapes of experimental drugs,9−11 including compounds with antibacterial activity.12,13 Inspired by these approaches, we set out to combine phenotypic screening with chemical proteomics to discover new anti-MRSA antibiotics and their interacting proteins.
Results
To this end, a focused library of 352 small molecules derived from our in-house drug discovery programs was constructed. These compounds were selected based on structural diversity, drug-like properties, and their lack of previous antibiotic testing. This compound set was first screened at 100 μM for antibacterial activity against MRSA (Figure 1A). This revealed 25 compounds that prevented bacterial growth (Supporting Data 1). Subsequently, the minimum inhibitory concentration (MIC) was determined for each of the 25 hits. Benzyl (4-(5-methoxy-2-oxo-1,3,4-oxadiazol-3(2H)-yl)-2-methylphenyl)-carbamate 1 was most potent with an MIC of 6.25 μM (2.2 μg/mL). Notably, 1 contains an oxadiazolone moiety previously shown to covalently react with catalytically active serine and cysteine residues in enzymes (Figure 1B).14−16
Figure 1.

(A) Flowchart from the initial screen to lead compound 3. (B) Proposed reaction mechanism of 1,3,4-oxadiazole-2-one derivatives toward reactive serine and cysteines. (C) General route for oxadiazolone synthesis. BioRender Premium with an academic license was used for Figure 1A.
To determine the structure–activity relationship and optimize the potency of 1, 61 derivatives were synthesized and tested for anti-MRSA activity (Tables S1–S6 and Supporting Data 2). This led to the identification of 2 as a simplified scaffold with comparable antibacterial activity. Subsequent systematic modification of 2 (Figure 1C) resulted in the discovery of N-(4-(5-methoxy-2-oxo-1,3,4-oxadiazol-3(2H)-yl)-2-methylphenyl)-3-phenoxy-benzamide 3 as our lead compound with a 16-fold improvement in potency against both MRSA USA300 (MIC = 0.8 μM, 0.3 μg/mL) and S. aureus ATCC 29213 (MIC = 1.6 μM) compared to hit 1.
Extended screening of 3 revealed it to be highly active against a variety of clinical isolates, including vancomycin-resistant strains (Table 1 and Supporting Data 3). Time-kill experiments also showed that 3 killed 99% of bacteria within 24 h, starting from a 106 CFU/mL inoculum (Figure 2A). Furthermore, 3 exhibits low cytotoxicity (Table S7: selectivity ratio HEK293T/MRSA USA300 = 10.5, HepG2/MRSA300 = 20.3) and is nonhemolytic (Figure S1).
Table 1. Minimum Inhibitory Concentration (MIC) of Compound 3, and Clinically Relevant Antibiotics against a Bacterial Panela.
| MIC (μM) |
|||||
|---|---|---|---|---|---|
| organism | strain | 3 | meropenem | vancomycin | daptomycin |
| S. aureus | MRSA USA300 | 0.8 | 1.1 | 1.4 | 1.2 |
| NY-155 (MRSA) | 0.8 | 9.1 | 0.7 | 1.2 | |
| MRSA131 | 1.6 | 2.3 | 0.7 | 1.2 | |
| COL (MRSA) | 1.6 | 293 | 1.4 | 2.4 | |
| 1.6 | ≤0.1 | 0.7 | 1.2 | ||
| SA MER (VISA) | 1.6 | 0.3 | 2.8 | 4.9 | |
| LIM3 (VISA) | 0.8 | ≤2.3 | 2.8 | 2.4 | |
| NRS126 (VISA) | 3.1 | 293 | 2.8 | 2.4 | |
| BR-VRSA | 1.6 | >293 | 88 | 1.2 | |
| VRSA-1 | 3.1 | 293 | 88 | 1.2 | |
| VRSA-2 | 0.8 | 293 | 88 | ≤0.6 | |
| Enterococcus faecium | >50 | ≤0.1 | 0.7 | 1.2 | |
| Gram-negative | >50 | ≤0.1–2.3 | >88 | >80 | |
MSSA: methicillin-susceptible S. aureus, VISA: vancomycin-intermediate S. aureus, and VRSA: vancomycin-resistant S. aureus.
Figure 2.
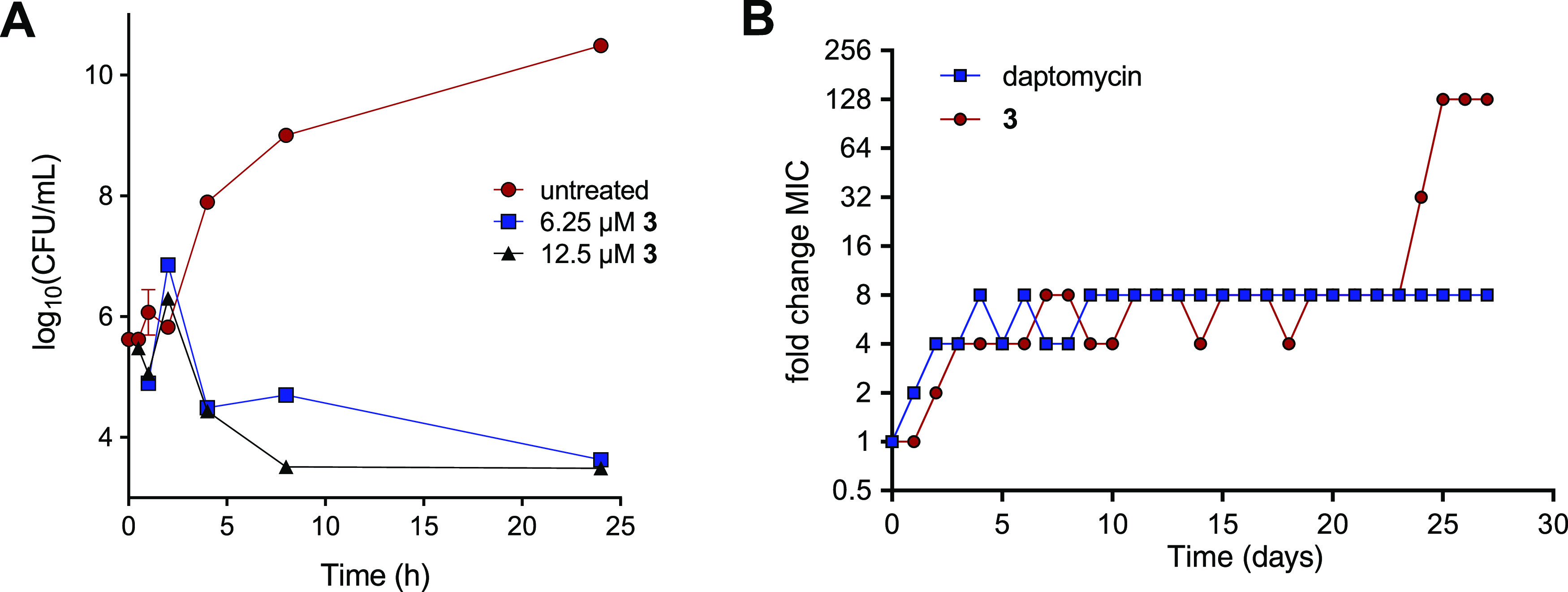
(A) Time-dependent killing by 3 of MRSA USA300. (B) Resistance development of MRSA USA300 against 3 and daptomycin during daily serial passaging with 0.25× MIC concentrations.
Next, we set out to generate strains resistant to 3 to investigate both the rate and mechanism of resistance development. The serial passage of MRSA USA300 in sub-MIC concentrations of 3 yielded resistant mutants after 4 weeks (Figures 2B and S2). In comparison, resistance to daptomycin, a clinically used lipopeptide antibiotic, emerged more slowly and did not exceed 8× MIC, a finding in line with previous reports.17,18 Notably, resistance toward 3 initially increased and then stabilized for several weeks before progressing to significantly higher values. This may indicate that multiple mutations are required to fully induce resistance, suggesting a polypharmacological MoA. Also of note, the passaged strains exhibiting resistance to 3 did not show cross-resistance to other antibiotic classes (Table S8).
Having established the potent anti-S. aureus activity of the oxadiazoloness, we set out to identify interaction partners using activity-based protein profiling (ABPP). Given the covalent mechanism ascribed to the oxadiazolones, we hypothesized that a strategically positioned ligation handle on 3 could be used to introduce a fluorescent or affinity tag (e.g., biotin) to visualize its interactions with proteins in living systems (Figure 3A). To this end, the meta-phenoxy group of 3 was substituted with an alkyne, resulting in activity-based probe 4 (Figure 3B). The antibacterial activity of 4 was confirmed in MRSA (MIC = 3.1 μM). The probe was subsequently used in an in situ competitive ABPP workflow (Figure 3A).19 To do so, MRSA cells at the exponential phase were treated with competitor 3 or dimethyl sulfoxide (DMSO), followed by labeling with probe 4. Bacteria were lysed and the probe-labeled proteins conjugated to a Cy5 fluorophore-azide via copper-catalyzed azide–alkyne click chemistry. This resulted in the clear labeling of several proteins by 4, of which most were dose-dependently outcompeted by 3 (Figure 3C). To identify the probe-labeled proteins, a biotin-azide reporter was also employed, allowing for affinity enrichment and identification of probe-labeled proteins by mass spectrometry (MS)-based proteomics.20 Around 30 proteins were found to be significantly enriched (P < 0.05, >2-fold enrichment) by probe treatment (Figure S3, Table S9, and Supporting Data 4A). Pretreatment with 3 significantly inhibited (P < 0.05, >2-fold inhibition) the labeling of 10 proteins by probe 4 (Figure 3D), suggesting that these proteins are interaction partners of oxadiazolone 3.
Figure 3.

(A) In situ competitive ABPP workflow on MRSA with using either sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or mass spectrometry read-out. (B) Activity-based probe 4. (C) Gel-based competitive ABPP over a concentration range of 3 versus 1 μM probe 4. (D) MS data inhibition plot comparing the labeled proteome of samples preincubated with inhibitor 3 (10 μM) followed by probe 4 labeling (3 μM) to solely probe-labeled samples. The vertical and horizontal threshold lines represent a log2 change of −1 and a log10(P value) of −1.3 (two-sided two-sample t-test, n = 3 independent experiments per group), respectively. Green dots indicate proteins that are probe targets, as defined in Table S9. BioRender Premium with an academic license was used for (A).
Among the MRSA proteins thus identified, the Fph proteins (B, C, E, H) were recently annotated as fluorophosphonate-binding hydrolases.21 FphB was found to be a fatty acid metabolizing virulence factor, while FphE activity has been used to phenotypically characterize MRSA.22 HZ1 is reported to have hydrolase activity (Table 2), while IB7 has putative thiolase activity,23 and FI2 is an uncharacterized protein. HH9 has recently been annotated as a lipase of negatively charged fatty acids.24 FabH is essential for bacterial fatty acid synthesis and has been explored as a drug target,26−28 while AdhE is an aldehyde–alcohol dehydrogenase, essential in facultative anaerobic organisms in anaerobic conditions.29,30 Both FabH and AdhE are known to metabolize substrates using an active site cysteine.
Table 2. List of Probe Targets Significantly Outcompeted by 3.
| # | uniprot ID | protein | description | sequence length (aa) | gene | essentiality | references |
|---|---|---|---|---|---|---|---|
| 1 | Q2FDS6 | FphE | uncharacterized hydrolase | 276 | SAUSA300_2518 | no | (21) and (22) |
| 2 | Q2FI93 | FabH | 3-oxoacyl-[acyl-carrier-protein] synthase 3 | 313 | fabH | yes | (25−28) |
| 3 | A0A0H2XJL0 | FphH | carboxylesterase | 246 | est | no | (21) |
| 4 | A0A0H2XHZ1 | HZ1 | putative lysophospholipase | 271 | SAUSA300_0070 | no | |
| 5 | A0A0H2XHD0 | FphC | hydrolase, α/β hydrolase fold family | 304 | SAUSA300_1194 | no | (21) |
| 6 | A0A0H2XHH9 | HH9 | S. aureus lipase 3 | 347 | SAUSA300_0641 | no | (24) |
| 7 | A0A0H2XJG5 | FphB | uncharacterized protein | 322 | SAUSA300_2473 | no | (21) |
| 8 | A0A0H2XFI2 | FI2 | uncharacterized protein | 275 | SAUSA300_0321 | no | |
| 9 | A0A0H2XIB7 | IB7 | acetyl-CoA c-acetyltransferase | 379 | vraB | no | (23) |
| 10 | A0A0H2XG10 | AdhE | aldehyde–alcohol dehydrogenase | 869 | adhE | no | (29−31) |
We next screened the probe-labeled proteome of nine transposon mutants of MRSA that lack the gene encoding one of the identified target proteins of 3 (Figure 4A,C). The labeling of AdhE, FphB, FphH, FI2, HZ1, and FphE, but not FphC and HH9, could be attributed to specific fluorescent bands on SDS-PAGE (Figure 4B). The lower resolution of gel-based ABPP or insufficient sensitivity compared to MS-based ABPP may explain why FphC and HH9 were not identified on the gel. Since FabH is essential for MRSA viability, no transposon mutant is available for this protein. Instead, we confirmed the identity of FabH on the gel by competitive ABPP using the selective FabH inhibitor Oxa2 (Figure S4).
Figure 4.

(A) Labeling of MRSA USA300 transposon mutants with 4. All mutants shown except FphE. (B) Wild type (WT) MRSA USA300 treated with probe 4. Bands annotated with corresponding proteins. (C) Labeling of the FphE transposon mutant.
To assess which target proteins were associated with the antibiotic effect, we hypothesized that the protein labeling profile of potent oxadiazolones (MIC ≤ 12.5 μM) would be different than that of analogues with weak activity (MIC > 50 μM). We, therefore, compared the interaction profile of three inactive derivatives (5–7) with that of three active compounds (1–3) in a competitive chemical proteomics assay (Figures 5A and S5 and Supporting Data 4B). Strong FphB labeling was observed upon treatment with 1, but not by the other compounds. F12, IB7, HH9, and HZ1 were not significantly labeled by bioactive oxadiazolone 3 but did show engagement with inactive compounds 5, 6, or 7. FphE and FphH were strongly labeled by all compounds at 10 μM. These observations, in combination with the viability of the transposon mutants, suggest that specific inhibition of FphB, IB7, HH9, F12, FphE, FphH, or HZ1 is not responsible for the antimicrobial activity of 3. FabH was significantly engaged, but not fully, by all compounds. Since the transposon mutant of FabH is not viable, and because Oxa2, a specific FabH inhibitor, can inhibit bacterial growth, this implies that partial inhibition of FabH activity could contribute to the bioactivity of the oxadiazolones. For both FphC and AdhE, however, labeling was observed with one or more of the active compounds and not by the inactive analogues (Figures 5B and S6 and Table S10).
Figure 5.

(A) Individual inhibition plots of active compounds 1, 2, and 3 and inactive compounds 5, 6, and 7. 3 was dosed at 1 μM, while 1, 2, and the inactive analogues were dosed at 10 μM. The vertical and horizontal threshold lines represent a log2 change of −1 and a log10(P value) of −1.3 (two-sided two-sample t-test, n = 3 independent experiments per group), respectively. Green dots indicate proteins that are probe targets, as defined in Table S9. (B) Venn diagram showing the overlap of >50% inhibited proteins between 3 and inactive analogues 5–7. (C) Relative general protein levels in strains resistant to 3 compared to WT. Each group was compared to WT using a two-sided two-sample t-test, n = 3 independent experiments per group. Statistical significance: ***P < 0.001; **P < 0.01; *P < 0.05; n.s. if P > 0.05. (D) Relative protein levels enriched by 4 in strains resistant to 3 (Figure 2B) compared to wild type. Each group was compared to WT using a two-sided two-sample t-test, n = 3 independent experiments per group.
Thus, our chemical proteomics data reveal that multiple targets (in particular, FphC, AdhE, and FabH) play a role in the observed antimicrobial activity of the oxadiazolones. We also assessed the sensitivity of the AdhE and FphC transposon mutants to inactive analogues 5–7. This revealed that both 6 and 7 showed increased antimicrobial activity in both transposon mutants (Tables 3 and S11), but not in a FphB transposon mutant (included as a negative control). Compound 5, which does not inhibit AdhE and very weakly inhibits FphC labeling (<20%), remained inactive against all transposon mutants. No direct synergy of FabH inhibition with any other single target could be concluded, as the antibacterial activity of Oxa2 was not potentiated on any of the transposon mutants (Table S11). While one cannot exclude the role of additional proteins, we interpret these data to mean that the combined engagement of FabH, FphC, AdhE, and, to some extent, FphE, is responsible for the antimicrobial activity observed for the oxadiazolones.
Table 3. MIC Values of MRSA USA300 Transposon Mutants of Target Proteins.
| MIC (μM) | |||
|---|---|---|---|
| inactive 5 | inactive 6 | inactive 7 | |
| MRSA USA300 WT | >50 | >50 | >50 |
| AdhE transposon mutant | >50 | 25 | 12.5 |
| FphC transposon mutant | >50 | 50 | 25 |
| FphB transposon mutant | >50 | >50 | >50 |
To test this hypothesis, we investigated whether the oxadiazolone targets were changed in two MRSA strains passaged to become resistant to 3 compared to WT MRSA. Using chemical (Figures 5C and S7 and Supporting Data 4C,D) and global proteomics (Figures 5D and S8 and Supporting Data 5), it was observed that AdhE and FphE engagement by probe 4 was significantly decreased, while protein abundance was upregulated in the two resistant strains. FphC engagement was also reduced, but to a lower extent. Interestingly, FabH protein levels were significantly increased in the resistant strains, which was accompanied by cross-resistance of these strains to FabH inhibitor Oxa2 (32× increase in MIC, Table S8). Taken together, these data suggest that the combined inhibition of FabH, FphC, FphE, and AdhE contributes to the antimicrobial activity of compound 3.
The two passaged MRSA strains exhibiting resistance to 3 were also screened for mutations in the genes that encode for the 10 target proteins (Supporting Data 6). This revealed a single point mutation in one of the resistant strains corresponding to Thr146Ile in the FabH protein. Of note, this is also the strain in which highly upregulated FabH levels were observed. Given the lack of other mutations in the strains resistant to 3, we concluded that genetic changes are not responsible for the high resistance developed and that the observed resistance must come from other mechanisms. To assess the stability of this resistance phenotype, the resistant strains were cultured in the absence of compound 3 for several days and then tested for susceptibility (Figure S9). Within a few days, the MIC dropped from 128× MIC to 8× MIC. This suggests that the observed resistance to 3 is inducible upon exposure to the compound and is lost in the absence of the compound.
Conclusions
To summarize, we here disclose oxadiazolones as a new chemotype with antibiotic activity against S. aureus strains including drug-resistant isolates. A medicinal chemistry program combined with chemical proteomics led to the identification of potent antibacterial compound 3 capable of interacting with multiple bacterial cysteine and serine hydrolases in a covalent manner. Three complementary lines of investigation point to FabH, FphC, AdhE, and to some extent FphE playing central roles in the antimicrobial activity of the oxadiazolones: (i) comparative chemical proteomics, (ii) gain of function in transposon mutants, and (iii) resistance-induced proteomic changes. FabH has previously been identified as a drug target, whereas the functions of AdhE and FphC have been less explored. Recent studies implicate AdhE as a virulence factor in Escherichia coli,31 while FphC is a predicted membrane-bound serine hydrolase of unknown function. We also cannot rule out that other factors, not detected by our chemical proteomics approach, might contribute to the antibacterial effect of 3. Interestingly, the resistance developed to 3 was transient, as it quickly diminished upon removal of the compound, and genomic screening of target proteins in the resistant strains identified only a single FabH mutation. This evidence suggests that mutations in key targets are not responsible for resistance to 3. Notably, this reversibility of resistance may be advantageous for the potential application of such compounds as antibiotics.
To conclude, our findings further highlight the value of synthetic compound libraries as an excellent source for antibiotic drug discovery complementary to natural products. By applying comparative and competitive chemical proteomics, using a new tailor-made activity-based probe with a strategically positioned ligation tag, we successfully highlighted the polypharmacological mode of action of the oxadiazolones and identified their targets in MRSA. Notably, a target-based approach alone would not have been able to uncover the mode of action of the oxadiazolones, thereby showcasing the power of chemical proteomics as a valuable chemical biology technique for antibiotic drug discovery. Future experiments are directed toward understanding the biological role of these targets, the nature of the resistance, and further optimization of the compounds as viable drug candidates.
Acknowledgments
Hans van der Elst and the Leiden University NMR facility are acknowledged for technical support. The authors thank Dr. Stephan M. Hacker for the critical reading of the manuscript. The lab of Prof. Matthew Bogyo is thanked for supplying the FphE transposon mutant, and the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) is thanked for supplying the remaining transposon mutants (NTML). Financial support was provided by the European Research Council (ERC consolidator grant to NIM, grant agreement no. 725523) and the NWO NACTAR program (project no. 16444).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10819.
SAR, cytotoxicity data, MIC values of selected compounds against resistant strains and transposon mutants, and list of enriched proteins (Tables S1–S11); additional data of the chemical proteomics, toxicity, and resistance experiments; and additional experimental details, materials, and methods, including synthetic procedures and NMR data of key compounds (Figures S1–S9) (PDB)
Supporting Data 1–6 (XLSX)
Author Contributions
∥ A.T.B. and I.K. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Tacconelli E.; Carrara E.; Savoldi A.; Harbarth S.; Mendelson M.; Monnet D. L.; Pulcini C.; Kahlmeter G.; Kluytmans J.; Carmeli Y.; Ouellette M.; Outterson K.; Patel J.; Cavaleri M.; Cox E. M.; Houchens C. R.; Grayson M. L.; Hansen P.; Singh N.; Theuretzbacher U.; Magrini N.; Aboderin A. O.; Al-Abri S. S.; Awang Jalil N.; Benzonana N.; Bhattacharya S.; Brink A. J.; Burkert F. R.; Cars O.; Cornaglia G.; Dyar O. J.; Friedrich A. W.; Gales A. C.; Gandra S.; Giske C. G.; Goff D. A.; Goossens H.; Gottlieb T.; Guzman Blanco M.; Hryniewicz W.; Kattula D.; Jinks T.; Kanj S. S.; Kerr L.; Kieny M. P.; Kim Y. S.; Kozlov R. S.; Labarca J.; Laxminarayan R.; Leder K.; Leibovici L.; Levy-Hara G.; Littman J.; Malhotra-Kumar S.; Manchanda V.; Moja L.; Ndoye B.; Pan A.; Paterson D. L.; Paul M.; Qiu H.; Ramon-Pardo P.; Rodríguez-Baño J.; Sanguinetti M.; Sengupta S.; Sharland M.; Si-Mehand M.; Silver L. L.; Song W.; Steinbakk M.; Thomsen J.; Thwaites G. E.; van der Meer J. W.; Van Kinh N.; Vega S.; Villegas M. V.; Wechsler-Fördös A.; Wertheim H. F. L.; Wesangula E.; Woodford N.; Yilmaz F. O.; Zorzet A. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- Lewis K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. 10.1016/j.cell.2020.02.056. [DOI] [PubMed] [Google Scholar]
- Brown E. D.; Wright G. D. Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- Murray C. J.; Ikuta K. S.; Sharara F.; Swetschinski L.; Robles Aguilar G.; Gray A.; Han C.; Bisignano C.; Rao P.; Wool E.; Johnson S. C.; Browne A. J.; Chipeta M. G.; Fell F.; Hackett S.; Haines-Woodhouse G.; Kashef Hamadani B. H.; Kumaran E. A. P.; McManigal B.; Agarwal R.; Akech S.; Albertson S.; Amuasi J.; Andrews J.; Aravkin A.; Ashley E.; Bailey F.; Baker S.; Basnyat B.; Bekker A.; Bender R.; Bethou A.; Bielicki J.; Boonkasidecha S.; Bukosia J.; Carvalheiro C.; Castañeda-Orjuela C.; Chansamouth V.; Chaurasia S.; Chiurchiù S.; Chowdhury F.; Cook A. J.; Cooper B.; Cressey T. R.; Criollo-Mora E.; Cunningham M.; Darboe S.; Day N. P. J.; De Luca M.; Dokova K.; Dramowski A.; Dunachie S. J.; Eckmanns T.; Eibach D.; Emami A.; Feasey N.; Fisher-Pearson N.; Forrest K.; Garrett D.; Gastmeier P.; Giref A. Z.; Greer R. C.; Gupta V.; Haller S.; Haselbeck A.; Hay S. I.; Holm M.; Hopkins S.; Iregbu K. C.; Jacobs J.; Jarovsky D.; Javanmardi F.; Khorana M.; Kissoon N.; Kobeissi E.; Kostyanev T.; Krapp F.; Krumkamp R.; Kumar A.; Kyu H. H.; Lim C.; Limmathurotsakul D.; Loftus M. J.; Lunn M.; Ma J.; Mturi N.; Munera-Huertas T.; Musicha P.; Mussi-Pinhata M. M.; Nakamura T.; Nanavati R.; Nangia S.; Newton P.; Ngoun C.; Novotney A.; Nwakanma D.; Obiero C. W.; Olivas-Martinez A.; Olliaro P.; Ooko E.; Ortiz-Brizuela E.; Peleg A. Y.; Perrone C.; Plakkal N.; Ponce-de-Leon A.; Raad M.; Ramdin T.; Riddell A.; Roberts T.; Robotham J. V.; Roca A.; Rudd K. E.; Russell N.; Schnall J.; Scott J. A. G.; Shivamallappa M.; Sifuentes-Osornio J.; Steenkeste N.; Stewardson A. J.; Stoeva T.; Tasak N.; Thaiprakong A.; Thwaites G.; Turner C.; Turner P.; van Doorn H. R.; Velaphi S.; Vongpradith A.; Vu H.; Walsh T.; Waner S.; Wangrangsimakul T.; Wozniak T.; Zheng P.; Sartorius B.; Lopez A. D.; Stergachis A.; Moore C.; Dolecek C.; Naghavi M. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brtz-Oesterhelt H.; Sass P. Postgenomic Strategies in Antibacterial Drug Discovery. Future Microbiol. 2010, 5, 1553–1579. 10.2217/fmb.10.119. [DOI] [PubMed] [Google Scholar]
- The Pew Charitable Trusts . A Scientific Roadmap for Antibiotic Discovery, 2016. www.pewtrusts.org/en/research-and-analysis/reports/2016/05/a-scientific-roadmap-for-antibiotic-discovery (accessed Dec 2, 2022).
- Aulner N.; Danckaert A.; Ihm J. E.; Shum D.; Shorte S. L. Next-Generation Phenotypic Screening in Early Drug Discovery for Infectious Diseases. Trends Parasitol. 2019, 35, 559–570. 10.1016/j.pt.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Swinney D. C. Phenotypic vs. Target-Based Drug Discovery for First-in-Class Medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- Van Esbroeck A. C. M.; Janssen A. P. A.; Cognetta A. B.; Ogasawara D.; Shpak G.; Van Der Kroeg M.; Kantae V.; Baggelaar M. P.; De Vrij F. M. S.; Deng H.; Allarà M.; Fezza F.; Lin Z.; Van Der Wel T.; Soethoudt M.; Mock E. D.; Den Dulk H.; Baak I. L.; Florea B. I.; Hendriks G.; De Petrocellis L.; Overkleeft H. S.; Hankemeier T.; De Zeeuw C. I.; Di Marzo V.; Maccarrone M.; Cravatt B. F.; Kushner S. A.; Van Der Stelt M. Activity-Based Protein Profiling Reveals off-Target Proteins of the FAAH Inhibitor BIA 10-2474. Science 2017, 356, 1084–1087. 10.1126/science.aaf7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S.; Heinzlmeir S.; Wilhelm M.; Polzer H.; Vick B.; Koenig P. A.; Reinecke M.; Ruprecht B.; Petzoldt S.; Meng C.; Zecha J.; Reiter K.; Qiao H.; Helm D.; Koch H.; Schoof M.; Canevari G.; Casale E.; Re Depaolini S.; Feuchtinger A.; Wu Z.; Schmidt T.; Rueckert L.; Becker W.; Huenges J.; Garz A. K.; Gohlke B. O.; Zolg D. P.; Kayser G.; Vooder T.; Preissner R.; Hahne H.; Tõnisson N.; Kramer K.; Götze K.; Bassermann F.; Schlegl J.; Ehrlich H. C.; Aiche S.; Walch A.; Greif P. A.; Schneider S.; Felder E. R.; Ruland J.; Médard G.; Jeremias I.; Spiekermann K.; Kuster B. The Target Landscape of Clinical Kinase Drugs. Science 2017, 358, eaan4368 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L.; Kemper E. K.; Suciu R. M.; Vinogradova E. V.; Backus K. M.; Horning B. D.; Paul T. A.; Ichu T. A.; Svensson R. U.; Olucha J.; Chang M. W.; Kok B. P.; Zhu Z.; Ihle N. T.; Dix M. M.; Jiang P.; Hayward M. M.; Saez E.; Shaw R. J.; Cravatt B. F. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171, 696.e23–709.e23. 10.1016/j.cell.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le P.; Kunold E.; Macsics R.; Rox K.; Jennings M. C.; Ugur I.; Reinecke M.; Chaves-Moreno D.; Hackl M. W.; Fetzer C.; Mandl F. A. M.; Lehmann J.; Korotkov V. S.; Hacker S. M.; Kuster B.; Antes I.; Pieper D. H.; Rohde M.; Wuest W. M.; Medina E.; Sieber S. A. Repurposing Human Kinase Inhibitors to Create an Antibiotic Active against Drug-Resistant Staphylococcus aureus, Persisters and Biofilms. Nat. Chem. 2020, 12, 145–158. 10.1038/s41557-019-0378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner I.; Shapiro J. A.; Hoßmann J.; Drechsel J.; Hacker S. M.; Rather P. N.; Pieper D. H.; Wuest W. M.; Sieber S. A. Broad Spectrum Antibiotic Xanthocillin X Effectively Kills Acinetobacter Baumannii via Dysregulation of Heme Biosynthesis. ACS Cent. Sci. 2021, 7, 488–498. 10.1021/acscentsci.0c01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. C.; Delorme V.; Bénarouche A.; Guy A.; Landry V.; Audebert S.; Pophillat M.; Camoin L.; Crauste C.; Galano J. M.; Durand T.; Brodin P.; Canaan S.; Cavalier J. F. Oxadiazolone Derivatives, New Promising Multi-Target Inhibitors against M. Tuberculosis. Bioorg. Chem. 2018, 81, 414–424. 10.1016/j.bioorg.2018.08.025. [DOI] [PubMed] [Google Scholar]
- Ben Ali Y.; Chahinian H.; Petry S.; Muller G.; Lebrun R.; Verger R.; Carrière F.; Mandrich L.; Rossi M.; Manco G.; Sarda L.; Abousalham A. Use of an Inhibitor to Identify Members of the Hormone-Sensitive Lipase Family. Biochemistry 2006, 45, 14183–14191. 10.1021/bi0613978. [DOI] [PubMed] [Google Scholar]
- Madani A.; Mallick I.; Guy A.; Crauste C.; Durand T.; Fourquet P.; Audebert S.; Camoin L.; Canaan S.; Cavalier J. F. Dissecting the Antibacterial Activity of Oxadiazolone-Core Derivatives against Mycobacterium abscessus. PLoS One 2020, 15, e0238178 10.1371/journal.pone.0238178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch M.; Gagetti P.; Davis J.; Ceriana P.; Errecalde L.; Corso A.; Rosato A. E. Daptomycin Resistance in Clinical MRSA Strains Is Associated with a High Biological Fitness Cost. Front. Microbiol. 2017, 8, 2303 10.3389/fmicb.2017.02303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igler C.; Rolff J.; Regoes R. Multi-Step vs. Single-Step Resistance Evolution under Different Drugs, Pharmacokinetics, and Treatment Regimens. Elife 2021, 10, e64116 10.7554/eLife.64116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooden E. J.; Florea B. I.; Deng H.; Baggelaar M. P.; Van Esbroeck A. C. M.; Zhou J.; Overkleeft H. S.; Van Der Stelt M. Mapping in Vivo Target Interaction Profiles of Covalent Inhibitors Using Chemical Proteomics with Label-Free Quantification. Nat. Protoc. 2018, 13, 752–767. 10.1038/nprot.2017.159. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Cox J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Lentz C. S.; Sheldon J. R.; Crawford L. A.; Cooper R.; Garland M.; Amieva M. R.; Weerapana E.; Skaar E. P.; Bogyo M. Identification of a S. aureus Virulence Factor by Activity-Based Protein Profiling (ABPP) Article. Nat. Chem. Biol. 2018, 14, 609–617. 10.1038/s41589-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Keller L. J.; Cordasco E.; Bogyo M.; Lentz C. S. Fluorescent Triazole Urea Activity-Based Probes for the Single-Cell Phenotypic Characterization of Staphylococcus aureus. Angew. Chem., Int. Ed. 2019, 58, 5643–5647. 10.1002/anie.201900511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriläinen G.; Poikela V.; Kursula P.; Wierenga R. K. The Thiolase Reaction Mechanism: The Importance of Asn316 and His348 for Stabilizing the Enolate Intermediate of the Claisen Condensation. Biochemistry 2009, 48, 11011–11025. 10.1021/bi901069h. [DOI] [PubMed] [Google Scholar]
- Kumar N. G.; Contaifer D.; Wijesinghe D. S.; Jefferson K. K. Staphylococcus aureus Lipase 3 (SAL3) Is a Surface-Associated Lipase That Hydrolyzes Short Chain Fatty Acids. PLoS One 2021, 16, e0258106 10.1371/journal.pone.0258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. Y.; Cronan J. E. β-Ketoacyl-Acyl Carrier Protein Synthase III (FabH) Is Essential for Bacterial Fatty Acid Synthesis. J. Biol. Chem. 2003, 278, 51494–51503. 10.1074/jbc.M308638200. [DOI] [PubMed] [Google Scholar]
- Wang J.; Kodali S.; Sang H. L.; Galgoci A.; Painter R.; Dorso K.; Racine F.; Motyl M.; Hernandez L.; Tinney E.; Colletti S. L.; Herath K.; Cummings R.; Salazar O.; González I.; Basilio A.; Vicente F.; Genilloud O.; Pelaez F.; Jayasuriya H.; Young K.; Cully D. F.; Singh S. B. Discovery of Platencin, a Dual FabF and FabH Inhibitor with in Vivo Antibiotic Properties. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7612–7616. 10.1073/pnas.0700746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Yang Y. S.; Fu J.; Zhu H. L. Novel FabH Inhibitors: A Patent and Article Literature Review (2000 2012). Expert Opin. Ther. Pat. 2012, 22, 1325–1336. 10.1517/13543776.2012.727798. [DOI] [PubMed] [Google Scholar]
- Wang J.; Ye X.; Yang X.; Cai Y.; Wang S.; Tang J.; Sachdeva M.; Qian Y.; Hu W.; Leeds J. A.; Yuan Y. Discovery of Novel Antibiotics as Covalent Inhibitors of Fatty Acid Synthesis. ACS Chem. Biol. 2020, 15, 1826–1834. 10.1021/acschembio.9b00982. [DOI] [PubMed] [Google Scholar]
- Fuchs S.; Pané-Farré J.; Kohler C.; Hecker M.; Engelmann S. Anaerobic Gene Expression in Staphylococcus aureus. J. Bacteriol. 2007, 189, 4275–4289. 10.1128/JB.00081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo J.; Zheng T.; Hon S.; Olson D. G.; Lynd L. R. The Bifunctional Alcohol and Aldehyde Dehydrogenase Gene, AdhE, Is Necessary for Ethanol Production in Clostridium Thermocellum and Thermoanaerobacterium saccharolyticum. J. Bacteriol. 2015, 197, 1386–1393. 10.1128/JB.02450-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham K. S. H.; Connolly J. P. R.; Ritchie J. M.; Wang D.; Gawthorne J. A.; Tahoun A.; Gally D. L.; Burgess K.; Burchmore R. J.; Smith B. O.; Beatson S. A.; Byron O.; Wolfe A. J.; Douce G. R.; Roe A. J. The Metabolic Enzyme AdhE Controls the Virulence of Escherichia ColiO157: H7. Mol. Microbiol. 2014, 93, 199–211. 10.1111/mmi.12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.