

Abstract
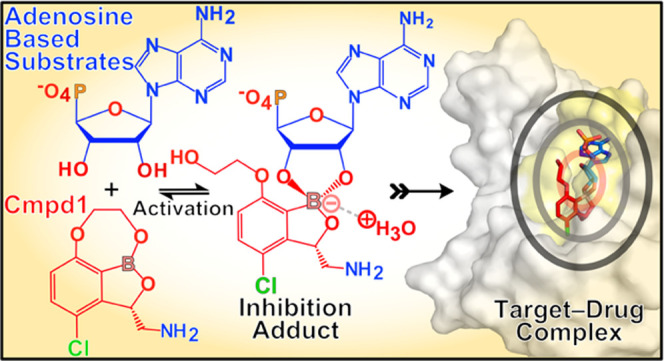
Prodrugs have little or no pharmacological activity and are converted to active drugs in the body by enzymes, metabolic reactions, or through human-controlled actions. However, prodrugs promoting their chemical bioconversion without any of these processes have not been reported before. Here, we present an enzyme-independent prodrug activation mechanism by boron-based compounds (benzoxaboroles) targeting leucyl-tRNA synthetase (LeuRS), including an antibiotic that recently has completed phase II clinical trials to cure tuberculosis. We combine nuclear magnetic resonance spectroscopy and X-ray crystallography with isothermal titration calorimetry to show that these benzoxaboroles do not bind directly to their drug target LeuRS, instead they are prodrugs that activate their bioconversion by forming a highly specific and reversible LeuRS inhibition adduct with ATP, AMP, or the terminal adenosine of the tRNALeu. We demonstrate how the oxaborole group of the prodrugs cyclizes with the adenosine ribose at physiological concentrations to form the active molecule. This bioconversion mechanism explains the remarkably good druglike properties of benzoxaboroles showing efficacy against radically different human pathogens and fully explains the mechanism of action of these compounds. Thus, this adenosine-dependent activation mechanism represents a novel concept in prodrug chemistry that can be applied to improve the solubility, permeability and metabolic stability of challenging drugs.
Introduction
Prodrugs have little or no pharmacological activity and carry a promoiety group to improve druglike properties such as solubility, permeability, transport, and stability toward enzymatic and metabolic degradation.1−3 Within the body, the prodrug promoiety group is modified, a phenomenon called bioconversion, to originate the active molecule. The bioconversion of the majority of prodrugs is achieved by enzymes; however, prodrug activation can also occur by metabolic reactions or controlled by human intervention through γ/X-ray photoactivation,4 electrical stimuli,5 or ultrasounds.6 Moreover, prodrugs can limit undesired toxic effects by specific delivery to targeted tissues or by time-delayed release and activation.3
Remarkably, prodrugs are rapidly evolving from something serendipitously discovered in the past to something rational and well-planned nowadays. This is reflected in the growing percentage of prodrugs that are approved by drug agencies year after year.7 Over the last decade, prodrugs represent 12% of new therapeutical compounds approved by the Food and Drug Administration (FDA),1 though this number is expected to grow exponentially. This is likely due to (1) the improvement of our knowledge on specific enzymes involved in prodrug modification and release of active drugs and (2) the fact that prodrug strategies are included in early stages of drug discovery, either as the original plan or as a back-up solution when derivatives fail to improve challenging drug properties. It is thus clear that prodrugs represent an emerging route for drug design; however, their success will rely on the continuous discovery of truly novel mechanisms of bioconversion.
Here, we report the discovery of a novel prodrug activation mechanism of boron-based compounds (aminomethyl benzoxaboroles) targeting leucyl-tRNA synthetase (LeuRS) that does not rely on any enzyme, metabolic modification, or human intervention to generate the active drug. LeuRS is an essential protein to ensure correct protein synthesis,8 and previous studies suggest that benzoxaboroles block the LeuRS editing site likely by an adenosine-dependent (ATP, AMP, or tRNALeu terminal adenosine) inhibition mechanism.9−15 However, the spatiotemporal and molecular bases of the formation of the covalent adenosine-based inhibition adduct were not known, despite the discovery of these compounds more than a decade ago.
Here, we perform a comprehensive biophysical and structural analysis to reveal that benzoxaboroles do not bind directly to LeuRS. Instead, these compounds are prodrugs that employ an enzyme-independent activation mechanism mediated by their oxaborole group that undergoes covalent cyclization with ribose hydroxyls of ATP, AMP, or the tRNALeu terminal adenosine base. At close-to-physiological concentrations of these biomolecules, prodrug activation is reversible and yields two adenosine-oxaborole adducts (diastereomers), one of which binds with potent nanomolar affinity to the LeuRS target. The active inhibition adduct is stabilized by an ion pair consisting of a positively charged water molecule (hydroxonium ion) and a negatively charged oxaborole group, which we invariably find in all cocrystal structures of pathogenic LeuRS with benzoxaboroles. This novel prodrug activation mechanism explains the remarkably good druglike properties of benzoxaboroles showing efficacy against radically different human pathogens and fully completes the mechanism of action of these compounds, some of which are undergoing clinical studies.
Results
Benzoxaboroles Do Not Bind to Their Target LeuRS
To investigate the molecular basis of the generation of the inhibition adduct formed by benzoxaboroles with adenosine-based nucleotides, we performed isothermal titration calorimetry (ITC) binding experiments with the recombinant Mycobacterium tuberculosis (TB) LeuRS editing domain and two different antituberculosis LeuRS inhibitors, Cmpd1 and Cmdp2 (Figure 1a,b). Notably, Cmpd1 (GSK3036656) has completed clinical studies (phase IIb) for the treatment of pulmonary multidrug-resistant tuberculosis. The direct titration of either of these compounds into the protein target did not show any detectable binding (Figure 1c), underlining the need for activation of these antituberculosis compounds to exert their potent inhibitory activity on LeuRS. When we performed equivalent experiments in the presence of adenosine-based molecules like AMP, we observed strong binding, with dissociation constant (Kd) values of 10.4 and 46 nM for Cmpd1 and Cmpd2, respectively (Figure 1d,e). These experiments place Cmpd1 as the most potent in vitro inhibitor of TB LeuRS to date, in comparison to previous benzoxaborole inhibitors13,16,17 as well as to other nonboron-based inhibitors of LeuRS.18 The increase in affinity of Cmpd1 compared to that of Cmpd2 is due to an increase in favorable binding enthalpy (Figure 1d,e), suggesting that Cmpd1 establishes additional polar interactions within the drug pocket. Our experiments demonstrate that these potent anti-TB compounds are not direct inhibitors of LeuRS; instead, they employ an inhibition mechanism that depends on the presence of adenosine-based biomolecules.
Figure 1.
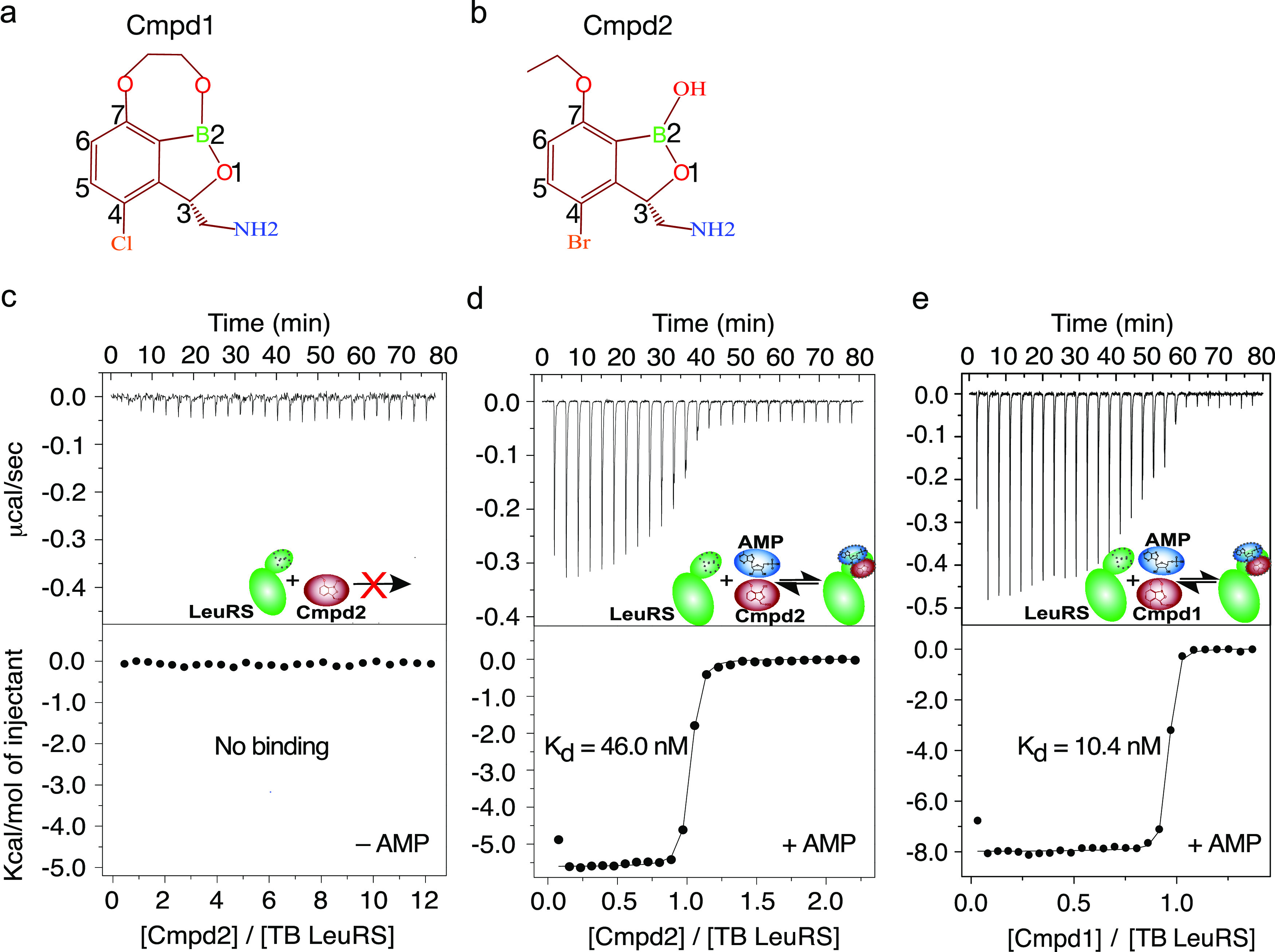
Cmpd1 and Cmpd2 bind to TB LeuRS via an adenosine-dependent mechanism. (a, b) Chemical structures of the two antituberculosis LeuRS inhibitors, Cmpd1 and Cmpd2. (c) ITC titration of compounds into TB LeuRS shows no binding. (d, e) ITC titrations of Cmpd2 and Cmpd1 into TB LeuRS in the presence of AMP, showing potent nanomolar affinities. AMP concentration was kept at physiological levels (10 mM).
Benzoxaborole Inhibitors of TB LeuRS Are Novel Prodrugs
To visualize the formation of the inhibition adduct, we used nuclear magnetic resonance (NMR) spectroscopy to study the conversion of Cmpd1 and Cmpd2 in the presence of ATP or AMP and in the absence of the protein target. We acquired one-dimensional 1H NMR spectra of free ATP, AMP, Cmpd1, Cmpd2, and equimolar mixtures of each compound with AMP or ATP at physiological concentrations (5 mM). The NMR spectra show that both Cmpd1 and Cmpd2 interact with AMP/ATP in the absence of TB LeuRS, leading to the formation of two different covalent adducts (diastereomers) per compound, differentiated by the stereochemistry of the boron atom, herein named as Adduct1a, Adduct1b, Adduct2a, and Adduct2b (Figures 2 and S1).
Figure 2.
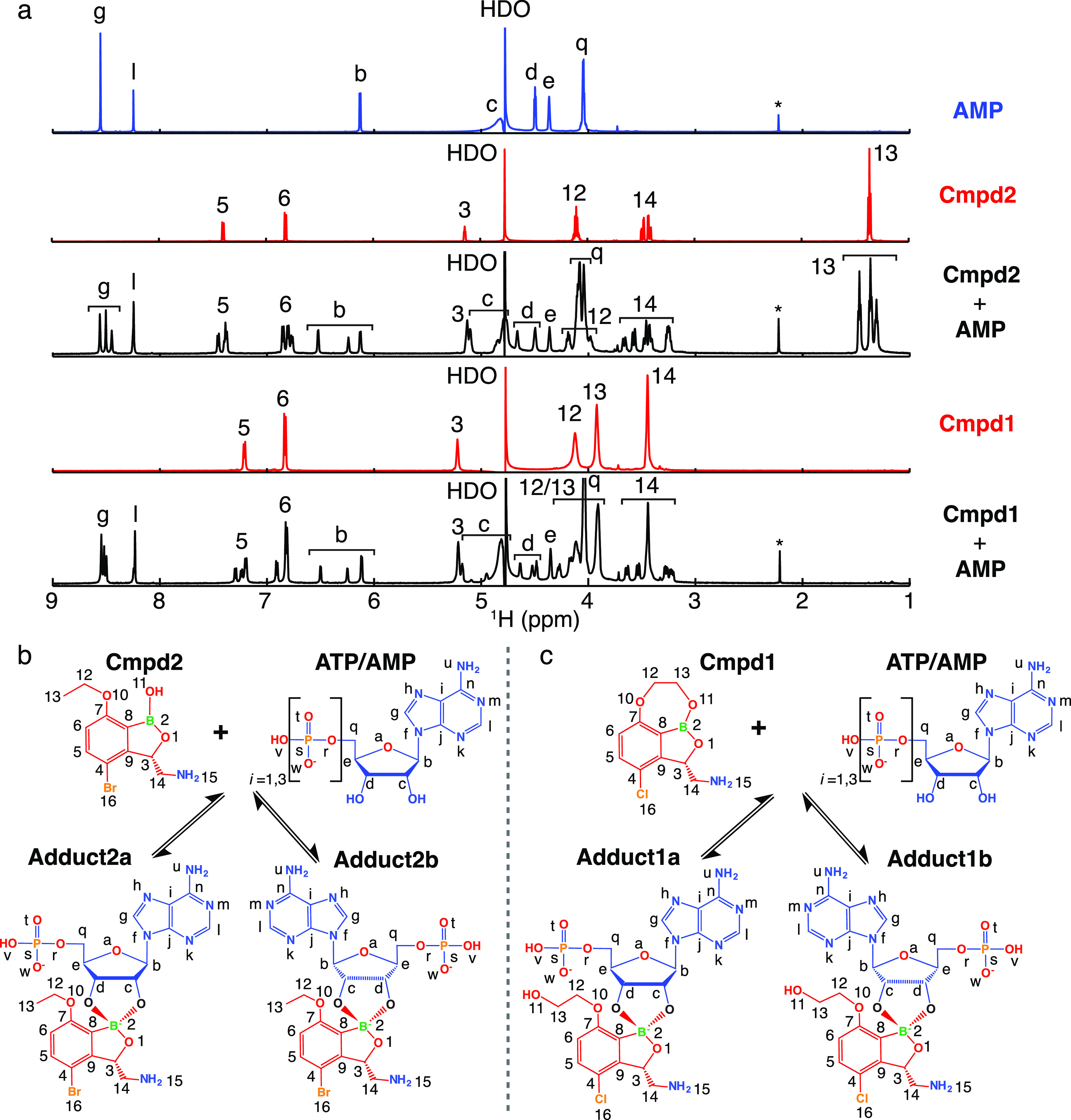
Benzoxaboroles are prodrugs that form covalent adducts with adenosine. (a) One-dimensional 1H NMR spectra of free AMP, Cmpd2, Cmpd1, and equimolar mixtures of each compound with AMP at physiological concentrations (5 mM). Labels indicate assignments of the resonances according to the annotations of the nuclei in panels b and c. (b, c) Prodrug activation mechanism of Cmpd2 and Cmpd1, respectively, with adenosine-based molecules leading to the formation of covalent adducts of types “a” and “b” due to the pseudosymmetry of ribose 2′ and 3′ hydroxyls.
The majority of the 1H nuclei give rise to different NMR resonances in the mixture corresponding to free AMP/ATP, free compound, and two different adducts with slightly different chemical shifts (Figure 2). The two adducts differ structurally by a 180° rotation of the adenosine ribose, indicating that the electrophilic boron can equally well attract the nucleophilic 2′- or 3′-hydroxyls of the adenosine ribose (Figures 2b,c and S2). This induces the transition of the boron atom from a neutral sp2 trigonal configuration to a negatively charged sp3 tetragonal configuration (Figure S2). Compared to Cmpd2, Cmpd1 contains an additional ring, which cyclizes the boron atom to carbon-7 (Figures 1a and 2c). Interestingly, the NMR data recorded for this compound show that the interaction with AMP/ATP causes the opening of this additional ring, as demonstrated by the strong similarity of the experimental chemical shifts in the mixtures of Cmpd1 and Cmpd2 with AMP (Figure 2a). Similar results are obtained with ATP, demonstrating that the number of phosphate groups does not affect the adduct formation with Cmpd1 and Cmpd2 (Figure S1).
Next, we investigated the binding affinity of each compound with adenosine nucleotides. From integration of the isolated resonance “b” of AMP/ATP (Figures 2a and S1), we determined the dissociation constants of the formation of the different adducts: 1.34 mM (Cmpd1–AMP), 1.09 mM (Cmpd2–AMP), 1.66 mM (Cmpd1–ATP), and 1.38 mM (Cmpd2–ATP). Thus, there is no significant difference in the dissociation constants for ATP compared to AMP, nor in the populations of adducts of type “a” compared to type “b” (Table S1). To further validate this finding, we carried out ITC experiments by directly titrating Cmpd1 into AMP (Figure 3a), allowing us to determine a dissociation constant of 2.8 mM, in good agreement with the NMR values. Analysis of 1H–1H NOESY spectra with different mixing times of Cmpd1 with AMP reveal that the formation of the covalent adducts is entirely reversible on the second time scale, with a direct but slower interconversion between the two adducts via hydrolysis of either the B–O2′ or B–O3′ bond followed by a rotation around the C2′–O2′ or C3′–O3′ bond, respectively (Figure S3).
Figure 3.
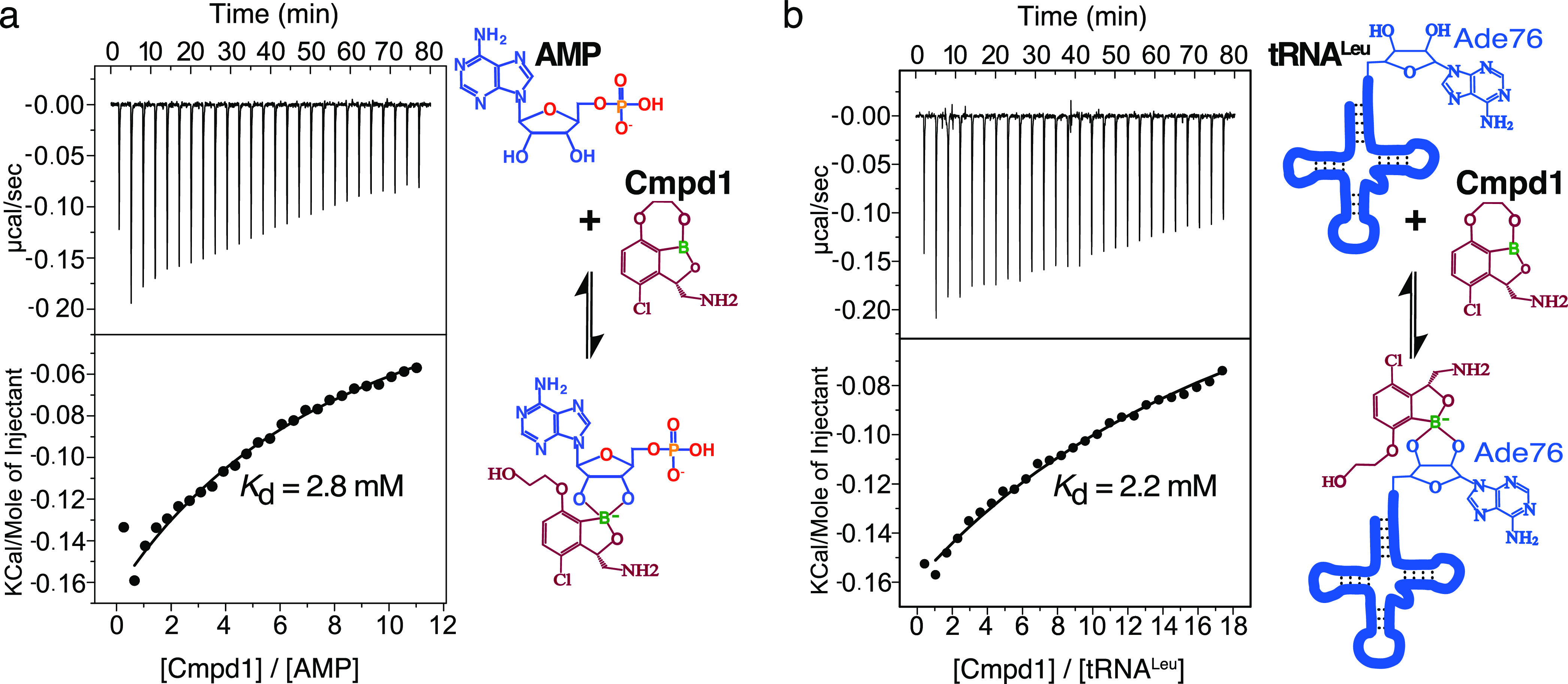
Determination of dissociation constants of covalent adducts. (a) ITC titration of Cmpd1 into AMP. (b) ITC titration of Cmpd1 into tRNALeu. Both AMP and tRNALeu show dissociation constants in the millimolar range, in agreement with the values determined from NMR.
Crystal structures have shown benzoxaboroles fused to the tRNALeu terminal adenosine bound into the LeuRS editing site. Therefore, we studied whether the enzyme-free prodrug activation mechanism observed with ATP/AMP also applies to tRNALeu. We carried out ITC experiments by titrating Cmpd1 into tRNALeu, showing that this compound binds directly to tRNALeu to form the inhibition adduct (Figure 3b). This explains why the tRNALeu is covalently fused to this type of benzoxaboroles in crystal structures of bacterial LeuRS.11,12 Moreover, the direct interaction of the tRNALeu and Cmpd1 could also be observed by NMR by employing an 15N, 13C adenosine-labeled tRNALeu sample (Figure S4). The determined dissociation constants by ITC and NMR were 2.2 and 1.0 mM, respectively, which are comparable to the binding affinities observed with ATP/AMP (Table S2). Collectively, these experiments demonstrate that benzoxaborole inhibitors of LeuRS activate with adenosine-based biomolecules using an enzyme-independent mechanism, leading to the formation of reversible covalent adducts with different structural and chemical drug properties. Thus, benzoxaborole inhibitors of LeuRS should be annotated as a new class of prodrugs.
Atomic Resolution Structure of TB LeuRS Bound to the Inhibition Adduct Formed by Cmpd1
To investigate how the different adducts bind to LeuRS and understand the structural basis of the inhibition mechanism, we performed crystallization studies with TB LeuRS, AMP, and Cmpd1. High quality crystals were obtained, allowing us to determine a crystal structure of TB LeuRS with Cmpd1 and AMP at 1.1 Å resolution (Figure 4a and Table S3). The atomic resolution structure confirms that Cmpd1 forms an adduct with AMP, and the high quality of the electron density maps allows us to unambiguously place the specific adduct that binds to the editing site of TB LeuRS (Figures 4b and S5). Our structure reveals that only one of the two adducts of Cmpd1 that we detected by NMR, Adduct1a, binds to TB LeuRS (Figure S6a,b). This is the same for Cmpd2 since only the equivalent adduct (Adduct2a, Figure 4d) is observed in the cocrystal structure with TB LeuRS.13 Moreover, in silico docking of adducts of type “b” into our TB LeuRS unbound crystal structure clearly shows steric clashes of the compound with several residues, including I440 and M441, indicating that their binding to the LeuRS drug pocket would not be favored (Figure S6c).
Figure 4.

Structural basis of the inhibition of TB LeuRS by active adducts “a”. (a) Global view of the crystal structure of the TB LeuRS editing domain in complex with Cmpd1 and AMP determined at 1.1 Å resolution. Crystals were obtained by mixing TB LeuRS protein with free AMP and Cmpd1. The protein is shown in green with the editing site (drug-binding pocket) in yellow. (b) Close view of the covalent adduct Cmpd1–AMP bound into the editing site of TB LeuRS. Cmpd1 and AMP are shown both in surface and stick representations colored in red and blue, respectively. The same color code is used in other panels. (c) Two views (rotated 45° with respect to each other) showing the main polar and hydrophobic interactions established by Adduct1a within the LeuRS binding pocket. Key protein residues are shown as yellow sticks and labeled. (d) Comparative analysis of the binding mode of adducts formed by Cmpd1 (left) and Cmpd2 (right), with additional Cmpd1 interactions highlighted as red, dashed lines. (e) Dissection of binding free energies into enthalpic and entropic contributions for the interaction of LeuRS with Cmpd1 and Cmpd2. Improved binding affinity of Cmpd1 compared to Cmpd2 is reflected in a more favorable binding enthalpy due to additional polar interactions being established in the complex with Cmpd1. The ring connecting the boron atom to carbon-7 in Cmpd1 opens up upon adduct formation giving rise to an oxy-ethanol group that establishes additional hydrogen bonds.
As observed in the NMR experiments, the crystal structure confirms the opening of the third ring of Cmpd1 to form Adduct1a and the naissance of an oxy-ethanol group that remains tightly bound into the editing site of TB LeuRS (Figure 4c). Importantly, the formation of Adduct1a and opening of the additional ring of Cmpd1 establishes novel interactions in the editing site of TB LeuRS compared to Cmpd2 (Figure 4c,d). Specifically, the newly founded hydroxyl group of Cmpd1 is caged between residues Thr337 and Arg449, where it adopts two alternative conformations enabling three additional hydrogen bonds with these residues (Figure 4c,d). This explains the higher affinity measured by ITC of Cmpd1 to TB LeuRS compared to other compounds like Cmpd2, arising from a more favorable binding enthalpy (ΔΔH = 3 kcal/mol) (Figure 4e). Importantly, this increase of in vitro affinity is translated into better antibacterial activity and in vivo oral efficacy,13,16,19 thereby explaining why Cmpd1 is the best-in-class anti-TB LeuRS inhibitor.
Structural Basis of the Formation of the Inhibition Adduct and Its Stabilization
In the drug pocket of the TB LeuRS–AMP–Cmpd1 structure, we observe a water molecule tightly bound (as judged by its B-factor) to the oxaborole group (Figures 5a and S7a). This water molecule fits very well into the small pocket formed by residues F335, T336, I440, and M441, where it forms hydrogen bonds with polar atoms of F335, I440, and M441. Furthermore, this water establishes optimal hydrogen bonds with the negatively charged oxaborole group of Cmpd1 (Figure 5a), strongly arguing that this water molecule is a positively charged hydroxonium ion (H3O+), as shown by its characteristic tetrahedral geometry and the number of hydrogen bonds (4) (Figure S7b), which likely remains bound upon adduct formation (Figure S2).
Figure 5.

Stabilization of prodrug adducts by a boron–hydroxonium ion pair is shared across pathogen phyla. (a) M. tuberculosis LeuRS in complex with the antituberculosis Cmpd1–adenosine adduct (Adduct1a) showing the hydroxonium ion bound in the editing site. Expanded view shows protein residues enabling key interactions with the hydroxonium ion (stick representation with atoms colored in yellow for carbon, red for oxygen, and blue for nitrogen). Hydrogen bonds are shown as green dashed lines. Highly conserved residues form an optimal cavity, shown as van der Waals surface representation, to accommodate the hydroxonium ion, shown as a red sphere. The same color code is used in all panels. (b) Cryptosporidium hominis LeuRS in complex with the antiparasitic AN6426–adenosine adduct bound to a hydroxonium ion (PDB: 5FOM) in the editing site (PDB: 5FOM). (c) Candida albicans LeuRS in complex with the antifungal Kerydin–adenosine adduct bound to a hydroxonium ion (PDB: 5AGJ). (d) Full-length Escherichia coli LeuRS in complex with tRNALeu and the broad spectrum antibacterial AN3365–adenosine 76 (tRNA) bound to a hydroxonium ion (PDB: 3ZJV). E. coli tRNALeu bases are shown in blue sticks, and the different LeuRS protein domains are labeled.
To further support this observation, we measured the binding of Cmpd1 to LeuRS in the presence of AMP at different pH values (Figure S8a–c). It is expected that at a more basic pH the stabilization of the hydroxonium ion within the protein complex (Figure S8d) would be less favored. The binding thermodynamics show a clear pH dependence with a binding enthalpy of −8 kcal/mol at pH 7.5 that decreases to −7.0 kcal/mol at pH 8.5 and to −5.5 kcal/mol at pH 9.5 (Figure S8e). Experiments at lower pH values were not possible due to protein instability. Our results suggest an important role for the hydroxonium ion in the stabilization of the adduct and therefore in the LeuRS inhibition mechanism. However, it cannot be excluded that other interactions established at the binding pocket depend on pH and therefore contribute to the observed effect.
To investigate whether this hydroxonium-mediated LeuRS inhibition mechanism is shared across different benzoxaborole inhibitors, we analyzed all cocrystal structures of benzoxaborole adducts bound to LeuRS, and their corresponding apo protein structures (Table S4). Inspection of all unbound LeuRS structures shows no water molecule at this position. Conversely, the equivalent water molecule and the four hydrogen bonds observed in the TB LeuRS–AMP–Cmpd1 complex were invariably found in all structures of LeuRS bound to benzoxaboroles, independently of the species (prokaryotic or eukaryotic). Remarkably, this hydroxonium ion is found in an identical position in LeuRS structures of (1) M. tuberculosis in complex with Cmpd2, (2) intestinal pathogen C. hominis in complex with AN642620 (Figure 5b), (3) fungal pathogen C. albicans in complex with Kerydin and AN3018 (Figure 5c),10,21 and (4) complexes of E. coli with benzoxaborole antibiotics such as AN3365 (Figure 5d), a clinical candidate for the treatment of complicated infections by Gram-negative bacteria,12 and Streptococcus pneumoniae in complex with antipneumococcal compound ZCL039.14 The only complex lacking this water molecule is the structure of human LeuRS with AN642622 (Figure S9 and Table S4), a 4-bromo analogue of Cmpd2 that displays a high selectivity index (>300) for TB LeuRS compared to human LeuRS.13
Altogether, our results support a prodrug activation mechanism consisting of a nucleophilic attack of one of the ribose hydroxyls on the boron atom followed by formation of a hydroxonium ion that is tightly bound to the LeuRS inhibition complex (Figure S2). This hydroxonium ion forms an electrostatic interaction (3.8 Å) with the negatively charged boron atom and stabilizes its tetrahedral sp3 configuration, as observed in the structures of TB LeuRS with Cmpd1 and Cmpd2 (Figure 5a), as well as other pathogens (Figure 5b–d). The hydroxonium ion is slightly closer to the 2′ ribose hydroxyl (3.7 Å) compared to the 3′ hydroxyl (>4 Å) (Table S4), suggesting that the nucleophilic attack prior to the formation of the inhibition Adduct1a might be favored for hydroxyl 3′, followed by cyclization by hydroxyl 2′, hydroxonium ion release and stabilization of the sp3 oxaborole group of the inhibition adduct (Figure S2). Moreover, our protein-wide analysis of LeuRS structures shows that the mechanism of formation of the inhibition adduct and stabilization by the hydroxonium ion are shared across different pathogenic species, both from the prokaryotic and eukaryotic phylum.
Cmpd1 Interacts with Several Ribose-Based Biomolecules
We have shown above that the benzoxaboroles interact with ATP and AMP involving specifically the two adjacent hydroxyl groups on the ribose moiety of the adenosine. However, other ribose-based biomolecules are present in bacteria and humans at similar concentrations to ATP (5–10 mM) including UDP-α-d-glucose (∼3 mM), d-ribose-5-phosphate (∼1 mM), UTP (∼8 mM), gluconolactone (∼1 mM), CTP (∼3 mM), GTP (∼5 mM), UDP-N-acetylglucosamine (∼9 mM), and d-glucose-6-phosphate (∼9 mM).23 We therefore investigated whether the benzoxaboroles can form covalent adducts also with these ribose-based biomolecules. Our NMR and ITC results show that Cmpd1 reacts with all the above-mentioned biomolecules and form two adducts with dissociation constants in the mM range, except for d-glucose-6-phosphate that has the hydroxyl groups on opposite sides of the ring structure (Figures 6 and S10 and S11). Interestingly, this is also the case for gluconolactone; however, this molecule was nevertheless shown to be reactive with Cmpd1 (Kd = 1.5 mM), although one diastereomer appears to be highly dominant as demonstrated by the NMR data (Figure S10). For all biomolecules reacting with Cmpd1, we performed ITC experiments by titrating Cmpd1 into TB LeuRS in the presence of the different biomolecules (Figure S12). Our results show that Cmpd1 is not able to bind to LeuRS in the presence of any of these biomolecules. This highlights the strict requirement of the adenosine moiety of ATP, ADP, AMP, or tRNALeu for the binding of the benzoxazoles to the protein target.
Figure 6.

Benzoxaboroles interact with ribose-based biomolecules. (a) One-dimensional 1H NMR spectra showing the resonance of the proton in position 5 of Cmpd1 in the absence (row 1) and presence (rows 2–5) of different biomolecules (1:1 molar ratio). (b–e) ITC titrations of Cmpd1 with different biomolecules (b, GTP; c, UDP-α-d-glucose; d, d-ribose-5-P; e, d-glucose-6-P). (f) Dissociation constants as determined by ITC for the interaction of Cmpd1 with different biomolecules. No binding was observed with d-glucose-6-P. ITC and NMR data for all biomolecules are shown in Figures S10 and S11.
Discussion
We report the first example of a prodrug that activates its bioconversion in an adenosine-dependent manner. The benzoxaborole inhibitors of pathogenic LeuRS activate their bioconversion by forming a highly specific and reversible LeuRS inhibition adduct with adenosine-based biomolecules, including ATP, AMP, or the tRNALeu terminal base. In addition, benzoxaboroles also form weak, reversible adducts with several ribose-based biomolecules; however, none of the tested adducts bind to TB LeuRS. We note that forward genetic studies with pathogens resistant to the benzoxaboroles validated LeuRS as the primary target;9−13 however, we cannot exclude the possibility that some of the identified ribose-based prodrug adducts inhibit other targets than LeuRS.
Collectively, our data supports a model where at physiological concentrations of ATP, AMP, tRNALeu, and other ribose-based biomolecules, the free benzoxaborole prodrug coexists with distinct covalent adducts formed by the oxaborole group, thereby yielding species with different chemical features that provide unique advantages for drug permeability, transport, and metabolic stability (Figure 7).13,16 Equilibria of the prodrug with adducts, which likely form extracellularly and intracellularly, facilitate drug permeability through membranes with very different physicochemical properties. This explains why similar benzoxaboroles penetrate remarkably well into completely different cell types, spanning from prokaryotic to eukaryotic intracellular parasites infecting human hosts, before exerting inhibitory activity on LeuRS.9,11−13,15,20
Figure 7.

Prodrug activation mechanism with adenosine-based biomolecules upon per os administration. Upon oral intake, Cmpd1 (GSK3036656) spontaneously forms adducts with adenosine-based biomolecules found in extracellular body fluids facilitating intracellular permeability through different host and pathogen membranes, drug transport, and provides stability toward metabolic degradation. The reversible nature of the prodrug activation mechanism allows the co-existence of a free compound and the two covalent adducts, one of which, Adduct1a, is the active molecule that specifically inhibits the cytoplasmic LeuRS target with nanomolar potency. CNT = concentrative nucleoside transporter; ENT = equilibrative nucleoside transporter.
Similar to nucleoside-based drugs, the highly hydrophilic nature of the ribose-based benzoxaborole adducts can facilitate cell uptake,24,25 likely mediated by concentrative nucleoside transporters and equilibrative nucleoside transporters,26,27 for which multiple isoforms exist,26 with the additional advantage that benzoxaborole adducts are reversible and do not rely on enzyme activation. Moreover, intracellular and extracellular ATP are highly abundant (5–10 mM) in many biological fluids,23,28 including saliva, which might enable rapid prodrug activation upon oral intake (Figure 7). This is in agreement with the high oral bioavailability (>80%) of these inhibitors.13,16,19 Furthermore, the strength and reversibility of adduct formation combined with the much higher cellular concentrations of the reacting biomolecules compared to the administered prodrug suggest that only small fractions of biomolecules are trapped in the adducts, thereby explaining the low toxicities.13,16,19
We note some similarities of these benzoxaborole prodrugs to a natural product, TM84, secreted by Agrobacterium radiobacter.29 Interestingly, TM84 is a direct LeuRS inhibitor that mimics adenosine fused to leucine and efficiently blocks the protein synthesis of phytopathogenic bacteria (Figure S9a,b).30,31 Another example with analogies to our prodrug mechanism is the Chinese herbal-based drug Halofuginone.32 This drug is an ATP-dependent, however non-covalent, inhibitor of another aminoacyl-tRNA synthetase (ProRS) (Figure S9c). We also note that other boron-based conjugates have been reported before, whose activation depends on metabolic or chemical reactions.33−36 For example, Evodiamine boronates contain a boron-based carrier that triggers prodrug bioconversion in the presence of reactive oxygen species, and the carrier is subsequently released to form the active drug. Conversely, in benzoxaborole prodrugs, the boron atom is not only driving the prodrug bioconversion but also playing an important role in the formation of the inhibition adduct that targets LeuRS. Moreover, compared to these previous conjugates, the benzoxaborole activation mechanism reported in this work does not require pH changes, reactive oxygen species or metabolic reactions.
More generally, this adenosine-dependent activation mechanism represents a novel prodrug concept in chemistry that can be used to improve molecules for example by incorporating the oxaborole moiety into the structure of other challenging drugs.
Materials and Methods
Sample preparation, NMR and ITC experiments and crystal structure determination are described in the Materials and Methods in the Supporting Information.
Acknowledgments
The authors thank the European Synchrotron Radiation Facility and the European Molecular Biology Laboratory in Grenoble for beamtime access and technical support and the CERMAV, Grenoble for access to the ITC instrument. Funding is acknowledged from the Agence National de la Recherche (ANR) through ANR JCJC RC18114CC NovoTargetParasite (to A.P.), ANR-20-AMRB-0003-01 AntibiOxaborole (to A.P.), ANR ScaffoldDisorder (to M.R.J. and A.P.), and the FINOVI foundation (to A.P. and M.R.J.). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 801728—PARAMIR to L.S.). This work used the platforms of the Grenoble Instruct-ERIC center (ISBG; UAR 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-0005-02) and GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). The Institut de Biologie Structurale acknowledges integration into the Interdisciplinary Research Institute of Grenoble. Crystallography programs used in this project were compiled and configured by SBGrid.37
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c04808.
Materials and Methods section, Figures S1–S13, and Tables S1–S4, describing crystallographic, NMR and ITC data, and parameters derived from structural analysis (PDF)
Accession Codes
The crystal structure of TB LeuRS in complex with the Cmpd1–AMP Adduct1a has been deposited in the Protein Data Bank in Europe (PDBe) with accession code 7PQK and DOI: http://doi.org/10.2210/pdb7PQK/pdb.
The authors declare no competing financial interest.
Supplementary Material
References
- Rautio J.; Meanwell N. A.; Di L.; Hageman M. J. The Expanding Role of Prodrugs in Contemporary Drug Design and Development. Nat. Rev. Drug Discovery 2018, 17, 559–587. 10.1038/nrd.2018.46. [DOI] [PubMed] [Google Scholar]
- Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Järvinen T.; Savolainen J. Prodrugs: Design and Clinical Applications. Nat. Rev. Drug Discovery 2008, 7, 255–270. 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- Stella V.; Borchardt R.; Hageman M.; Oliyai R.; Maag H.; Tilley J.. Prodrugs: Challenges and Rewards; Springer-Verlag: New York, 2007. [Google Scholar]
- Geng J.; Zhang Y.; Gao Q.; Neumann K.; Dong H.; Porter H.; Potter M.; Ren H.; Argyle D.; Bradley M. Switching on Prodrugs Using Radiotherapy. Nat. Chem. 2021, 13, 805–810. 10.1038/s41557-021-00711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman D. J.; González-Fernández E.; Clavadetscher J.; Tucker L.; Staderini M.; Mount A. R.; Murray A. F.; Bradley M. Electrodrugs: An Electrochemical Prodrug Activation Strategy. Chem. Commun. 2018, 54, 9242–9245. 10.1039/C8CC04151B. [DOI] [PubMed] [Google Scholar]
- Bezagu M.; Clarhaut J.; Renoux B.; Monti F.; Tanter M.; Tabeling P.; Cossy J.; Couture O.; Papot S.; Arseniyadis S. In Situ Targeted Activation of an Anticancer Agent Using Ultrasound-Triggered Release of Composite Droplets. Eur. J. Med. Chem. 2017, 142, 2–7. 10.1016/j.ejmech.2017.03.057. [DOI] [PubMed] [Google Scholar]
- Karaman R.Prodrugs Design: A New Era; Nova Science Publishers Inc, 2014. [Google Scholar]
- Ibba M.; Soll D. Aminoacyl-TRNA Synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Rock F. L.; Mao W.; Yaremchuk A.; Tukalo M.; Crépin T.; Zhou H.; Zhang Y.-K.; Hernandez V.; Akama T.; Baker S. J.; Plattner J. J.; Shapiro L.; Martinis S. A.; Benkovic S. J.; Cusack S.; Alley M. R. K. An Antifungal Agent Inhibits an Aminoacyl-tRNA Synthetase by Trapping tRNA in the Editing Site. Science 2007, 316, 1759–1761. 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- Seiradake E.; Mao W.; Hernandez V.; Baker S. J.; Plattner J. J.; Alley M. R. K.; Cusack S. Crystal Structures of the Human and Fungal Cytosolic Leucyl-tRNA Synthetase Editing Domains: A Structural Basis for the Rational Design of Antifungal Benzoxaboroles. J. Mol. Biol. 2009, 390, 196–207. 10.1016/j.jmb.2009.04.073. [DOI] [PubMed] [Google Scholar]
- Palencia A.; Crépin T.; Vu M. T.; Lincecum T. L.; Martinis S. A.; Cusack S. Structural Dynamics of the Aminoacylation and Proofreading Functional Cycle of Bacterial Leucyl-tRNA Synthetase. Nat. Struct. Mol. Biol. 2012, 19, 677–684. 10.1038/nsmb.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez V.; Crépin T.; Palencia A.; Cusack S.; Akama T.; Baker S. J.; Bu W.; Feng L.; Freund Y. R.; Liu L.; Meewan M.; Mohan M.; Mao W.; Rock F. L.; Sexton H.; Sheoran A.; Zhang Y.; Zhang Y.-K.; Zhou Y.; Nieman J. A.; Anugula M. R.; Keramane E. M.; Savariraj K.; Reddy D. S.; Sharma R.; Subedi R.; Singh R.; O’Leary A.; Simon N. L.; De Marsh P. L.; Mushtaq S.; Warner M.; Livermore D. M.; Alley M. R. K.; Plattner J. J. Discovery of a Novel Class of Boron-Based Antibacterials with Activity against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2013, 57, 1394–1403. 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia A.; Li X.; Bu W.; Choi W.; Ding C. Z.; Easom E. E.; Feng L.; Hernandez V.; Houston P.; Liu L.; Meewan M.; Mohan M.; Rock F. L.; Sexton H.; Zhang S.; Zhou Y.; Wan B.; Wang Y.; Franzblau S. G.; Woolhiser L.; Gruppo V.; Lenaerts A. J.; O’Malley T.; Parish T.; Cooper C. B.; Waters M. G.; Ma Z.; Ioerger T. R.; Sacchettini J. C.; Rullas J.; Angulo-Barturen I.; Pérez-Herrán E.; Mendoza A.; Barros D.; Cusack S.; Plattner J. J.; Alley M. R. K. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium Tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. 10.1128/AAC.01339-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.-H.; Liu R.-J.; Fang Z.-P.; Zhang J.; Ding Y.-Y.; Tan M.; Wang M.; Pan W.; Zhou H.-C.; Wang E.-D. Discovery of a Potent Benzoxaborole-Based Anti-Pneumococcal Agent Targeting Leucyl-tRNA Synthetase. Sci. Rep. 2013, 3, 2475 10.1038/srep02475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoiki E.; Palencia A.; Guo D.; Ahyong V.; Dong C.; Li X.; Hernandez V. S.; Zhang Y.-K.; Choi W.; Gut J.; Legac J.; Cooper R.; Alley M. R. K.; Freund Y. R.; DeRisi J.; Cusack S.; Rosenthal P. J.; Anti-Malarial Benzoxaboroles Target P. Falciparum Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 4886–4895. 10.1128/AAC.00820-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Hernandez V.; Rock F. L.; Choi W.; Mak Y. S. L.; Mohan M.; Mao W.; Zhou Y.; Easom E. E.; Plattner J. J.; Zou W.; Pérez-Herrán E.; Giordano I.; Mendoza-Losana A.; Alemparte C.; Rullas J.; Angulo-Barturen I.; Crouch S.; Ortega F.; Barros D.; Alley M. R. K. Discovery of a Potent and Specific M. Tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-Chloro-7-(2-Hydroxyethoxy)Benzo[c][1,2]Oxaborol-1(3H)-Ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed] [Google Scholar]
- Lukarska M.; Palencia A.. Aminoacyl-tRNA Synthetases as Drug Targets. In The Enzymes: Biology of Aminoacyl-tRNA Synthetases; Academic Press, 2020; pp 321–350. [DOI] [PubMed] [Google Scholar]
- Kovalenko O. P.; Volynets G. P.; Rybak M. Y.; Starosyla S. A.; Gudzera O. I.; Lukashov S. S.; Bdzhola V. G.; Yarmoluk S. M.; Boshoff H. I.; Tukalo M. A. Dual-Target Inhibitors of Mycobacterial Aminoacyl-tRNA Synthetases among N-Benzylidene-N′-Thiazol-2-Yl-Hydrazines. MedChemComm 2019, 10, 2161–2169. 10.1039/C9MD00347A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenero D.; Derimanov G.; Carlton A.; Tonkyn J.; Davies M.; Cozens S.; Gresham S.; Gaudion A.; Puri A.; Muliaditan M.; Rullas-Trincado J.; Mendoza-Losana A.; Skingsley A.; Barros-Aguirre D. First-Time-in-Human Study and Prediction of Early Bactericidal Activity for GSK3036656, a Potent Leucyl-tRNA Synthetase Inhibitor for Tuberculosis Treatment. Antimicrob. Agents Chemother 2019, 63, e00240 10.1128/AAC.00240-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia A.; Liu R.-J.; Lukarska M.; Gut J.; Bougdour A.; Touquet B.; Wang E.-D.; Li X.; Alley M. R. K.; Freund Y. R.; Rosenthal P. J.; Hakimi M.-A.; Cusack S. Cryptosporidium and Toxoplasma Parasites Are Inhibited by a Benzoxaborole Targeting Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 5817–5827. 10.1128/AAC.00873-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Palencia A.; Seiradake E.; Ghaemi Z.; Cusack S.; Luthey-Schulten Z.; Martinis S. Analysis of the Resistance Mechanism of a Benzoxaborole Inhibitor Reveals Insight into the Leucyl-tRNA Synthetase Editing Mechanism. ACS Chem. Biol. 2015, 10, 2277–2285. 10.1021/acschembio.5b00291. [DOI] [PubMed] [Google Scholar]
- Liu R.-J.; Long T.; Li H.; Zhao J.; Li J.; Wang M.; Palencia A.; Lin J.; Cusack S.; Wang E.-D. Molecular Basis of the Multifaceted Functions of Human Leucyl-tRNA Synthetase in Protein Synthesis and Beyond. Nucleic Acids Res. 2020, 48, 4946–4959. 10.1093/nar/gkaa189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; Van Dien S. J.; Rabinowitz J. D. Absolute Metabolite Concentrations and Implied Enzyme Active Site Occupancy in Escherichia Coli. Nat. Chem. Biol. 2009, 5, 593–599. 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the Development of Nucleoside and Nucleotide Analogues for Cancer and Viral Diseases. Nat. Rev. Drug Discovery 2013, 12, 447–464. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- Sinokrot H.; Smerat T.; Najjar A.; Karaman R. Advanced Prodrug Strategies in Nucleoside and Non-Nucleoside Antiviral Agents: A Review of the Recent Five Years. Molecules 2017, 22, 1736 10.3390/molecules22101736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright N. J.; Lee S.-Y. Toward a Molecular Basis of Cellular Nucleoside Transport in Humans. Chem. Rev. 2021, 121, 5336–5358. 10.1021/acs.chemrev.0c00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J. D.; Yao S. Y. M.; Baldwin J. M.; Cass C. E.; Baldwin S. A. The Human Concentrative and Equilibrative Nucleoside Transporter Families, SLC28 and SLC29. Mol. Aspects Med. 2013, 34, 529–547. 10.1016/j.mam.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Gribble F. M.; Loussouarn G.; Tucker S. J.; Zhao C.; Nichols C. G.; Ashcroft F. M. A Novel Method for Measurement of Submembrane ATP Concentration. J. Biol. Chem. 2000, 275, 30046–30049. 10.1074/jbc.M001010200. [DOI] [PubMed] [Google Scholar]
- Roberts W. P.; Tate M. E.; Kerr A. Agrocin 84 Is a 6-N-Phosphoramidate of an Adenine Nucleotide Analogue. Nature 1977, 265, 379–381. 10.1038/265379a0. [DOI] [PubMed] [Google Scholar]
- Reader J. S.; Ordoukhanian P. T.; Kim J.-G.; de Crécy-Lagard V.; Hwang I.; Farrand S.; Schimmel P. Major Biocontrol of Plant Tumors Targets tRNA Synthetase. Science 2005, 309, 1533 10.1126/science.1116841. [DOI] [PubMed] [Google Scholar]
- Chopra S.; Palencia A.; Virus C.; Tripathy A.; Temple B. R.; Velazquez-Campoy A.; Cusack S.; Reader J. S. Plant Tumour Biocontrol Agent Employs a tRNA-Dependent Mechanism to Inhibit Leucyl-tRNA Synthetase. Nat. Commun. 2013, 4, 1417 10.1038/ncomms2421. [DOI] [PubMed] [Google Scholar]
- Keller T. L.; Zocco D.; Sundrud M. S.; Hendrick M.; Edenius M.; Yum J.; Kim Y.-J.; Lee H.-K.; Cortese J. F.; Wirth D. F.; Dignam J. D.; Rao A.; Yeo C.-Y.; Mazitschek R.; Whitman M. Halofuginone and Other Febrifugine Derivatives Inhibit Prolyl-tRNA Synthetase. Nat. Chem. Biol. 2012, 8, 311–317. 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslah H.; Skarbek C.; Gourson C.; Plamont M.-A.; Pethe S.; Jullien L.; Le Saux T.; Labruère R. In-Cell Generation of Anticancer Phenanthridine Through Bioorthogonal Cyclization in Antitumor Prodrug Development. Angew. Chem., Int. Ed. 2021, 60, 24043–24047. 10.1002/anie.202110041. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Zheng J.; Xiao L.; Shang T.; Cai Y.; Li Y.; Xu Y.; Chen X.; Liu Y.; Yang B. Construction of a Phenylboronic Acid-Functionalized Nano-Prodrug for pH-Responsive Emodin Delivery and Antibacterial Activity. ACS Omega 2021, 6, 8672–8679. 10.1021/acsomega.1c00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Gao Y.; Chen T.; Xu Y.; Lu W.; Yu J.; Xiao Y.; Liu S. Reduction-Triggered Release of CPT from Acid-Degradable Polymeric Prodrug Micelles Bearing Boronate Ester Bonds with Enhanced Cellular Uptake. ACS Biomater. Sci. Eng. 2017, 3, 3364–3375. 10.1021/acsbiomaterials.7b00618. [DOI] [PubMed] [Google Scholar]
- Li X.; Wu S.; Dong G.; Chen S.; Ma Z.; Liu D.; Sheng C. Natural Product Evodiamine with Borate Trigger Unit: Discovery of Potent Antitumor Agents against Colon Cancer. ACS Med. Chem. Lett. 2020, 11, 439–444. 10.1021/acsmedchemlett.9b00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin A.; Eisenbraun B.; Key J.; Sanschagrin P. C.; Timony M. A.; Ottaviano M.; Sliz P. Collaboration Gets the Most out of Software. eLife 2013, 2, e01456 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.