

Abstract
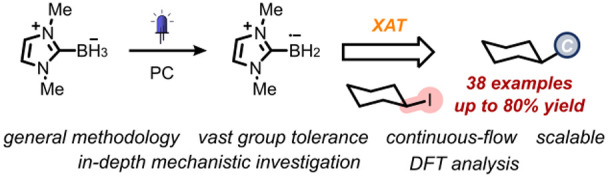
Herein, we present a comprehensive study on the use of N-heterocyclic carbene (NHC)-ligated boryl radicals to enable C(sp3)–C(sp3) bond formation under visible-light irradiation via Halogen-Atom Transfer (XAT). The methodology relies on the use of an acridinium dye to generate the boron-centered radicals from the corresponding NHC-ligated boranes via single-electron transfer (SET) and deprotonation. These boryl radicals subsequently engage with alkyl halides in an XAT step, delivering the desired nucleophilic alkyl radicals. The present XAT strategy is very mild and accommodates a broad scope of alkyl halides, including medicinally relevant compounds and biologically active molecules. The key role of NHC-ligated boryl radicals in the operative reaction mechanism has been elucidated through a combination of experimental, spectroscopic, and computational studies. This methodology stands as a significant advancement in the chemistry of NHC-ligated boryl radicals, which had long been restricted to radical reductions, enabling C–C bond formation under visible-light photoredox conditions.
Introduction
The possibility to exploit photonic energy in organic synthetic endeavors has dramatically impacted the way chemists assemble molecules. In particular, photocatalysis has enabled a convenient entry to open-shell intermediates,1 spurring the development of efficient manifolds for the generation of C-,2 N-,3 and O-4 centered radicals, as well as halogen radicals,5 which can be subsequently used to forge new chemical bonds.6 In contrast, boron-based congeners have long remained in obscurity,7 mainly due to the intrinsic difficulties associated with the handling of these highly electron-deficient and unstable intermediates. However, ligated boryl radicals (LBRs),8 i.e. boron-centered radicals where the boron atom is coordinated with a Lewis base, are more stable and provide a suitable entry for use in radical chemistry (Figure 1A).9,10
Figure 1.
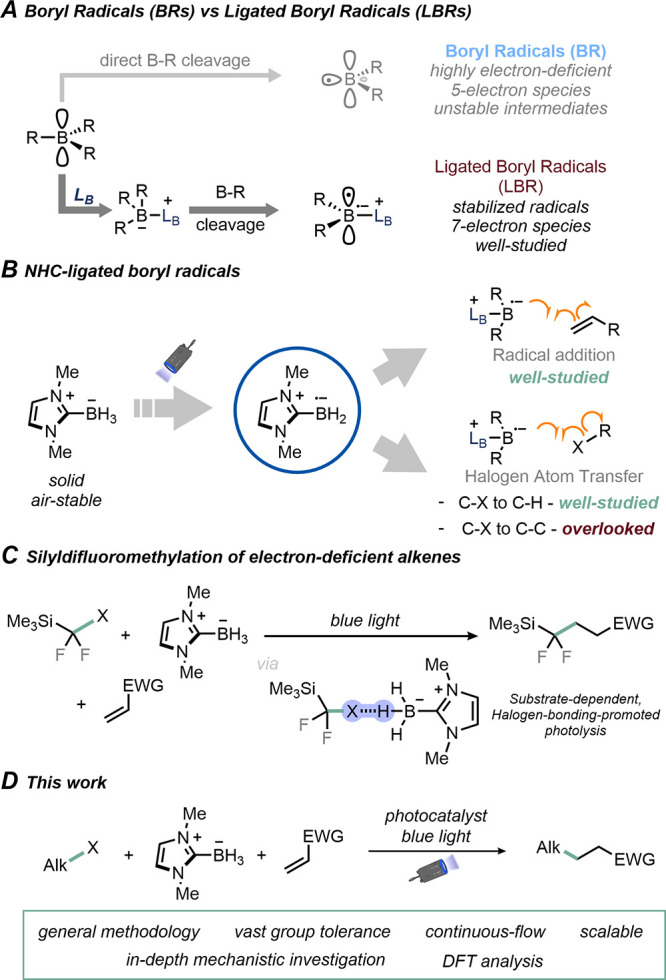
(A) Boryl radicals (BRs) versus ligated boryl radicals (LBRs). (B) NHC boranes as a bench-stable source of LBRs. (C) Silyldifluoromethylation of electron-deficient alkenes. (D) This work. LB: Lewis base.
In particular, N-heterocyclic carbene-based (NHC) boranes are emerging as a convenient source of LBRs: NHC boranes are stable crystalline compounds, and a diverse array of NHCs can be ligated to boranes allowing the LBR properties to be fine-tuned.11 Notably, these boranes can be uniquely paired with photocatalysis to generate the targeted boron-centered radicals.12 Herein, a photocatalyst absorbs visible light and engages with the ligated borane in a single-electron transfer step which, upon deprotonation, generates the corresponding boryl radical. Although these boron-centered radicals have attracted mainly interest from the synthetic community as nucleophilic radicals,13,14 they have also been used in the role of halogen-atom transfer (XAT) agents.
In the latter scenario, the halogen-affinity of the LBR is exploited for the homolytic activation of a C–X bond to yield carbon-centered radicals. However, this manifold has been so far mainly used to reduce C–X bonds into the corresponding C–H bonds via a radical chain mechanism.11,12,15 Surprisingly, boryl radicals have been largely overlooked for the construction of C–C bonds (Figure 1B). In an early example, the radical silyldifluoromethylation of electron-deficient alkenes was reported.16 Herein, a very specific interaction, based on halogen-bonding between the substrate and an NHC borane, was needed to trigger the desired C–X bond photolysis and subsequently initiate the radical chain mechanism sustained by the LBR (Figure 1C). Inspired by this report, we questioned whether it would be possible to realize a more general strategy to generate the pivotal ligated boryl radical. Such a pathway might allow the engagement of a broader array of substrates, ultimately leading to a general approach for C–C bond formation. Moreover, succeeding in this challenge would provide a cheap, tunable, and sustainable alternative for other XAT-based approaches17 using silyl18 and α-aminoalkyl radicals.19 To this end, we disclose our results regarding the development of a mild and broadly applicable protocol for C(sp3)–C(sp3) bond formation using photoinduced XAT by NHC-ligated boryl radicals under blue-light irradiation (Figure 1D). The key role of the NHC-ligated boryl radicals in the operative reaction mechanism has been uncovered through a combination of experimental, spectroscopic, and computational studies.
Results and Discussion
At the outset of our investigations, we recognized the importance of finding the ideal combination of ligated borane and photocatalyst to establish an efficient and competent system to promote the desired reactivity. In detail, the photocatalyst would absorb the visible light and subsequently generate the LBR via a SET followed by deprotonation.12 The resulting boryl radical would be ultimately entrusted with the XAT step.
We immediately realized that the success of our plan hinges on (i) the redox potentials of the borane and the photocatalyst and (ii) the halogen-affinity of the resulting LBR. Based on literature and our experimental data (see Section 11 in the Supporting Information), we identified NHC-ligated borane B1 as an ideal candidate for our purposes: in fact, its oxidation potential (Epa(B1•+/B1)) is +0.89 V vs SCE,20 which suggests that this approach should be feasible in combination with routinely used photoredox catalysts. Other ligated boranes tested (see Section 11 in the Supporting Information) were found to have an exceedingly high redox potential, thus preventing the formation of the desired ligated boryl radical.
Next, we started our investigation by screening different photoredox catalysts that possess a higher excited state reduction potential (E(PC*/PCred)) than that of B1 (Table 1). Our experiments revealed that, when a degassed CH3CN solution of 2a (0.1 M), 1a (2 equiv), and B1 (1 equiv) was irradiated with blue light (λ = 456 nm, 12 h, rt) in the presence of 2 mol % of the organic photocatalysts PC1 or PC2, product 3 could be obtained in 43–44% GC yield (Table 1, entries 1 and 2). In contrast, Ru(bpy)3(PF6)2 (PC3) gave worse results (Table 1, entry 3).
Table 1. Optimization of Conditionsa.


GC yields are given using biphenyl as external standard.
Next, we screened the effect of the solvent on the transformation and noticed that protic reaction mixtures boosted the reaction yield (Table 1, entry 4: up to 68% in CH3CN/H2O 9:1). Fine-tuning the ratio of the reagents, the photocatalyst loading, and reaction time allowed an excellent 79% yield to be obtained for the targeted hydroalkylation (Table 1, entries 5–6). Several control experiments revealed that excluding light, PC1, or B1 did not lead to any product formation (Table 1, entries 7–8). Moreover, 3 was not produced at elevated temperatures either (Table 1, entry 9). The exclusion of molecular oxygen appeared to be crucial as air-equilibrated conditions led to a significantly reduced yield (Table 1, entry 10: 37%), while O2-saturation shut down reactivity (Table 1, entry 11). It is important to mention that, when direct UV-A light irradiation (Kessil lamp, λ = 390 nm, full intensity) was used, product 3 was formed in a 75% assay yield, without any photocatalyst added.
Using the optimized set of conditions (Table 1, entry 6), we next evaluated the scope of the visible-light induced hydroalkylation protocol (Figure 2).
Figure 2.

Substrate scope for LBRs-mediated XAT under visible-light irradiation. For secondary and tertiary organic halides: 2 (0.5 mmol), 1 (2 equiv), B1 (1.2 equiv) in CH3CN/H2O 9:1 (5 mL) in the presence of PC1 (5 mol %), 3 h. For primary organic halides: 2 (2 equiv), 1 (0.5 mmol), B1 (1.2 equiv) in CH3CN/H2O 9:1 (5 mL) in the presence of PC1 (5 mol %), 12 h. Solutions were bubbled with N2 (5 min) prior to irradiation (λ = 456 nm). a Reaction time: 18 h. b Solvent: ethyl acetate. c See Supporting Information for further details. d NaI (2 equiv) was added to the reaction mixture. brsm: based on remaining starting material.
Hereto, dimethyl maleate was combined with several alkyl iodides, and we found that the expected products were obtained in all cases (4–23). Notably, the acid-sensitive acetal function is well tolerated under our optimized conditions (8, 80%). Also iodides of medicinally relevant nitrogen-containing scaffolds, including Boc-protected azetidine, pyrrolidine, and piperidine, were competent reaction partners, allowing isolation of the corresponding adducts in good yields (9–12, 57–73%). In a similar vein, oxygen- and sulfur-containing alkyl iodides could be engaged in the reaction protocol (13–15, 40–77%). Next, we employed 1-iodoadamantane as a model for tertiary alkyl iodides, and we found that the hydroalkylated product was obtained in very good yield (16, 79%). The presence of a free carboxylic acid slightly reduced the reaction efficiency; however, the targeted compound could still be accessed in synthetically useful quantities (17, 56%).
Finally, we focused on primary alkyl iodides, which are interesting yet more challenging to engage in the reaction protocol. A slight modification of the reaction conditions (see GP4 in the Supporting Information, including an inverted organic halide/olefin ratio and extended light exposure) resulted in complete conversion and yielded the compounds 18–23 in satisfactory yields (40–57%). Of note are the unprotected aliphatic alcohols (20) and easily oxidizable phenols (23).
With respect to the SOMOphile scope, we found that different olefins could successfully take part in the transformation (3, 24–30). The product of our benchmark reaction (3) was obtained in 73% yield. Notably, when using phenyl vinyl sulfone (2c), we were able to trap stabilized benzyl radicals, while the reaction with dimethyl maleate did not afford the expected product. Also, norbornenone, N-phenyl acrylamide, methyl acrylate, and diethyl vinyl phosphonate could be engaged as SOMOphiles in the reaction protocol, showing its tolerance toward a wide variety of functional groups, such as ketones, amides, esters, and phosphonates (27–30, 40–59% yield). The applicability of our visible-light photocatalytic hydroalkylation process was also demonstrated by the fact that several derivatives of biologically active molecules could be readily modified; these include densely functionalized compounds, such as derivatives of sugars (i.e., α-d-glucofuranose and α-l-sorbofuranose), caffeine, cyclouridine, and even a dipeptide (31−36, 43–76%).
While alkyl bromides were not reactive under our original reaction conditions, we found that simple addition of NaI (2 equiv) could obviate this issue by generating the corresponding alkyl iodide in situ via SN2. With this operationally facile approach, we could subject various primary alkyl bromides to the hydroalkylation strategy and obtain the targeted compounds in decent yields (37–40, 44–50% yield).
Finally, we also successfully translated our batch protocol to a fast and scalable continuous-flow process, which should enable a fast transition between medicinal and process chemistry (see GP6 described in the Supporting Information).21 In flow, we were able to prepare compounds 3, 35, and 36 in good yields requiring only 30 min of light exposure. We also exploited the continuous-flow technology to scale our benchmark reaction up to 5 mmol (78% yield of the isolated product, Figure 3), corresponding to a productivity of 21 g d–1 of compound 3.
Figure 3.

Translation to continuous-flow conditions (see GP6 in the Supporting Information) and scale-up.
To gain insight into the reaction mechanism, we performed a series of experimental and computational studies. In particular, we identified two crucial steps to be investigated, i.e. (i) the generation of the ligated boryl radical22 and (ii) the occurrence of a radical chain process. To reveal the presence of the ligated boryl radical, we recorded an EPR spectrum of a deoxygenated benzene solution of PC1 (0.05 M) and B1 (0.05 M) containing phenyl N-tert-butylnitrone (PBN, 0.0125 M) as a spin trap. Prior to irradiation no signal was observed; however, after continuous irradiation for 15 min (λ = 460 nm) we clearly saw the appearance of two distinct features: one can be attributed to the reduced photocatalyst (acridine radical),23 while the second feature is derived from the trapping of the ligated boryl radical with PBN (Figure 4a; for further details, see Section 6.1 in the Supporting Information).15a The assignment is further supported by the observation of the adduct of the LBR with PBN (PBN*NHCBH2•) in HRMS (Figure S5). These experiments unequivocally show the formation of the desired LBR under photoredox conditions. Direct observation of the ligated boryl radical via EPR was unsuccessful due to its short lifetime.24 As further evidence, we also tracked the interaction between PC1 and B1 via UV–vis spectroscopy. In particular, when a deoxygenated CH3CN solution (10–5 M) of PC1 and B1 was irradiated at 456 nm, we observed that the former was directly converted into a species absorbing in the 450–700 nm range (two isosbestic points were discerned; see Figure 4b). We propose this species to be the acridine radical generated upon single-electron reduction by B1, based on a comparison with the literature for a similar compound.25
Figure 4.
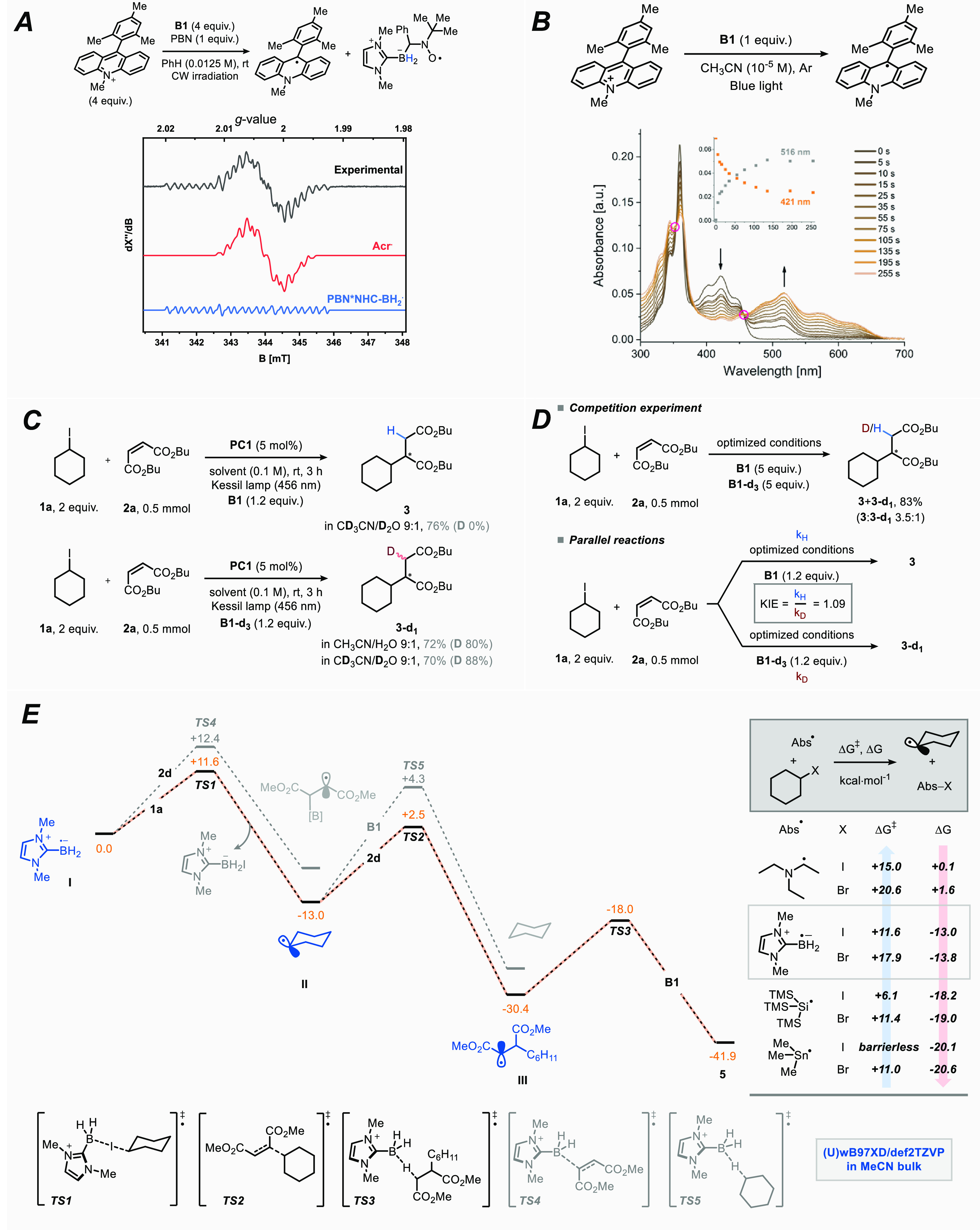
(A) Electron paramagnetic resonance (EPR) spectrum (black) obtained upon irradiation (λ = 460 nm) of a deoxygenated 0.0125 M benzene solution of phenyl N-tert-butylnitrone containing PC1 (0.05 M) and B1 (0.05 M). Simulated profiles for acridine radical generated from PC1 upon SET (red) and of the PBN*NHCBH2• (blue) are also shown (see Section 6.1 in the Supporting Information for experimental and simulation parameters). (B) UV–vis spectra of a deoxygenated CH3CN solution of PC1 and B1 (both 10–5 M) irradiated over 4.25 min. (C) Deuteration experiments. (D) Determination of the kinetic isotope effect (KIE). (E) Computational investigation.
In order to get some insights into the radical chain mechanism, we conducted experiments with deuterium-labeled substrates, and the results are collected in Figure 4c. The deuterium incorporation was calculated via 1H NMR on purified products. Overall, these experiments showed that deuterium incorporation (product 3-d1) was only observed when deuterated borane B1-d3 was exploited as an XAT agent, while the use of B1 resulted in the formation of product 3 exclusively.
These results reveal that the hydroalkylated product is obtained upon HAT from another molecule of B1 rather than the solvent, thus pointing toward a radical chain mechanism. We next wondered if the latter could be the rate-determining step of the transformation; therefore, we set off to evaluate the kinetic isotope effect (KIE) of the reaction (Figure 4d). First, we measured the KIE through a competition experiment. Hereto, we performed our benchmark reaction in the presence of an equimolar mixture of B1 and B1-d3 (5 equiv each) and a KIE value of 3.5 was found. However, when we calculated this value with the parallel reactions method by performing independent reactions under optimized conditions, one containing B1 and one containing B1-d3, we found a KIE of only 1.09. Taken together, these experiments suggest that the HAT step is not involved in the rate-determining step of the reaction.26 Finally, we determined a quantum yield of 2%. Such a modest value is in accordance with a process being supported by either short-lived radical chain propagations, an inefficient initiation process27 or decomposition of the photocatalyst (further mechanistic insights are reported in Section 6 of the Supporting Information).28
We also performed a computational investigation intended to model the entire reaction profile through the simulation of all the key steps, including some possible parasitic pathways. Thus, we adopted DFT at the ωB97xD/def2TZVP level of theory to optimize the relevant stationary points, also including the effect of the solvent through an implicit model (Figure 4e; see also Section 10 of the Supporting Information for further details). We started by considering LBR I reacting with iodocyclohexane 1a through TS1 (ΔG‡ = +11.6 kcal·mol–1) to afford cyclohexyl radical II. This nucleophilic radical (II) adds subsequently onto dimethyl maleate 2b through TS2 (ΔG‡ = +15.5 kcal·mol–1) to deliver radical adduct III. Next, the targeted hydroalkylated product 5 is formed through reaction of III with NHC-ligated borane B1 via TS3 (ΔG‡ = +12.4 kcal·mol–1). Notably, all these steps are exergonic in nature, with ΔG values in the −11.9 to −17.4 kcal·mol–1 range.
In this intricate ballet of fleeting radical intermediates, we realized that a careful balance between the XAT, the radical addition, and the final HAT steps was crucial to avoid parasitic reaction pathways.11a,11b,15b Accordingly, we also evaluated the possibility for these intermediates to undergo competitive, yet nonproductive pathways, including the direct addition of LBR I onto dimethyl maleate 2b with formation of a new B–C bond (through TS4) and the reduction of the cyclohexyl radical II to cyclohexane (through TS5). However, our computational analysis revealed that both steps occur with higher activation energies and less negative energy gains compared to those describing the desired process. Intrigued by the lack of reactivity of organic bromides in our reaction, we also computed ΔG and ΔG‡ for the XAT step by LBR I for bromocyclohexane.
By comparing the results with those obtained for the iodo analogue 1a, it seems that the difference in reactivity can be mainly attributed to kinetic factors (ΔG‡R-I = +11.6 kcal·mol–1 vs ΔG‡R-Br = +17.9 kcal·mol–1), as the process shows similar driving forces for both halides (ΔGR-I = −13.0 kcal·mol–1 vs ΔGR-Br = −13.8 kcal·mol–1).
Finally, we were interested in comparing quantitatively LBR I with other commonly used halogen abstractors, including α-aminoalkyl and (tris(trimethylsilyl)silyl radicals and a conventional tin-based XAT reagent (Me3Sn•). As depicted in Figure 4e, a clear trend emerges. On the one hand, the XAT step shows very low activation energies in the case of metalloidyl radicals (i.e., R3Si• and R3Sn•; for the tin-based abstractor it is barrierless), while LBR I and the α-aminoalkyl radical display significant ΔG‡ values (+11.6 and 15.0 kcal·mol–1, respectively). On the other hand, from a thermodynamic point of view, the XAT process is highly exergonic for metalloidyl radicals, moderately exergonic for LBR I, and essentially thermoneutral for the α-amino radical. In the latter case, the formation of an iminium ion resulting from the elimination of iodide was invoked as the driving force for the whole process.19a
With both experimental and computational insights considered together, a mechanistic scenario is proposed in Figure 5. PC1 absorbs light resulting in formation of the corresponding highly oxidizing excited state (E(PC*/PCred) = +2.06 V vs SCE).29 This excited state can react with B1 (Epa(B1•+/B1) = +0.89 V vs (SCE) via single-electron transfer to afford LBR I upon deprotonation. The latter intermediate is entrusted with the desired XAT step, which is expected to be relatively fast (k ≈ 108 M–1 s–1),30 thus yielding the alkyl radical II. This radical can be subsequently trapped by the electron-poor olefin to give adduct III. The observed inhibition effect of O2 (Table 1, entries 10–11) can be explained by taking into account that LBR I is known to react even faster with molecular oxygen (k > 108 M–1 s–1),30 thus confiscating this crucial intermediate for the XAT event. Alternatively, it has been reported that molecular oxygen can also quench the excited state of PC1 at diffusion controlled rates to form singlet oxygen (k = 2 × 109 M–1 s–1).31 Next, as shown by the deuterium labeling experiments, III abstracts a hydrogen atom from B1 in a polarity-matched step to give product 3 and subsequently kicks off the chain propagation.32
Figure 5.

Proposed mechanism for the photoinduced XAT by N-heterocarbene boryl radicals for C–C bond formation. Redox potentials are reported vs SCE.
Conclusions
In conclusion, we have shown that N-heterocyclic carbene (NHC) borane B1 is an efficient XAT agent to sustain the radical hydroalkylation of olefins. This is a significant advancement to previous reports where NHC-ligated boryl radicals were mainly used as radical chain carriers for radical reductions. Our method shows remarkable generality, robustness, and versatility, as it does not rely on any interaction between the ligated borane and the organic halide to generate nucleophilic alkyl radicals under visible light irradiation. Due to the mild reaction conditions, it is applicable to a vast array of substrates, including biologically active compounds. And finally, continuous-flow technology can be exploited to accelerate and scale our methodology.
A detailed experimental and spectroscopic mechanistic investigation describes the key role of the NHC-based boryl radicals in the operative reaction mechanism. This is further corroborated by computational analysis, indicating that the described process is the most favorable one with respect to possible competing pathways.
While NHC-ligated boryl radicals have been only recently exploited in radical chemistry, this investigation represents an important step toward the appreciation and the exploitation of the properties and the reactivity of these boryl species. Hence, we are confident that this work will stimulate further research into the use of LBRs for radical-based synthetic transformations.
Acknowledgments
T.W. has received support from the China Scholarship Council (CSC) for her PhD studies. L.C. acknowledges European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 101023615 (project name: HAT-TRICK). Calculations were carried out at the CINECA Supercomputer Center (Italy), with computer time granted by ISCRA projects (codes: HP10CIH7D5 and HP10CXZVO3).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10444.
Experimental procedures, characterization data of synthesized compounds, copies of NMR spectra, complete mechanistic investigation. The primary NMR FID files for starting materials, compounds 3–40, as well as optimized geometries of species displayed in Figure 4e, IRC and PES profiles are available in the FigShare repository at https://www.doi.org/10.21942/uva.20459517. (PDF)
Author Contributions
∥ T.W. and L.C. contributed equally to this work.
China Scholarship Council (CSC) and European Union’s Horizon 2020 research and innovation program
The authors declare no competing financial interest.
Supplementary Material
References
- a Stephenson C.; Yoon T.; MacMillan D. W. C.. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018. 10.1002/9783527674145. [DOI] [Google Scholar]; b Capaldo L.; Ravelli D.; Fagnoni M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C-H Bonds Elaboration. Chem. Rev. 2022, 122 (2), 1875–1924. 10.1021/acs.chemrev.1c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chan A. Y.; Perry I. B.; Bissonnette N. B.; Buksh B. F.; Edwards G. A.; Frye L. I.; Garry O. L.; Lavagnino M. N.; Li B. X.; Liang Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122 (2), 1485–1542. 10.1021/acs.chemrev.1c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Holmberg-Douglas N.; Nicewicz D. A. Photoredox-Catalyzed C-H Functionalization Reactions. Chem. Rev. 2022, 122 (2), 1925–2016. 10.1021/acs.chemrev.1c00311. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Murray P. R. D.; Cox J. H.; Chiappini N. D.; Roos C. B.; McLoughlin E. A.; Hejna B. G.; Nguyen S. T.; Ripberger H. H.; Ganley J. M.; Tsui E.; et al. Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis. Chem. Rev. 2022, 122 (2), 2017–2291. 10.1021/acs.chemrev.1c00374. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Witzel S.; Hashmi A. S. K.; Xie J. Light in Gold Catalysis. Chem. Rev. 2021, 121 (14), 8868–8925. 10.1021/acs.chemrev.0c00841. [DOI] [PubMed] [Google Scholar]
- Huang C.-Y.; Li J.; Li C.-J. Photocatalytic C(sp3) Radical Generation via C–H, C–C, and C–X Bond Cleavage. Chem. Sci. 2022, 13 (19), 5465–5504. 10.1039/D2SC00202G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkäs M. D. Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS Catal. 2017, 7 (8), 4999–5022. 10.1021/acscatal.7b01385. [DOI] [Google Scholar]
- Chang L.; An Q.; Duan L.; Feng K.; Zuo Z. Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122 (2), 2429–2486. 10.1021/acs.chemrev.1c00256. [DOI] [PubMed] [Google Scholar]
- Bonciolini S.; Noël T.; Capaldo L. Synthetic Applications of Photocatalyzed Halogen-Radical Mediated Hydrogen Atom Transfer for C–H Bond Functionalization. Eur. J. Org. Chem. 2022, 2022, e202200417 10.1002/ejoc.202200417. [DOI] [Google Scholar]
- Noël T.; Zysman-Colman E. The Promise and Pitfalls of Photocatalysis for Organic Synthesis. Chem. Catal. 2022, 2 (3), 468–476. 10.1016/j.checat.2021.12.015. [DOI] [Google Scholar]
- a Su Y.; Kinjo R. Boron-Containing Radical Species. Coord. Chem. Rev. 2017, 352, 346–378. 10.1016/j.ccr.2017.09.019. [DOI] [Google Scholar]; b Renaud P.Boron in Radical Chemistry. In Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd.: Chichester, U.K., 2012. 10.1002/9781119953678.rad020. [DOI] [Google Scholar]
- Capaldo L.; Noël T.; Ravelli D. Photocatalytic Generation of Ligated Boryl Radicals from Tertiary Amine-Borane Complexes: An Emerging Tool in Organic Synthesis. Chem. Catal. 2022, 2 (5), 957–966. 10.1016/j.checat.2022.03.005. [DOI] [Google Scholar]
- For representative works on ligated boryl radicals (LBRs), see:; a Baban J. A.; Roberts B. P. An Electron Spin Resonance Study of Phosphine-Boryl Radicals; Their Structures and Reactions with Alkyl Halides. J. Chem. Soc. Perk. T. 1984, 10, 1717. 10.1039/p29840001717. [DOI] [Google Scholar]; b Baban J. A.; Roberts B. P. An e.s.r. Study of Amine-Boryl Radicals (R3N–BH2•) in Solution. J. Chem. Soc., Chem. Commun. 1983, 21, 1224–1226. 10.1039/C39830001224. [DOI] [Google Scholar]; c Baban J. A.; Marti V. P. J.; Roberts B. P. Spin-Trapping of Ligated Boryl Radicals. Tetrahedron Lett. 1985, 26 (10), 1349–1352. 10.1016/S0040-4039(00)94891-3. [DOI] [Google Scholar]; d Baban J. A.; Marti V. P. J.; Roberts B. P. Ligated Boryl Radicals. Part 2. Electron Spin Resonance Studies of Trialkylamine–Boryl Radicals. J. Chem. Soc. Perk. T. 2 1985, 11, 1723–1733. 10.1039/P29850001723. [DOI] [Google Scholar]; e Lu D.; Wu C.; Li P. Synergistic Effects of Lewis Bases and Substituents on the Electronic Structure and Reactivity of Boryl Radicals. Chem.—Eur. J. 2014, 20 (6), 1630–1637. 10.1002/chem.201303705. [DOI] [PubMed] [Google Scholar]; f Paul V.; Roberts B. P. Polarity Reversal Catalysis of Hydrogen Atom Abstraction Reactions. J. Chem. Soc., Chem. Commun. 1987, 17, 1322–1324. 10.1039/c39870001322. [DOI] [Google Scholar]; g Paul V.; Roberts B. P. Homolytic Reactions of Ligated Boranes. Part 10. Electron Spin Resonance Studies of Radicals Derived from Ligated Arylboranes. J. Chem. Soc., Perk. T. 2 1988, 10, 1895–1901. 10.1039/p29880001895. [DOI] [Google Scholar]; h Paul V.; Roberts B. P. Homolytic Reactions of Ligated Boranes. Part 8. Electron Spin Resonance Studies of Radicals Derived from Ligated Alkylboranes. J. Chem. Soc., Perk. T. 2 1988, 7, 1183–1193. 10.1039/p29880001183. [DOI] [Google Scholar]; i Baban J. A.; Roberts B. P. Homolytic Reactions of Ligated Boranes. Part 9. Overall Addition of Alkanes to Electron-Deficient Alkenes by a Radical Chain Mechanism. J. Chem. Soc., Perk. T. 2 1988, 7, 1195–1200. 10.1039/p29880001195. [DOI] [Google Scholar]
- For recent synthetic application of LBRs, see:; a Zhang Z.-Q.; Sang Y.-Q.; Wang C.-Q.; Dai P.; Xue X.-S.; Piper J. L.; Peng Z.-H.; Ma J.-A.; Zhang F.-G.; Wu J. Difluoromethylation of Unactivated Alkenes Using Freon-22 through Tertiary Amine-Borane-Triggered Halogen Atom Transfer. J. Am. Chem. Soc. 2022, 144 (31), 14288–14296. 10.1021/jacs.2c05356. [DOI] [PubMed] [Google Scholar]; b Lei G.; Xu M.; Chang R.; Funes-Ardoiz I.; Ye J. Hydroalkylation of Unactivated Olefins via Visible-Light-Driven Dual Hydrogen Atom Transfer Catalysis. J. Am. Chem. Soc. 2021, 143 (29), 11251–11261. 10.1021/jacs.1c05852. [DOI] [PubMed] [Google Scholar]; c Kim J. H.; Constantin T.; Simonetti M.; Llaveria J.; Sheikh N. S.; Leonori D. A Radical Approach for the Selective C–H Borylation of Azines. Nature 2021, 595 (7869), 677–683. 10.1038/s41586-021-03637-6. [DOI] [PubMed] [Google Scholar]
- a Ueng S.-H.; Solovyev A.; Yuan X.; Geib S. J.; Fensterbank L.; Lacôte E.; Malacria M.; Newcomb M.; Walton J. C.; Curran D. P. N -Heterocyclic Carbene Boryl Radicals: A New Class of Boron-Centered Radical. J. Am. Chem. Soc. 2009, 131 (31), 11256–11262. 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]; b Ueng S.-H.; Makhlouf Brahmi M.; Derat E.; Fensterbank L.; Lacôte E.; Malacria M.; Curran D. P. Complexes of Borane and N-Heterocyclic Carbenes: A New Class of Radical Hydrogen Atom Donor. J. Am. Chem. Soc. 2008, 130 (31), 10082–10083. 10.1021/ja804150k. [DOI] [PubMed] [Google Scholar]; c Curran D. P.; Solovyev A.; Makhlouf Brahmi M.; Fensterbank L.; Malacria M.; Lacôte E. Synthesis and Reactions of N-Heterocyclic Carbene Boranes. Angew. Chem., Int. Ed. 2011, 50 (44), 10294–10317. 10.1002/anie.201102717. [DOI] [PubMed] [Google Scholar]
- Taniguchi T. Advances in Chemistry of N-Heterocyclic Carbene Boryl Radicals. Chem. Soc. Rev. 2021, 50 (16), 8995–9021. 10.1039/D1CS00385B. [DOI] [PubMed] [Google Scholar]
- For reviews about radical addition of LBRs onto unsaturated systems, see:; a Lai D.; Ghosh S.; Hajra A. Light-Induced Borylation: Developments and Mechanistic Insights. Org. Biomol. Chem. 2021, 19 (20), 4397–4428. 10.1039/D1OB00323B. [DOI] [PubMed] [Google Scholar]; b Taniguchi T. Boryl Radical Addition to Multiple Bonds in Organic Synthesis. Eur. J. Org. Chem. 2019, 2019 (37), 6308–6319. 10.1002/ejoc.201901010. [DOI] [Google Scholar]
- For selected works about radical addition of boryl radicals onto unsaturated systems, see:; a Zhu C.; Dong J.; Liu X.; Gao L.; Zhao Y.; Xie J.; Li S.; Zhu C. Photoredox-Controlled β-Regioselective Radical Hydroboration of Activated Alkenes with NHC-Boranes. Angew. Chem., Int. Ed. 2020, 59 (31), 12817–12821. 10.1002/anie.202005749. [DOI] [PubMed] [Google Scholar]; b Li G.; Huang G.; Sun R.; Curran D. P.; Dai W. Regioselective Radical Borylation of α,β-Unsaturated Esters and Related Compounds by Visible Light Irradiation with an Organic Photocatalyst. Org. Lett. 2021, 23 (11), 4353–4357. 10.1021/acs.orglett.1c01270. [DOI] [PubMed] [Google Scholar]; c Qi J.; Zhang F.; Jin J.; Zhao Q.; Li B.; Liu L.; Wang Y. New Radical Borylation Pathways for Organoboron Synthesis Enabled by Photoredox Catalysis. Angew. Chem., Int. Ed. 2020, 59 (31), 12876–12884. 10.1002/anie.201915619. [DOI] [PubMed] [Google Scholar]; d Xia P. J.; Song D.; Ye Z. P.; Hu Y. Z.; Xiao J. A.; Xiang H. Y.; Chen X. Q.; Yang H. Photoinduced Single-Electron Transfer as an Enabling Principle in the Radical Borylation of Alkenes with NHC–Borane. Angew. Chem., Int. Ed. 2020, 59 (17), 6706–6710. 10.1002/anie.201913398. [DOI] [PubMed] [Google Scholar]; e Xu W.; Jiang H.; Leng J.; Ong H. W.; Wu J. Visible-Light-Induced Selective Defluoroborylation of Polyfluoroarenes, Gem-Difluoroalkenes, and Trifluoromethylalkenes. Angew. Chem., Int. Ed. 2020, 59 (10), 4009–4016. 10.1002/anie.201911819. [DOI] [PubMed] [Google Scholar]; f Zhou N.; Yuan X. A.; Zhao Y.; Xie J.; Zhu C. Synergistic Photoredox Catalysis and Organocatalysis for Inverse Hydroboration of Imines. Angew. Chem., Int. Ed. 2018, 57 (15), 3990–3994. 10.1002/anie.201800421. [DOI] [PubMed] [Google Scholar]; g Shi Q.; Xu M.; Chang R.; Ramanathan D.; Peñin B.; Funes-Ardoiz I.; Ye J. Visible-Light Mediated Catalytic Asymmetric Radical Deuteration at Non-Benzylic Positions. Nat. Commun. 2022, 13 (1), 4453. 10.1038/s41467-022-32238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pan X.; Lacôte E.; Lalevée J.; Curran D. P. Polarity Reversal Catalysis in Radical Reductions of Halides by N-Heterocyclic Carbene Boranes. J. Am. Chem. Soc. 2012, 134 (12), 5669–5674. 10.1021/ja300416f. [DOI] [PubMed] [Google Scholar]; b Ueng S.-H.; Fensterbank L.; Lacôte E.; Malacria M.; Curran D. P. Radical Reductions of Alkyl Halides Bearing Electron Withdrawing Groups with N-Heterocyclic Carbene Boranes. Org. Biomol. Chem. 2011, 9 (9), 3415–3420. 10.1039/c0ob01075h. [DOI] [PubMed] [Google Scholar]
- Supranovich V. I.; Levin V. v.; Struchkova M. I.; Korlyukov A. A.; Dilman A. D. Radical Silyldifluoromethylation of Electron-Deficient Alkenes. Org. Lett. 2017, 19 (12), 3215–3218. 10.1021/acs.orglett.7b01334. [DOI] [PubMed] [Google Scholar]
- Juliá F.; Constantin T.; Leonori D. Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis. Chem. Rev. 2022, 122 (2), 2292–2352. 10.1021/acs.chemrev.1c00558. [DOI] [PubMed] [Google Scholar]
- a Le C.; Chen T. Q.; Liang T.; Zhang P.; MacMillan D. W. C. A Radical Approach to the Copper Oxidative Addition Problem: Trifluoromethylation of Bromoarenes. Science 2018, 360 (6392), 1010–1014. 10.1126/science.aat4133. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lovett G. H.; Chen S.; Xue X.-S.; Houk K. N.; MacMillan D. W. C. Open-Shell Fluorination of Alkyl Bromides: Unexpected Selectivity in a Silyl Radical-Mediated Chain Process. J. Am. Chem. Soc. 2019, 141 (51), 20031–20036. 10.1021/jacs.9b11434. [DOI] [PubMed] [Google Scholar]; c Smith R. T.; Zhang X.; Rincón J. A.; Agejas J.; Mateos C.; Barberis M.; García-Cerrada S.; de Frutos O.; MacMillan D. W. C. Metallaphotoredox-Catalyzed Cross-Electrophile C(sp3)–(Csp3) Coupling of Aliphatic Bromides. J. Am. Chem. Soc. 2018, 140 (50), 17433–17438. 10.1021/jacs.8b12025. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Brill Z. G.; Ritts C. B.; Mansoor U. F.; Sciammetta N. Continuous Flow Enables Metallaphotoredox Catalysis in a Medicinal Chemistry Setting: Accelerated Optimization and Library Execution of a Reductive Coupling between Benzylic Chlorides and Aryl Bromides. Org. Lett. 2020, 22 (2), 410–416. 10.1021/acs.orglett.9b04117. [DOI] [PubMed] [Google Scholar]; e Devery J. J.; Nguyen J. D.; Dai C.; Stephenson C. R. J. Light-Mediated Reductive Debromination of Unactivated Alkyl and Aryl Bromides. ACS Catal. 2016, 6 (9), 5962–5967. 10.1021/acscatal.6b01914. [DOI] [Google Scholar]; f Zhang P.; Le C.; MacMillan D. W. C. Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc. 2016, 138 (26), 8084–8087. 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]; g ElMarrouni A.; Ritts C. B.; Balsells J. Silyl-Mediated Photoredox-Catalyzed Giese Reaction: Addition of Non-Activated Alkyl Bromides. Chem. Sci. 2018, 9 (32), 6639–6646. 10.1039/C8SC02253D. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Dong J.; Lyu X.; Wang Z.; Wang X.; Song H.; Liu Y.; Wang Q. Visible-Light-Mediated Minisci C–H Alkylation of Heteroarenes with Unactivated Alkyl Halides Using O2 as an Oxidant. Chem. Sci. 2019, 10 (4), 976–982. 10.1039/C8SC04892D. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Sakai H. A.; Liu W.; Le C.; MacMillan D. W. C. Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc. 2020, 142 (27), 11691–11697. 10.1021/jacs.0c04812. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Luridiana A.; Mazzarella D.; Capaldo L.; Rincón J. A.; García-Losada P.; Mateos C.; Frederick M. O.; Nuño M.; Jan Buma W.; Noël T. The Merger of Benzophenone HAT Photocatalysis and Silyl Radical-Induced XAT Enables Both Nickel-Catalyzed Cross-Electrophile Coupling and 1,2-Dicarbofunctionalization of Olefins. ACS Catal. 2022, 12 (18), 11216–11225. 10.1021/acscatal.2c03805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Constantin T.; Zanini M.; Regni A.; Sheikh N. S.; Juliá F.; Leonori D. Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367 (6481), 1021–1026. 10.1126/science.aba2419. [DOI] [PubMed] [Google Scholar]; b Constantin T.; Juliá F.; Sheikh N. S.; Leonori D. A Case of Chain Propagation: α-Aminoalkyl Radicals as Initiators for Aryl Radical Chemistry. Chem. Sci. 2020, 11 (47), 12822–12828. 10.1039/D0SC04387G. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhao H.; McMillan A. J.; Constantin T.; Mykura R. C.; Juliá F.; Leonori D. Merging Halogen-Atom Transfer (XAT) and Cobalt Catalysis to Override E2-Selectivity in the Elimination of Alkyl Halides: A Mild Route toward Contra -Thermodynamic Olefins. J. Am. Chem. Soc. 2021, 143 (36), 14806–14813. 10.1021/jacs.1c06768. [DOI] [PubMed] [Google Scholar]; d Caiger L.; Sinton C.; Constantin T.; Douglas J. J.; Sheikh N. S.; Juliá F.; Leonori D. Radical Hydroxymethylation of Alkyl Iodides Using Formaldehyde as a C1 Synthon. Chem. Sci. 2021, 12 (31), 10448–10454. 10.1039/D1SC03083C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J.; Zhang F.; Jin J.; Zhao Q.; Li B.; Liu L.; Wang Y. New Radical Borylation Pathways for Organoboron Synthesis Enabled by Photoredox Catalysis. Angew. Chem., Int. Ed. 2020, 59 (31), 12876–12884. 10.1002/anie.201915619. [DOI] [PubMed] [Google Scholar]
- a Wan T.; Wen Z.; Laudadio G.; Capaldo L.; Lammers R.; Rincón J. A.; García-Losada P.; Mateos C.; Frederick M. O.; Broersma R.; Noël T. Accelerated and Scalable C(sp3)–H Amination via Decatungstate Photocatalysis Using a Flow Photoreactor Equipped with High-Intensity LEDs. ACS Centr. Sci. 2022, 8 (1), 51–56. 10.1021/acscentsci.1c01109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Buglioni L.; Raymenants F.; Slattery A.; Zondag S. D. A.; Noël T. Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry. Chem. Rev. 2022, 122 (2), 2752–2906. 10.1021/acs.chemrev.1c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mazzarella D.; Pulcinella A.; Bovy L.; Broersma R.; Noël T. Rapid and Direct Photocatalytic C(sp3)–H Acylation and Arylation in Flow. Angew. Chem. In. Ed. 2021, 60 (39), 21277–21282. 10.1002/anie.202108987. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sambiagio C.; Noël T. Flow Photochemistry: Shine Some Light on Those Tubes!. Trends Chem. 2020, 2 (2), 92–106. 10.1016/j.trechm.2019.09.003. [DOI] [Google Scholar]; e Rehm T. H. Flow Photochemistry as a Tool in Organic Synthesis. Chem.—Eur. J. 2020, 26 (71), 16952–16974. 10.1002/chem.202000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Si X.; Rominger F.; Hashmi A. S. K. Visible-Light-Induced Radical Carbo-Cyclization/gem-Diborylation through Triplet Energy Transfer between a Gold Catalyst and Aryl Iodides. J. Am. Chem. Soc. 2020, 142 (23), 10485–10493. 10.1021/jacs.0c03197. [DOI] [PubMed] [Google Scholar]
- Tsudaka T.; Kotani H.; Ohkubo K.; Nakagawa T.; Tkachenko N. v.; Lemmetyinen H.; Fukuzumi S. Photoinduced Electron Transfer in 9-Substituted 10-Methylacridinium Ions. Chem.—Eur. J. 2017, 23 (6), 1306–1317. 10.1002/chem.201604527. [DOI] [PubMed] [Google Scholar]
- Walton J. C.; Brahmi M. M.; Fensterbank L.; Lacôte E.; Malacria M.; Chu Q.; Ueng S.-H.; Solovyev A.; Curran D. P. EPR Studies of the Generation, Structure, and Reactivity of N-Heterocyclic Carbene Borane Radicals. J. Am. Chem. Soc. 2010, 132 (7), 2350–2358. 10.1021/ja909502q. [DOI] [PubMed] [Google Scholar]
- MacKenzie I. A.; Wang L.; Onuska N. P. R.; Williams O. F.; Begam K.; Moran A. M.; Dunietz B. D.; Nicewicz D. A. Discovery and Characterization of an Acridine Radical Photoreductant. Nature 2020, 580 (7801), 76–80. 10.1038/s41586-020-2131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons E. M.; Hartwig J. F. On the Interpretation of Deuterium Kinetic Isotope Effects in C–H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed. 2012, 51 (13), 3066–3072. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- Pitre S. P.; McTiernan C. D.; Vine W.; DiPucchio R.; Grenier M.; Scaiano J. C. Visible-Light Actinometry and Intermittent Illumination as Convenient Tools to Study Ru(bpy)3Cl2 Mediated Photoredox Transformations. Sci. Rep.-UK 2015, 5 (1), 16397. 10.1038/srep16397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlili A.; Lakhdar S. Acridinium Salts and Cyanoarenes as Powerful Photocatalysts: Opportunities in Organic Synthesis. Angew. Chem., Int. Ed. 2021, 60 (36), 19526–19549. 10.1002/anie.202102262. [DOI] [PubMed] [Google Scholar]
- Joshi-Pangu A.; Lévesque F.; Roth H. G.; Oliver S. V.; Campeau L.-C.; Nicewicz N.; DiRocco D. A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81 (16), 7244–7249. 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]
- Pan X.; Vallet A.-L.; Schweizer S.; Dahbi K.; Delpech B.; Blanchard N.; Graff B.; Geib S. J.; Curran D. P.; Lalevée J.; Lacôte E. Mechanistic and Preparative Studies of Radical Chain Homolytic Substitution Reactions of N-Heterocyclic Carbene Boranes and Disulfides. J. Am. Chem. Soc. 2013, 135 (28), 10484–10491. 10.1021/ja403627k. [DOI] [PubMed] [Google Scholar]
- Benniston A. C.; Harriman A.; Li P.; Rostron J. P.; van Ramesdonk H. J.; Groeneveld M. M.; Zhang H.; Verhoeven J. W. Charge Shift and Triplet State Formation in the 9-Mesityl-10-Methylacridinium Cation. J. Am. Chem. Soc. 2005, 127 (46), 16054–16064. 10.1021/ja052967e. [DOI] [PubMed] [Google Scholar]
- When monitoring the reaction over time, we detected N,N-dimethylimidazolium iodide (via 1H NMR) and boric acid (via 11B-NMR). These byproducts are formed upon decomposition of NHC-BH2I in an aqueous environment as reported in the literature (ref (15b), see also Section 6.8 in the Supporting Information). Notably, when the reaction was performed in dry C6D6, the formation of NHC-BH2I could be observed via 11B-NMR spectroscopy (see Section 6.8 in the Supporting Information), which supports again the occurrence of the XAT step.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.