

Abstract

Intermolecular cyclopropanation of mono-, di-, and trisubstituted olefins with α-bromo-β-ketoesters and α-bromomalonates under organophotocatalysis is reported. The reaction displays broad functional group tolerance, including substrates bearing acids, alcohols, halides, ethers, ketones, nitriles, esters, amides, carbamates, silanes, stannanes, boronic esters, as well as arenes, and furnishes highly substituted cyclopropanes. The transformation may be performed in the presence of air and moisture with 0.5 mol % of a benzothiazinoquinoxaline as organophotocatalyst. Mechanistic investigations, involving Stern–Volmer quenching, quantum yield determination, and deuteration experiments, are carried out, and a catalytic cycle for the transformation is discussed.
Cyclopropanes are found in natural products, pharmaceuticals, agrochemicals, and fragrances and constitute useful synthetic building blocks.1 Their high strain energy and unique bonding pattern makes them distinct, with reactivity not observed for other carbocycles.2 A variety of classic cyclopropanation methods have been devised: metal-catalyzed reactions with diazoalkanes,3 Simmons–Smith,4 Kulinkovich,5 as well as Corey–Chaykovsky6 reactions, and carbanion alkylations.7 Additional approaches employ noble metals or photocatalysis.8 Herein, we report the first intermolecular organophotocatalyzed cyclopropanation of unactivated olefins with α-bromo-β-ketoesters and α-bromomalonates (Figure 1).
Figure 1.

Intermolecular organophotocatalyzed cyclopropanation of unactivated olefins.
Photochemically driven transformations have recently been the focus of intense investigations.9 Ir or Ru complexes as photocatalysts have enabled the application of visible-light photocatalysis in the service of synthesis. Parallel approaches, involving organophotocatalysts, have been investigated.10 Polyaromatics have been used as dyes, and their application in organophotocatalysis is rapidly increasing.11
In connection with our interest in olefin functionalization12 and photocatalysis,13 we set out to develop approaches to substituted cyclopropanes. Simple cyclopropanes can be synthesized alkylatively from β-ketoesters and vic-dibromides via γ-bromo-β-ketoesters.7a,14 Stephenson has accessed γ-bromomalonates directly from olefins through intermolecular atom-transfer radical addition (ATRA) of α-bromomalonates with LiBr and Ru(bpy)3Cl2 or Ir[(dF(CF3)-ppy)2(dtbbpy)]PF6 (Figure 2).15 Tokuyama subsequently examined intramolecular cyclopropanations of α-bromo-β-ketoesters in the presence of 2,6-lutidine, LiBr, and Ru(bpy)3Cl2.16 However, direct intermolecular cyclopropanation of α-bromo-β-ketoesters with unactivated olefins remains elusive.
Figure 2.

Approaches toward γ-bromomalonates and cyclopropanes.
As part of a study on semipinacol rearrangements, Ohmiya and Nagao proposed reduction of bromomalonates by benzophenothiazine photocatalysts.17 We hypothesized benzophenothiazines could also effect reduction of bromoketoesters, leading to cyclopropanation. However, when we subjected α-bromo-β-ketoester 1a and phenylbutene 2a to N-phenyl benzophenothiazine (PC-3) (10 mol %) and 20 mol % LiBF4, along with 2.0 equiv 2,6-lutidine under an inert atmosphere in MeCN (0.4 M), cyclopropane 3a was isolated in merely 24% yield (Table 1, Entry 1). Our analysis of the UV–vis spectrum of PC-3 revealed poor absorbance (ε446 nm < 100 M–1 cm–1).
Table 1. Optimization of Reaction Conditionsa.

| Entry | Deviation from std. conditions | Yield of 3a (%)b |
|---|---|---|
| 1 | 10 mol % PC-3, 20 mol % LiBF4 2.0 equiv 2,6-lutidine, Ar | 24 |
| 2 | 10 mol % PC-1, 20 mol % LiBF4 2.0 equiv 2,6-lutidine, Ar | 43 |
| 3 | none | 97 |
| 4 | Ar | 98 |
| 5 | Ru(bpy)3Cl2 instead of PC-1 | 6 |
| 6 | PC-2 instead of PC-1 | 62 |
| 7 | without PC-1 | 0 |
| 8 | dark, 40 °C | 0 |
| 9 | without 2,6-lutidine | 0 |

Reaction temperature rises to 40 °C.
Yields obtained by 1H NMR (mesitylene as internal standard).
In identifying reducing photocatalysts with larger extinction coefficients in the blue-light range, a study of the cyclophosphinylation reaction of diaryl diynes caught our attention.18 Therein, Xu examined and characterized dyes electro- and photochemically. Among these, the two benzothiazinoquinoxaline derivatives, PC-1 and PC-2, stood out based on their red-shifted absorption maxima, increased absorbance in the blue-light range (ε446 nm > 3000 M–1 cm–1), and suitable reduction potential. We hypothesized they could be appropriate catalysts for cyclopropanation of unactivated olefins with α-bromo-β-ketoesters. To the best of our knowledge, they have not been further investigated as photocatalysts.
Subjecting 1a and 2a to 10 mol % PC-1, 20 mol % LiBF4, and 2.0 equiv 2,6-lutidine under argon in MeCN (0.4 M) (Table 1, Entry 2) gave cyclopropane 3a in 43% yield. Reaction optimization (see SI) revealed the benefits of additional equivalents of base and, surprisingly, indicated that the presence of Li salt is detrimental. Catalyst loading was reduced to 0.5 mol % without decreasing product formation. Under optimized conditions (0.5 mol % PC-1, 4.0 equiv 2,6-lutidine in MeCN (0.4 M), Entry 3), 3a was formed in 97% yield. Notably, in contrast to previous reports, this transformation is not sensitive to air, giving identical yields under ambient atmosphere (Entry 4). For comparison, when the reaction was conducted with Ru(bpy)3Cl2 under otherwise identical conditions, 3a was produced in 6% yield (Entry 5). Photocatalyst PC-2 also furnished the product, albeit in lower yield than PC-1 (Entry 6). The reaction did not provide product in the absence of organophotocatalyst or light, and 2a was fully recovered (Entries 7 and 8). Reaction without 2,6-lutidine led to formation of complex mixtures (Entry 9, see SI).
We investigated the substrate scope with α-bromo-β-ketoesters next (Figure 3). A variety of γ-substituted α-bromoacetoacetates were employed, including bromoacetoacetate along with ethyl, iso-propyl, and phenethyl ketones 1a–d, affording cyclopropanes 3a–d in 56–80% yield from 4-phenylbutene. tert-Butyl ester 1e gave 3e in 62% yield. Next, a collection of alkene substrates was examined. Naphthyl and anisole adducts 3f and 3g were isolated in 73% and 72% yield, respectively. Electron-poor and -rich aryl groups were tolerated, leading to 3h–m in 56–86% yield. Primary alcohol 3n and alkyl chloride 3o were obtained in 55% and 84% yield, respectively. Notably, the transformation tolerates O-H protic functional groups, which under conditions involving carbenes instead favor O-H insertion.193p–t were accessed in 58–87% yield.
Figure 3.
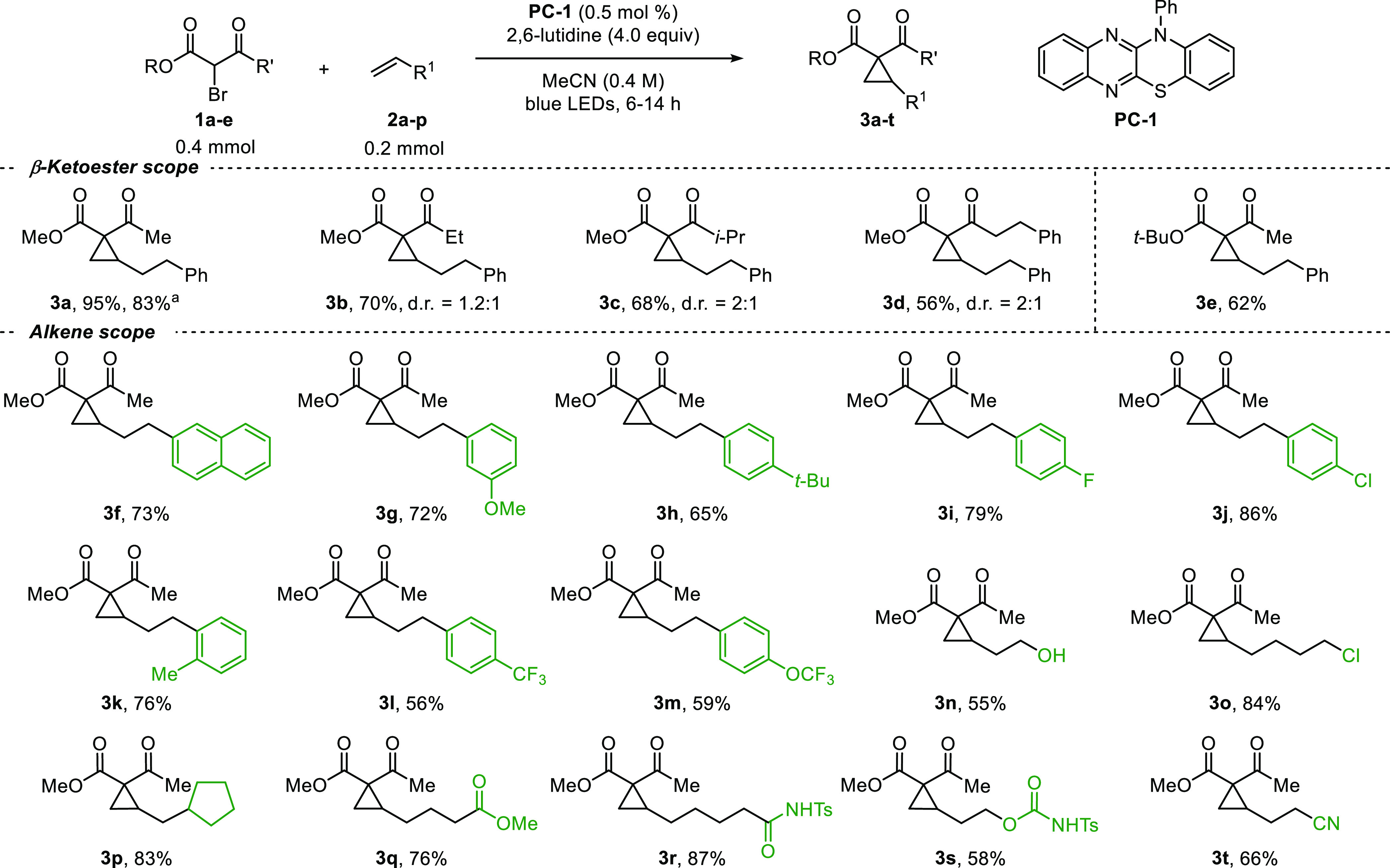
Substrate scope. d.r. = 1:1 (1H NMR analysis of unpurified reaction mixture) unless otherwise indicated. aConducted on 2.0 mmol scale.
The rather modest diastereoselectivity in the cyclopropanation prompted us to examine esters that may induce diastereocontrol. When 2,6-di-tert-butylphenyl ester 4 was subjected to reaction conditions, product cyclopropane 6a was obtained in 61% yield and 9:1 d.r. The use of 4 constituted a general solution to render the cyclopropanation studied herein diastereoselective (Figure 4), and cyclopropanes 6b–f were obtained in 45–71% yield and 7:1 to 10:1 d.r. It had previously been shown that 2,6-di-tert-butylphenyl cyclopropanoates may be saponified to remove the auxiliary.20
Figure 4.

Substrate scope. d.r. determined by 1H NMR analysis of unpurified reaction mixture.
We set out to explore the generality of the cyclopropanation reaction. When dimethyl bromomalonate (7) and 2a were subjected to conditions optimized for α-bromo-β-ketoesters, only γ-bromomalonate (the ATRA product) was observed (see SI). Intriguingly, upon addition of 10 mol % LiBF4, cyclopropane product 9a was obtained in 40% yield. Gratifyingly, use of 0.5 mol % PC-1, 1.0 equiv LiBF4, and 4.0 equiv 2,6-lutidine in MeCN afforded cyclopropane 9a in 82% yield. Interestingly, contrary to bromoketoesters, use of PC-2 instead of PC-1 led to product formation in higher yield (95%). The use of other Li salts gave analogous results. However, use of n-Bu4NBF4 gave only the ATRA product (see SI). In contrast to previous results with α-bromo-β-ketoesters 1a–e, these findings implicate an important and beneficial role for Li+ with bromomalonates.
Next, we investigated the functional group tolerance of this cyclopropanation (Figure 5). One previous report for the cyclopropanation of terminal olefins with diethyl bromomalonate uses a heterogeneous, porous, polymeric photocatalyst.21 We showcase that the transformation with organophotocatalyst PC-2 is tolerant of a wide range of functionalities, terminal, 1,1-disubstituted, 1,2-disubstituted, and trisubstituted olefins. To this end cyclopropanes 9a–k were obtained in 44–93% yield. Vinyl stannane 8l and vinyl boronic pinacol ester 8m were converted to cyclopropanes 9l and 9m in 73% and 53% yield, respectively, giving cyclopropanes that could be elaborated via metal-catalyzed cross-coupling reactions.22 Next, effects of alkene substitution patterns were investigated. 1,1-Disubstituted olefin 8n gave 9n in 78% yield. E- and Z-oct-2-ene separately furnished 9o in 64–65% yield and 1:1 d.r. Trisubstituted olefins were well tolerated, with 9p and 9q isolated in 80% and 51% yield, respectively. 9r was accessed from 1-decene in 91–94% yield and converted to the natural products cis- and trans-olibanic acid in 72% overall yield. Finally, 9a was also produced on a 2.0 mmol scale in 81% yield, and 9r was prepared on a 15 mmol scale in 85% yield.
Figure 5.

Substrate scope. a2.0 mmol scale. bReaction conducted in sulfolane. cReaction time = 8 h. d15 mmol scale with 0.1 mol % PC-2, 20 mol % LiBF4, [1-decene] = 2 M.
To gain an understanding of the mechanism of this transformation, a series of experiments were conducted after devising a mechanistic construct (Figure 6). Initial investigations focused on the absorbance characteristics of PC-1 and PC-2 (Figure 7, A).
Figure 6.

Mechanistic proposal. Lutidine = 2,6-lutidine.
Figure 7.

Mechanistic investigations. A, UV–vis absorbance of PC-1 and PC-2 in MeCN (12 μM). B, Cyclic voltammetry and Stern–Volmer relationship between PC-1* and 1a, 2a, and 2,6-lutidine; fluorescence spectra recorded in MeCN (12 μM PC-1). C, Radical scavenger experiment carried out with 1.0 equiv of TEMPO. D, Quantum yield determined on 1.0 mmol scale; 8% conversion after 120 min. E, Equilibrium involving β-dicarbonyl compounds and D2O (1.0 equiv) in d3-MeCN (0.4 M) at 40 °C.
Both photocatalysts show strong absorption in the blue-light range, with absorbance maxima of 412 and 414 nm, respectively. The UV–vis spectra of PC-1 and PC-2, with concentrations corresponding to 0.5 mol % catalyst loading (c = 1 mM), showed absorbance above 3.0 (no transmittance detected) for λ < 457 nm. The blue LEDs employed in the transformation emit >90% of light between 410 and 510 nm. Thus, almost the entire spectrum of wavelengths emitted by the blue LED photoreactor can be absorbed by PC-1 or PC-2 (Figure 7, A). Next, the fate of the excited-state photocatalyst was examined (Figure 7, B). Stern–Volmer relationship studies showed fluorescence quenching by α-bromo-β-ketoester 1a, while no quenching by 2,6-lutidine or 2a was observed. Combining those results with cyclic voltammetry (CV) measurements of PC-1 (E1/2*(PC•+/PC*) = −1.43 V vs SCE) and α-bromo-β-ketoester 1a (Ep/2 (B/B•–) = −1.03 V vs SCE), we concluded that reduction of 1a is feasible, leading to C-centered radical 10a (Figure 6). When the reaction was carried out in the presence of 1.0 equiv of TEMPO, no product formation was observed (Figure 7, C), which is consistent with proposed radical pathways and previous studies on ATRA.15a,23 Two possible mechanistic pathways can be devised following addition of 10a to the olefin. First, radical propagation via bromide abstraction by secondary alkyl radical 10b would form secondary alkyl bromide 10c (X = Br) and another β-ketoester radical 10a (Figure 6, D). Alternatively, oxidized photocatalyst PC-1•+ (E1/2(PC•+/PC) = 1.06 V vs SCE) may effect oxidation of 10b (E1/2(C+/C•) = 0.47 V vs SCE)15b,24 to a carbocation which could then be trapped by bromide or PC-1. Previously, it has been suggested that thiazine photocatalysts may form adducts with cations.17b,25
Atom-transfer radical additions leading to γ-bromomalonates have been proposed to proceed via radical propagation in previous studies.15a,23 As discussed by Yoon, a light-on-light-off experiment is insufficient to assess radical chain processes,23 prompting us to determine the quantum yield.
For the reaction with bromoketoester 1a and phenylbutene 2a (1.0 mmol scale), after 120 min, 8% conversion (see SI) of alkene was observed, and the quantum yield was calculated as Φ = 0.012, which is consistent with a closed photoredox catalytic cycle (Figure 7, D).
We noted that LiBF4 was necessary to effect cyclopropanation with α-bromomalonates, whereas it was detrimental in the reaction with α-bromo-β-ketoesters. For the latter, addition of LiBF4 resulted in lower yields (34% vs 95%, see SI) and increased decomposition of 1a. To effect cyclopropanation, enolization of intermediate 10c by the base (2,6-lutidine) is necessary (Figure 6, E). We hypothesize enolization of intermediate malonate (pKa = 13) is facilitated by Lewis acidic Li+, which is not required for enolization of intermediate acetoacetate 10c (pKa = 11). To study enolization of 10c, deuteration experiments were conducted in d3-MeCN with 1.0 equiv of D2O (Figure 7, E). Dimethyl methylmalonate and methyl methylacetoacetate were chosen as models for 10c. Equilibration (66% deuterium incorporation) in the presence of LiBF4 and 2,6-lutidine was complete in 9 and 5 min for malonates and acetoacetates, respectively. In the absence of LiBF4, only acetoacetates reached equilibrium, albeit only after 2 h. These experiments support formation of an enolate under reaction conditions and account for the difference in reactivity observed between malonates, which require Li+, and acetoacetates, which do not. Additionally, in the presence of base, bromoketoester 1a undergoes decomposition, while no such effect was observed for bromomalonates. We suggest that with Li+ a larger fraction of starting material 1a is enolized, leading to increased decomposition. Thus, a fine balance between sufficient enolization of intermediate 10c to effect cyclization and minimal enolization of starting material 1a is required (Figure 7, E).
In conclusion, we have developed an intermolecular cyclopropanation of unactivated alkenes with α-bromomalonates and, for the first time, α-bromo-β-ketoesters under organophotocatalytic conditions. The transformation shows broad functional group tolerance and is amenable to terminal as well as highly substituted olefins. We demonstrated that structurally similar substrates, α-bromomalonates and α-bromo-β-ketoesters, show distinctly different behavior in this transformation. While Li+ is beneficial for α-bromomalonates, it is not only superfluous for α-bromo-β-ketoesters but, in fact, detrimental. Salient features of the transformation are its air tolerance and the successful application of benzothiazinoquinoxalines PC-1 and PC-2 as organophotocatalysts. More broadly, further exploration of this remarkable class of modular photocatalysts may lead to novel reaction discovery.
Acknowledgments
This work was funded by the European Research Council (OLECAT, Grant ID 833540). We are grateful to Dr. M.-O. Ebert, R. Frankenstein, S. Burkhardt, and R. Arnold for support with low- and high-temperature NMR experiments and time series, Dr. E. Meister for support with fluorescence spectroscopy, C.A. Bärtschi, C. Marro, and P. Trüssel for the maintenance and construction of the photoreactor, and M.K. Bogdos for support with CV measurements (all ETH Zurich). W.M. Amberg is grateful for partial support with funding from the SSCI (Scholarship Fund of the Swiss Chemical Industry).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c11680.
Experimental details; NMR and CV data (PDF)
Author Contributions
‡ D.M.F., H.L., and W.M.A. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Wong H. N. C.; Hon M. Y.; Tse C. W.; Yip Y. C.; Tanko J.; Hudlicky T. Use of cyclopropanes and their derivatives in organic synthesis. Chem. Rev. 1989, 89, 165–198. 10.1021/cr00091a005. [DOI] [Google Scholar]; b Carson C. A.; Kerr M. A. Heterocycles from cyclopropanes: applications in natural product synthesis. Chem. Soc. Rev. 2009, 38, 3051. 10.1039/b901245c. [DOI] [PubMed] [Google Scholar]; c Tang P.; Qin Y. Recent Applications of Cyclopropane-Based Strategies to Natural Product Synthesis. Synthesis 2012, 44, 2969–2984. 10.1055/s-0032-1317011. [DOI] [Google Scholar]; d Cavitt M. A.; Phun L. H.; France S. Intramolecular donor–acceptor cyclopropane ring-opening cyclizations. Chem. Soc. Rev. 2014, 43, 804–818. 10.1039/C3CS60238A. [DOI] [PubMed] [Google Scholar]; e Dowd P.; Zhang W. Free radical-mediated ring expansion and related annulations. Chem. Rev. 1993, 93, 2091–2115. 10.1021/cr00022a007. [DOI] [Google Scholar]; f Faust R. Fascinating Natural and Artificial Cyclopropane Architectures. Angew. Chem., Int. Ed. 2001, 40, 2251–2253. 10.1002/1521-3773(20010618)40:12<2251::AID-ANIE2251>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; g Wessjohann L. A.; Brandt W.; Thiemann T. Biosynthesis and Metabolism of Cyclopropane Rings in Natural Compounds. Chem. Rev. 2003, 103, 1625–1648. 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]; h Talele T. T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]; i Schröder F. Present and Future of Cyclopropanations in Fragrance Chemistry. Chemistry & Biodiversity 2014, 11, 1734–1751. 10.1002/cbdv.201400041. [DOI] [PubMed] [Google Scholar]
- Ebner C.; Carreira E. M. Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev. 2017, 117, 11651–11679. 10.1021/acs.chemrev.6b00798. [DOI] [PubMed] [Google Scholar]
- a Silberrad O.; Roy C. S. XXIV.—Gradual decomposition of ethyl diazoacetate. J. Chem. Soc., Trans. 1906, 89, 179–182. 10.1039/CT9068900179. [DOI] [Google Scholar]; b Paulissen R.; Reimlinger H.; Hayez E.; Hubert A. J.; Teyssié P. Transition metal catalysed reactions of diazocompounds - II insertion in the hydroxylic bond. Tetrahedron Lett. 1973, 14, 2233–2236. 10.1016/S0040-4039(01)87603-6. [DOI] [Google Scholar]; c Moser W. R. Mechanism of the copper-catalyzed addition of diazoalkanes to olefins. II. Electronic effects. J. Am. Chem. Soc. 1969, 91, 1141–1146. 10.1021/ja01033a018. [DOI] [Google Scholar]; d Paulissen R.; Hubert A. J.; Teyssie P. Transition metal catalysed cyclopropanation of olefin. Tetrahedron Lett. 1972, 13, 1465–1466. 10.1016/S0040-4039(01)84656-6. [DOI] [Google Scholar]; e Padwa A.; Austin D. J.; Hornbuckle S. F.; Semones M. A.; Doyle M. P.; Protopopova M. N. Control of chemoselectivity in catalytic carbenoid reactions. Dirhodium(II) ligand effects on relative reactivities. J. Am. Chem. Soc. 1992, 114, 1874–1876. 10.1021/ja00031a048. [DOI] [Google Scholar]; f Padwa A.; Krumpe K. E. Application of Intramolecular Carbenoid Reactions in Organic Synthesis. Tetrahedron 1992, 48, 5385–5453. 10.1016/S0040-4020(01)88298-3. [DOI] [Google Scholar]; g Doyle M. P.; Forbes D. C. Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–936. 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; h Doyle M. P. Perspective on Dirhodium Carboxamidates as Catalysts. J. Org. Chem. 2006, 71, 9253–9260. 10.1021/jo061411m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Simmons H. E.; Smith R. D. A New Synthesis of Cyclopropanes from Olefins. J. Am. Chem. Soc. 1958, 80, 5323–5324. 10.1021/ja01552a080. [DOI] [Google Scholar]; b Simmons H. E.; Smith R. D. A New Synthesis of Cyclopropanes. J. Am. Chem. Soc. 1959, 81, 4256–4264. 10.1021/ja01525a036. [DOI] [Google Scholar]; c Winstein S.; Sonnenberg J. Homoconjugation and Homoaromaticity. III.1 The 3-Bicyclo [3.1.0]hexyl System2,3. J. Am. Chem. Soc. 1961, 83, 3235–3244. 10.1021/ja01476a016. [DOI] [Google Scholar]; d Dauben W. G.; Berezin G. H. The Preparation of Cyclic Cyclopropylcarbinols. J. Am. Chem. Soc. 1963, 85, 468–472. 10.1021/ja00887a023. [DOI] [Google Scholar]; e Dauben W. G.; Ashcraft A. C. The Total Synthesis of (±)-Thujopsene. J. Am. Chem. Soc. 1963, 85, 3673–3676. 10.1021/ja00905a032. [DOI] [Google Scholar]; f Poulter C. D.; Friedrich E. C.; Winstein S. Stereochemistry of the methylene iodide-zinc-copper couple methylenation of cyclic allylic alcohols. J. Am. Chem. Soc. 1969, 91, 6892–6894. 10.1021/ja01052a083. [DOI] [Google Scholar]; g Lorenz J. C.; Long J.; Yang Z.; Xue S.; Xie Y.; Shi Y. A Novel Class of Tunable Zinc Reagents (RXZnCH2) for Efficient Cyclopropanation of Olefins. J. Org. Chem. 2004, 69, 327–334. 10.1021/jo030312v. [DOI] [PubMed] [Google Scholar]
- a Kulinkovich O. G. S. S. V.; Vasilevskii D. A.; Pritytskaya T. S. Reaction of Ethylmagnesium Bromide with Esters of Carboxylic-Acids in the Presence of Tetraisopropoxytitanium. Zh. Org. Khim. 1989, 25, 2244–2245. [Google Scholar]; b Kulinkovich O. G.; Sviridov S. V.; Vasilevski D. A. Titanium(IV) Isopropoxide-Catalyzed Formation of 1-Substituted Cyclopropanols in the Reaction of Ethylmagnesium Bromide with Methyl Alkanecarboxylates. Synthesis 1991, 1991, 234–234. 10.1055/s-1991-26431. [DOI] [Google Scholar]; c Haym I.; Brimble M. A. The Kulinkovich hydroxycyclopropanation reaction in natural product synthesis. Org. Biomol. Chem. 2012, 10, 7649. 10.1039/c2ob26082d. [DOI] [PubMed] [Google Scholar]; d Chaplinski V.; De Meijere A. A Versatile New Preparation of Cyclopropylamines from Acid Dialkylamides. Angew. Chem., Int. Ed. 1996, 35, 413–414. 10.1002/anie.199604131. [DOI] [Google Scholar]; e Bertus P.; Szymoniak J. New and easy route to primary cyclopropylamines from nitriles. Chem. Commun. 2001, 1792–1793. 10.1039/b105293b. [DOI] [PubMed] [Google Scholar]
- a Corey E. J.; Chaykovsky M. Dimethylsulfoxonium Methylide. J. Am. Chem. Soc. 1962, 84, 867–868. 10.1021/ja00864a040. [DOI] [Google Scholar]; b Corey E. J.; Chaykovsky M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. 10.1021/ja01084a034. [DOI] [Google Scholar]
- a Perkin W. H. Ueber die Einwirkung von Aethylenbromid auf Malonsäureäther. Ber. Dtsch. Chem. Ges. 1884, 17, 54–59. 10.1002/cber.18840170111. [DOI] [Google Scholar]; b Pirrung M. C.; Dunlap S. E.; Trinks U. P. Ethylene Biosynthesis part 10. Synthesis and study of racemic, (1R, 2S)-, and (1S, 2R)-1-Amino-2-(hydroxymethyl)cyclopropanecarboxylic Acid. Helv. Chim. Acta 1989, 72, 1301–1310. 10.1002/hlca.19890720618. [DOI] [Google Scholar]
- a Strickler H.; Davis J. B.; Ohloff G. N. Zur Cyclisierung von Dehydrolinalylacetat in Gegenwart von Zinkchlorid. Helv. Chim. Acta 1976, 59, 1328–1332. 10.1002/hlca.19760590435. [DOI] [Google Scholar]; b Bruneau C. Electrophilic Activation and Cycloisomerization of Enynes: A New Route to Functional Cyclopropanes. Angew. Chem., Int. Ed. 2005, 44, 2328–2334. 10.1002/anie.200462568. [DOI] [PubMed] [Google Scholar]; c Mainetti E.; Mouries V.; Fensterbank L.; Malacria M.; Marco-Contelles J. The Effect of a Hydroxy Protecting Group on the PtCl2-Catalyzed Cyclization of Dienynes–A Novel, Efficient, and Selective Synthesis of Carbocycles. Angew. Chem., Int. Ed. 2002, 41, 2132–2135. 10.1002/1521-3773(20020617)41:12<2132::AID-ANIE2132>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; d Lemière G.; Gandon V.; Cariou K.; Hours A.; Fukuyama T.; Dhimane A.-L.; Fensterbank L.; Malacria M. Generation and Trapping of Cyclopentenylidene Gold Species: Four Pathways to Polycyclic Compounds. J. Am. Chem. Soc. 2009, 131, 2993–3006. 10.1021/ja808872u. [DOI] [PubMed] [Google Scholar]; e Shi X.; Gorin D. J.; Toste F. D. Synthesis of 2-Cyclopentenones by Gold(I)-Catalyzed Rautenstrauch Rearrangement. J. Am. Chem. Soc. 2005, 127, 5802–5803. 10.1021/ja051689g. [DOI] [PubMed] [Google Scholar]; f Witham C. A.; Mauleón P.; Shapiro N. D.; Sherry B. D.; Toste F. D. Gold(I)-Catalyzed Oxidative Rearrangements. J. Am. Chem. Soc. 2007, 129, 5838–5839. 10.1021/ja071231+. [DOI] [PubMed] [Google Scholar]; g Mamane V.; Gress T.; Krause H.; Fürstner A. Platinum- and Gold-Catalyzed Cycloisomerization Reactions of Hydroxylated Enynes. J. Am. Chem. Soc. 2004, 126, 8654–8655. 10.1021/ja048094q. [DOI] [PubMed] [Google Scholar]; h Amberg W. M.; Carreira E. M. Enantioselective Total Synthesis of (+)-Aberrarone. J. Am. Chem. Soc. 2022, 144, 15475–15479. 10.1021/jacs.2c07150. [DOI] [PubMed] [Google Scholar]; i Herraiz A. G.; Suero M. G. A Transition-Metal-Free & DiazoFree Styrene Cyclopropanation. Chem. Sci. 2019, 10, 9374–9379. 10.1039/C9SC02749A. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Li P.; Zhao J.; Shi L.; Wang J.; Shi X.; Li F. Iodine-catalyzed diazoactivation to access radical reactivity. Nat. Commun. 2018, 9, 1972. 10.1038/s41467-018-04331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Phelan J. P.; Lang S. B.; Compton J. S.; Kelly C. B.; Dykstra R.; Gutierrez O.; Molander G. A. Redox-neutral photocatalytic cyclopropanation via radical/polar crossover. J. Am. Chem. Soc. 2018, 140, 8037–8047. 10.1021/jacs.8b05243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; Macmillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; c Narayanam J. M. R.; Stephenson C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]; d Genzink M. J.; Kidd J. B.; Swords W. B.; Yoon T. P. Chiral Photocatalyst Structures in Asymmetric Photochemical Synthesis. Chem. Rev. 2022, 122, 1654–1716. 10.1021/acs.chemrev.1c00467. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chan A. Y.; Perry I. B.; Bissonnette N. B.; Buksh B. F.; Edwards G. A.; Frye L. I.; Garry O. L.; Lavagnino M. N.; Li B. X.; Liang Y.; Mao E.; Millet A.; Oakley J. V.; Reed N. L.; Sakai H. A.; Seath C. P.; Macmillan D. W. C. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. 10.1021/acs.chemrev.1c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/C8CS00054A. [DOI] [PubMed] [Google Scholar]; g König B. Photocatalysis in Organic Synthesis - Past, Present, and Future. Eur. J. Org. Chem. 2017, 2017, 1979–1981. 10.1002/ejoc.201700420. [DOI] [Google Scholar]; h Tucker J. W.; Stephenson C. R. J. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]; i Kärkäs M. D.; Matsuura B. S.; Stephenson C. R. Enchained by visible light–mediated photoredox catalysis. Science 2015, 349, 1285–1286. 10.1126/science.aad0193. [DOI] [PubMed] [Google Scholar]
- Zeitler K. Photoredox Catalysis with Visible Light. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. 10.1002/anie.200904056. [DOI] [PubMed] [Google Scholar]
- Bortolato T.; Cuadros S.; Simionato G.; Dell’Amico L. The advent and development of organophotoredox catalysis. Chem. Commun. 2022, 58, 1263–1283. 10.1039/D1CC05850A. [DOI] [PubMed] [Google Scholar]
- a Fischer D. M.; Balkenhohl M.; Carreira E. M. Cobalt-Catalyzed Cyclization of Unsaturated N-Acyl Sulfonamides: a Diverted Mukaiyama Hydration Reaction. JACS Au 2022, 2, 1071–1077. 10.1021/jacsau.2c00186. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Waser J.; Carreira E. M. Catalytic hydrohydrazination of a wide range of alkenes with a simple mn complex. Angew. Chem., Int. Ed. Engl. 2004, 43, 4099–4102. 10.1002/anie.200460811. [DOI] [PubMed] [Google Scholar]; c Waser J.; Carreira E. M. Convenient Synthesis of Alkylhydrazides by the Cobalt-Catalyzed Hydrohydrazination Reaction of Olefins and Azodicarboxylates. J. Am. Chem. Soc. 2004, 126, 5676–5677. 10.1021/ja048698u. [DOI] [PubMed] [Google Scholar]; d Waser J.; González-Gómez J. C.; Nambu H.; Huber P.; Carreira E. M. Cobalt-Catalyzed Hydrohydrazination of Dienes and Enynes: Access to Allylic and Propargylic Hydrazides. Org. Lett. 2005, 7, 4249–4252. 10.1021/ol0517473. [DOI] [PubMed] [Google Scholar]; e Waser J.; Nambu H.; Carreira E. M. Cobalt-Catalyzed Hydroazidation of Olefins: Convenient Access to Alkyl Azides. J. Am. Chem. Soc. 2005, 127, 8294–8295. 10.1021/ja052164r. [DOI] [PubMed] [Google Scholar]; f Gaspar B.; Waser J.; Carreira E. Cobalt-Catalyzed Synthesis of Tertiary Azides from α,α-Disubstituted Olefins under Mild Conditions Using Commercially Available Reagents. Synthesis 2007, 2007, 3839–3845. 10.1055/s-2007-1000817. [DOI] [Google Scholar]
- a Gerken V. C.; Carreira E. M. Carbon Nitride Photoredox Catalysis Enables the Generation of the Dioxolanyl Radical for Conjugate Addition Reactions. ACS Catal. 2022, 12, 10787–10792. 10.1021/acscatal.2c03229. [DOI] [Google Scholar]; b Rössler S. L.; Jelier B. J.; Tripet P. F.; Shemet A.; Jeschke G.; Togni A.; Carreira E. M. Pyridyl Radical Cation for C–H Amination of Arenes. Angew. Chem., Int. Ed. 2019, 58, 526–531. 10.1002/anie.201810261. [DOI] [PubMed] [Google Scholar]
- Freer P. C.; Perkin W. H. LXXXIV.—The synthetical formation of closed carbon - chains. Part I (continued). The action of ethylene bromide on the sodium-derivatives of ethylic acetoacetate, benzoylacetate, and acetonedicarboxylate. J. Chem. Soc., Trans. 1887, 51, 820–849. 10.1039/CT8875100820. [DOI] [Google Scholar]
- a Wallentin C.-J.; Nguyen J. D.; Finkbeiner P.; Stephenson C. R. J. Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]; b Nguyen J. D.; Tucker J. W.; Konieczynska M. D.; Stephenson C. R. J. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]; For one of the isolated products, it was shown that in a second separate step a cyclopropane was formed upon heating to 115 °C under basic conditions (Cs2CO3) for 24 h in DMF.; c Furst L.; Matsuura B. S.; Narayanam J. M. R.; Tucker J. W.; Stephenson C. R. J. Visible Light-Mediated Intermolecular C–H Functionalization of Electron-Rich Heterocycles with Malonates. Org. Lett. 2010, 12, 3104–3107. 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]
- Tokuyama reports a singular example of an intermolecular reaction, namely hexenol undergoing cyclopropanation with an α-bromo-β-ketoester under metal photoredox catalysis.; Ide K.; Furuta M.; Tokuyama H. Photoredox-catalyzed intramolecular cyclopropanation of alkenes with α-bromo-β-keto esters. Org. Biomol. Chem. 2021, 19, 9172–9176. 10.1039/D1OB01733K. [DOI] [PubMed] [Google Scholar]
- a Nakagawa M.; Matsuki Y.; Nagao K.; Ohmiya H. A Triple Photoredox/Cobalt/Brønsted Acid Catalysis Enabling Markovnikov Hydroalkoxylation of Unactivated Alkenes. J. Am. Chem. Soc. 2022, 144, 7953–7959. 10.1021/jacs.2c00527. [DOI] [PubMed] [Google Scholar]; b Kodo T.; Nagao K.; Ohmiya H. Organophotoredox-catalyzed semipinacol rearrangement via radical-polar crossover. Nat. Commun. 2022, 13, 2684. 10.1038/s41467-022-30395-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Jiao M.-J.; Feng Z.-T.; Wang X.-Z.; Xu G.-Q.; Xu P.-F. Design, Synthesis, and Application of Highly Reducing Organic Visible-Light Photocatalysts. Org. Lett. 2018, 20, 5700–5704. 10.1021/acs.orglett.8b02420. [DOI] [PubMed] [Google Scholar]
- Moore E. J.; Steck V.; Bajaj P.; Fasan R. Chemoselective Cyclopropanation over Carbene Y–H Insertion Catalyzed by an Engineered Carbene Transferase. J. Org. Chem. 2018, 83, 7480–7490. 10.1021/acs.joc.8b00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Häner R.; Maetzke T.; Seebach D. Generation and Reactions of Lithiatedtert-Butyl and 2,6-Di(tert-butyl)-4-methylphenyl Cyclopropanecarboxylates. Helv. Chim. Acta 1986, 69, 1655–1665. 10.1002/hlca.19860690720. [DOI] [Google Scholar]; b Gassman P. G.; Lumb J. T.; Zalar F. V. The Cleavage of Nonenolizable Ketones. J. Am. Chem. Soc. 1967, 89, 946–952. 10.1021/ja00980a036. [DOI] [Google Scholar]; c Evans D. A.; Woerpel K. A.; Hinman M. M.; Faul M. M. Bis(oxazolines) as chiral ligands in metal-catalyzed asymmetric reactions. Catalytic, asymmetric cyclopropanation of olefins. J. Am. Chem. Soc. 1991, 113, 726–728. 10.1021/ja00002a080. [DOI] [Google Scholar]
- a Wang R.; Zhou C.; Huang X.; Wu J.-Y.; Zhang X. Phenylphenothiazine-Based Porous Organic Polymers as Visible-Light Heterogeneous Photocatalysts for Switchable Bromoalkylation and Cyclopropanation of Unactivated Terminal Alkenes. ACS Sustainable Chem. Eng. 2022, 10, 4650–4659. 10.1021/acssuschemeng.2c00054. [DOI] [Google Scholar]; b Huang H.-L.; Xu J.; Fan Y.-X.; Su Q.-Q.; Du J.-Y.; Zhang R.-F.; Wang Y.-L.; Hu H.; Gao F. Visible-Light-Induced Difunctionalization of Alkenyl Ketones with α-Carbonyl Alkyl Bromide: Concomitant Installation of C–C Bonds. J. Org. Chem. 2022, 87, 14093. 10.1021/acs.joc.2c01682. [DOI] [PubMed] [Google Scholar]
- a De Meijere A.; Khlebnikov A. F.; Sünnemann H. W.; Frank D.; Rauch K.; Yufit D. S. Convenient Access to Various 1-Cyclopropylcyclopropane Derivatives. Eur. J. Org. Chem. 2010, 2010, 3295–3301. 10.1002/ejoc.201000209. [DOI] [Google Scholar]; b Wiedemann S.; Rauch K.; Savchenko A.; Marek I.; De Meijere A. Convenient Route to 2-(Trialkylstannyl)cyclopropylamines and Their Application in Palladium-Catalyzed Cross-Coupling Reactions. Eur. J. Org. Chem. 2004, 2004, 631–635. 10.1002/ejoc.200300454. [DOI] [Google Scholar]
- Cismesia M. A.; Yoon T. P. Characterizing chain processes in visible light photoredox catalysis. Chemical Science 2015, 6, 5426–5434. 10.1039/C5SC02185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner D. D. M.; Houmam A.; Rømming C.; Skattebøl L.; Barre L.; Hammerich O.; Søtofte I.; Langstrom B. Redox properties of free radicals. Acta Chem. Scand. 1998, 52, 377–384. 10.3891/acta.chem.scand.52-0377. [DOI] [Google Scholar]
- Shibutani S.; Nagao K.; Ohmiya H. Organophotoredox-Catalyzed Three-Component Coupling of Heteroatom Nucleophiles, Alkenes, and Aliphatic Redox Active Esters. Org. Lett. 2021, 23, 1798–1803. 10.1021/acs.orglett.1c00211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.