

Abstract
Despite the potency of most first-line anti-cancer drugs, non-adherence to these drug regimens remains high and is attributable to the prevalence of “off-target” drug effects that result in serious adverse events (SAEs) like hair loss, nausea, vomiting, and diarrhea. Some anti-cancer drugs are converted by liver uridine 5’-diphospho-glucuronosyltransferases through homeostatic host metabolism to form drug-glucuronide conjugates. These sugar-conjugated metabolites are generally inactive and can be safely excreted via the biliary system into the gastrointestinal tract. However, β-glucuronidase (βGUS) enzymes expressed by commensal gut bacteria can remove the glucuronic acid moiety, producing the reactivated drug and triggering dose-limiting side effects. Small-molecule βGUS inhibitors may reduce this drug-induced gut toxicity, allowing patients to complete their full course of treatment. Herein, we report the discovery of novel chemical series of βGUS inhibitors by structure-based virtual high-throughput screening (vHTS). We developed homology models for βGUS and applied them to large-scale vHTS against nearly 400,000 compounds within the chemical libraries of the National Center for Advancing Translational Sciences at the National Institutes of Health. From the vHTS results, we cherrypicked 291 compounds via a multifactor prioritization procedure, providing 69 diverse compounds that exhibited positive inhibitory activity in a follow-up βGUS biochemical assay in vitro. Our findings correspond to a hit rate of 24% and could inform the successful downstream development of a therapeutic adjunct that targets the human microbiome to prevent SAEs associated with first-line, standard-of-care anti-cancer drugs.
Introduction
In the age of data-driven medicine, drug development benefits from analysis of post-market surveillance data, such that problematic use of existing drugs may inspire new design strategies to optimize the use of these therapeutics1–3. This is especially relevant in cancer, a spectrum of diseases for which pharmacological intervention may be within the first line of therapeutic remediation. Most anti-cancer treatment modalities act through promoting death of rapidly dividing cells, such that they may suppress tumorigenesis but can also generate significant “off-target” effects from loss of epithelial cell volume across a patient’s organ systems4–6. Phenotypes emerging from this widespread apoptosis may include nausea and vomiting, loss of appetite, hair loss, and the potential for development of an immunocompromised state, given the high replication rates of innate and adaptive immune cells7–9. Therefore, non-adherence to anti-cancer treatment remains high among neoplastic patients, as patients seek to avoid severe side effects of their drug regimens10–12. This problem speaks to the ongoing need for new therapeutic strategies that may allow for suppression of these “off-target” effects, consequently improving the therapeutic index of anti-cancer drugs by reducing dose-limiting toxicity.
Enteric processing of anti-cancer drugs to urine-excretable metabolites forms non-toxic drug-glucuronides by uridine 5’-diphospho-glucuronosyltransferase (UGT)-mediated metabolism in the liver. In turn, β-glucuronidase enzymes (GUS) among commensal gut microbiota act on the drug-glucuronides to cleave their sugar moieties for nutrition, releasing reactivated drug molecules in the presence of vulnerable enterocytes. Consequently, the microbiome-dependent reactivation of small-molecule anti-cancer drugs in the presence of vulnerable enterocytes may be responsible for gastrointestinal (GI) serious, adverse events (SAEs), reducing patient adherence to their anti-cancer treatment regimens13–15. Indeed, this pattern of drug metabolism and pharmacokinetics (DMPK) is associated with at least 279 unique strains of GUS within the diverse microbial flora of the human gut16. We note that though humans also contain an endemic analog of GUS that is structurally heterogeneous to those in their microbiomes, the high density of bacterial GUS analogs renders the human analog minimally active, compared to that within resident gut bacteria17. For the remainder of this article, we will distinguish bacterial GUS and human GUS analogs as βGUS and hGUS, respectively.
As Ervin et al. discussed, a βGUS-inhibitory prodrug may increase the safety and tolerability of first-line anti-cancer agents: these investigators unpacked the metabolic fate of the anti-neoplastic agent regorafenib in the liver and GI tract. In turn, they found that raloxifene, a drug approved by the United States Food and Drug Administration for the treatment and prevention of osteoporosis in post-menopausal women18, is a potential inhibitor of the βGUS enzymes that re-activate regorafenib in the GI tract. Herein, the results of this study suggested that delivering a therapeutic adjunct capable of βGUS blockade could potentially reduce the burden of patient SAEs associated with small-molecule anti-cancer therapy19.
The mechanism of action (MOA) of βGUS in cleaving glucuronic acid moieties from glucuronidated compounds is well-described within the relevant biochemical literature and resembles that classically implicated in peptic ulceration upon excessive patient exposure to nonsteroidal, anti-inflammatory drugs (NSAIDs)20. Therefore, given that this article does not seek to probe the mechanistic details of the βGUS enzyme’s function, we refer readers with an interest in deep discussion of this information to the works of Wallace et al., Ervin et al., and Awolade et al., who present a thorough description of βGUS MOA21–23. These authors also describe the protein biochemistry that underlies the nature of modulatory effects on this class of enzymes; we present the relevance and validation of this information—within the scope of the present study—in an ad hoc fashion throughout our manuscript, as we focus on the application of in silico high-throughput screening (HTS) as a method of novel discovery for precision inhibitors of homologous βGUS analogs, rather than on the granularity of the established biochemistry that underlies these interactions. Indeed, these interactions are critical to understanding the relevance of our efforts to GI physiology but are most relevant to this study as validating information for our models.
Orthologs of βGUS exhibit a wide range of catalytic efficiencies for glucuronide substrates, driven by key structural differences among homologous enzymes16,24–26. Despite this structural and functional heterogeneity, previous drug screening efforts have focused solely on characterizing the βGUS ortholog encoded by gut commensal E. coli. This is a major weakness of the existing literature because most βGUS orthologs share < 25% amino acid identity with E. coli GUS16. Therefore, the goal of the present study was to develop a robust HTS platform that can identify inhibitors of common βGUS analogs in the human microbiome. While broad-spectrum antibiotics, such as cephalosporins, fluoroquinolones, and tetracyclines, offer the ability to suppress microbial action on glucuronidated drugs, these drugs can also suppress the beneficial activity of the gut microbiome27–29. Therefore, since such modulation could generate SAEs similar to—or potentially in augmentation to—those resulting from exposure to anti-cancer therapies30, a precision inhibition strategy that affects only βGUS, while maintaining the beneficial activity of the human microbiome, is critical to the development of a drug within this space. This necessary precision specifies a difficult drug design task, which requires consideration of a very large set of potential inhibitors to identify agents with the most optimal activity profiles.
Virtual high-throughput drug screening (vHTS) has emerged as an efficient method of targeted drug discovery, as it allows for simulation of a drug’s mode of binding to a specified target and thereby generates binding energy parameters that may act as predictive hypotheses of efficacy31–33. When the structure of a target is available or feasibly predictable, this technique is an efficient method of prioritizing compounds for further interrogation as potential hits: though a standard HTS approach allows for fast, combinatorial screening of compounds of interest towards a specific target, the combinatorial dimensionality of this technique results in significant material and labor costs and has low efficiency34. Therefore, in contrast to the typical <1% hit rate that most HTS panels achieve, vHTS—with in vitro follow-up testing of only compounds with the highest predicted activity—can achieve nearly an order of magnitude more success, as evidenced by hit rates ~3–4% and significantly less material costs in the interrogation of promising hits35.
Therefore, given the well-defined target rationale of this drug design task—as well as the challenge of establishing precision blockades of βGUS—we established a first-in-kind vHTS platform to discover inhibitors of βGUS and identify hits towards the development of a potential therapeutic adjunct to reduce the frequency of SAEs associated with first-line anti-cancer drugs. As we describe in the remainder of this manuscript, this screen harnessed nearly 400,000 compounds across the chemical libraries of the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH)36. We identified novel inhibitors with diverse structural scaffolds and single-digit μM potency in our βGUS biochemical assay.
Methods
1. Identification of a representative βGUS ortholog
To develop a vHTS platform for discovering inhibitors of βGUS, we first identified an ortholog of βGUS against which we could screen the NCATS chemical library. Herein, we focused our vHTS efforts on “H11G11-BG,” a representative βGUS ortholog implicated in the processing of regorafenib-glucuronide, a glucuronide metabolite of the tyrosine kinase inhibitor regorafenib used to treat metastatic colorectal cancer, hepatocellular carcinomas, and gastrointestinal stromal tumors17,21,25. The H11G11-BG protein was first identified as a βGUS gene encoded by the human fecal metagenome17.
We isolated the H11G11-BG genetic sequence (NCBI Accession: CBJ55484) and purified the recombinant protein for biochemical hit validation, per the procedure of Wallace et al22. Briefly, we codon optimized the nucleotide sequence for translation in E. coli and inserted the gene into a pLIC-His vector with ampicillin resistance and linkage to an N-terminal 6x-Histidine affinity tag for downstream purification. We then transformed competent cells (E. coli BL21-DE3), grew them to an OD600 of 0.6, and induced gene expression with IPTG. Then, we centrifuged the cells and resuspended them in buffer. Resuspended cells were supplemented with protease inhibitors and lysozyme, and were sonicated for cell lysis. Following an additional centrifugation to pellet cell debris, the cell lysate (supernatant) was collected and poured onto a column pre-loaded with 5 mL His-NTA resin. The cell lysate was washed to remove non-specific proteins. H11G11-BG was eluted and the resulting fractions collected and combined. We employed size exclusion as a final purification step and eluted protein isolate into 20 mM HEPES, pH 7.4, and 50 mM NaCl for biochemical activity assays. Then, we assessed purity by SDS-PAGE gel separation and gel filtration elution (>90% purity); we determined protein concentration using a spectrophotometer at a 280 nm absorption setting, using a standard combination of molecular weight, extinction coefficient, and Beer-Lambert Law calibration data to calculate concentration. The 754-residue amino acid (AA) sequence of H11G11-BG is cataloged in Universal Protein resource (UniProt) (https://www.uniprot.org/)37 as D5GU7138.
2. Homology modeling with MOE
The 2019 version of Molecular Operating Environment (MOE) (https://www.chemcomp.com/Products.htm)39 was used to generate a single-template homology model of our βGUS ortholog. To guide the selection of a template for homology modeling, we queried the H11G11-BG FASTA sequence in the Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi)40; this search gave the closest relatives of H11G11-BG homologs are β-glucuronidases and β-galactosidases. Nine potential templates were identified using an arbitrary benchmark of 80% sequence identity as the cutoff. In combination with the query results from sequence alignments in MOE based on the data from the most recent versions of the Protein Data Bank (PDB) (https://www.rcsb.org/)41 and UniProt (as were available when we completed this study in Summer 2019), the top five hits with the lowest p-value from two-sample similarity testing were selected and further curated. A point accepted mutation (PAM) matrix was generated to identify the best template chain with the highest residue similarity to our sequence of H11G11-BG, the lowest p-value and best sequence coverage of the homology model. We selected 6MVH (a flavin mononucleotide-binding β-glucuronidase isolated from Roseburia hominis; https://www.rcsb.org/structure/6MVH)42 as the most optimal template for βGUS and generated the homology model followed by force field-based energy refinement. 6MVH presented 58.5% sequence identity and 73% positive matching to our query sequence H11G11-BG, which is nearly double the benchmark 30% necessary for the confident generation of a predictive quaternary structure43.
3. Homology modeling with I-TASSER
The sequence of H11G11-BG was used as the input in the Iterative Threading ASSEmbly Refinement (I-TASSER) server, hosted by the Yang Zhang laboratory at University of Michigan (https://zhanglab.ccmb.med.umich.edu/I-TASSER/)44 to generate a threaded model through sequence alignment of the target sequence with available structures from PDB and UniProt as we describe above. Unlike MOE, I-TASSER generated the homology model based on multi-domain alignment — in this case the H11G11-BG homology model was derived from six domain-specific glucuronidase and glycosidase templates. The generated homology model was evaluated in I-TASSER with the following quantitative metrics: I-TASSER’s confidence score (C-score) for the model’s active site was 0.02, per a scale of C-score ϵ [−2, 5] (5 is most confident45), and the template modeling score (TM) was 72 ± 11%, per a range of TM ϵ [0, 100]% (100% is a perfect match between a model and an ensemble of guide structures).
4. Molecular docking
We undertook a systematic search of the βGUS literature to determine the importance of developing a holo representation of our target structure. Therein, we downloaded a β-(D)-glucuronic acid substrate molecule from the library of available βGUS ligands in PDB. Next, in MOE, we docked this molecule to our homology model to generate the appropriate target structure against which we could screen candidate inhibitors in a substrate-dependent manner. Then, to generate an optimal binding pose for application in vHTS, we curated a library of potential inhibitors, so we could select a template from among these compounds. To do this, we first reflected on previous in vitro assays that our group at NCATS had attempted to discover a battery of βGUS inhibitors. We downloaded the ~35 structure files of all compounds that we previously screened against other non-H11G11-BG βGUS enzymes and which demonstrated inhibitory activity (IC50 < 20 μM, efficacy < −50%, curve class (CC) ϵ [−1.1, −1.2, −1.3, −1.4]) and loaded them as a library into MOE. We then harmonized this set of compounds with the co-crystalized ligands we queried from other βGUS orthologs in PDB and enabled MOE to dock these compounds to our holo target structure.
Docking studies against NCATS chemical collections were conducted using the homology model derived from I-TASSER, given its quantified strength and its gestalt construction from six similar targets. Prior to molecular modeling and docking, the 3D structure of H11G11-BG was prepared using the “Structure Preparation” module in MOE. All hydrogens were added to the structure with standard protonation state. Finally, the modeled structure was energy-minimized using the QuickPrep module in the MOE program. The default parameter was selected, and the gridbox was defined by the center of the active sit with 4.5 Å pocket extension. Library compound conformations were generated on-the-fly during MOE docking. Triangle Matcher and London dG scoring function were used in initial docking pose placement, and Rigid Receptor and GBVI/WSA dG scoring function were used in final pose refinement.
5. vHTS
Both our homology models appeared to provide a high degree of confidence (per the reasonable sequence identity to the template structure, C-score, and TM values above). Because we sought to perform largescale drug screening, we decided to move forward with only the I-TASSER model for our drug discovery analysis. To prepare our target structure accordingly, we employed version 3.3.1 of the OEDocking package, which is available through OpenEye Scientific Software (https://www.eyesopen.com/oedocking)46. Using the MakeReceptor method within this package, we defined a gridbox to include the key pharmacophoric residues within the configuration of the I-TASSER homology model that resulted from the most energetically favorable docking of substrate and a known βGUS inhibitor to the target’s active site (please see further discussion of the constraining residues we identified in “Results and Discussion”). We removed the substrate molecule itself before screening to avoid steric penalties from artifactual binding patterns that could result in collisions between the ligand and substrate molecules. Then, from the command line, we harmonized the 399,423 compounds within the NCATS chemical libraries with our docking collection of ligands we extracted from PDB and the most active compounds from our previous βGUS assays. The compound library was pre-filtered by drug-likeness filters based on Lipinski’s rule of five to eliminate obviously unwanted moieties. We executed vHTS through the FRED method within OEDocking, harnessing a high-performance computing cluster on Biowulf, a NIH server capable of supercomputing47.
To facilitate “quality control” and consensus prioritization of our resultant vHTS hits, we first rank-ordered hits by their predicted binding energies. We then removed all compounds resembling broad-spectrum antibiotics, such as cephalosporins, fluoroquinolones, and tetracyclines, given our interest in discovering precision βGUS inhibitors and the relevance of this goal to the immunological manifestations of neoplasia, as we describe in the “Introduction” section of this manuscript. Additionally, we removed lower-ranking compounds with bulky moieties, given our concerns about potential steric hinderance at the inhibitor binding site upon re-docking to our holo target model. We also parsed compounds with problematic chemistry, such as hits with several cyclopropane functionalities (given their associated angle strain and instability) and hits with trifluorinated aromatic moieties (given the strong electron-withdrawing effects of these groups and their potential impacts on efficacy). We eliminated other hits with functionalities that are strongly associated with cytotoxicity on an ad hoc basis, per the expertise of our study team. More details about the relevance of our cherry-picking approach are available in the “Results and Discussion” section of this manuscript.
6. Biochemical validation assay
The GUS biochemical assay was developed and miniaturized for high-throughput screening using 4MUG (4-Methylumbelliferyl-β-D-glucuronide hydrate, from Sigma, M9130) as the substrate. Three microliter per well of assay buffer solution (0.67 nM purified GUS enzyme, 400 μM 4MUG, 50 mM HEPES at pH 7.5 and 0.01% Triton X-100) was dispensed into solid-bottom black 1536-well, assay plates (Greiner, solid white medium-binding plates) with Aurora Discovery BioRAPTR Flying Reagent Dispenser (FRD; Beckton Coulter). Then 23 nL of compound solution was transferred to the assay plate using the Kalypsys pin tool (Waco Automation, San Diego, CA). Following compound transfer, 1 μL of 4MUG assay solution was added to the assay plate yielding a total assay volume of 4 μL/well, with the final reagent concentration of 0.5 nM GUS and 100 μM 4MUG. After 1 hr incubation at room temperature, the fluorescence signal was detected by a ViewLux (PerkinElmer) with 340 nm excitation filter and 450 nm emission filter. RFU was used as fluorescence expression. Inhibitor 9 was used as a control compound for data normalization.
Results and Discussion
1. Structural analysis of βGUS homology models
Upon studying the homology model of the highest rank from MOE, we identified the putative binding site in this model, per the co-localization of key active site residues for βGUS, harmonically identified by Wallace et al. as Tyr433, Tyr437, Phe448, Glu464, Lys563, Arg56422. Indeed, the observation of these residues or those of similar biochemical profile at these positions validated the strength of this homology model. Also supportive of the consistency of our approach with existing knowledge on βGUS was our observation that—upon alignment—the primary sequences of 6MVH and our homology model were highly similar for the 441 residues of closest adjacency to the template active site. However, for the remaining 313 residues of 6MVH, the homology model is strikingly dominated by extraneous alpha helices and turns that are not in keeping with the selected template. While these structures likely account for empirical reductions in similarity between our template and our homology model, we noted that they appear insignificant in the scope of a downstream vHTS task, as this mode of compound screening considers rigid docking of hit candidates only to a specified ligand binding site. Therefore, though this rationale may appear to neglect the dynamism and the structural importance of non-active site domains within our target, we assert that the high similarity of 441 residues near the βGUS active site could—within the framework allowed by vHTS, which requires specification of a granular region of a target structure against which to screen compounds34—provide us hints at the dynamism that occurs upon ligand binding at the site where this flexibility may most significantly affect efficacy of competitive inhibition. However, we note—as is standard across most of the vHTS literature31,32,34—that the drug discovery task we report in this manuscript involves thoroughly rigid docking (i.e., we are unaware of an existing computing package that can simulate domain transpositions robustly). Finally, to confirm that this homology model was of sufficiently high quality to warrant vHTS, we generated a Ramachandran plot to note potential steric strain within the target structure. From MOE’s Ramachandran plot (available in the Supplementary Figure 1 to this manuscript), we found that our model produced an acceptably low number of residues (12) with outlying levels of angle strain. Then, we annotated the outliers on the ribbon structure of our homology model to observe the location of these residues. In doing this, we noted that most of these residues fell within a variable section of the target (i.e., in a loop or other functionally dynamic domain), suggesting that the few loci with excessive strain in our structure are of little consequence to the robustness of our model, as these regions may adopt allowable conformations to mitigate the strain in a true, functional in vivo system. Nonetheless, we noted that that action site of our homology model—as well as its βGUS template—appears to be surrounded by several loops. While this suggests that the active site may be dynamic, we again affirm that our vHTS investigation is not compromised by this structural motif as vHTS employs rigid docking.
In analyzing the quality of our I-TASSER model beyond the quantification we provide in “Methods,” we found that the strength of this model was apparent from the high degree of conservation between the active site residues that the I-TASSER server identified within a putative binding site and the active site residues as reported by Wallace and colleagues. Further reflecting on the results returned by the I-TASSER server, we noted that this putative active site falls within the domain of a βGUS analog from Ruminococcus gnavus (6MVG; https://www.rcsb.org/structure/6MVG)48.
Visualizations of MOE and I-TASSER homology models that we generated for H11G11-BG are available within Figure 1, which shows the structural similarities and differences between these models.
2. Docking analysis of known βGUS inhibitors
Upon prioritization of our docking hits, we observed that the compound with the lowest MOE docking score was a sulfonated glucose molecule; however, given that this compound closely mimics βGUS substrate—and presents with small surface area around the βGUS active site—we decided to probe compounds with larger surface area and slightly higher binding energy differentials in selecting a template binding mode. Doing this, we determined that the lowest binding energy ligand that also met our requirements for coverage of key residues was 1-((6,8-dimethyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-1-(2-hydroxyethyl)-3-(4-hydroxyphenyl)thiourea, a ligand we extracted from PDB and that corresponds to a known inhibitor of Escherichia coli βGUS (5CZK; https://www.rcsb.org/structure/5czk)49. Re-docking this ligand to our holo target structure and using MOE’s Protonate3D functionality to obtain the least sterically-encumbered conformation for all target residues with rotatable bonds, we captured the resulting binding pose as our template for vHTS. We noted that this pose appears to maintain the key interactions for our βGUS target. For our readers’ review, we include a visualization of this pose as Figure 2 within this manuscript. We generated Figure 2 via the structure visualization software PyMOL50.
Key pharmacophoric residues that we identified from our docking analysis included the following list of AAs; we specified ligand interactions with bolded residues as constraints in the vHTS procedure we describe in “Methods.”
Glu464 (This residue allows H-bonding & nucleophilic attack, depending on the ambient pH of the target-ligand ensemble.)
Tyr433 (Given the relative positioning of phenyl rings within this residue and within our template inhibitor, meta-stabilization via π-π stacking is possible if rotation of the Tyr R group places its phenyl ring parallel to the candidate’s phenyl ring. Indeed, we observed this positioning in our MOE and MakeReceptor visualizations.)
Tyr437 (This residue allows for stabilization of the target-ligand assembly via H-bonding to the residue’s free hydroxy group.)
Lys563 (This residue allows for stabilization of the target-ligand assembly via H-bonding to its charged R group.)
Arg564 (This residue allows for stabilization of the target-ligand assembly via H-bonding to its charged R group.)
Phe448 (This residue appears to provide steric hinderance to inhibitor binding in the apo configuration of βGUS, but substrate binding facilitates its retrograde movement, such that this configurational change allows ligands to access the inhibitor binding site that lies adjacent to the target’s active site.)
3. Discovery of novel inhibitors and their binding modes through vHTS
The top-ranked 2,500 low-binding energy hits from NIH Biowulf cluster were retrieved and energy-minimized within the βGUS active site. From the hit list, we noted some replication of the same compounds which have already been presented in PDB co-crystal structures or identified as HTS screening hits, as we expected. After removing the redundant hits and the promiscuous compounds with potential undesirable functionalities, cytotoxic compounds, and PAINS motif as we describe in “Methods”, we selected 1,000 triaged vHTS hits (please see examples of these compounds in our Supplement) for multi-parameter assessment. Structural clustering was performed for these 1,000 compounds and the binding models of all clusters and singletons were visually inspected. Finally, a total of 291 compounds were cherrypicked manually based on the predicted binding model, ranking score, representative structural cluster, and stock availability of the compounds for testing. We compiled a library of structure files for these 291 compounds, imputing them to MOE for re-docking to our homology model to ensure that they maintained the key interactions we specified in the creation of our vHTS gridbox. In doing this, we noted that our prioritized hits interacted with the key stabilizing and nucleophilic residues we identified within the βGUS active site and demonstrated extensive surface area coverage of this domain, as several top-ranking compounds contained multiple six-membered rings. This motif promoted jutting of the ligand towards the solvent side of the target, as appears common with many of the effectual ligands for homologous strains of GUS that we found in PDB’s ligand library. By visual inspection, we also noticed the recurrence of three (3) distinct motifs, at large, within our high-ranking cherry-picks: a pyrazole group, a benzoimidazole derivative, and multiple electron-donating halogens substituted and distributed across component benzyl rings of these ligands.
Nonetheless, we did not observe the presence of a π-π stacking interaction with Tyr433 across the optimal binding poses of our top-scoring candidate inhibitors. This might be the outcome of flexible, rotatable bonds at the residue-aligned sites within the ligand structures; alternatively, this phenomenon could have resulted from an inbuilt limitation in the sensitivity of detecting a π-π stacking within OpenEye’s constraint recognition system, as the presence of several atoms in appropriate proximity and approximately parallel to Tyr433 might have triggered faulty recognition of this interaction in the generation of our list of hits. Nonetheless, we maintained our list of 291 top-ranking cherry-picks without prejudice, to evaluate their comparative performance in biochemical validation assays in vitro.
In analyzing the results of the biochemical vHTS hit validation assay and its counter screen that we describe in “Methods,” we identified 69 out of our 291 vHTS hits to be confirmed as active (i.e., CC ≠ 4 and CC < 0), giving a 24% hit rate for our drug discovery exercise. Among these potent compounds, we observed few singleton hits. Additionally, we uncovered 13 potent compounds with activity heuristically defined as high-quality actives for future probe development (i.e., IC50 < 10 μM, efficacy < −50%)51. A visual summary of our protocol—as well as the numerical results arising from its execution—is available in Figure 3.
In Figure 4, we present the structure, binding mode, and dose-response behavior of the top-three hits from our high-quality shortlist. We noted adherence of the binding modes among these compounds to our core criteria in selecting inhibitor templates through our docking experiments, which served as preliminary proof-of-concept of the efficacy of our approach and a further “sanity check” in confirming the validity of our results.
Because we assert that our 69 top hits cluster among representative chemical series (and therefore present few singletons), here, we discuss the most potent compounds within each of three highest-ranking series.
As Figure 4 shows, NCGC00253873—our top hit and a representative compound of the most potent chemical series within our results—shows maintenance of key interactions with the “constraining” residues that we specified in our vHTS gridbox when we re-docked it to our I-TASSER homology model. It demonstrates IC50 = 3.8 μM for the physiologically-relevant strain of βGUS that we employed in our biochemical assay, with efficacy = −71%, CC = −1.2, and a clearly-defined, logistic dose-response curve. This compound is an analog of the RUC-2 inhibitor—a variant of the series of RUC-4 inhibitors (RUC-4i), which have garnered recent interest for their anti-thrombogenic inhibition of glycoprotein IIb/IIIa52–54. To date, RUC-4i has completed a phase I randomized, controlled trial of dose tolerability (https://clinicaltrials.gov/ct2/show/NCT03844191?term=ruc-4&draw=2&rank=2)55 and is now in the beginning stages of a phase II open label trial for pharmacodynamic and pharmacokinetic characterization of efficacy in reducing disease severity among patients with sinus tachycardia-elevation myocardial infarction (https://clinicaltrials.gov/ct2/show/NCT04284995?term=ruc-4&draw=2&rank=1)56. Therefore, this result might provide an early signal for a drug repurposing opportunity within the therapeutic space we describe in this manuscript. Additionally, our finding may be especially timely for further evaluation, given the present interest in treatments of coagulopathy57–59 like the RUC-4i series.
A representative of our second highest-ranking series—5-(2,3-dichlorophenyl)-N-(3-(oxazolo[4,5-b]pyridine-2-yl)phenyl)furan-2-carboxamide (NCGC00099510)—showed IC50 = 9.6 μM for βGUS, with logistic dose-response behavior, CC = −1.2, and efficacy = −70%. A third series-representative hit compound we present in Figure 4—4-(cyclopropylmethyl)-N-phenyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-e][1,4]diazepin-8-amine (NCGC00411059)—also demonstrated logistic dose-response behavior, with IC50 = 11.1 μM, efficacy = −72%, and CC = −2.2. The inclusion of this compound in our list of top-ranking compounds speaks to the power of vHTS in providing an additional layer of “quality control” to standard HTS workflows, as, in our former non-virtual, HTS probes of βGUS analogs similar to H11G11-BG, we screened this compound against βGUS in triplicate and with a seven-point titration series to develop a full dose-response curve, but we did not observe its bioactivity. Therefore, we had de-prioritized NCGC00411059 from future consideration as a potential βGUS inhibitor, but the strength of our vHTS results now suggest to us that we should re-examine this candidate as it may have more therapeutic potential than our previous assessments of its activity revealed. We regarded the observation of compound efficacy in our biochemical βGUS assay as powerful, since the significantly reduced scale of this screening compared to that of solely testing the NCATS chemical libraries through an in vitro platform60 suggests that these results may be viewed as more definitive than would otherwise arise from a standard HTS workflow, given our ability to leverage virtual screening as a preliminary high-pass filter for compound efficacy and our integration of chemical logic at each stage of our discovery process. Furthermore, the situation we present of newfound observations of compound activity speaks to the power of vHTS in rescuing “false negatives” from a high-throughput drug discovery platform and therefore increasing its sensitivity. This is a pattern that is only scarcely observed in previous literature61, but we believe it to be an important attribute of the vHTS approach and therefore a notable aspect of the results we present.
We again note that nearly all of our screening hits demonstrated common, salient chemotypes, such that our 69 actives presented with few singleton hits. The screening data from our βGUS biochemical assay for these compounds are available within our Supplement.
Finally, in importing our bioactive hits to MOE and re-docking them to our I-TASSER target model, we observed that these compounds all exhibited mixed-mode inhibition, as they bound to our βGUS homolog at a location adjacent to the active site while still maintaining interactions with key active site residues, as with the inhibitors we display in Figure 4. These observations foment the conclusions we describe above and present an interesting exploration of a new, substrate-dependent mode of βGUS inhibition that allowed us to discover several prospectively inhibitory series of precise therapeutic adjuncts. We believe that the results we present here represent a major stride in evaluation of the druggability of a target implicated in adverse reactions to high-use, first-line anti-cancer drugs, as we obtained a 24% hit rate resulting from our drug discovery experiments. Given that most assays performed under HTS conditions result in hit rates ~1%, this suggests that our approach to discovery of a βGUS inhibitor presents with nearly 23-fold enrichment over a similar method of non-virtual drug discovery.
It is important to note that visual inspection of the binding mode and multi-parameter hit assessment procedure are very important components of virtual drug discovery. While the “automated” components of our drug discovery process provided the edifice upon which we could specify the minimum biochemical specifications to define a “hit” (and thereby quickly eliminate the majority of our large screening library from further consideration), the procedure for ensuring practicality and efficiency of our platform (i.e., our prioritization of the most relevant hits for downstream testing) relied on our ability to curate expert knowledge. vHTS is a hypothesis-generation tool that allowed us several machine-defined actives to consider manually for signals of efficacy. However, the strength of our platform—as evidenced by its high hit rate and inbuilt βGUS activity—is holistically associated to our power to identify the most promising hypotheses of drug activity using multi-parameter prioritization procedure (binding model examination, ranking scores, clustering analysis, and promiscuity assessmen) from our vHTS results and our biochemical reasoning. This information allowed for us to observe activity for a high percentage of our prioritized predictions within our validation assay. Therefore, we believe that our hit rate is neither artifact nor artificially inflated. Instead, it speaks to our selection of powerful heuristics for prioritizing vHTS hypotheses. We similarly affirm that the best application of multi-parameter assessment to vHTS results is neither arbitrary nor deterministic, as the use of strong conceptual logic and ad hoc decision-making in evaluation of each listing on an automated bioactives list speaks to a design-oriented approach to drug discovery.
Conclusions
Standard of care (SOC) for patients with non-excisable neoplasia is prescription of an anti-cancer drug regimen; although most small-molecule anti-cancer drugs are well-established and demonstrate potency for their targets, patient non-adherence to these medications due to their associated SAEs remains a significant barrier to their efficacy. Gut commensal βGUS enzymes can reverse compound inactivation catalyzed by host Phase II glucuronidation by cleaving drug-glucuronide conjugates and forming toxic levels of reactivated drug in the gastrointestinal tract. Thus, microbiota βGUS has been implicated as a novel drug target to prevent the gastrointestinal toxicities of existing therapeutics, including the anti-cancer drugs irinotecan and regorafenib20–22,62–64. However, as the gut microbiome provides immunological and digestive benefits to neoplastic patients who are often immunocompromised and present with iatrogenic epithelial cytopenia in their GI tracts, broad-spectrum antibiotics have a minimal therapeutic index for this population. This suggests that there is a clinical unmet need for a precision, βGUS-inhibitory adjunct therapy to be delivered alongside SOC anti-cancer drugs to ensure that neoplastic patients can better tolerate their treatment. Such a therapy could maximize the therapeutic value of existing anti-cancer agents, while simultaneously minimizing the safety risks of these drugs.
The work we present in this manuscript demonstrates a holistic, multilayered, and first-in-kind attempt at systematic discovery of hits for a βGUS inhibitor, which we attempted via the powerful approach of vHTS with downstream biochemical assay validation in vitro. The results that we present from analysis of ~400,000 compounds suggest success of this approach, as they centered around a hit rate that is 23-fold enriched compared to standard experimental HTS drug screening initiatives, 69 hits that map to common scaffolds for βGUS inhibitor design (with 13 hits demonstrating high-quality bioactivity), and an opportunity to consider drug repurposing with a class of agents that is of increasing interest for its potential application in treating coagulopathy.
In the future, we hope to further develop our βGUS drug design platform by more detailed interrogation of druggable sites within our target model. While, in the work we present here, we identified candidates for inhibitor design through probes of candidate binding near the βGUS active site, we are curious to attempt our methods towards discovery of potential allosteric modulators of this target and, if an allosteric binding site appears plausible, to test the compounds we identified as hits from our current work to identify the potential for multimodal inhibition of this target. We also plan to further develop our testing platform of top candidates from our list of validated hits, as this will allow us to continue prioritizing our 69 hits. We are currently considering the development of biomarker discovery studies for βGUS inhibition and may consider testing our compounds in more representative in vitro systems, such as 3D human tissue culture, to better simulate their effects on the diverse microenvironments of human gut. We are also interested in developing appropriate mechanistic and enzyme kinetics probes to confirm our hypotheses on the modes of hit action that we developed through docking experiments and interpretation of the signals from our biochemical assay. Indeed, if the results that we present here hold true in these future screens, we believe that our platform could have strong potential in catalyzing the pace of drug discovery for a precision therapeutic that has application to improving health outcomes for millions of prospective patients65.
Supplementary Material
Acknowledgements
This work was supported by the NCATS Intramural Research Program. APC has also been supported by awards U54TR02243-02 from NCATS and R21HD105304 from NIH’s National Institute of Child Health and Development. The content of this manuscript is solely the responsibility of the authors and does not represent the official views of the NIH.
Appendix
Figure 1:
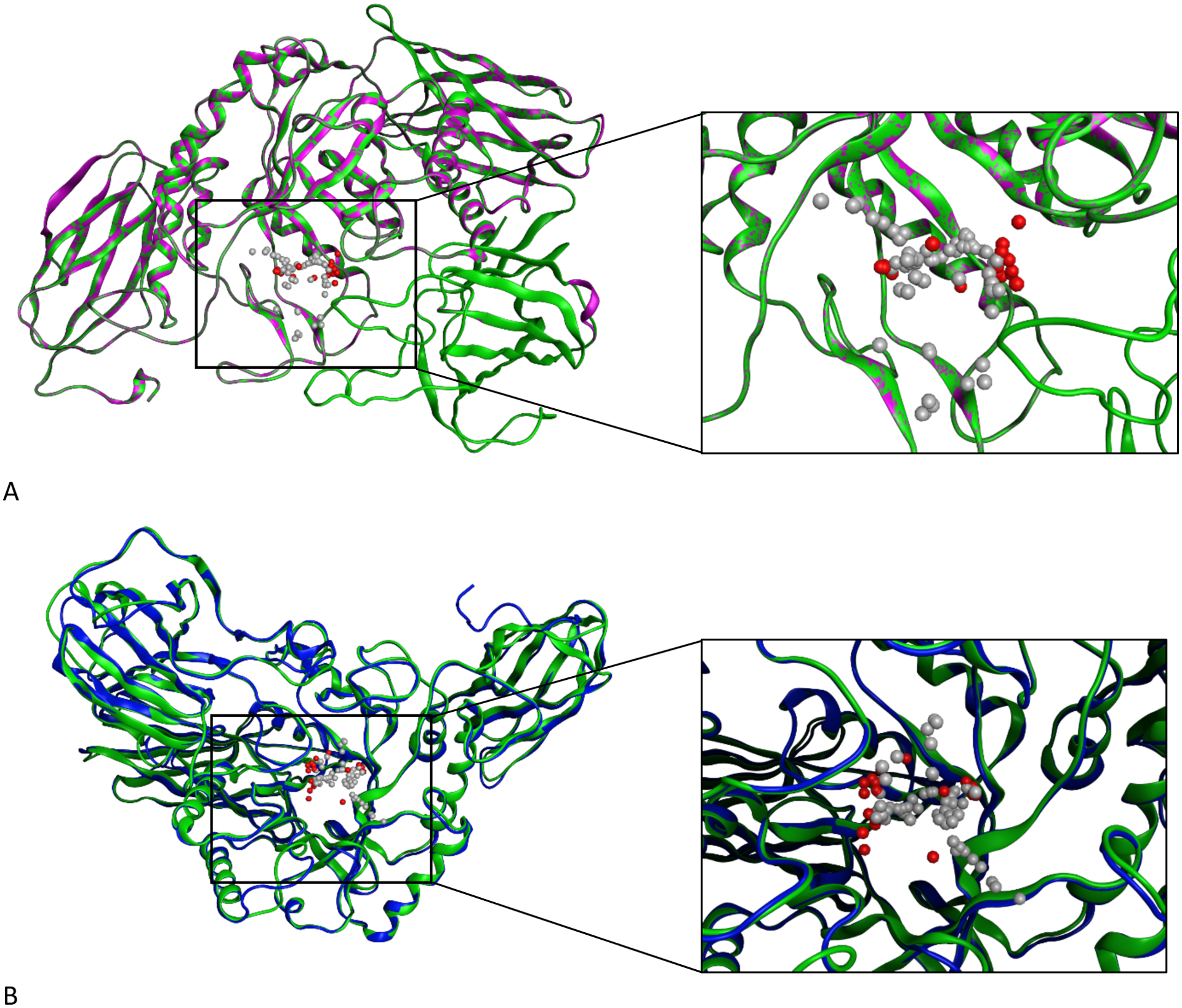
Homology models for ßGUS using both MOE and the I-TASSER server. Panel A shows the single-template homology model we generated in MOE with an enlarged view of the model’s predicted active site (green: MOE model, purple: 6MVH template). Panel B shows the threaded model we obtained from I-TASSER, superimposed on the homology model we generated in MOE and with an enlarged and residue-labeled view of the models’ predicted active sites when occupied by an analog of glucose (blue: I-TASSER model, green: MOE model). The gray and red space-filling spheres are hydrophobic and hydrophilic cavities predicted by the SiteFinder module in MOE.
Figure 2:
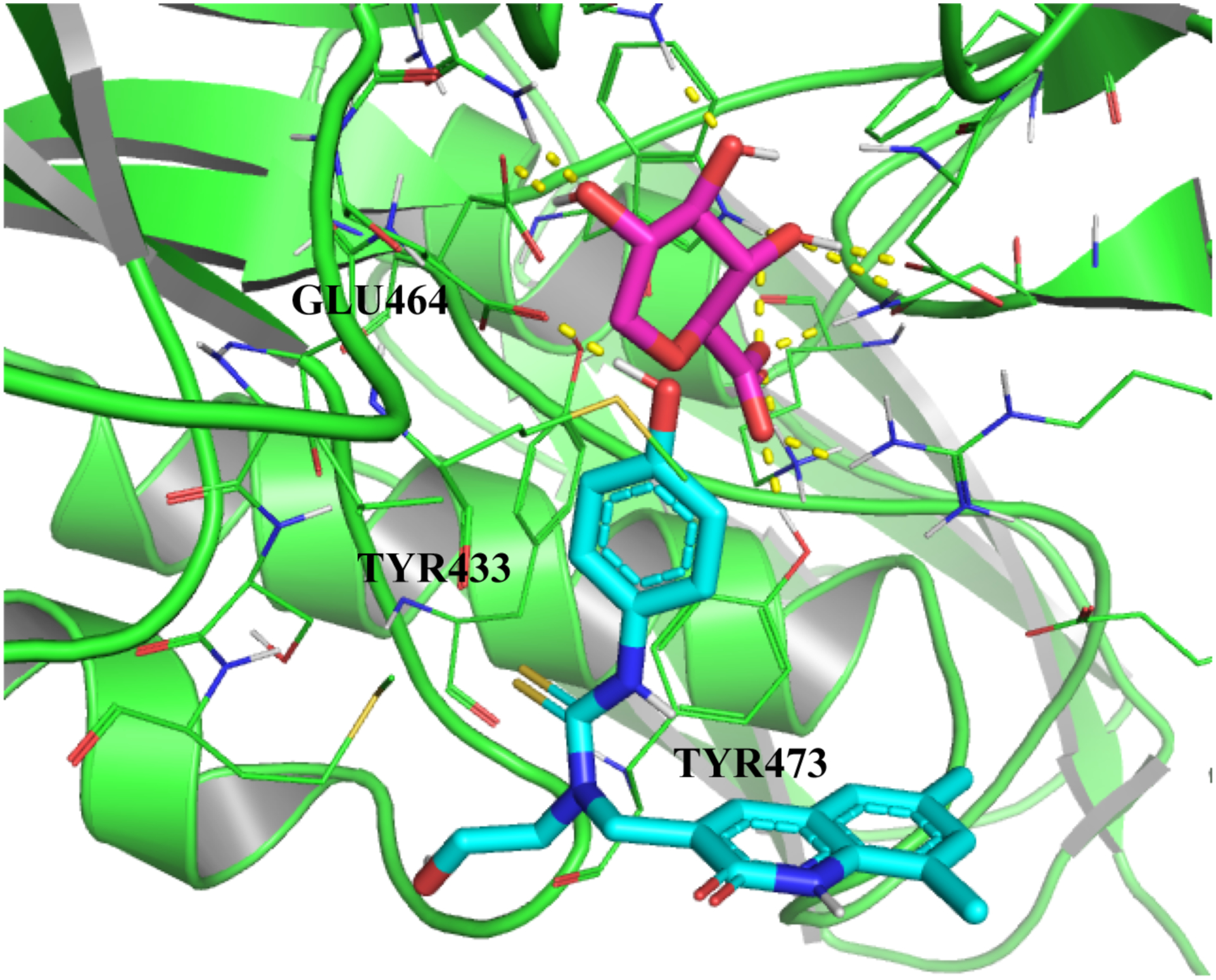
From our docking experiments, we developed a holo template of ßGUS bound to 1-((6,8-dimethyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-1-(2-hydroxyethyl)-3-(4-hydroxyphenyl)thiourea for development of our gridbox and subsequent VS. This template inhibitor appears to exhibit mixed-mode inhibition, as its most favorable binding pose occupies a site tangent to the ßGUS active site but maintains several of the key interactions that we would expect to observe under competitive inhibition. Constraining residues that we specified in our gridbox are labeled, and the substrate glucose analog is colored purple and the template inhibitor—with its solvent side exposed—is colored cyan. Hydrogen bonding interactions are indicated with dashed yellow lines.
Figure 3:
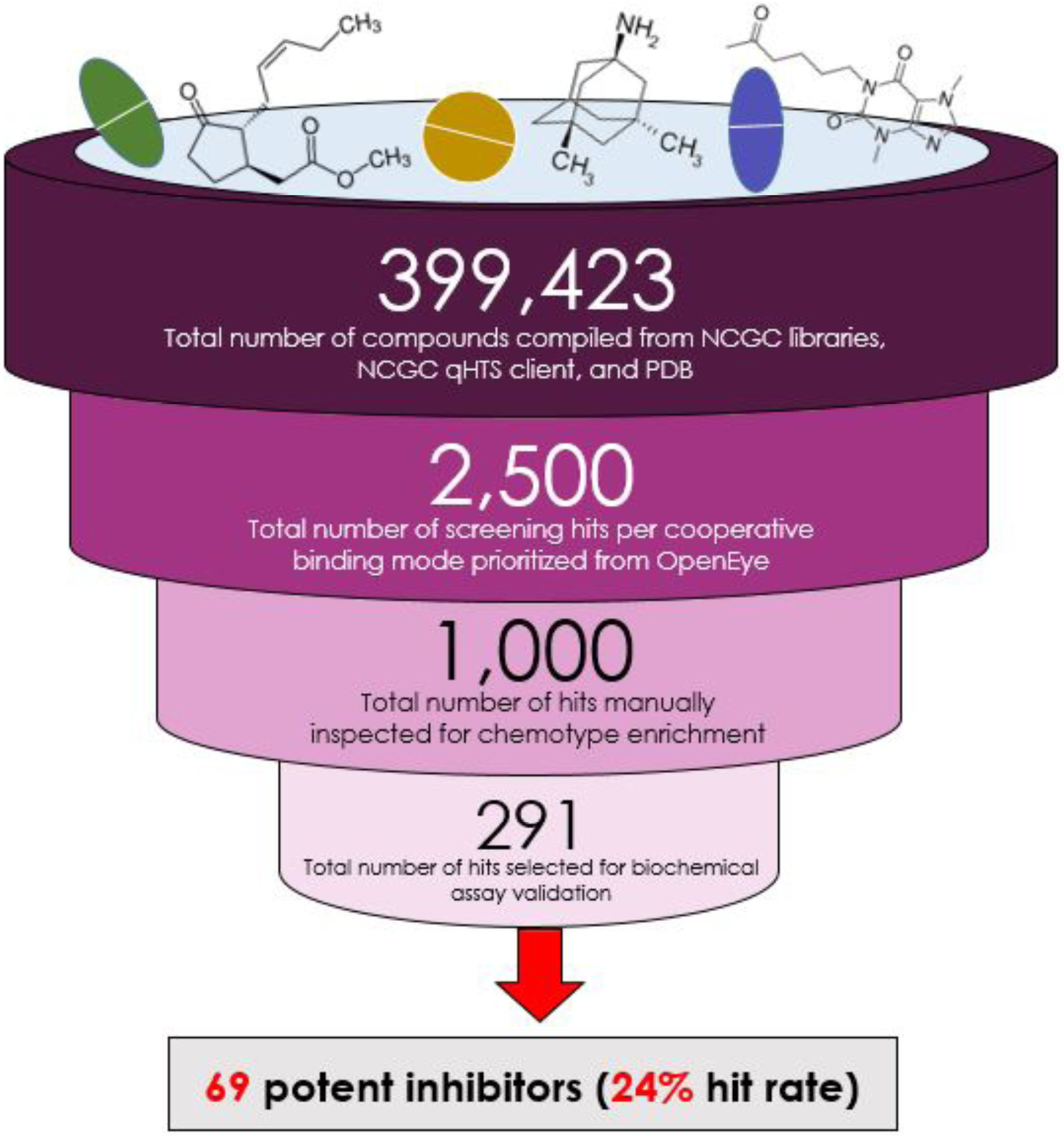
A visual summary of the workflow we employed to discover ßGUS inhibitors via vHTS and a validating biochemical assay, accompanied by quantification of this platform that supports our claims of systematic and high-throughput drug screening, sensitive heuristics for prioritization of vHTS hypotheses, and a high hit rate.
Figure 4:
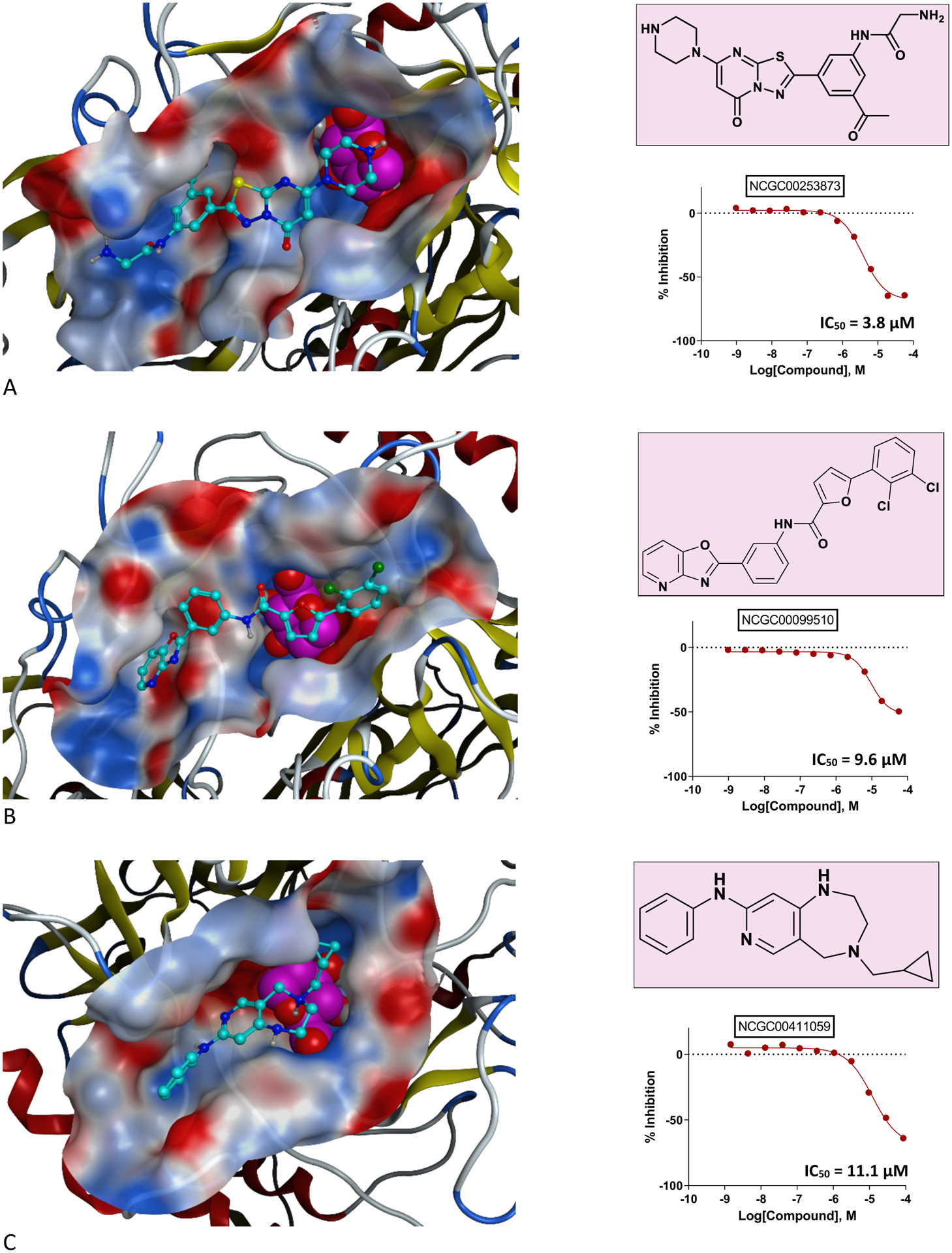
Representatives of chemical series among our top validated vHTS hits include RUC-2i (Panel A) and 5-(2,3-dichlorophenyl)-N-(3-(oxazolo[4,5-b]pyridine-2-yl)phenyl)furan-2-carboxamide (Panel B). Panel C provides an example of false negative rescue of 4-(cyclopropylmethyl)-N-phenyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-e][1,4]diazepin-8-amine via VS. In each delineation of inhibitor binding mode, the glucose is colored in purple with space-filled representation, the ligand is colored cyan, and the binding pocket is shown as the electrostatic surface.
Footnotes
Conflicts of Interest
We declare no competing interests relevant to the execution and outcomes of this study. JH and BDW have been employees of Symberix, Inc.
Data and Software Availability
All protein structures underlying the development of our homology models are publicly available on PDB; links to these structure files are available at the first mention of each protein in-text. PyMOL was used to prepare all chemical visualizations and is available through the link cited in-text and provided in the “References” section of this manuscript. This research employed packaged chemical computing software (MOE, OpenEye) purchased by and licensed to NCATS; links to these platforms and details of their versions are available through their first mentions in-text. Our Ramachandran plot and HTS data for the hits described in “Results and Discussion” are available as Supplementary Information to this manuscript; we provide this information as image and spreadsheet files, respectively. Readers interested in accessing NCATS’s in-house chemical libraries for collaborative research should contact Dr. Shen at the email address above.
References
- (1).Pulley JM; Rhoads JP; Jerome RN; Challa AP; Erreger KB; Joly MM; Lavieri RR; Perry KE; Zaleski NM; Shirey-Rice JK; Aronoff DM Using What We Already Have: Uncovering New Drug Repurposing Strategies in Existing Omics Data. Annu. Rev. Pharmacol. Toxicol 2019. 10.1146/annurev-pharmtox-010919-023537. [DOI] [PubMed] [Google Scholar]
- (2).Challa AP; Lavieri RR; Lewis JT; Zaleski NM; Shirey-Rice JK; Harris PA; Aronoff DM; Pulley JM Systematically Prioritizing Candidates in Genome-Based Drug Repurposing. Assay Drug Dev. Technol 2019, 17 (8), 352–363. 10.1089/adt.2019.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Challa AP; Madu CO; Lu Y BRCA 1/2 Tumors and Gene Expression Therapy for Breast Cancer Development and Metastasis. Oncomedicine 2017, 2, 132–137. 10.7150/oncm.20393. [DOI] [Google Scholar]
- (4).Gospodarowicz M; Trypuc J; D’Cruz A; Khader J; Omar S; Knaul F Cancer Services and the Comprehensive Cancer Center. In Cancer: Disease Control Priorities, Third Edition (Volume 3); Gelband H, Jha P, Sankaranarayanan R, Horton S, Eds.; The International Bank for Reconstruction and Development / The World Bank: Washington (DC), 2015. [PubMed] [Google Scholar]
- (5).Shewach DS; Kuchta RD Introduction to Cancer Chemotherapeutics. Chem. Rev 2009, 109 (7), 2859–2861. 10.1021/cr900208x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Information, N. C. for B.; Pike, U. S. N. L. of M. 8600 R.; MD, B.; Usa, 20894. How Does Chemotherapy Work?; Institute for Quality and Efficiency in Health Care (IQWiG), 2019. [Google Scholar]
- (7).Nurgali K; Jagoe RT; Abalo R Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol 2018, 9. 10.3389/fphar.2018.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pearce A; Haas M; Viney R; Pearson S-A; Haywood P; Brown C; Ward R Incidence and Severity of Self-Reported Chemotherapy Side Effects in Routine Care: A Prospective Cohort Study. PLoS ONE 2017, 12 (10). 10.1371/journal.pone.0184360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).de Boer-Dennert M; de Wit R; Schmitz PI; Djontono J; v Beurden V; Stoter G; Verweij J Patient Perceptions of the Side-Effects of Chemotherapy: The Influence of 5HT3 Antagonists. Br. J. Cancer 1997, 76 (8), 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).GEYNISMAN DM; WICKERSHAM KE Adherence to Targeted Oral Anticancer Medications. Discov. Med 2013, 15 (83), 231–241. [PMC free article] [PubMed] [Google Scholar]
- (11).daCosta DiBonaventura M; Copher R; Basurto E; Faria C; Lorenzo R Patient Preferences and Treatment Adherence Among Women Diagnosed with Metastatic Breast Cancer. Am. Health Drug Benefits 2014, 7 (7), 386–396. [PMC free article] [PubMed] [Google Scholar]
- (12).McCue DA; Lohr LK; Pick AM Improving Adherence to Oral Cancer Therapy in Clinical Practice. Pharmacother. J. Hum. Pharmacol. Drug Ther 2014, 34 (5), 481–494. 10.1002/phar.1399. [DOI] [PubMed] [Google Scholar]
- (13).Allain EP; Rouleau M; Lévesque E; Guillemette C Emerging Roles for UDP-Glucuronosyltransferases in Drug Resistance and Cancer Progression. Br. J. Cancer 2020, 122 (9), 1277–1287. 10.1038/s41416-019-0722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cummings J; Ethell BT; Jardine L; Boyd G; Macpherson JS; Burchell B; Smyth JF; Jodrell DI Glucuronidation as a Mechanism of Intrinsic Drug Resistance in Human Colon Cancer: Reversal of Resistance by Food Additives. Cancer Res. 2003, 63 (23), 8443–8450. [PubMed] [Google Scholar]
- (15).Hu DG; Mackenzie PI; Lu L; Meech R; McKinnon RA Induction of Human UDP-Glucuronosyltransferase 2B7 Gene Expression by Cytotoxic Anticancer Drugs in Liver Cancer HepG2 Cells. Drug Metab. Dispos 2015, 43 (5), 660–668. 10.1124/dmd.114.062380. [DOI] [PubMed] [Google Scholar]
- (16).Pollet RM; D’Agostino EH; Walton WG; Xu Y; Little MS; Biernat KA; Pellock SJ; Patterson LM; Creekmore BC; Isenberg HN; Bahethi RR; Bhatt AP; Liu J; Gharaibeh RZ; Redinbo MR An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25 (7), 967–977.e5. 10.1016/j.str.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gloux K; Berteau O; Oumami HE; Béguet F; Leclerc M; Doré J A Metagenomic β-Glucuronidase Uncovers a Core Adaptive Function of the Human Intestinal Microbiome. Proc. Natl. Acad. Sci 2011, 108 (Supplement 1), 4539–4546. 10.1073/pnas.1000066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Raloxifene Package Insert.
- (19).Chen S; Yueh M-F; Bigo C; Barbier O; Wang K; Karin M; Nguyen N; Tukey RH Intestinal Glucuronidation Protects against Chemotherapy-Induced Toxicity by Irinotecan (CPT-11). Proc. Natl. Acad. Sci. U. S. A 2013, 110 (47), 19143–19148. 10.1073/pnas.1319123110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).LoGuidice A; Wallace BD; Bendel L; Redinbo MR; Boelsterli UA Pharmacologic Targeting of Bacterial β-Glucuronidase Alleviates Nonsteroidal Anti-Inflammatory Drug-Induced Enteropathy in Mice. J. Pharmacol. Exp. Ther 2012, 341 (2), 447–454. 10.1124/jpet.111.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ervin SM; Hanley RP; Lim L; Walton WG; Pearce KH; Bhatt AP; James LI; Redinbo MR Targeting Regorafenib-Induced Toxicity through Inhibition of Gut Microbial β-Glucuronidases. ACS Chem. Biol 2019, 14 (12), 2737–2744. 10.1021/acschembio.9b00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wallace BD; Roberts AB; Pollet RM; Ingle JD; Biernat KA; Pellock SJ; Venkatesh MK; Guthrie L; O’Neal SK; Robinson SJ; Dollinger M; Figueroa E; McShane SR; Cohen RD; Jin J; Frye SV; Zamboni WC; Pepe-Ranney C; Mani S; Kelly L; Redinbo MR Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol 2015, 22 (9), 1238–1249. 10.1016/j.chembiol.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Awolade P; Cele N; Kerru N; Gummidi L; Oluwakemi E; Singh P Therapeutic Significance of β-Glucuronidase Activity and Its Inhibitors: A Review. Eur. J. Med. Chem 2020, 187, 111921. 10.1016/j.ejmech.2019.111921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Biernat KA; Pellock SJ; Bhatt AP; Bivins MM; Walton WG; Tran BNT; Wei L; Snider MC; Cesmat AP; Tripathy A; Erie DA; Redinbo MR Structure, Function, and Inhibition of Drug Reactivating Human Gut Microbial β-Glucuronidases. Sci. Rep 2019, 9 (1), 825. 10.1038/s41598-018-36069-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dashnyam P; Mudududdla R; Hsieh T-J; Lin T-C; Lin H-Y; Chen P-Y; Hsu C-Y; Lin C-H β-Glucuronidases of Opportunistic Bacteria Are the Major Contributors to Xenobiotic-Induced Toxicity in the Gut. Sci. Rep 2018, 8. 10.1038/s41598-018-34678-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pellock SJ; Walton WG; Biernat KA; Torres-Rivera D; Creekmore BC; Xu Y; Liu J; Tripathy A; Stewart LJ; Redinbo MR Three Structurally and Functionally Distinct β-Glucuronidases from the Human Gut Microbe Bacteroides Uniformis. J. Biol. Chem 2018, 293 (48), 18559–18573. 10.1074/jbc.RA118.005414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bhalodi AA; van Engelen TSR; Virk HS; Wiersinga WJ Impact of Antimicrobial Therapy on the Gut Microbiome. J. Antimicrob. Chemother 2019, 74 (Suppl 1), i6–i15. 10.1093/jac/dky530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Marchant J When Antibiotics Turn Toxic. Nature 2018, 555 (7697), 431–433. 10.1038/d41586-018-03267-5. [DOI] [PubMed] [Google Scholar]
- (29).Francino MP Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol 2016, 6. 10.3389/fmicb.2015.01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cully M Antibiotics Alter the Gut Microbiome and Host Health. Nat. Res 2019. 10.1038/d42859-019-00019-x. [DOI] [Google Scholar]
- (31).Bajorath J Integration of Virtual and High-Throughput Screening. Nat. Rev. Drug Discov 2002, 1 (11), 882–894. 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- (32).Zoete V; Grosdidier A; Michielin O Docking, Virtual High Throughput Screening and in Silico Fragment-Based Drug Design. J. Cell. Mol. Med 2009, 13 (2), 238–248. 10.1111/j.1582-4934.2008.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hu X; Zhang Y-Q; Lee OW; Liu L; Tang M; Lai K; Boxer MB; Hall MD; Shen M Discovery of Novel Inhibitors of Human Galactokinase by Virtual Screening. J. Comput. Aided Mol. Des 2019, 33 (4), 405–417. 10.1007/s10822-019-00190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Seifert MHJ; Wolf K; Vitt D Virtual High-Throughput in Silico Screening. BIOSILICO 2003, 1 (4), 143–149. 10.1016/S1478-5382(03)02359-X. [DOI] [Google Scholar]
- (35).Virtual Screening: An Alternative or Complement to High Throughput Screening?: Proceedings of the Workshop ‘New Approaches in Drug Design and Discovery’, Special Topic ‘Virtual Screening’, Schloß Rauischholzhausen, Germany, March 15–18, 1999; Klebe G, Ed.; Springer Netherlands, 2002. 10.1007/0-306-46883-2. [DOI] [Google Scholar]
- (36).Compound Management https://ncats.nih.gov/preclinical/core/compound (accessed 2020 – 07 −07).
- (37).UniProt Consortium. UniProt: A Hub for Protein Information. Nucleic Acids Res. 2015, 43 (Database issue), D204–212. 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).h11g11-bg - H11G11-BG protein - uncultured bacterium - h11g11-bg gene & protein https://www.uniprot.org/uniprot/D5GU71 (accessed 2020 −07 −07).
- (39).Molecular Operating Environment (MOE) | MOEsaic | PSILO https://www.chemcomp.com/Products.htm (accessed 2020 −07 −07).
- (40).BLAST: Basic Local Alignment Search Tool https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed 2020 −07 −07).
- (41).Bank, R. P. D. RCSB PDB: Homepage https://www.rcsb.org/ (accessed 2020 −03 −03).
- (42).Bank, R. P. D. RCSB PDB - 6MVH: Crystal structure of FMN-binding beta-glucuronidase from Roseburia hominis https://www.rcsb.org/structure/6MVH (accessed 2020 −07 −07).
- (43).Xiang Z Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci 2006, 7 (3), 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).I-TASSER server for protein structure and function prediction https://zhanglab.ccmb.med.umich.edu/I-TASSER/ (accessed 2020 −07 −07).
- (45).Yang J; Zhang Y Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinforma. Ed. Board Andreas Baxevanis Al 2015, 52, 5.8.1–5.815. 10.1002/0471250953.bi0508s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Software, O. S. OEDocking Software | Molecular Docking Tools | Fred Docking https://www.eyesopen.com/oedocking (accessed 2020 −07 −07).
- (47).NIH HPC Systems https://hpc.nih.gov/systems/ (accessed 2020 −07 −07).
- (48).Bank, R. P. D. RCSB PDB - 6MVG: Crystal structure of FMN-binding beta-glucuronidase from Ruminococcus gnavus https://www.rcsb.org/structure/6MVG (accessed 2020 −07 −07).
- (49).Bank, R. P. D. RCSB PDB - 5CZK: Structure of E. coli beta-glucuronidase bound with a novel, potent inhibitor 1-((6,8-dimethyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-1-(2-hydroxyethyl)-3-(4-hydroxyphenyl)thiourea https://www.rcsb.org/structure/5czk (accessed 2020 −07 −07).
- (50).PyMOL | pymol.org https://pymol.org/2/ (accessed 2021 −08 −13).
- (51).Hughes JP; Rees S; Kalindjian SB; Philpott KL Principles of Early Drug Discovery. Br. J. Pharmacol 2011, 162 (6), 1239–1249. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Li J; Vootukuri S; Shang Y; Negri A; Jiang J; Nedelman M; Diacovo TG; Filizola M; Thomas CJ; Coller BS RUC-4: A Novel AIIbβ3 Antagonist for Pre-Hospital Therapy of Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol 2014, 34 (10), 2321–2329. 10.1161/ATVBAHA.114.303724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Vootukuri S; Li J; Nedelman M; Thomas C; Jiang J-K; Babayeva M; Coller BS Preclinical Studies of RUC-4, a Novel Platelet AIIbβ3 Antagonist, in Non-Human Primates and with Human Platelets. J. Clin. Transl. Sci 2019, 3 (2–3), 65–74. 10.1017/cts.2019.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).CeleCor Therapeutics Announces Positive First-in-Human Data of RUC-4, a Novel Subcutaneous Platelet GPIIb/IIIa Inhibitor, Presented at TCT 2019 | Business Wire https://www.businesswire.com/news/home/20190925005021/en/CeleCor-Therapeutics-Announces-Positive-First-in-Human-Data-RUC-4 (accessed 2020 −07 −07).
- (55).A Randomized Phase 1 Dose-Escalation Study of Subcutaneously(SC) Administered RUC-4 - Full Text View - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03844191 (accessed 2020 −07 −07).
- (56).A Phase 2 Open Label Study to Assess the PK/PD Properties of RUC-4 in Patients With a ST-elevation Myocardial Infarction - Full Text View - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04284995 (accessed 2020 −07 −07).
- (57).COVID-19–associated coagulopathy https://www.the-hospitalist.org/hospitalist/article/221875/coronavirus-updates/covid-19-associated-coagulopathy (accessed 2020 −07 −07).
- (58).Becker RC COVID-19 Update: Covid-19-Associated Coagulopathy. J. Thromb. Thrombolysis 2020, 1–14. 10.1007/s11239-020-02134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Connors JM; Levy JH COVID-19 and Its Implications for Thrombosis and Anticoagulation. Blood 2020, 135 (23), 2033–2040. 10.1182/blood.2020006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Assay Development & Screening https://ncats.nih.gov/preclinical/drugdev/assay (accessed 2020 −07 −07).
- (61).Dahlin JL; Walters MA The Essential Roles of Chemistry in High-Throughput Screening Triage. Future Med. Chem 2014, 6 (11), 1265–1290. 10.4155/fmc.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Taylor MR; Flannigan KL; Rahim H; Mohamud A; Lewis IA; Hirota SA; Greenway SC Vancomycin Relieves Mycophenolate Mofetil–Induced Gastrointestinal Toxicity by Eliminating Gut Bacterial β-Glucuronidase Activity. Sci. Adv 2019, 5 (8), eaax2358. 10.1126/sciadv.aax2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Saitta KS; Zhang C; Lee KK; Fujimoto K; Redinbo MR; Boelsterli UA Bacterial β-Glucuronidase Inhibition Protects Mice against Enteropathy Induced by Indomethacin, Ketoprofen or Diclofenac: Mode of Action and Pharmacokinetics. Xenobiotica Fate Foreign Compd. Biol. Syst 2014, 44 (1), 28–35. 10.3109/00498254.2013.811314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bhatt AP; Pellock SJ; Biernat KA; Walton WG; Wallace BD; Creekmore BC; Letertre MM; Swann JR; Wilson ID; Roques JR; Darr DB; Bailey ST; Montgomery SA; Roach JM; Azcarate-Peril MA; Sartor RB; Gharaibeh RZ; Bultman SJ; Redinbo MR Targeted Inhibition of Gut Bacterial β-Glucuronidase Activity Enhances Anticancer Drug Efficacy. Proc. Natl. Acad. Sci 2020, 117 (13), 7374–7381. 10.1073/pnas.1918095117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2020. CA. Cancer J. Clin 2020, 70 (1), 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All protein structures underlying the development of our homology models are publicly available on PDB; links to these structure files are available at the first mention of each protein in-text. PyMOL was used to prepare all chemical visualizations and is available through the link cited in-text and provided in the “References” section of this manuscript. This research employed packaged chemical computing software (MOE, OpenEye) purchased by and licensed to NCATS; links to these platforms and details of their versions are available through their first mentions in-text. Our Ramachandran plot and HTS data for the hits described in “Results and Discussion” are available as Supplementary Information to this manuscript; we provide this information as image and spreadsheet files, respectively. Readers interested in accessing NCATS’s in-house chemical libraries for collaborative research should contact Dr. Shen at the email address above.