

Abstract
The flavivirids (family Flaviviridae) are a group of positive-sense RNA viruses that include well-documented agents of human disease. Despite their importance and ubiquity, the timescale of flavivirid evolution is uncertain. An ancient origin, spanning millions of years, is supported by their presence in both vertebrates and invertebrates and by the identification of a flavivirus-derived endogenous viral element in the peach blossom jellyfish genome (Craspedacusta sowerbii, phylum Cnidaria), implying that the flaviviruses arose early in the evolution of the Metazoa. To date, however, no exogenous flavivirid sequences have been identified in these hosts. To help resolve the antiquity of the Flaviviridae, we mined publicly available transcriptome data across the Metazoa. From this, we expanded the diversity within the family through the identification of 32 novel viral sequences and extended the host range of the pestiviruses to include amphibians, reptiles, and ray-finned fish. Through co-phylogenetic analysis we found cross-species transmission to be the predominate macroevolutionary event across the non-vectored flavivirid genera (median, 68 per cent), including a cross-species transmission event between bats and rodents, although long-term virus–host co-divergence was still a regular occurrence (median, 23 per cent). Notably, we discovered flavivirus-like sequences in basal metazoan species, including the first associated with Cnidaria. This sequence formed a basal lineage to the genus Flavivirus and was closer to arthropod and crustacean flaviviruses than those in the tamanavirus group, which includes a variety of invertebrate and vertebrate viruses. Combined, these data attest to an ancient origin of the flaviviruses, likely close to the emergence of the metazoans 750–800 million years ago.
Keywords: Flaviviridae, Flavivirus, Pestivirus, Hepacivirus, virus discovery, Metazoa, phylogeny
1. Introduction
The flavivirids (family Flaviviridae) are a group of positive-sense single-stranded RNA viruses comprising the genera Flavivirus, Pestivirus, Pegivirus, and Hepacivirus. These viruses include well-documented agents of human and livestock disease, including dengue virus, hepatitis C virus, yellow fever virus, Zika virus, and Bovine viral diarrhea virus 1. Reflecting their regular occurrence as pathogens, our understanding of flavivirid biology is necessarily skewed towards a subset of metazoan hosts, particularly those known to experience overt disease or act as reservoirs for these viruses, impeding our ability to understand the evolutionary history of this family. Currently available data suggest that all established genera, with the exception of the genus Flavivirus, are vertebrate-infecting viruses and do not require an arthropod vector for transmission (Simmonds et al. 2017).
The genus Flavivirus can itself be divided into four groups defined by phylogenetic position and host range: the (i) mosquito-borne flaviviruses, (ii) tick-borne flaviviruses, (iii) insect-specific flaviviruses, and (iv) vertebrate-specific flaviviruses, also known as the ‘no known vector’ flaviviruses (Blitvich and Firth 2017; Simmonds et al. 2017). A wide diversity of more divergent ‘flavi-like’ viruses have also been identified, including a group associated with crustaceans and decapods, as well as the tamanaviruses (after Tamana bat virus), which contains viruses from a broad range of vertebrate and invertebrate species (Price 1978; Geoghegan et al. 2018; Shi et al. 2018; Skoge et al. 2018; Parry et al. 2019; Le Lay et al. 2020; Soto et al. 2020; Costa et al. 2021). Another clade of related flavi-like viruses was recently identified in free-living parasitic flatworms (order Tricladida) (Dheilly et al. 2022).
Metagenomic surveys have identified flavivirid sequences with diverse genome structures, straying from the single 9–13 kb polyprotein that previously appeared to be canonical for the family. This expanded diversity includes a group of novel, predominantly arthropod-associated viruses—the jingmenviruses—that are both segmented and perhaps multicomponent (Qin et al. 2014; Ladner et al. 2016; Shi et al. 2016; Simmonds et al. 2017). Metagenomic studies have also expanded the host range of hepaci-, pesti- and pegiviruses in non-mammalian hosts, including the discovery of hepaci- and pegiviruses in birds (Goldberg et al. 2019; Porter et al. 2020; Chang, Rose, and Holmes 2021; Zhang et al. 2022), hepaci- and pesti-like viruses in cartilaginous fish (Chondrichthyes) (Shi et al. 2018), and hepaciviruses in reptiles and bony fish (Osteichthyes) (Shi et al. 2018; Porter et al. 2020; Costa et al. 2022; Harding et al. 2022).
The identification of flavivirid sequences in marine invertebrate and basal vertebrate lineages has led to suggestions that the evolution of the Flaviviridae may follow that of the metazoans through virus–host co-divergence over timescales of hundreds of millions of years (Shi et al. 2018; Bamford et al. 2022; Lensink, Yiqiao, and Lequime 2022). This, in turn, has stimulated questions regarding their host range and mode of transmission, while the complex evolutionary history of the flaviviruses and related sequences has been highlighted by their broad host range and sequence diversity. For example, the large phylogenetic gap between the cartilaginous fish and mammalian pestiviruses suggests that related viruses in bony fish, amphibians, reptiles, and birds exist but have yet to be sampled. The identification of flaviviruses in freshwater and marine crustaceans and a flavivirus-derived endogenous viral element (EVE) in the peach blossom jellyfish genome (Craspedacusta sowerbii, phylum Cnidaria) (Bamford et al. 2022) points towards an aquatic origin for the flaviviruses and highlights their long evolutionary association with the Metazoa. In particular, the cnidarian EVE suggests the existence of exogenous cnidarian flaviviruses. These are of importance for understanding the evolution of the Flaviviridae, as cnidarians, which include jellyfish, sea anemones, and corals, are an early branching lineage of the metazoans thought to have originated 700 million years ago (Erwin 2015). The phylogeny of the Metazoa can itself be divided into two major groups: those with bilateral body symmetry, the bilaterians, which comprise 99 per cent of all animal species, and, basal to them, the non-bilaterians, which include all the early diverging metazoan lineages—the Cnidaria, Placozoa, Porifera, and Ctenophora. Because non-bilaterians lack the body plan and circulatory system of vertebrates, it is possible that viruses in these hosts use an alternate mode of cell-to-cell transmission. To date, however, no flavivirids have been identified in these early diverging metazoan phyla.
Transcriptome mining is a proven method of virus discovery that leverages previous investment in metagenomics (Greninger 2018; Parry et al. 2019; Grimwood et al. 2021; Iwamoto et al. 2021; Miller et al. 2021; Paraskevopoulou et al. 2021; Dheilly et al. 2022; Edgar et al. 2022; Mifsud et al. 2022; Olendraite, Brown, and Firth 2022). To understand the host range of flavivirid sequences throughout the Metazoa and hence more accurately determine the age of the Flaviviridae, we used the Serratus RNA-dependent RNA polymerase (RdRp) search (https://www.serratus.io/explorer/rdrp) to mine the Sequence Read Archive (SRA) database for novel flavivirid sequences. To supplement this analysis, total RNA-sequencing data of the tunicate Botrylloides leachii was generated and screened to identify additional flavivirid sequences.
2. Methods
2.1. Screening of SRAs for flavivirid-like sequences
The Serratus RdRp search and palmID analysis suite (Babaian and Edgar 2022; Edgar et al. 2022) were used to identify datasets within the SRA (as of May 2022) that contain signatures of novel flavivirid-like sequences. This search was limited to the family Flaviviridae with a threshold score of ≥50 (for an explanation of the Serratus classifier score, see https://github.com/ababaian/serratus/wiki/.summary-Reports). The de novo transcriptome assemblies available at the National Center for Biotechnology Information (NCBI) Transcriptome Shotgun Assembly (TSA) Database (https://www.ncbi.nlm.nih.gov/genbank/tsa/) (as of June 2021) were also screened using the translated Basic Local Alignment Search Tool algorithm (TBLASTN) under default scoring parameters and the BLOSUM45 matrix. Amino acid sequences from representatives of the four Flaviviridae genera along with the related jingmenviruses were used as queries for the palmID and TSA database searches (Supplementary Table S1a). All novel virus sequences discovered were then used as queries in further SRA and TSA searches. The SRA and TSA search range was limited to Eukaryotes (NCBI taxonomic identifier (taxid 2759)), excluding the Viridiplantae (taxid 33090). Invertebrate datasets were limited to aquatic species as terrestrial invertebrate SRAs have been previously examined (Paraskevopoulou et al. 2021).
2.2. Tunicate collection, RNA extraction, and metagenomic next-generation sequencing
The tunicate B. leachii was collected by divers wearing surgical gloves at 0.5–3 m depth at the pier pilings in Chowder Bay, Sydney, Australia (site description in Marzinelli (2012)), on 24 November 2021. Sections of colonies were detached from the substratum using sterile tweezers, which were rinsed in 80 per cent ethanol between samples and brought to the surface, where they were placed in sterile cryogenic tubes. Samples were stored in liquid nitrogen on-site and then transferred to a −80°C freezer until extraction. Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) as previously described in the study by Geoghegan et al. (2021). These libraries were constructed using the Truseq Total RNA Library Preparation Protocol (Illumina). Host ribosomal RNA was depleted with the Ribo-Zero Plus Kit (Illumina), and paired-end sequencing (150 bp) was performed on the NovaSeq 6000 platform (Illlumina). Library construction and metatranscriptomic sequencing were performed by the Australian Genome Research Facility.
2.3. Identification of novel flavivirid genomes
Raw FASTQ files for all libraries that contained flavivirid-like sequences were obtained through the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home). Adapter removal and quality trimming were conducted using Trimmomatic (v0.38) with parameters SLIDINGWINDOW:4:5, LEADING:5, TRAILING:5, and MINLEN:25 (Bolger, Lohse, and Usadel 2014). To recover full-length virus sequences, raw reads were assembled de novo into contigs using MEGAHIT (v1.2.9) (Li et al. 2015). The assembled contigs were then compared to the NCBI non-redundant protein database (as of August 2021) and a custom Flaviviridae protein database using Diamond BLASTx (v2.0.9) with an E-value threshold of 1 × 10−5 (Buchfink, Xie, and Huson 2015). To identify highly divergent sequences, a custom Flaviviridae protein database was regularly updated with the novel viruses identified.
2.4. Genome extension and annotation
Sequence reads were mapped onto virus-like contigs using Bbmap (v37.98), and areas of heterogeneous coverage were manually checked using Geneious (v11.0.9) (Kearse et al. 2012; Bushnell 2014). Where possible, the extremities of contigs were manually extended and re-submitted to read mapping until the contig appeared complete or no overhanging extremities were observed. Sequences of vector origin were detected using VecScreen (https://www.ncbi.nlm.nih.gov/tools/vecscreen/) and removed. Contig abundances were calculated using the RNA-Seq by Expectation Maximization software (v1.3.0) (Li and Dewey 2011). GetORF from EMBOSS (v6.6.0) was used to predict open reading frames (ORFs) (Rice, Longden, and Bleasby 2000). To annotate protein functional domains, the InterProScan software package (v5.56) was used with the TIGRFAMs (v15.0), SFLD (v4.0), PANTHER (v15.0), SuperFamily (v1.75), PROSITE (v2022_01), CDD (v3.18), Pfam (v34.0), SMART (v7.1), PRINTS (v42.0), and CATH-Gene3D databases (v4.3.0) (Jones et al. 2014). Genome diagrams were constructed using a manually curated selection of predicted functional domains and visualized using gggenomes (Hackl and Ankenbrand, 2022).
2.5. Detection of endogenous virus elements
To screen for EVEs within the viral-like contigs, the putative virus-like nucleotide sequence was compared to the corresponding host genome (where available) and a subset of the whole-genome shotgun contig database (as of October 2022) using the TBLASTN algorithm with an E-value cutoff of 1 × 10–20. In addition, the virus-like sequences were checked for host gene contamination using the contamination function implemented in CheckV (v0.8.1) (Nayfach et al. 2021). All EVEs were removed from subsequent analyses.
2.6. Assessment of library composition
Taxonomic identification for the contigs assembled for each library was obtained by aligning them to the custom NCBI nt database using the KMA aligner and the CCMetagen program (Clausen, Aarestrup, and Lund 2018; Marcelino et al. 2020). In the case of the cigar comb jelly flavivirus, where raw reads are not publicly available, contigs from the corresponding TSA (GHXY01000001:GHXY01366104) were used as input. Virus abundance was calculated by counting the number of nucleotides matching the reference sequence with an additional correction for template length (the default parameter in KMA). Krona graphs were created using the KMA and CCMetagen methods and further edited in Adobe Illustrator (https://www.adobe.com) (Clausen, Aarestrup, and Lund 2018; Marcelino et al., 2020).
The virus sequences identified in this study were named using a combination of the host common name—if known—and the appropriate Flaviviridae genera (e.g. Harrimaniidae flavivirus). Virus–host assignments were made using a combination of host/virus abundance measurements and phylogenetic analyses. Where <80 per cent of host abundance was associated with the target species of the library, the possibility of alternative hosts was considered. In this case, the other organisms comprising this library were examined to determine if they might represent the source of the virus sequence. For instance, given the known host range of the flavivirids, it is more likely that these sequences are derived from metazoan species than from bacteria, fungi, or archaea. As such, metazoan species were given greater weighting when assigning putative virus–host assignments. Where host assignment proved difficult to assign with accuracy, the suffix ‘associated’ was added to the host name to signify this (e.g. digyalum oweni-associated virus). Where the taxonomic position of a virus was ambiguous, the suffix ‘-like’ was used (e.g. African cichlid flavi-like virus).
2.7. Phylogenetic analysis
The phylogenetic trees of the putative flavivirid sequence identified here were inferred using a maximum likelihood approach. Translated virus contigs were aligned with known flavivirid protein sequences from NCBI/GenBank using MAFFT (v7.402) employing the generalized affine gap cost algorithm (Katoh and Standley 2013; Sayers et al. 2021). Poorly aligned regions were removed using trimAl (v1.2) with a gap threshold ranging from 0.7 to 0.9 and a variable conserve value (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009). All phylogenetic trees were estimated using IQ-TREE2. Branch support was calculated using 1,000 bootstrap replicates with the UFBoot2 algorithm and an implementation of the SH-like approximate likelihood ratio test within IQ-TREE2 (Guindon et al. 2010; Hoang et al. 2017). Selection of the best-fit model of amino acid substitution was determined using the Akaike information criterion (AIC), the corrected AIC, and the Bayesian information criterion with the ModelFinder function in IQ-TREE 2 (Kalyaanamoorthy et al. 2017; Minh et al. 2020). The trimming methods, alignment lengths, and phylogenetic models chosen in this analysis are outlined in Supplementary Table S1b. Phylogenetic trees were annotated using the R packages phytools (v1.0–3) and ggtree (v3.3.0.9) and further edited in Adobe Illustrator (https://www.adobe.com) (Revell 2012; Yu et al. 2017).
2.8. Assessment of cross-species virus transmission
To visualize the relative occurrence of cross-species transmission and virus–host co-divergence across the Flaviviridae, we analysed the co-phylogenetic relationship between viruses and their hosts. Host cladograms were created using the phyloT software, a phylogenetic tree generator based on NCBI taxonomy (http://phylot.biobyte.de/). Virus–host associations were obtained from the NCBI virus database (Brister et al. 2015; Hatcher et al. 2017) and the Virus–Host database (release 213) (Mihara et al. 2016) (accessed 14 September 2022). Tanglegrams that graphically represent the correspondence between host and virus trees were created using the R packages phytools (1.0–3) (Revell 2012) and ape (v5.6–2) (Paradis and Schliep 2019). The virus phylogenies used in the co-phylogenies were constructed as described earlier. The relative frequencies of cross-species transmission versus virus–host co-divergence were quantified using the Jane package, which employs a maximum parsimony approach to establish the best ‘map’ of the virus phylogeny onto the host phylogeny (Conow et al. 2010). The cost of duplication, host jumping, and extinction event types were set to 1.0, while the cost of virus–host co-divergence was set to zero as it was considered the null event. The number of generations and the population size were set to 100. Jane was chosen over its successor eMPRess (Santichaivekin et al. 2020), as it allows a virus to be associated with multiple host species and handles polytomies (Santichaivekin et al. 2020). For a multi-host virus, each association was represented as a polytomy in the virus phylogeny. A co-phylogenetic analysis of the genus Flavivirus was not conducted as vector-borne viruses with both invertebrate and invertebrate hosts are problematic to incorporate into analyses of this kind.
3. Results
Screening of transcriptomes revealed the presence of flavivirid-like sequences in 154 sequencing libraries within the SRA and TSA databases as well as one newly generated sequencing library from tunicates. The assembly and mining of these sequencing libraries identified 32 novel virus-like sequences, which were subsequently assigned as hepaci-like (20), flavivirus-like (7), pesti-like (4), and unclassified flavivirial-like sequences (1) (Table 1, Fig. 1). These virus-like sequences were predominately found in metazoan transcriptomes belonging to aquatic species (amphibians, bony fish, cnidarians, comb jellies, crustaceans, and hemichordates), although some were also found in land-dwelling vertebrates (birds, primates, and rodents). One virus-like sequence was assembled from a non-metazoan, alveolate library. No pegi-like virus sequences were found. We now examine each genus in turn.
Table 1.
Summary table of the viruses discovered in this study.
| Virus name | Abbreviation | Virus classification | Sample organism | SRA/TSA | Sequence length (bp) | % Std abundance | Closest Blast Hit (BLASTx) | % Ident (BLASTx) | E-value (BLASTx) | Tissue | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| African cichlid flavi-like virus | AfciFV | Flavi-like | Cyprichromis microlepidotus | SRR9688165 | 10,364 | 0.01 | Salmon flavivirus (QJU12405.1) | 38 | 0 | Lower pharyngeal jaw | El Taher et al. (2021) |
| Chowder bay tunicate–associated flavi-like virus | CbtuFV | Flavi-like | B. leachii | PRJEB57836 | 10,455 | 0.001 | Flavivirus sp.(QOQ37358.1) | 29 | 1.00E−80 | Whole body | NA |
| Cigar comb jelly flavi-like virus | CcjeFV | Flavi-like | Beroe forskalii | TSA:GHXY01073473.1 | 346 | NA | Salmon flavivirus (QJU12405.1) | 36 | 2.77E−07 | Whole body | Schultz et al. (2020) |
| Cigar comb jelly flavi-like virus | CcjeFV | Flavi-like | B. forskalii | TSA:GHXY01284671.1 | 226 | NA | Salmon flavivirus (QJU12405.1) | 51 | 2.66E−29 | Whole body | Schultz et al. (2020) |
| Cnidaria flavivirus | CnidFV | Flavivirus | Eunicea flexuosa | SRR12876665 | 10,729 | 0.004 | Kadam virus (YP 009345035.1) | 28 | 2.47E−312 | Mixed | Prada and Hellberg (2021) |
| Harrimaniidae flavivirus | HarFV | Flavivirus | Harrimaniidae sp | SRR1695462 | 10,951 | 0.003 | Rio Bravo virus (NP 620,044.1) | 28 | 2.89E−278 | Mixed | Cannon et al. (2014) |
| Photeros flavivirus | PhoFV | Flavivirus | Photeros sp | SRR10811635 | 11,290 | 0.003 | Crangon crangon flavivirus (QCH00713.1) | 38 | 0 | Whole body | Hensley et al. (2019) |
| Sea-firefly flavivirus | SefiFV | Flavivirus | Vargula hilgendorfii | DRR171178 | 10,930 | 0.016 | Crangon crangon flavivirus (QCH00713.1) | 37 | 0 | Whole body | Bessho-Uehara et al. (2020) |
| Brown Julie hepacivirus | BrjuHV | Hepacivirus | Julidochromis dickfeldi | SRR9688628 | 2160 | 0.0007 | Guangxi houndshark hepacivirus (AVM87259.1) | 41 | 1.08E−140 | Liver | El Taher et al. (2021) |
| Brown Julie hepacivirus | BrjuHV | Hepacivirus | J. dickfeldi | SRR9688628 | 2990 | 0.001 | Rodent hepacivirus (AGC52826.1) | 43 | 1.08E−111 | Liver | El Taher et al. (2021) |
| Cape dune mole-rat hepacivirus | CdmrHV | Hepacivirus | Bathyergus suillus | SRR2141210 | 6224 | 0.002 | Bat hepacivirus (QQM18105.1) | 42 | 0 | Mixed | Davies et al. (2015) |
| Catfish hepacivirus | CatfHV | Hepacivirus | G. macromaculatus | SRR6426015 | 9865 | 0.005 | Guangxi houndshark hepacivirus (AVM87259.1) | 32 | 0 | Liver | Jiang et al. (2019) |
| Caudopunctatus cichlid hepacivirus | CaciHV | Hepacivirus | Neolamprologus toae | SRR9680779 | 7165 | 0.009 | Guangxi houndshark hepacivirus (AVM87256.1) | 34 | 4.49E−284 | Liver | El Taher et al. (2021) |
| Caudopunctatus cichlid hepacivirus | CaciHV | Hepacivirus | N. toae | SRR9680779 | 2385 | 0.003 | Rodent hepacivirus (AGC52826.1) | 43 | 2.96E−116 | Liver | El Taher et al. (2021) |
| Crab-eating macaque hepacivirus | CremHV | Hepacivirus | Macaca fascicularis | SRR6032465 | 9855 | 0.031 | Guereza hepacivirus (YP 009325369.1) | 57 | 0 | Whole blood | Speranza et al. (2018) |
| Cyprichromis hepacivirus | CyprHV | Hepacivirus | Cyprichromis sp. ‘dwarf jumbo’ AB-2019 | SRR9688149 | 8678 | 0.012 | Guangxi houndshark hepacivirus (AVM87256.1) | 33 | 0 | Liver | El Taher et al. (2021) |
| Dewindti cichlid hepacivirus | DeciHV | Hepacivirus | Aulonocranus dewindti | SRR9689044 | 1583 | 0.0009 | Rodent hepacvirus (ATP66829.1) | 50 | 5.08E−127 | Liver | El Taher et al. (2021) |
| Dewindti cichlid hepacivirus | DeciHV | Hepacivirus | A. dewindti | SRR9689044 | 1345 | 0.0005 | Hepacivirus L (YP 009679023.1) | 44 | 4.95E−99 | Liver | El Taher et al. (2021) |
| Featherfin cichlid hepacivirus | FeciHV | Hepacivirus | Cyathopharynx furcifer | SRR9680031 | 1070 | 0.0004 | Nanhai ghost shark hepacivirus 1 (AVM87556.1) | 44 | 3.73E−71 | Liver | El Taher et al. (2021) |
| Freshwater butterflyfish hepacivirus | FrbuHV | Hepacivirus | Pantodon buchholzi | SRR5997808 | 8422 | 0.004 | Nanhai ghost shark hepacivirus 1 (AVM87556.1) | 29 | 4.40E−275 | Viscera mixture | Sun et al. (2016) |
| Gold head compressiceps hepacivirus | GhcoHV | Hepacivirus | Altolamprologus compressiceps | SRR9688991 | 2553 | 0.002 | Guangxi houndshark hepacivirus (AVM87259.1) | 43 | 1.00E−176 | Liver | El Taher et al. (2021) |
| Gold head compressiceps hepacivirus | GhcoHV | Hepacivirus | A. compressiceps | SRR9688991 | 1603 | 0.002 | Hepacivirus L (YP 009679023.1) | 43 | 3.59E−97 | Liver | El Taher et al. (2021) |
| Greater mouse-eared bat hepacivirus | GmebHV | Hepacivirus | Myotis myotis | SRR2063918 | 3300 | 0.046 | Rodent hepacivirus (QLM02863.1) | 71 | 0 | Pharyngeal and anal swab | Wu et al. (2012) |
| Greater mouse-eared bat hepacivirus | GmebHV | Hepacivirus | M. myotis | SRR2063918 | 2981 | 0.046 | Rodent hepacivirus (QLM02863.1) | 63 | 0 | Pharyngeal and anal swab | Wu et al. (2012) |
| Neolamprologus buescheri hepacivirus | NebuHV | Hepacivirus | N. buescheri | SRR9681059 | 2105 | 0.002 | Wenling shark virus (YP 009179227.1) | 45 | 4.57E−74 | Liver | El Taher et al. (2021) |
| Neolamprologus buescheri hepacivirus | NebuHV | Hepacivirus | N. buescheri | SRR9681059 | 2294 | 0.002 | Rodent hepacvirus (ATP66829.1) | 40 | 1.32E−144 | Liver | El Taher et al. (2021) |
| Neolamprologus savoryi hepacivirus | NesaHV | Hepacivirus | N. savoryi | SRR9680861 | 5451 | 0.007 | Wenling shark virus (YP 009179227.1) | 45 | 2.95E−93 | Liver | El Taher et al. (2021) |
| Neolamprologus savoryi hepacivirus | NesaHV | Hepacivirus | N. savoryi | SRR9680861 | 1223 | 0.001 | Wenling shark virus (YP 009179227.1) | 34 | 9.32E−231 | Liver | El Taher et al. (2021) |
| Northern treeshrew hepacivirus | NotrHV | Hepacivirus | Tupaia belangeri | DRR155087 | 9208 | 0.00003 | Hepacivirus P (YP 009553586.1) | 56 | 0 | Liver | Sanada et al. (2019) |
| Variabilichromis moorii hepacivirus | VamoHV | Hepacivirus | V. moorii | SRR9689131 | 4358 | 0.006 | Guangxi houndshark hepacivirus (AVM87259.1) | 36 | 1.82E−231 | Liver | El Taher et al. (2021) |
| Neolamprologus savoryi hepacivirus | VamoHV | Hepacivirus | V. moorii | SRR9689131 | 2857 | 0.004 | Rodent hepacivirus (AGC52826.1) | 44 | 4.71E−113 | Liver | El Taher et al. (2021) |
| Viviparous lizard hepacivirus | ViliHV | Hepacivirus | Zootoca vivipara | SRR11262332 | 11,862 | 0.001 | Duck hepacivirus (QKT21547.1) | 27 | 9.26E−263 | Whole body | Yurchenko, Recknagel, and Elmer (2020) |
| White sucker hepacivirus | WhsuHV | Hepacivirus | Catostomus commersonii | SRR2912516 | 10,120 | 0.001 | Guangxi houndshark hepacivirus (AVM87256.1) | 31 | 0 | Liver | Hahn et al. (2016) |
| Xenotilapia hepacivirus | XenoHV | Hepacivirus | Xenotilapia sp. ‘spilopterus north’ AB-2019 | SRR9670143 | 2596 | 0.003 | Rodent hepacivirus (QLM02868.1) | 40 | 3.32E−118 | Liver | El Taher et al. (2021) |
| Xenotilapia hepacivirus | XenoHV | Hepacivirus | Xenotilapia sp. ‘spilopterus north’ AB-2019 | SRR9670143 | 4117 | 0.002 | Guangxi houndshark hepacivirus (AVM87256.1) | 35 | 6.73E−211 | Liver | El Taher et al. (2021) |
| Zebra finch hepacivirus 1 | ZefiHV1 | Hepacivirus | Taeniopygia guttata | SRR7031415 | 2262 | 0.0001 | Duck hepacivirus (QDF44087.1) | 52 | 2.74E−180 | Liver | NA |
| Zebra finch hepacivirus 1 | ZefiHV1 | Hepacivirus | T. guttata | SRR7031415 | 3396 | 0.0001 | Jogalong virus (QHD25538.1) | 49 | 0 | Ovary | NA |
| Zebra finch hepacivirus 2 | ZefiHV2 | Hepacivirus | T. guttata | SRR6896649 | 1245 | 0.0002 | Jogalong virus (QHD25538.1) | 51 | 3.85E−67 | Liver | Biederman et al. (2018) |
| Zebra finch hepacivirus 2 | ZefiHV2 | Hepacivirus | T. guttata | SRR6896649 | 1842 | 0.0001 | Jogalong virus (QHD25538.1) | 53 | 1.64E−209 | Ovary | Biederman et al. (2018) |
| Cayenne caecilian pestivirus | CacaPV | Pestivirus | Typhlonectes compressicauda | SRR5591415, SRR5591441 | 1391 | 0.0000 | Bovine viral diarrhoea virus 2 (AAC64056.1) | 58 | 1.59E−128 | Kidney | Torres-Sánchez et al. (2019) |
| Frog pestivirus | FrogPV | Pestivirus | L. catesbeianus | SRR5810423 | 15,334 | 0.012 | Classical swine fever virus (NP 777,506) | 28 | 1.35E−256 | Ventral skin | Eskew et al. (2018) |
| Glass knifefish pestivirus | GlknPV | Pestivirus | Eigenmannia virescens | SRR6675258 | 14,199 | 0.013 | Pronghorn antelope pestivirus (YP 009026415.1) | 29 | 4.28E−232 | Muscle | Thompson et al. (2018) |
| Transcaucasian sand viper pestivirus | TrsaPV | Pestivirus | Vipera transcaucasiana | SRR12802473 | 3054 | 0.0001 | Bamboo rat pestivirus (UMO75502.1) | 25 | 6.51E−50 | Venom gland | Xie et al. (2022) |
| Transcaucasian sand viper pestivirus | TrsaPV | Pestivirus | V. transcaucasiana | SRR12802473 | 2120 | 0.0001 | Atypical porcine pestivirus (QBJ01589.1) | 53 | 1.65E−188 | Venom gland | Xie et al. (2022) |
| Digyalum oweni-associated virus | DiowV | Unclassifed pesti-like virus | Digyalum oweni | SRR9888042 | 3715 | 0.0003 | Xinzhou spider virus 3 (YP 009254746.1) | 24 | 4.09E−13 | Individual gut cell | Janouškovec et al. (2019) |
| Digyalum oweni-associated virus | DiowV | Unclassifed pesti-like virus | Digyalum oweni | SRR9888042 | 4594 | 0.0002 | Soybean thrips virus 4 (QPZ88419.1) | 25 | 8.94E−15 | Individual gut cell | Janouškovec et al. (2019) |
Figure 1.
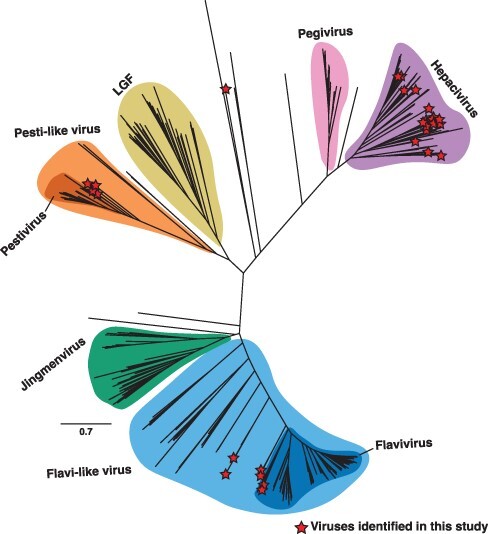
Phylogeny of the Flaviviridae. Unrooted maximum likelihood phylogenetic tree of the flavivirid sequences based on the conserved amino acid in the RdRp (NS5). All branches are scaled according to the number of amino acid substitutions per site. Established genera and notable clades that are yet to be ratified by ICTV are highlighted. Novel virus sequences identified in this study are displayed with a red star. LGF refers to the ‘large genome flaviviruses’.
3.1. Genus Flavivirus
We identified seven putative flavi-like virus sequences, including cnidaria flavivirus (CnidFV) and cigar comb jelly flavi-like virus (CcjeFV) in libraries of the early diverging metazoan phyla Cnidaria and Ctenophora, harrimaniidae flavivirus (HarFV) in an acorn worm (Enteropneusta), photeros flavivirus (PhoFV) and sea-firefly flavivirus (SefiFV) in marine ostracods, Chowder Bay tunicate–associated flavivirus in tunicates (CbtuFV), and African cichlid flavivirus (AfciFV) in a cichlid fish (Fig. 2). For all but one of these sequences (CcjeFV), complete genome sequences ranging in length from 10,364 to 11,290 nucleotides were assembled. CcjeFV consists of two partial RdRp fragments, 346 and 226 bp in length.
Figure 2.
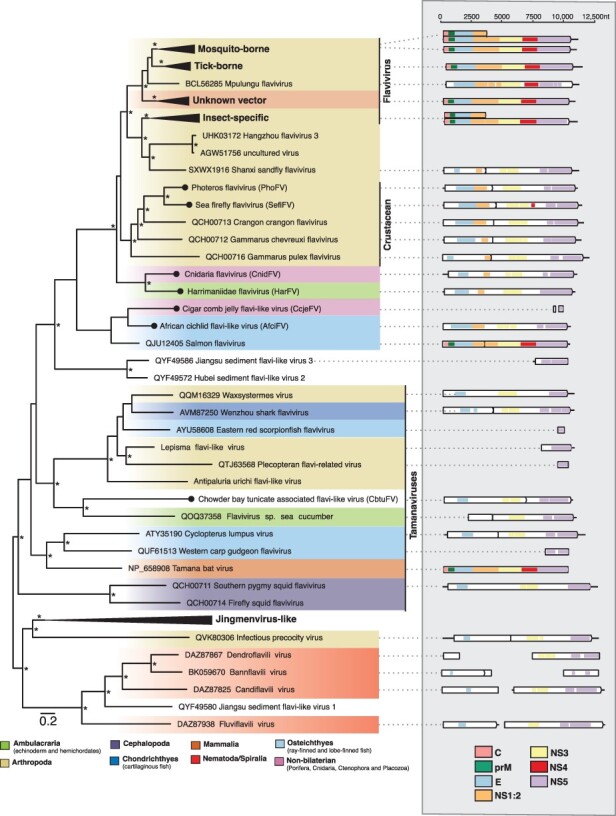
Phylogenetic relationships of the flavi-like viruses identified in this study. (Left) Phylogenetic relationships of the flavi- and jingmenviruses. ML phylogenetic trees based on the conserved amino acid in the RdRp (NS5) show the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. Branches are highlighted to represent host clade (Ambulacraria = green, Arthropoda = khaki, Cephalopoda = purple, Chondrichthyes = light blue, Mammalia = orange, Nematoda/Spiralia = red, Osteichthyes = dark blue, non-bilaterian = light purple). All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support where SH-aLRT ≥ 80 per cent and UFboot ≥ 95 per cent. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny. The data underlying this figure and the definitions of acronyms used are presented in Supplementary Table S2.
A range of genome structures was observed and found to be largely consistent with those found in this genus. For example, PhoFV and SefiFV, like the other viruses identified in marine crustaceans, are predicted to contain a programmed −1 ribosomal frameshift on a ‘slippery’ heptanucleotide sequence downstream of the NS1 region (Rhys, Sassan, and Williams 2019) (Fig. 2, Supplementary Fig. S1). However, CbtuFV was predicted to contain two ORFs, with the NS4/5 region encoded on the second ORF, although no ‘slippery’ heptanucleotide motifs could be detected (Fig. 2). The remaining full-length sequences were predicted to contain a single ORF. Virus domains consistent with this genus were detected across all sequences (Fig. 2).
Phylogenetic analyses of the conserved NS5 region place the ostracod sequences (PhoFV and SefiFV) within a larger diversity of marine crustacean flaviviruses. Two sequences, CnidFV and HarFV, fell basal to all classified members of the genus Flavivirus along with the crustacean flaviviruses (Fig. 2). Notably, these sequences appear closer in phylogenetic position and amino acid identity to tick, insect-specific, and crustacean flaviviruses than those viruses in the more divergent tamanavirus clade. The flavivirus-derived EVEs identified in the Cnidaria fell into approximately the same phylogenetic location as CnidFV and SefiFV (Supplementary Fig. S2). CcjeFV and AfciFV were placed phylogenetically with salmon flavivirus (QJU12405.1), although unlike salmon flavivirus, AfciFV consists of a single ORF.
3.2. Genus Pestivirus
We identified four pesti-like virus sequences in amphibians, reptiles, and bony fish (Table 1). Two full genomes—glass knifefish pestivirus (GlknPV) and frog pestivirus (FrogPV)—were recovered, ranging from 14,199 to 15,334bp in length, in addition to two partial genomes, Transcaucasian sand viper pestivirus (FrogPV) and Cayenne caecilian pestivirus (CacaPV) (Fig. 3). These sequences exhibit more sequence similarity with mammalian pestiviruses than those associated with cartilaginous fish, with an average of 28 per cent versus 24 per cent amino acid identity across the complete polyprotein. This is reflected in the phylogenetic positioning of the novel pesti-like viruses based on the conserved NS5 region (Fig. 3). The newly identified reptile and amphibian pesti-like virus sequences, FrogPV and CacaPV, form a sister group to those found in rodents, bats, and pigs, while the sequence discovered in fish, GlknPV, fell basal to this group but remained as a sister group to those viruses from cartilaginous fish (Shi et al. 2018). The topology of the pestivirus phylogeny varied depending on whether the NS3 or NS5 domains were used in the analysis. In particular, FrogPV formed a sister lineage to the known pestiviruses in a phylogeny based on the NS3 region (Fig. 3, Supplementary Fig. S3).
Figure 3.
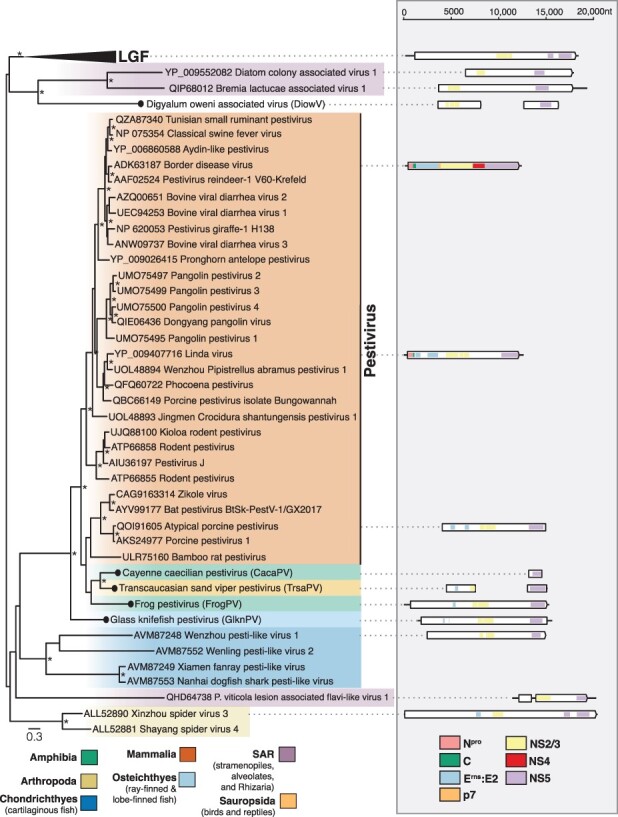
Phylogenetic relationships of the pesti-like viruses identified in this study. (Left) Phylogenetic relationships of the pestiviruses and unclassified relatives. ML phylogenetic trees based on the conserved amino acid in the RdRp (NS5) show the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. The colour scheme is as found in Fig. 2, with the following exceptions, Amphibia = green, Sauropsida = light orange, SAR = light purple. All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support where SH-aLRT ≥ 80 per cent and UFboot ≥ 95 per cent. LGF refers to the ‘large genome flaviviruses’. Non-novel sequences without NCBI accession were obtained from Wu et al. (2020). (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny. The data underlying this figure and the definitions of acronyms used are presented in Supplementary Table S2.
3.3. Genus Hepacivirus
We identified 20 novel hepacivirus sequences, of which 14 were found in ray-finned fish (Actinopterygii), expanding on the two hepaciviruses previously identified in this group (Fig. 4). The remaining sequences (n = 6) add to the known diversity of bat, avian, primate, rodent, and treeshrew hepaciviruses (Fig. 4). Of the novel hepaciviruses, five complete genomes were assembled, ranging from 9,208 to 11,862bp in length (Fig. 4). Partial genome sequences containing at least the NS3 and NS5 domains were assembled for the remaining sequences, with the exception of the featherfin cichlid hepacivirus, for which only the NS5 region could be assembled (Fig. 4). Of note, greater mouse-eared bat hepacivirus (GmebHV) was assembled from a library generated for the analysis of bat viromes (Wu et al. 2012) and shares 70 per cent amino acid identity with rodent hepacivirus (QLM02863.1).
Figure 4.
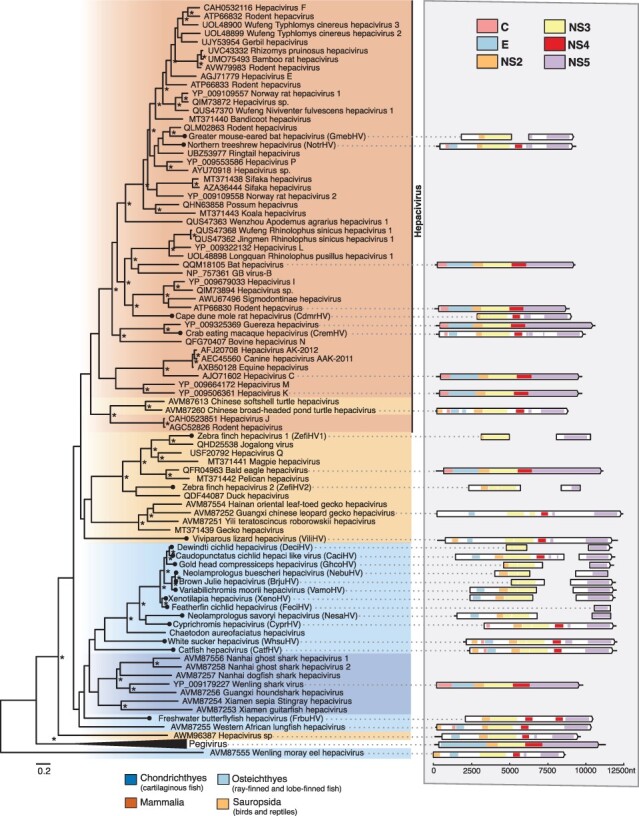
Phylogenetic relationships of the hepaciviruses viruses identified in this study. (Left) Phylogenetic relationships of the ‘pegi-hepaci’ clade. ML phylogenetic trees based on the conserved amino acid in the RdRp (NS5) show the topological position of virus-like sequences discovered in this study (black circles) in the context of their closest relatives. The colour scheme is as found in Fig. 2, with the following exception, Sauropsida = light orange. All branches are scaled to the number of amino acid substitutions per site, and trees were midpoint rooted for clarity only. An asterisk indicates node support where SH-aLRT ≥ 80 per cent and UFboot ≥ 95 per cent. (Right) Genomic organization of the virus sequences identified in this study and representative species used in the phylogeny. The data underlying this figure and the definitions of acronyms used are presented in Supplementary Table S2.
3.4. An unclassified flavivirid-like virus
In addition to the viruses that fell within established genera, we identified a partial flavi-like virus sequence termed digyalum oweni-associated virus (DiowV) in D. oweni, a species of parasitic protist belonging to the phylum Apicomplexa. Two contigs were assembled from this library, 3689 and 4577 bp in length and predicted to contain the NS3 and NS5 domains, respectively (Fig. 3). DiowV shares the greatest amino acid identity (24 per cent) with the Xinzhou spider virus 3 (YP_009254746) among other large genome flaviviruses (LGF). When included in the ‘pesti-LGF’ tree, DiowV, along with diatom colony–associated virus 1 (YP_009552082) and bremia lactucae–associated virus 1 (QIP68012), forms a sister group to the LGF. However, in the family-wide tree, these sequences, along with Snake River alfalfa virus (ON669064), fall outside of the ‘pesti-LGF’ lineage and basal to the ‘pegi-hepaci’ group, although these branches receive poor bootstrap support (Fig. 1).
3.5. Genetic composition of sequencing libraries
Metagenomic sequencing libraries are often comprised of organisms in addition to the target host, which can complicate virus–host assignment. To quantify the composition of these libraries and improve virus–host assignments, we utilized the KMA and CCMetagen tools (Fig. 5). For 20 of the libraries, over 80 per cent of eukaryotic contigs were assigned to the expected target host of the sequencing library (median, 90 per cent; range, 0–98 per cent). In the case of the E. flexuosa (family Plexauridae) library in which CnidFV was assembled, a genus of unicellular microalgae, Symbiodinium (phylum Dinoflagellata), represented 64 per cent of all contigs (Fig. 5). In this library, soft corals (order Alcyonacea, phylum Cnidaria), which include E. flexuosa, represented 63 per cent of metazoan abundance, while tunicates and bony fish represented 13 and 10 per cent of abundance, respectively. Despite Plexauridae comprising 60 per cent of cnidarian abundance, other soft coral families were also detected, including the Ellisellidae, Nephtheidae, Acanthogorgiidae, and Nidaliidae, each representing ∼10 per cent of cnidarian abundance. Likewise, the tunicate library from which CbtuFV was assembled comprised reads belonging to various marine organisms, including Bryozoa, Cnidaria, and crustaceans, representing an average of 8 per cent abundance each.
Figure 5.
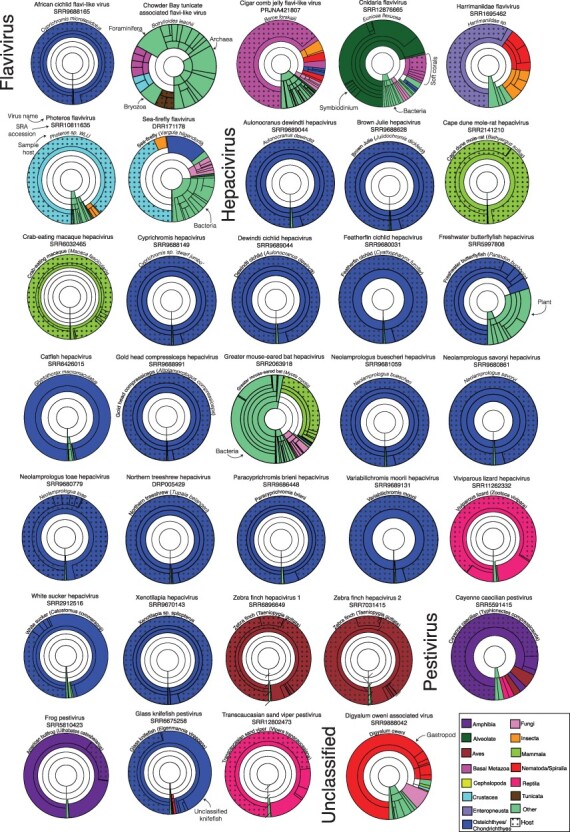
Taxonomic assignments of contigs in sequencing libraries. Each Krona graph illustrates the relative abundance of taxa in a metatranscriptome at varying taxonomic levels. For clarity, a maximum depth of five taxonomic levels was chosen for each graph. The library SRA accession number, host species, and the corresponding virus of interest are annotated above each graph. Segments are highlighted based on the species’ taxonomic grouping. Dots have been used to signify where contigs have been taxonomically assigned within the same family as the host species. Contigs without any matches in the database are not shown.
Contigs belonging to catfish (order Siluriformes) comprised 95 per cent of the Glyptothorax macromaculatus library from which catfish hepacivirus (CatfHV) was assembled, although it is uncertain to which family of catfish this sample belonged. Likewise, the American bullfrog (Lithobates catesbeianus) transcriptome comprised 60 per cent contigs associated with fork-tongued frogs (Dicroglossidae) and 17 per cent associated with true frogs (Ranidae), including L. catesbeianus. No host-associated contigs were detected in the D. oweni library in which DiowV was assembled. Instead, 64 per cent of the library is composed of contigs associated with marine gastropod molluscs.
3.6. Long-term virus–host evolutionary relationships
To examine the frequency of four macroevolutionary events (i.e. co-divergence, duplication, host-switching, and extinction) among the Flaviviridae, we estimated co-phylogenies to quantify the evolutionary relationship between the ‘pegi-hepaci’ and pestivirus clades and their hosts (Fig. 6; members of the genus Flavivirus were excluded because of the high frequency of vector-borne viruses). In accordance with earlier studies (Geoghegan, Duchêne, and Holmes 2017), this analysis revealed that cross-species transmission was the most common evolutionary event across the ‘pegi-hepaci’ and pestivirus clades, representing 65 and 71 per cent of events, respectively (Supplementary Fig. S4). Two viruses, GmebHV and freshwater butterflyfish hepacivirus (FrbuHV) identified in this study, present notable exceptions (Fig. 6). GmebHV is distinct from known bat hepaciviruses (Hepacivirus K, Hepacivirus L, and Hepacivirus M), and instead groups with those viruses found in rodents, shrews, sloths, and raccoons (Fig. 4). FrbuHV, along with Western African lungfish hepacivirus and Wenling moray eel hepacivirus, fell basal to those viruses identified in cartilaginous fish.
Figure 6.
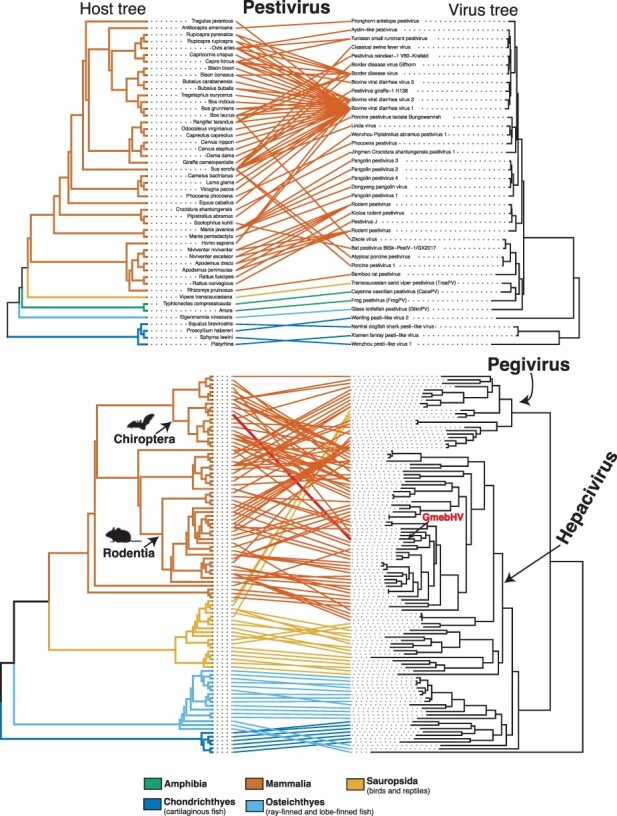
Tanglegram of rooted phylogenetic trees for representative virus groups and their hosts. Branches of the host tree (left) and lines are coloured to represent the host clade. The colour scheme is as found in Fig. 2, with the following exceptions, Amphibia = green, Sauropsida = light orange. All branches on the virus tree are scaled to the number of amino acid substitutions per site, and both trees were midpoint rooted for clarity only. Greater mouse-eared bat hepacivirus (GmebHV) is highlighted in red. Images were obtained from http://phylopic.org under Public Domain Dedication. Supplementary Fig. S5 provides the names of the hosts and viruses for the ‘pegi-hepaci’ co-phylogeny.
Importantly, despite the widespread occurrence of cross-species transmission, virus–host co-divergence was also predicted to have occurred relatively frequently across the ‘pegi-hepaci’ and pestivirus clades, representing 22 and 23 per cent of all events, respectively. For these groups, duplication events were more uncommon, representing 10 and 6 per cent of total events (Supplementary Fig. S4). Extinction events were rarely predicted, representing 4 per cent of events in the ‘pegi-hepaci’ clade, while no extinction events were detected in the Pestivirus co-phylogeny.
4. Discussion
Through transcriptome mining, we identified 32 novel flavivirid sequences across the Metazoa, including the first flavivirus-like sequences in non-bilaterians, pestivirus-like sequences in amphibians, reptiles, and bony fish, as well as a range of vertebrate hepaciviruses. Hence, this work provides further evidence of the long-term associations between the Flaviviridae and Metazoa and highlights the vast number of viruses that remain undiscovered.
The Cnidaria are a primitive and basal phylum of Metazoa. Based on the identification of a flavivirus-like sequence in a cnidarian sample (CnidFV), we suggest that the origins of this group of viruses likely extend much further back in time than previous estimates and are closer to the emergence of the metazoans 750–800 million years ago (Erwin 2015). This conclusion is supported by the earlier finding of a flavivirus-derived EVE in the Cnidaria (Bamford et al. 2022). Notably, CnidFV and the cnidarian EVE are more closely related to members of the genus Flavivirus than are the tamana/flavi-like viruses, suggesting that these groups, including the jingmenviruses, are evolutionarily distinct (Bamford et al. 2022). As such, we suggest that the tamana/flavi-like viruses should be given a distinct taxonomic classification within the Flaviviridae. However, it is clear that it is difficult to fully resolve the evolutionary history of the flavivirids with our current understanding of their diversity, although it appears that the origins of this group lie in aquatic environments (Lensink, Yiqiao, and Lequime 2022).
It is important to note that host assignment of the non-bilaterian flaviviruses is tentative as these sequences are extremely divergent and have only rarely been sampled. Due to the detection of several cnidarian species in addition to the target species, the octocoral E. flexuosa in library SRR12876665, we have assigned the resulting virus sequence as cnidaria flavivirus (CnidFV). The high abundance of Symbiodinium in this library is unsurprising given that the octocoral-Symbiodinium mutualism is well known (van de Water, Allemand, and Ferrier-Pagès 2018). However, the phylogenetic placement of this virus with those found in a marine acorn worm suggests that it is more likely associated with E. flexuosa than Symbiodinium. While CnidFV and the peach blossom jellyfish EVE are relatively closely related to each other, there is substantial genetic divergence between these sequences. This has been previously observed with crocidura pestivirus and a Crocidura EVE and may reflect divergent evolution since the historic endogenization event (Li et al. 2022).
The discovery of Wenzhou pesti-like virus 1, Wenling pesti-like virus 2, Xiamen fanray pesti-like virus, and Nanhai dogfish shark pesti-like virus in cartilaginous fish marked the expansion of the pestiviruses from warm-blooded mammals to basal vertebrate species, suggesting that these viruses infect a range of vertebrate lineages (Shi et al. 2018). For the first time, we identified pesti-like viruses in reptiles, amphibians, and bony fish, extending the host range of these viruses to encompass all vertebrate classes with the exception of Aves. The deep evolutionary association between pestiviruses and vertebrates is further reflected in the clear pattern of pestivirus–host co-divergence among the viruses identified in this study. As a result, we anticipate that novel pestiviruses will be found infecting a wider diversity of vertebrates and that their known host range largely reflects where sampling efforts have been directed to date. Additionally, frog pestivirus was identified in the ventral skin of the American bullfrog, although other species of frog were detected in this library. Within the study in which this library was generated, the American bullfrog appeared resistant to the fungal pathogen Batrachochytrium dendrobatidis (Bd) (Eskew et al. 2018). Co-infection with Bd and ranaviruses is frequently observed in frogs, but whether the interactions between these pathogens are antagonistic or facilitative is currently unclear (Bosch et al. 2020). If Bd and pestiviruses are found to commonly co-infect frogs, future efforts should be directed towards studying their interactions.
We identified 20 novel hepacivirus sequences, among which a clade of cichlid-associated hepacivirus sequences is notable. This clade was derived from a study of Lake Tanganyika, a freshwater lake shared by Tanzania, the Democratic Republic of the Congo, Burundi, and Zambia that is known for its high diversity of endemic cichlid species (Koblmüller et al. 2008; El Taher et al. 2021). Importantly, the fish and reptile hepaciviruses identified in this study were predominately associated with samples of liver tissue, suggesting that hepatotropism is likely a universal feature of these viruses across vertebrates (Smith et al. 2016).
Bats and rodents harbour a large diversity of hepaciviruses and are thought to have played an important role in their global spread and broader evolutionary history (Epstein et al. 2010; Drexler et al. 2013; Kapoor et al. 2013; Quan et al. 2013; de Souza et al. 2019; Bletsa et al. 2021). We identified GmebHV, which falls within a clade of rodent, sloth, and raccoon hepaciviruses. The clear relatedness between GmebHV and rodent hepacivirus (QLM02863), combined with evidence from our co-evolutionary analyses, suggests that this sequence might represent a cross-species transmission event between bats and rodents. Similarly, ancestral state reconstructions have previously shown that cross-species transmission from rodents is likely the source of the sloth and ringtail hepaciviruses (Moreira-Soto et al. 2020; Jo et al. 2022). In this case, we cannot resolve the direction of virus transmission with any certainty or whether other species are involved.
In very broad terms, we find that the hepaci-, pesti-, and pegiviruses cluster with the phylogeny of their hosts, with the relevant frequent cross-species virus transmission events only occurring within host classes (i.e. Mammalia, Sauropsida, and Chondrichthyes). The exceptions were FrbuHV, Western African lungfish hepacivirus, and Wenling moray eel hepacivirus that fell basal to those viruses identified in cartilaginous fish (although the phylogenetic position of these viruses should be treated with caution as the relevant nodes have weak bootstrap support; Fig. 4, Fig. 6). The clear phylogenetically defined barriers between host classes may reflect differences in receptor binding and cell entry mechanisms among distantly related hosts (Parrish et al. 2008). Host ecology also likely contributes to these barriers, particularly as physical separation means that fewer cross-species virus transmission events are expected to occur between marine and land vertebrates than among land vertebrates (Luis et al. 2015; French et al. 2022). Together, this suggests that more cross-species transmission occurs among closely related hosts, which may have also resulted in the apparent loss of co-divergence signal within relatively well-sampled taxonomic groups (e.g. mammals). At deeper taxonomic levels, we found clear evidence for virus–host co-divergence, particularly in lower vertebrates, which is consistent with previous findings (Hartlage, Cullen, and Kapoor 2016; Geoghegan, Duchêne, and Holmes 2017; Shi et al. 2018; Porter et al. 2020). However, it is also apparent that the results of our co-phylogenetic analysis are influenced by the sample of virus diversity used and will likely change as more viruses are identified. In addition, virus phylogenies were estimated using RdRp (NS5) alone. It is possible that differences in the phylogenies of the entire polyprotein or NS3 region would produce different estimates of the frequencies of co-divergence and host jumping.
Wenling moray eel hepacivirus (AVM87555) forms a sister group to the ‘pegi-hepaci’ lineage, although this may be artefactual due to recombination or extreme rate variation (Porter et al. 2020). If the position of the Wenling moray eel hepacivirus is correct, this suggests that a common ancestor of the ‘pegi-hepaci’ lineage may have existed in an aquatic environment. This notion is supported by the recent finding of ‘pegi-hepaci’ derived EVE in a marine mollusc (Bamford et al. 2022). The apparent lack of pegiviruses in aquatic vertebrate and invertebrate species in this study does not equate to their absence in these organisms due to the current depth of SRA libraries available.
Another notable observation from this study was the identification of a flavivirus in non-bilaterians, which raises additional questions on the ancestral mode of flavivirus transmission. Non-bilaterians lack the circulatory system of vertebrates, suggesting that an alternative mode of cell-to-cell virus transmission may exist in these animals (Bamford et al. 2022).
In sum, through a broad-scale survey of publicly available and newly generated transcriptome data, we revealed a wide diversity of flavivirid sequences in undersampled metazoan species. In doing so, we provide additional information for an ancient origin of the flaviviruses, likely closer to the emergence of the metazoans some 750–800 million years ago, and hence for the long-term association between the Flaviviridae and the Metazoa as a whole.
Supplementary Material
Acknowledgements
We thank A.H. McGrath for assistance in field sampling. We acknowledge the University of Sydney’s high-performance computing cluster, Artemis, for providing the computing resources used for this study. This work would not be possible without the sequencing data that have been generously shared, and we are grateful to the NCBI SRA team for their management of this important resource.
Contributor Information
Jonathon C O Mifsud, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney NSW 2006, Australia.
Vincenzo A Costa, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney NSW 2006, Australia.
Mary E Petrone, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney NSW 2006, Australia.
Ezequiel M Marzinelli, School of Life and Environmental Sciences, The University of Sydney, Sydney NSW 2006, Australia; Sydney Institute of Marine Science, 19 Chowder Bay Rd, Mosman, NSW 2088, Australia; Singapore Centre for Environmental Life Sciences Engineering, Nanyang Technological University, Singapore 637551 Singapore.
Edward C Holmes, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney NSW 2006, Australia.
Erin Harvey, Sydney Institute for Infectious Diseases, School of Medical Sciences, The University of Sydney, Sydney NSW 2006, Australia.
Data availability
All tunicate sequence reads are available on the NCBI SRA under BioProject PRJEB57836. All viral genomes and corresponding sequences assembled in this study have been deposited in the European Nucleotide Archive at EMBL-EBI and GenBank under BioProject PRJEB57836. The sequences, alignments, phylogenetic trees, and the custom Flaviviridae database generated in this study are available at https://github.com/JonathonMifsud/Transcriptome_mining_extends_the_host_range_of_the_Flaviviridae_to_non-bilaterians.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
E.C.H. is supported by an National Health and Medical Research Council Australian Laureate Fellowship (FL170100022). J.C.O.M. is supported by the Australian Government’s Research Training Program Scholarship. E.M.M. received funding from the Australian Research Council (DP180104041).
Conflict of interest:
None declared
References
- Babaian A., and Edgar R. (2022) ‘Ribovirus Classification by a Polymerase Barcode Sequence’, PeerJ, 10: e14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford C. G. G. et al. (2022) ‘Comparative Analysis of Genome-encoded Viral Sequences Reveals the Evolutionary History of Flavivirids (Family Flaviviridae)’, Virus Evolution, 8: veac085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessho-Uehara M. et al. (2020) ‘Kleptoprotein Bioluminescence: Parapriacanthus Fish Obtain Luciferase from Ostracod Prey’, Science Advances, 6: eaax4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederman M. K. et al. (2018) ‘Discovery of the First Germline-restricted Gene by Subtractive Transcriptomic Analysis in the Zebra Finch, Taeniopygia guttata’, Current Biology, 28: 1620–27.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bletsa M. et al. (2021) ‘Molecular Detection and Genomic Characterization of Diverse Hepaciviruses in African Rodents’, Virus Evolution, 7: veab036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitvich B. J., and Firth A. E. (2017) ‘A Review of Flaviviruses That Have No Known Arthropod Vector’, Viruses, 9: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics, 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch J. et al. (2020) ‘Single Infection with Batrachochytrium dendrobatidis or Ranavirus Does Not Increase Probability of Co-infection in a Montane Community of Amphibians’, Scientific Reports, 10: 21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister J. R. et al. (2015) ‘NCBI Viral Genomes Resource’, Nucleic Acids Research, 43: 571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Bushnell B. (2014) BBMap: A Fast, Accurate, Splice-aware Aligner. Berkeley: Lawrence Berkeley National Lab (LBNL). [Google Scholar]
- Cannon J. T. et al. (2014) ‘Phylogenomic Resolution of the Hemichordate and Echinoderm Clade’, Current Biology, 24: 2827–32. [DOI] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W.-S., Rose K., and Holmes E. C. (2021) ‘Meta-transcriptomic Analysis of the Virome and Microbiome of the Invasive Indian Myna (Acridotheres tristis) in Australia’, One Health, 13: 100360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen P. T. L. C., Aarestrup F. M., and Lund O. (2018) ‘Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA’, BMC Bioinformatics, 19: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conow C. et al. (2010) ‘Jane: A New Tool for the Cophylogeny Reconstruction Problem’, Algorithms for Molecular Biology, 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V. A. et al. (2021) ‘Metagenomic Sequencing Reveals a Lack of Virus Exchange between Native and Invasive Freshwater Fish across the Murray–Darling Basin, Australia’, Virus Evolution, 7: veab034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V. A. et al. (2022) ‘Limited Cross-species Virus Transmission in a Spatially Restricted Coral Reef Fish Community’, bioRxiv, 2022.05.17.492384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. T. et al. (2015) ‘Family Wide Molecular Adaptations to Underground Life in African Mole-rats Revealed by Phylogenomic Analysis’, Molecular Biology and Evolution, 32: 3089–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza W. M. et al. (2019) ‘A Novel Hepacivirus in Wild Rodents from South America’, Viruses, 11: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheilly N. M. et al. (2022) ‘A World of Viruses Nested within Parasites: Unraveling Viral Diversity within Parasitic Flatworms (Platyhelminthes)’, Microbiology Spectrum, 10: e00138–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler J. F. et al. (2013) ‘Evidence for Novel Hepaciviruses in Rodents’, PLoS Pathogens, 9: e1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. et al. (2022) ‘Petabase-scale Sequence Alignment Catalyses Viral Discovery’, Nature, 602: 142–7. [DOI] [PubMed] [Google Scholar]
- El Taher A. et al. (2021) ‘Gene Expression Dynamics during Rapid Organismal Diversification in African Cichlid Fishes’, Nature Ecology Evolution, 5: 243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J. H. et al. (2010) ‘Identification of GBV-D, a Novel GB-like Flavivirus from Old World Frugivorous Bats (Pteropus giganteus) in Bangladesh’, PLoS Pathogens, 6: e1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin D. H. (2015) ‘Early Metazoan Life: Divergence, Environment and Ecology’, Philosophical Transactions of the Royal Society B: Biological Sciences, 370: 20150036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskew E. A. et al. (2018) ‘Gene Expression Differs in Susceptible and Resistant Amphibians Exposed to Batrachochytrium dendrobatidis’, Royal Society Open Science, 5: 170910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French R. K. et al. (2022) ‘Host Phylogeny Shapes Viral Transmission Networks in an Island Ecosystem’, bioRxiv, 2022.10.04.510907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L. et al. (2018) ‘Hidden Diversity and Evolution of Viruses in Market Fish’, Virus Evolution, 4: vey031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L. et al. (2021) ‘Virome Composition in Marine Fish Revealed by Meta-transcriptomics’, Virus Evolution, 7: veab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L., Duchêne S., and Holmes E. C. (2017) ‘Comparative Analysis Estimates the Relative Frequencies of Co-divergence and Cross-species Transmission within Viral Families’, PLoS Pathogens, 13: e1006215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg T. L. et al. (2019) ‘Multidecade Mortality and a Homolog of Hepatitis C Virus in Bald Eagles (Haliaeetus leucocephalus), the National Bird of the USA’, Scientific Reports, 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger A. L. (2018) ‘A Decade of RNA Virus Metagenomics Is (Not) Enough’, Virus Research, 244: 218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood R. et al. (2021) ‘A Novel Rubi-like Virus in the Pacific Electric Ray (Tetronarce californica) Reveals the Complex Evolutionary History of the Matonaviridae’, Viruses, 13: 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Hackl T., and Ankenbrand M. J. (2022) ‘Gggenomes: A Grammar of Graphics for Comparative Genomics’, R package version 0.9.5.9000. <https://github.com/thackl/gggenomes>.
- Hahn C. M. et al. (2016) ‘Transcriptome Discovery in Non-model Wild Fish Species for the Development of Quantitative Transcript Abundance Assays’, Comparative Biochemistry and Physiology. Part D, Genomics & Proteomics, 20: 27–40. [DOI] [PubMed] [Google Scholar]
- Harding E. F. et al. (2022) ‘Revealing the Uncharacterised Diversity of Amphibian and Reptile Viruses’, ISME Communications, 2: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlage A. S., Cullen J. M., and Kapoor A. (2016) ‘The Strange, Expanding World of Animal Hepaciviruses’, Annual Review of Virology, 3: 53–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E. L. et al. (2017) ‘Virus Variation Resource–improved Response to Emergent Viral Outbreaks’, Nucleic Acids Research, 45: D482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley N. M. et al. (2019) ‘Phenotypic Evolution Shaped by Current Enzyme Function in the Bioluminescent Courtship Signals of Sea Fireflies’, Proceedings of the Royal Society B, 286: 20182621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2017) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M. et al. (2021) ‘Identification of Novel Avian and Mammalian Deltaviruses Provides New Insights into Deltavirus Evolution’, Virus Evolution, 7: veab003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janouškovec J. et al. (2019) ‘Apicomplexan-like Parasites Are Polyphyletic and Widely but Selectively Dependent on Cryptic Plastid Organelles’, Elife, 8: e49662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W. et al. (2019) ‘Insights into Body Size Evolution: A Comparative Transcriptome Study on Three Species of Asian Sisoridae Catfish’, International Journal of Molecular Sciences, 20: 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo W. K. et al. (2022) ‘Natural Co-infection of Divergent Hepatitis B and C Virus Homologues in Carnivores’, Transboundary and Emerging Diseases, 69: 195–203. [DOI] [PubMed] [Google Scholar]
- Jones P. et al. (2014) ‘InterProScan 5: Genome-scale Protein Function Classification’, Bioinformatics, 30: 1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A. et al. (2013) ‘Identification of Rodent Homologs of Hepatitis C Virus and Pegiviruses’, mBio, 4: e00216–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics, 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblmüller S. et al. (2008) ‘Age and Spread of the Haplochromine Cichlid Fishes in Africa’, Molecular Phylogenetics and Evolution, 49: 153–69. [DOI] [PubMed] [Google Scholar]
- Ladner J. T. et al. (2016) ‘A Multicomponent Animal Virus Isolated from Mosquitoes’, Cell Host & Microbe, 20: 357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay C. et al. (2020) ‘Unmapped RNA Virus Diversity in Termites and Their Symbionts’, Viruses, 12: 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink M. J., Li Y., and Lequime S. (2022) ‘Aquatic Flaviviruses’, Journal of Virology, 96: e00439–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., and Dewey C. N. (2011) ‘RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome’, BMC Bioinformatics, 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015) ‘MEGAHIT: An Ultra-fast Single-node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics, 31: 1674–6. [DOI] [PubMed] [Google Scholar]
- Li Y. et al. (2022) ‘Endogenous Viral Elements in Shrew Genomes Provide Insights into Pestivirus Ancient History’, Molecular Biology and Evolution, 39: msac190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis A. D. et al. (2015) ‘Network Analysis of Host–virus Communities in Bats and Rodents Reveals Determinants of Cross-species Transmission’, Ecology Letters, 18: 1153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelino V. R. et al. (2020) ‘CCMetagen: Comprehensive and Accurate Identification of Eukaryotes and Prokaryotes in Metagenomic Data’, Genome Biology, 21: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzinelli E. M. (2012) ‘Artificial Structures Influence Fouling on Habitat-forming Kelps’, Biofouling, 28: 339–49. [DOI] [PubMed] [Google Scholar]
- Mifsud J. C. O. et al. (2022) ‘Transcriptome Mining Expands Knowledge of RNA Viruses across the Plant Kingdom’, Journal of Virology, 96: e00260–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara T. et al. (2016) ‘Linking Virus Genomes with Host Taxonomy’, Viruses, 8: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. K. et al. (2021) ‘Slippery When Wet: Cross-species Transmission of Divergent Coronaviruses in Bony and Jawless Fish and the Evolutionary History of the Coronaviridae’, Virus Evolution, 7: veab050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B. Q. et al. (2020) ‘IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era’, Molecular Biology and Evolution, 37: 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira-Soto A. et al. (2020) ‘Cross-order Host Switches of Hepatitis C-related Viruses Illustrated by a Novel Hepacivirus from Sloths’, Virus Evolution, 6: veaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S. et al. (2021) ‘CheckV Assesses the Quality and Completeness of Metagenome-assembled Viral Genomes’, Nature Biotechnology, 39: 578–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olendraite I., Brown K., and Firth A. E. (2022) ‘Identification of RNA Virus-derived RdRp Sequences in Publicly Available Transcriptomic Datasets’, bioRxiv, 2022.10.18.512700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E., and Schliep K. (2019) ‘Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R’, Bioinformatics, 35: 526–8. [DOI] [PubMed] [Google Scholar]
- Paraskevopoulou S. et al. (2021) ‘Viromics of Extant Insect Orders Unveil the Evolution of the Flavi-like Superfamily’, Virus Evolution, 7: veab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish C. R. et al. (2008) ‘Cross-species Virus Transmission and the Emergence of New Epidemic Diseases’, Microbiology and Molecular Biology Reviews, 72: 457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry R., Asgar S., and Williams B. R. G. (2019) ‘Discovery of Novel Crustacean and Cephalopod Flaviviruses: Insights into the Evolution and Circulation of Flaviviruses between Marine Invertebrate and Vertebrate Hosts’, Journal of Virology, 93: e00432–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A. F. et al. (2020) ‘Novel Hepaci- and Pegi-like Viruses in Native Australian Wildlife and Non-human Primates’, Virus Evolution, 6: veaa064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada C., and Hellberg M. E. (2021) ‘Speciation-by-depth on Coral Reefs: Sympatric Divergence with Gene Flow or Cryptic Transient Isolation?’, Journal of Evolutionary Biology, 34: 128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. L. (1978) ‘Isolation of Rio Bravo and a Hitherto Undescribed Agent, Tamana Bat Virus, from Insectivorous Bats in Trinidad, with Serological Evidence of Infection in Bats and Man’, American Journal of Tropical Medicine and Hygiene, 27: 153–61. [DOI] [PubMed] [Google Scholar]
- Qin X.-C. et al. (2014) ‘A Tick-borne Segmented RNA Virus Contains Genome Segments Derived from Unsegmented Viral Ancestors’, Proceedings of the National Academy of Sciences of the United States of America, 111: 6744–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan P.-L. et al. (2013) ‘Bats Are a Major Natural Reservoir for Hepaciviruses and Pegiviruses’, Proceedings of the National Academy of Sciences of the United States of America, 110: 8194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell L. J. (2012) ‘Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things)’, Methods in Ecology and Evolution, 3: 217–23. [Google Scholar]
- Rice P., Longden I., and Bleasby A. (2000) ‘EMBOSS: The European Molecular Biology Open Software Suite’, Trends in Genetics, 16: 276–7. [DOI] [PubMed] [Google Scholar]
- Sanada T. et al. (2019) ‘Construction of Complete Tupaia belangeri Transcriptome Database by Whole-genome and Comprehensive RNA Sequencing’, Scientific Reports, 9: 12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santichaivekin S. et al. (2020) ‘eMPRess: A Systematic Cophylogeny Reconciliation Tool’, Bioinformatics, 16: 2481–2. [DOI] [PubMed] [Google Scholar]
- Sayers E. W. et al. (2021) ‘GenBank’, Nucleic Acids Research, 49: D92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D. T. et al. (2020) ‘Conserved Novel ORFs in the Mitochondrial Genome of the Ctenophore Beroe forskalii’, PeerJ, 8: e8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses’, Journal of Virology, 90: 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2018) ‘The Evolutionary History of Vertebrate RNA Viruses’, Nature, 556: 197–202. [DOI] [PubMed] [Google Scholar]
- Simmonds P. et al. (2017) ‘ICTV Virus Taxonomy Profile: Flaviviridae’, The Journal of General Virology, 98: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoge R. H. et al. (2018) ‘New Virus of the Family Flaviviridae Detected in Lumpfish (Cyclopterus lumpus)’, Archives of Virology, 163: 679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. B. et al. (2016) ‘Proposed Update to the Taxonomy of the Genera Hepacivirus and Pegivirus within the Flaviviridae Family’, The Journal of General Virology, 97: 2894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto E. et al. (2020) ‘First Isolation of a Novel Aquatic Flavivirus from Chinook Salmon (Oncorhynchus tshawytscha) and Its in Vivo Replication in a Piscine Animal Model’, Journal of Virology, 94: e00337–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speranza E. et al. (2018) ‘A Conserved Transcriptional Response to Intranasal Ebola Virus Exposure in Nonhuman Primates Prior to Onset of Fever’, Science Translational Medicine, 10: eaaq1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. et al. (2016) ‘Fish-T1K (Transcriptomes of 1,000 Fishes) Project: Large-scale Transcriptome Data for Fish Evolution Studies’, GigaScience, 5: s13742-016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. et al. (2018) ‘Rapid Evolution of a Voltage-gated Sodium Channel Gene in a Lineage of Electric Fish Leads to a Persistent Sodium Current’, PLoS Biology, 16: e2004892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Sánchez M. et al. (2019) ‘Multi-tissue Transcriptomes of Caecilian Amphibians Highlight Incomplete Knowledge of Vertebrate Gene Families’, DNA Research: An International Journal for Rapid Publication of Reports on Genes and Genomes, 26: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Water J. A., Allemand D., and Ferrier-Pagès C. (2018) ‘Host-microbe interactions in octocoral holobionts - recent advances and perspectives’, Microbiome, 6: 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2012) ‘Virome Analysis for Identification of Novel Mammalian Viruses in Bat Species from Chinese Provinces’, Journal of Virology, 86: 10999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. et al. (2020) ‘Abundant and Diverse RNA Viruses in Insects Revealed by RNA-seq Analysis: Ecological and Evolutionary Implications’, mSystems, 5: e00039–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B. et al. (2022) ‘Dynamic Genetic Differentiation Drives the Widespread Structural and Functional Convergent Evolution of Snake Venom Proteinaceous Toxins’, BMC Biology, 20: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ‘Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36. [Google Scholar]
- Yurchenko A. A., Recknagel H., and Elmer K. R. (2020) ‘Chromosome-level Assembly of the Common Lizard (Zootoca vivipara) Genome’, Genome Biology and Evolution, 12: 1953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-L. et al. (2022) ‘A Highly Divergent Hepacivirus Identified in Domestic Ducks Further Reveals the Genetic Diversity of Hepaciviruses’, Viruses, 14: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All tunicate sequence reads are available on the NCBI SRA under BioProject PRJEB57836. All viral genomes and corresponding sequences assembled in this study have been deposited in the European Nucleotide Archive at EMBL-EBI and GenBank under BioProject PRJEB57836. The sequences, alignments, phylogenetic trees, and the custom Flaviviridae database generated in this study are available at https://github.com/JonathonMifsud/Transcriptome_mining_extends_the_host_range_of_the_Flaviviridae_to_non-bilaterians.