

Abstract
Site-specific incorporation of unnatural amino acids (Uaas) in living cells relies on engineered aminoacyl-tRNA synthetase/tRNA pairs borrowed from a distant domain of life. Such heterologous suppressor tRNAs often show poor intrinsic activity, presumably due to suboptimal interaction with a non-native translation system. This limitation can be addressed in E. coli using directed evolution. However, no suitable selection system is currently available to do the same in mammalian cells. Here we report virus-assisted directed evolution of tRNAs (VADER) in mammalian cells, which employs a double-sieve selection scheme to facilitate single-step enrichment of active-yet-orthogonal tRNA mutants from naïve libraries. Using VADER, we developed improved mutants of M. mazei pyrrolysyl-tRNA, as well as a bacterial tyrosyl tRNA. We also show that the higher activity of the most efficient mutant pyrrolysyl-tRNA is specific for mammalian cells, alluding to an improved interaction with the unique mammalian translation apparatus.
Introduction
Site-specific incorporation of unnatural amino acids (Uaas) into proteins in mammalian cells holds much potential to enable both basic science as well as biotechnology applications.1–4 Central to this technology is a nonsense-suppressing aminoacyl-tRNA synthetase (aaRS)/tRNA pair which is engineered to charge the Uaa of interest without cross-reacting with any of its host counterparts. Such “orthogonal” aaRS/tRNA pairs are typically imported into the host cell from a different domain of life.1–3 The performance of the heterologous suppressor tRNA is often suboptimal in the new host, given that it must directly interact with a foreign translation system. Indeed, several studies have confirmed that Uaa incorporation efficiency in mammalian cells is limited by the poor performance of the heterologous suppressor tRNAs, which must be massively overexpressed to achieve acceptable levels of Uaa incorporation efficiency.5–7 The ability to overcome suboptimal tRNA performance will be beneficial to improve the robustness of the Uaa mutagenesis technology in mammalian cells, and to enable advanced applications.
The biology of tRNAs is complex and multifaceted (Supplementary Figure 1), involving expression, processing, post-transcriptional modifications, cellular stability, interaction with the cognate aaRS and the components of the translational system (e.g., elongation factors and the ribosome), etc.8 How these different facets of tRNA biology contribute to the poor performance of foreign suppressor tRNAs is poorly understood, which makes it challenging to develop better variants through rational design. Although a semi-rational approach was recently used with some success to improve the activity of pyrrolysyl-tRNA in mammalian cells,9 it is challenging to generalize such strategies for tRNA engineering. Directed evolution has been used with much success to develop improved orthogonal suppressor tRNA mutants in E. coli.10–19 This was made possible by the development of clever selection systems, which enables the enrichment of active and orthogonal suppressor tRNA mutants from large synthetic libraries. The ability to perform analogous tRNA evolution in mammalian cells has the potential to yield improved suppression systems, but no suitable platform is currently available. It is important to perform such directed evolution experiments in mammalian cells, instead of in lower organisms, where directed evolution is better established, to ensure that the tRNA mutants are selected based on their improved interactions with the unique mammalian translation system.
Existing directed evolution strategies in mammalian cells largely rely on stable integration of the target gene in a cell line, followed by the creation of sequence diversity through untargeted or targeted random mutagenesis.20–22 This approach is unsuitable for tRNA evolution for two reasons: A) To ensure a clear genotype-phenotype connection, it is essential to have no more than a single genomically integrated mutant tRNA gene per cell. However, it has been shown that a large number of tRNA genes (>100) are needed per cell to achieve detectable Uaa incorporation efficiency;5,7 B) The low mutagenic frequency associated with these strategies is poorly suited for tRNA evolution, given its small size (<100 bp). One particular challenge arises when diversifying the stem regions of a tRNA, which are most frequently targeted for directed evolution: any mutation must be accompanied by a matching mutation on the other side to retain base-pairing, the loss of which compromises tRNA secondary structure and activity. Capturing such rich sequence diversity within the small tRNA gene is practically only feasible using synthetic site-saturation mutagenesis libraries. To enrich suppressor tRNA variants that are orthogonal and active in mammalian cells from such synthetic libraries, we need the following: i) controlled delivery of the library members into mammalian cells, such that each cell receives a single variant; ii) a selection scheme that enriches the active tRNA mutants, and removes cross-reactive ones; and iii) the ability to identify the surviving mutants. We envisioned achieving these requirements by coupling the activity of the suppressor tRNA to the replication of a mammalian virus: i) encoding the library of tRNA variants in the virus genome would enable its controlled delivery to mammalian cells; ii) inserting a nonsense codon in an essential virus protein would render viral replication dependent on the activity of the suppressor tRNA, facilitating selective amplification of virions encoding active tRNA variants; and iii) the enriched tRNA sequences can be readily retrieved by isolating and sequencing the genome of the freshly amplified progeny virus. Some examples of virus-assisted directed evolution approaches in mammalian cells have been recently reported.20,23,24 However, these approaches use either natural or engineered error-prone replication of the virus genome to introduce sequence diversity, which is unsuitable for tRNA evolution due to the reasons described above. In addition, tRNA evolution demands simultaneous optimization of two distinct aspects: improving Uaa-incorporation activity, and suppressing potential cross-reactivity with the host aaRSs. No strategy currently exists to enable such sophisticated selection in mammalian cells.
We recently reported the feasibility of producing adeno-associated virus (AAV2) in mammalian cells, site-specifically incorporating Uaas into its capsid through amber suppression.25,26 In this system, successful AAV2 production is dependent on the activity of the suppressor aaRS/tRNA pair, providing an attractive platform for virus-assisted directed evolution of tRNA (VADER) in mammalian cells. AAV2 also offers additional advantages such as the lack of known pathogenicity, and a small genome that is amenable to facile manipulations.27 Here we report the development of an optimized double-sieve selection scheme (Figure 1a) for VADER that facilitates efficient one-step enrichment of active and orthogonal tRNA variants from naïve synthetic libraries. Using VADER, we developed mutants of M. mazei pyrrolysyl tRNA, which show significantly improved activity at lower expression levels. Furthermore, coupling VADER with next-generation sequencing (NGS) analysis provided a global view of the evolutionary landscape, where performance of each tRNA mutant in the entire library can be simultaneously tracked. We also show that the improved activity of the evolved tRNAPyl is host-specific, indicating that it likely stems from a better ‘fit’ with the unique translation apparatus of the mammalian cells. Finally, we demonstrate the generality of our approach by developing improved mutants of the E. coli tyrosyl-tRNA, using a ‘single-sieve’ variant of NGS-coupled VADER that lacks the negative selection step.
Figure 1. The VADER selection scheme.

a, Mammalian cells are infected with AAV2 encoding the tRNA library at low MOI. Plasmids encoding TAG-mutant of Cap, other genetic components needed for AAV replication, and the cognate aaRS are provided in trans by transfection in the presence of a suitable azido-Uaa. Active and orthogonal tRNA mutants facilitate generation of packaged progeny AAV2 incorporating the Uaa into their capsid, which are isolated by chemoselective biotin conjugation followed by streptavidin pulldown. b, Two AAV2 vectors, encoding i) E. coli tRNATyr and EGFP (Tyr-EGFP), and ii) tRNAPyl and mCherry (Pyl-mCherry), were mixed in a 104:1 ratio and subjected to the VADER selection scheme using MbPylRS and its substrate AzK. FACS analysis of the surviving population shows a >30,000-fold cumulative enrichment of Pyl-mCherry. Data shown as mean ± s.d. (n = 3 independent experiments)
Results
Development and validation of the VADER selection system
Successful directed evolution relies on the delivery of no more than a single library member per cell. However, we have previously shown that the presence of a large number of tRNA genes per cell is required to observe detectable levels of nonsense suppression,5–7 posing a paradoxical challenge for developing VADER. We envisioned a solution to this challenge that takes advantage of the natural replication of the AAV2 genome in mammalian cells in the presence of its Rep gene (encoding multiple proteins that assist in viral replication) and additional helper genes from adenovirus (AdHelper), which would generate numerous copies of the incoming viral genome.27 We demonstrated that such replication is associated with a remarkable enhancement in transgene expression from the AAV2 genome by infecting HEK293T cells with an mCherry-encoding AAV2 in the presence or absence of Rep+AdHelper (provided in trans; Supplementary Figure 2).
Next, we evaluated if such amplification can produce sufficient levels of suppressor tRNA from a single incoming virion to support the expression of TAG-inactivated capsid gene and the production of progeny virus. HEK293T cells were infected with AAV2 encoding an mCherry reporter and an M. mazei pyrrolysyl-tRNA amber suppressor tRNA (tRNACUAPyl) at a low multiplicity of infection (MOI). Cells were then transfected with plasmids encoding the following components: i) Rep and Cap-454-TAG (the Cap gene of AAV2 encodes three overlapping capsid proteins, VP1–3; 454-TAG inactivates all three);25,26,28 ii) AdHelper genes; and iii) wild-type pyrrolysyl-tRNA synthetase from M. barkeri (MbPylRS). We observed progeny virus production only in the presence of the Uaa azido-lysine (AzK; Supplementary Figure 3), a substrate for MbPylRS. Efficiency of virus production was ~6% of an identical experiment, where wild-type Cap gene was used instead (Supplementary Figure 3). These experiments validate the basis of VADER: the ability to couple AAV2 replication to the activity of a suppressor tRNA encoded in its genome.
Synthetic tRNA libraries can harbor variants capable of cross-reacting with host aaRSs, so an additional strategy is necessary to remove such cross-reactive tRNA variants. We envisioned a novel approach to selectively enrich virions that encode active and orthogonal tRNAs based on their ability to incorporate a chemically unique Uaa into the AAV2 capsid, while the cross-reactive ones would incorporate a canonical amino acid instead. If a Uaa with a bioorthogonal conjugation handle is used here, the resulting desired virus population can be isolated by chemoselective attachment of a biotin, followed by streptavidin enrichment (Figure 1a). We have previously demonstrated successful AzK incorporation and bioorthogonal modification of the AAV2 capsid.26 Using a photo-cleavable DBCO-biotin conjugate, we were indeed able to isolate AAV2–454-AzK following this strategy (Supplementary Figure 4). Together, the VADER selection scheme (Figure 1a) should enable the enrichment of an active and orthogonal tRNA population from a synthetic mutant library.
To show that an active suppressor tRNA can be enriched from a defined mixture of active and inactive tRNAs using VADER, we generated two AAV2 vectors; one encoding a tRNACUATyr (E. coli tyrosyl tRNA)29 and an EGFP, and another encoding tRNACUAPyl and an mCherry (Figure 1b, Supplementary Figure 5). Packaged Pyl-mCherry and Tyr-EGFP virus were mixed in a 1:104 ratio and subjected to the optimized VADER selection scheme (Figure 1a), using MbPylRS and its substrate AzK in step 1. MbPylRS charges AzK only to tRNAPyl, and not tRNATyr;30 Pyl-mCherry virus should thus undergo enrichment relative to Tyr-EGFP. Indeed, FACS analyses revealed that the former is enriched approximately 280- and 130-fold after selective amplification and bioorthogonal capture steps, respectively, providing a >30,000-fold cumulative enrichment in a single round of selection (Figure 1b; Supplementary Figure 6).
Application of VADER to generate improved tRNA variants
Next, we sought to use VADER for the directed evolution of tRNAPyl, one of the most popular platforms for mammalian genetic code expansion (Figure 2a).1–3,31 Its acceptor (A) and T stems were chosen for diversification, as these regions typically interact with the components of the translation system.32 We created four different site-saturation mutant libraries covering both stems (Figure 2a), excluding the first base pair in the A-stem that is important for binding PylRS.33 Each library was introduced into the AAV packaging plasmid through restriction cloning, with >100-fold sequence coverage, and packaged into AAV2 particles. Each library was subjected to the VADER scheme, and 30–50 surviving clones from each were characterized by sequencing. Base-pairing in these stem regions is important to maintain tRNA secondary structure and activity, but such fully base-paired sequences constitute a small fraction of the input tRNA library (Supplementary Figure 7). We were encouraged to find an enrichment of such fully base-paired sequences within the selected population, possibly indicating the successful selection of functional variants (Supplementary Figure 7; Supplementary Table 1, 2). Interestingly, sequenced selectants from the T-stem libraries revealed several wild-type or very similar mutants, while those from the A-stem libraries were more divergent (Supplementary Figure 7; Supplementary Table 1, 2). We evaluated the activity of each unique fully base-paired tRNA mutant isolated from the selection by co-transfecting them into HEK293T cells with MbPylRS and an EGFP-39TAG reporter in the presence and absence of 1 mM AzK. All tested mutants facilitated AzK-dependent expression of EGFP-39TAG, measured relative to the expression of a wild-type mCherry reporter encoded in the tRNA-plasmid (Figure 2d), confirming the selection of active and orthogonal mutants through VADER.
Figure 2. Directed evolution of tRNAPyl.

a, The sequences randomized to create four different libraries (A1, A2, T1, T2) of tRNAPyl are highlighted in four different colors. b, Degree of base pairing observed in the enriched mutants (through NGS analysis) upon the selection of each library. c, For each library, the wild-type sequence is shown on top and analysis of the 1% most-enriched sequences (n = 41) is shown below, revealing the relative abundance of each base at each randomized position. d, Efficiency of TAG suppression for unique tRNAPyl selectants measured using the EGFP-39TAG reporter. The tRNA encoded in the pAAV plasmid (also harboring a wild-type mCherry reporter) was co-transfected into HEK293T cells with MbPylRS and EGFP-39TAG in the presence or absence of 1 mM AzK. Expression of EGFP-39TAG was measured in cell-free extract, normalized relative to wild-type mCherry expression and plotted as a percentage of the normalized activity of wild-type tRNAPyl. Data shown as mean ± s.d. (n = 6 independent experiments for hits A1-GGG/CCC, A1-GGG/UCC, A1-AGG/CCU, A1-GCU/AGC, A2-AGG/UCU, A2-AAC/GUU, A2-ACU/AGU, A2-GGG/GCC, A2-ACU/GGU, A2-GUG/UGC, and T2-AG/UGCU; n = 8 independent experiments for hits A1-GGC/GCC, A1-GGG/CUC, A2-GGG/UCC, A2-GGG/UCU, and T1-CUG/CAG; n = 10 independent experiments for hits A1-GGG/CCC, A2-AGC/GCU, and A2-UGG/UCA; n = 18 independent experiments for A2-GGG/CCU; and n = 4 independent experiments for all others). *sequences identified through NGS; †enriched sequences containing a G:G mispair.
To gain a more comprehensive look into the selection process, we used next-generation sequencing (NGS) to assess the composition of each of the four input libraries, as well as their selection output after VADER performed in duplicate. Gratifyingly, we were able to identify all possible mutants in each library and evaluate their relative abundance (Supplementary Figure 8). If a tRNA mutant is more active, it would facilitate higher Cap-TAG expression, and the production of more progeny virus. Consequently, the degree of enrichment of each tRNA mutant upon selection, which can be calculated from its normalized abundance before and after the selection (by NGS; Supplementary Figure 9), should correlate with its activity in mammalian cells. Indeed, we found that the strongly enriched mutants (top 0.2% – 1%) from each library were heavily base-paired (Figure 2b), a hallmark of functional tRNAs. Additionally, for the individually characterized tRNA mutants (Figure 2d), we found a strong correlation between their relative enrichment and the observed suppression efficiency (Supplementary Figure 10), further confirming that the degree of enrichment factor upon selection is a reliable indicator of tRNA performance in mammalian cells. We identified several highly enriched sequences from each library that were not identified by sequencing individual surviving clones. We synthesized several such mutants and characterized their activity relative to the wild-type tRNAPyl (indicated by a * in Figure 2d). Cumulative characterization of all sequences revealed many A-stem mutants with significantly higher activity than the wild-type tRNAPyl (Figure 2d), with the most active variant (tRNAPyl -A2.1) showing a 3-fold improvement. In contrast, the T-stem libraries yielded fewer active variants (Supplementary Figure 9), and none showed higher activity than the wild-type tRNAPyl (Figure 2d).
NGS-coupled VADER provides a global view of the selection process, where the performance of each mutant in mammalian cells can be tracked across the entire library (Supplementary Figure 9). In addition to identifying individual top-performers, it revealed many additional nuances. For example, mispairing within tRNA stem regions is generally detrimental for its function, and such sequences identified from a traditional selection output would be typically ignored as artifacts. However, we identified sequences containing G:G mispairs in both A-stem libraries that were highly enriched upon selection, suggesting that these are legitimate hits. We tested two such mutants (indicated with a † in Figure 2d), both of which were active and orthogonal; the sequence GGG/GCC from A2 library actually showed 50% higher activity than WT-tRNAPyl. Additionally, the global enrichment profile of each library (Supplementary Figure 9) revealed that a large fraction of the A-stem libraries showed significant enrichment, indicating remarkable sequence plasticity of this region. In contrast, a much smaller fraction of the T-stem libraries showed significant enrichment, highlighting its limited tolerance for sequence alterations. Finally, analyses of all highly enriched (top 1%) sequences from the NGS-coupled VADER revealed interesting trends (Figure 2c). For example, most enriched mutants from the A-stem libraries show a sequence preference that is distinct from the wild-type tRNAPyl: these are significantly more G:C rich, with a strong preference for G residues at positions 2–7, and pyrimidines on the 3′ side of the A-stem. Characterization of individual clones corroborates that these sequence features are associated with enhanced activity. In contrast, the best-performing mutants from the T-stem libraries largely mirrored the wild-type sequence. Poor understanding of foreign tRNA biology in mammalian cells currently limits our ability to shed light on the mechanistic underpinnings of these observations. However, high-throughput characterization of large tRNA mutant libraries in this way can create an empirical knowledge base that will aid the design of improved variants.
Further characterization of tRNAPyl-A2.1
More active tRNAPyl mutants identified through VADER can improve the robustness and scope of the Uaa mutagenesis technology in mammalian cells using the popular pyrrolysyl platform. To this end, we further benchmarked the performance of the most efficient mutant, tRNAPyl-A2.1, against its WT counterpart. Even though this mutant was selected on the basis of incorporating AzK into the AAV2 capsid, we expected it to maintain its enhanced activity for other Uaas genetically encoded using the pyrrolysyl pair. Indeed, in addition to AzK (Figure 3c), tRNAPyl-A2.1 also facilitated improved incorporation of Nε-acetyllysine (AcK; Figure 3b, d), diazirine-lysine (DiazK), and strained cyclooctyne-L-lysine (SCOK), using previously established PylRS mutants (Supplementary Figure 11).34–36 Furthermore, A2.1 facilitated incorporation of AcK – a weak substrate – into two different sites in the reporter, whereas the expression of the same reporter using WT tRNAPyl was near background level (Figure 3d). We also constructed opal and ochre (UGA and UAA) suppressor variants of tRNAPyl-A2.1, each of which exhibited significantly higher activity relative to WT-tRNAPyl (Figure 3e). In fact, the opal suppression efficiency of A2.1 was on par with that of amber suppression using WT-tRNAPyl (Figure 3c, 3e). This will be particularly beneficial for site-specific incorporation of two distinct Uaas, which requires suppression of two different nonsense codons, and has been limited by the poor efficiency of non-UAG nonsense codons by the WT-tRNAPyl.30,37 We further validated the enhanced activity of tRNAPyl-A2.1 by incorporating AzK into the full-length human epidermal growth factor receptor (EGFR) at a significantly higher efficiency (Figure 3f). We also compared the activity of tRNAPyl-A2.1 with two tRNAPyl mutants (tRNAM15 and tRNAC15; Supplementary Figure 12), which were developed using a semi-rational approach for improved activity in mammalian cells.9 A2.1 and M15 facilitated similar levels of AzK incorporation into the EGFP-39TAG reporter, whereas the activity of C15 was significantly lower.
Figure 3. Characterization of tRNAPyl-A2.1 activity.

a, Sequences of wild-type (WT) and A2.1. b, Structures of AcK and AzK. c, Expression of EGFP-39TAG using the WT or A2.1 tRNA with MbPylRS, in the presence (+) and absence (−) of AzK. d, Expression of EGFP-39TAG and EGFP-39TAG-151TAG using the WT or A2.1 tRNA with AcK-selective MbPylRS, in the presence and absence of AcK. e, Expression of EGFP-39TGA and EGFP-39TAA using tRNAUCAPyl and tRNAUUAPyl (for both WT and A2.1), respectively, and MbPylRS in the presence and absence of AzK. f, Expression of EGFR-127TAG (with a C-terminal EGFP fusion) using the WT or A2.1 tRNA with MbPylRS, in the presence and absence of AzK. g, Mammalian cell-optimized baculovirus (BacMam) vectors and the scheme of the experiment for h-i. h, Expression of EGFP-39TAG as increasing MOI of tRNA-BacMam vector is used. i, Expression of mCherry in the same experiments as h shows equivalent delivery of both tRNAs, which increases proportionately with higher MOI. Expression of the fluorescent reporters is measured in HEK293T cell-free extract. Also see Supplementary Figure 13 for the corresponding images. For c-f and h, EGFP-39TAG expression is reported relative to wild-type EGFP expression from the same vector. Data shown as mean ± s.d. (n = 3 independent experiments in 3c, 3d, 3e, 3h, and 3i; n = 12 independent experiments in 3f).
So far, we tested the activities of tRNAPyl variants by transiently transfecting the appropriate plasmids. However, transient transfection delivers a large number of plasmids into cells, resulting in an overexpression of the encoded genes.7 We have shown in the past that such overexpression can confound the comparison between two systems in mammalian cells: the performance of the more active counterpart can get saturated at higher expression, allowing the weaker variant to ‘catch up’.7 To compare the activity across varying expression levels, we previously developed a mammalian-optimized baculoviral (BacMam) delivery vector, which enables convenient tuning of transgene expression by simply altering the virus-to-cell ratio.7 This controlled delivery system was used to further compare the activity of tRNAPyl-A2.1 to its WT counterpart. We used two BacMam vectors in this assay: one that encodes MbPylRS and EGFP-39TAG, and another encoding four copies of the tRNAPyl (either wild-type or A2.1) as well as a wild-type mCherry reporter (Figure 3g). The former was used at a constant MOI, while the latter was gradually increased from an MOI of 0.3 to 10 (Figure 3g). The mCherry and the EGFP signals represent the amount of tRNA-encoding vectors delivered, and the resulting UAG-suppression activity, respectively. The EGFP signal was normalized relative to cells infected with a separate BacMam vector encoding a WT-EGFP reporter at the same MOI. The mCherry expression levels increased linearly with increasing amount of tRNA-vector used, and signals from both tRNA vectors were comparable (Figure 3i, Supplementary Figure 13). When the tRNA-encoding vectors were delivered at low MOIs of 1 and 2, EGFP-39TAG expression (relative to WT-EGFP) facilitated by A2.1 was 2% and 7%, respectively, whereas the same for WT-tRNA was undetectable and 1% (Figure 3h, Supplementary Figure 13). Above MOI 5, A2.1 activity became saturated at ~50% reporter expression, while the WT-tRNA activity continued to close the efficiency-gap. These observations provide a more nuanced view of their relative activities, where A2.1 shows significantly higher benefit under copy-number limited scenarios. This property is expected to be particularly beneficial for advanced applications of the Uaa technology, where there are practical limits on the number of tRNA copies that can be delivered (e.g., in vivo applications,38 development of stable cell lines,5 etc.).
The origin of the improved activity of tRNAPyl-A2.1
As discussed earlier, the connection between the poor performance of foreign tRNAs and various facets of tRNA biology remains unclear. It also limits our ability to comprehend the molecular basis of the improvement achieved through directed evolution. Indeed, many orthogonal tRNAs have been evolved in E. coli, but the underlying mechanism for their improved activity is not understood.10–19 A mutant tRNA might exhibit higher activity simply due to higher cellular abundance (e.g., due to higher stability). We used Northern blot analysis to show that, upon transient transfection of HEK293T cells, A2.1 is actually expressed at a lower level relative to WT-tRNAPyl (Figure 4a), suggesting that it is intrinsically more active.
Figure 4. Origin of the improved activity of tRNAPyl-A2.1.
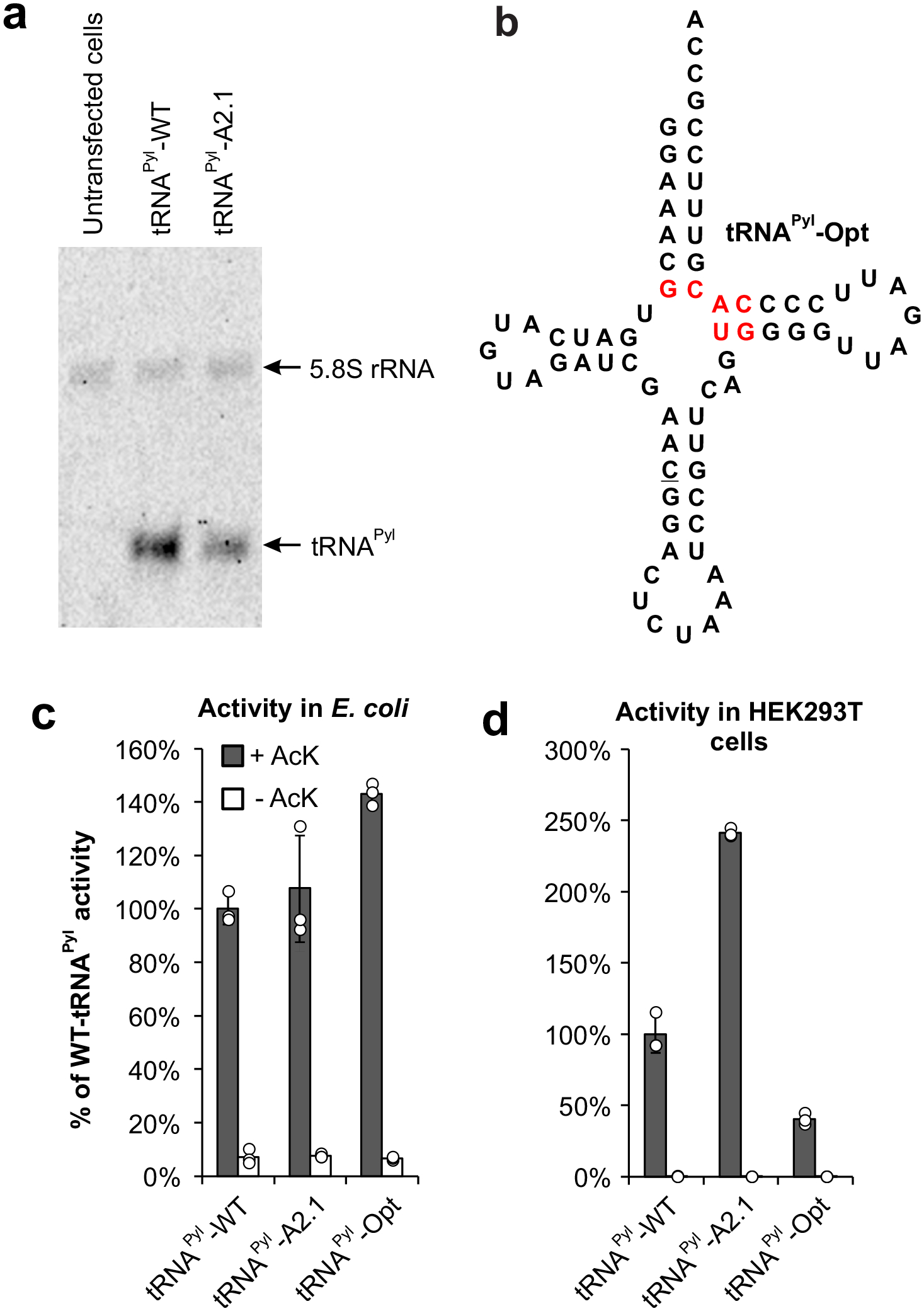
a, Northern blot analysis shows lower expression of A2.1 relative to WT, when plasmids encoding a tRNAPyl and a wild-type mCherry reporter was transfected into HEK293T cells. The mCherry reporter was used to ensure comparable transfection efficiency. b, Sequence of tRNAPyl-Opt (optimized in E. coli) showing key mutations in red. c, Activity of tRNAPyl-WT, A2.1, and Opt in E. coli measured using the sfGFP-151-TAG reporter. d, Activity of tRNAPyl-WT, A2.1, and Opt in HEK293T cells measured using the EGFP-39-TAG reporter. For both c and d, a mutant MbPylRS selective for AcK was used and the activities were measured in the presence and the absence of AcK. Expression of the fluorescent reporter was measured in cell-free extract and reported as the % of WT-tRNAPyl activity. Data shown as mean ± s.d. (n = 3 independent experiments).
Due to its universal orthogonality, the pyrrolysyl pair can be used for Uaa mutagenesis in both E. coli and mammalian cells. A tRNAPyl mutant (tRNAPyl-Opt; Figure 4b) was previously developed in E. coli that shows modest improvement in activity relative to tRNAPyl-WT.39,40 We wondered how tRNAPyl-A2.1 and tRNAPyl-Opt, which have been engineered for higher activity in mammalian cells and E. coli, respectively, would perform in a different host cell. If the higher activity can be transferred to different host, it would indicate that a factor intrinsic to this pair (e.g., PylRS-tRNAPyl interaction or tRNA stability) underlies the improvement. Alternatively, if the improvement is specific for a particular host, this would suggest that an improved interaction with the translation system of the particular host leads to the higher activity. We compared the activities of tRNAPyl WT, A2.1 and Opt in both E. coli and HEK293T cells using the same MbPylRS mutant (which charges AcK).34 As expected, A2.1 and Opt showed higher activity than WT in HEK293T cells (Figure 4d), and E. coli (Figure 4c), respectively. However, the activity of A2.1 in E. coli was comparable with WT, while Opt showed a significantly weaker activity in mammalian cells relative to WT. These observations strongly suggest that the enhanced activity of A2.1 originates from an improved interaction with the unique translation system of mammalian cells. It also underscores the importance of performing tRNA evolution directly in the target host to ensure proper ‘fit’ with its unique translation machinery. This is in contrast with the paradigm in aaRS engineering, where enhanced mutants developed in E. coli are routinely imported into mammalian cells with the retention of function.29,41,42
Directed evolution of tRNATyr using a single-step scheme
To further validate the generality of this platform, we sought to apply VADER for the directed evolution of E. coli tyrosyl tRNA, which was the first system to be used for Uaa mutagenesis in mammalian cells.43–45 The E. coli tyrosyl-tRNA synthetase (EcTyrRS)/tRNA pair has been engineered to genetically encode numerous useful Uaas,29,41 and has facilitated many novel applications, including dual Uaa mutagenesis in mammalian cells,30,46 and large-scale production of Uaa-modified therapeutic proteins.4,47 Such applications will significantly benefit from improved variants of the tRNATyr. We first confirmed that the tRNATyr is compatible with the VADER selection system. When a defined mixture of two AAV2 vectors encoding either tRNATyr or tRNAPyl was subjected to the selective amplification step of VADER, in the presence of an established polyspecific EcTyrRS mutant that charges O-methyltyrosine (OMeY),29 the tRNATyr-encoding virus was significantly enriched in the output (Supplementary Figure 14). However, the use of the azide-bearing Uaa p-azidophenylalanine (pAzF) instead of OMeY, which can also be charged with the same polyspecific EcTyrRS,29 failed to generate packaged virus, possibly due to perturbations to the virus capsid. Although the absence of an azide-Uaa precludes the 2nd-step of VADER involving bioorthogonal capture, which is designed to remove the cross-reactive tRNA population, we devised an alternative scheme to achieve the same goal (Figure 5a). We reasoned that active yet orthogonal tRNA mutants can be identified by their conditional enrichment upon performing the selective amplification step of VADER in the presence of the cognate synthetase, but not in its absence.
Figure 5. Directed evolution of tRNATyr.

a, A modified VADER scheme lacking the bioorthogonal capture step. Instead, the selective amplification is performed either in the presence or the absence of the cognate synthetase. Active and orthogonal tRNA mutants are identified by their selective enrichment in the presence of the cognate aaRS. b, The randomization scheme to create the E. coli tRNATyr mutant library. c, The consensus corresponding to 172 homologous bacterial tRNATyr sequences, which was used to guide the tRNATyr randomization scheme. d, Sequence of the tRNATyr mutants exhibiting the highest average enrichment following the VADER scheme shown in panel a in the presence of EcTyrRS. e, Observed enrichment of each mutant in the tRNATyr library upon subjecting them to the VADER selection scheme either in the presence of a cognate EcTyrRS mutant that charge OMeY, or MbPylRS (does not recognize tRNATyr). Each selection was performed in duplicate, and the normalized enrichment factors observed from each were plotted against each other. Nine out of the ten most enriched sequences in the presence of EcTyrRS did not show strong enrichment when MbPylRS was used instead (shown in green; sequences shown in panel d); the cross-reactive mutant is shown in red. f, Efficiency of TAG suppression for these nine tRNATyr mutants using the EGFP-39TAG. The tRNA encoded in the pAAV plasmid (also harboring a wild-type mCherry reporter) was co-transfected into HEK293T cells with OMeY-selective EcTyrRS and EGFP-39TAG in the presence or absence of 1 mM OMeY. Expression of EGFP-39TAG was measured in cell-free extract, normalized relative to wild-type mCherry expression and plotted as a percentage of the normalized activity of wild-type tRNATyr. Data shown as mean ± s.d. (n = 3 independent experiments).
A library of approximately 20,000 mutants was generated by randomizing different bases in the acceptor stem (Figure 5b), guided by the consensus sequence from 172 different bacterial tRNATyr homologs48 (Figure 5c) to retain conserved sequence elements. This library was cloned into the AAV2 packaging vector with >100-fold sequence coverage and packaged into AAV2 virions. This library was subjected to the selective amplification step of VADER, either in the presence of the cognate EcTyrRS mutant that charge OMeY, or a non-cognate MbPylRS (Figure 5a). Each selection was performed in duplicate and enrichment factor for each tRNATyr mutant from these replicates were plotted against each other. We were encouraged to find that nine out of the ten tRNATyr mutants that exhibit the highest enrichment in the presence of EcTyrRS (Figure 5d) did not show strong enrichment when the non-cognate MbPylRS was used instead (Figure 5e, Supplementary Table 5). These nine promising tRNATyr sequences were synthesized and tested by co-transfecting them into HEK293T cells with OMeY-selective EcTyrRS and EGFP-39TAG in the presence and absence of 1 mM OMeY (Figure 5f). Many of these mutants demonstrated significantly higher activity relative to wild-type-tRNATyr, with the most active one showing an approximately three-fold improvement. Further evolution of this tRNA, as well as detailed characterization of the improved mutant isolated here are currently in progress. Success of this alternative VADER scheme, lacking the bioorthogonal capture step, further highlights the versatility of this platform for directed evolution of tRNAs and beyond.
Discussion
In summary, we have developed a general strategy to systematically address the poor performance of suppressor tRNAs in mammalian cells using a novel virus-assisted directed evolution platform. Coupling VADER with NGS facilitated a global analysis of how each member of our synthetic tRNA mutant libraries perform in mammalian cells, revealing sequence traits that positively or negatively affect their performance. Application of this technology to synthetic libraries of tRNAPyl revealed a high degree of evolvability of its A-stem, but not the T-stem, and led to the identification of many A-stem mutants with improved activities. The most active mutant (A2.1) was found to express at a lower level relative to tRNAPyl-WT, pointing at its higher intrinsic activity. We also established an assay to compare the activities of different tRNA mutants across a range of different expression levels by controlled delivery using a baculovirus vector, which revealed a significantly higher benefit of A2.1 at lower expression levels. We also showed that the enhanced tRNAPyl activity achieved through directed evolution is host-specific, suggesting that a better interaction with the non-native host translational apparatus likely underlies their improved performance. Finally, we developed improved mutants of E. coli tRNACUATyr, by subjecting an acceptor-stem library to a modified VADER scheme lacking the bioorthogonal capture step. These results suggest that it should be possible to extend the application of VADER to other suppressor tRNAs commonly used for genetic code expansion in mammalian cells. Additionally, it should be possible to adapt this platform for the directed evolution of other biological machinery for Uaa mutagenesis and beyond.
Methods
Cell culture.
HEK293T cells (ATCC, catalog number CRL-3216) were maintained at 37 °C and 5% CO2 in DMEM-high glucose (HyClone) supplemented with penicillin/streptomycin (HyClone, final concentration of 100 U/mL penicillin and 100 μg/mL streptomycin) and 10% fetal bovine serum (Corning). All references to DMEM below refer to the complete medium described here.
Statistical methods.
For assessing EGFP reporter expression in HEK293T cells, the mean of at least three independent experiments was reported, and error bars represent s.d. For evaluating the enrichment factors at each selection step, three independent experiments were performed, and for each sample three independent measurements (FACS) were made and the average of these values were reported (error represents s.d.).
General cloning
For all cloning, the E. coli TOP10 strain was used for transformation and plasmid propagation and bacteria were grown using LB for solid and liquid culture. All PCR reactions were carried out using Phusion Hot Start II DNA Polymerase (Thermo Scientific) according to the manufacturer’s protocol. Restriction enzymes and T4 DNA ligase were from New England Biolabs (NEB). All DNA oligos were purchased from Integrated DNA Technologies (IDT). Sanger sequencing was performed by Eton Bioscience.
Unnatural amino acids
Azido-lysine (AzK) was purchased from Iris Biotech GMBH (Germany).
Nε-acetyllysine (AcK) was purchased from Bachem.
Diazirine-lysine (DiazK) was purchased from Sirius Fine Chemicals (Germany).
Strained cyclooctyne-L-lysine (SCOK) was purchased from Sirius Fine Chemicals.
O-Methyl-L-tyrosine (OMeY) was purchased from Thermo Scientific.
Plasmids
The AAV2 production plasmids pHelper and pAAV-RC2 were purchased from Cell Biolabs. pIDTSmart-RC2(T454TAG)-MbPylRS has been previously described.26 pIDTSmart-PytR plasmids with anticodons for TAG, TGA, and TAA have been previously described.30 pAcBac1-EGFP has been previously described.7,50
pAAV-ITR-PytR-GFP was generated by amplifying the U6-tRNA cassette from pIDTSmart-PytR7 with primers tRNA-AAV-F and R and inserting into the MluI site of the AAV cargo plasmid pAAV-GFP (purchased from Cell Biolabs). A KpnI site was added before the U6 promoter using primer tRNA-Amp-F. pAAV-ITR-EcYtR-GFP was constructed in an analogous manner by amplifying the U6-tRNA cassette from pIDTSmart-YtR; pAAV-ITR-PytR-mCherry was generated by replacing the GFP reporter in pAAV-ITR-PytR-GFP; and pAAV-ITR-EcYtR-mCherry was generated by replacing the GFP reporter in pAAV-ITR-EcYtR-GFP.
The pAAV-ITR-GFP library cloning vector was generated by cutting pAAV-ITR-PytR-GFP at two NdeI sites flanking the tRNA and ligating the resulting vector back together. This vector retains the library cloning sites but lacks a tRNA which could cause background issues during library cloning.
pIDTSmart-1xPytR-evolved was generated by amplifying the best hit tRNA (Ac2.1 GGG/CCU) from selection using primers tRNA-Amp-F and R and cloning this insert into the pIDTSmart-PytR backbone using AvrII and NheI. Anticodons were mutated using site-directed mutagenesis.
pIDTSmart-MbPylRS, pIDTSmart-AcKRS3, pIDTSmart-HpRS, pIDTSmart-AbKRS, and pIDTSmart-OMeYRS were generated by cloning each synthetase into the pIDTSmart backbone at the AvrII and NheI sites.
All primer sequences can be found in Supplementary Table 3.
Library generation
Pyrrolysyl tRNA libraries were generated by site-saturation mutagenesis using pAAV-PytR-GFP as a template. For acceptor stem libraries, the 5′ and 3′ pieces of the tRNA were first amplified using primer pairs tRNA-Amp-F + AccLib-Short-R and AccLib-Short-F + tRNA-Amp-R respectively. Randomized bases were then added by reamplifying each fragment, replacing AccLib-Short-F and R with AccLib1 or AccLib2-NNN-F and R. Finally, the fragments were joined by overlap extension PCR and then amplified using tRNA-Amp-F and R, digested with KpnI and NcoI, and ligated into the pAAV-ITR-GFP library vector which had been digested with the same enzymes. Each ligation used ~1 μg each of vector and insert. T stem library generation was similar, but for each library, only one primer containing randomized nucleotides was used. All primer sequences can be found in Table S5. The Tyrosyl tRNA library was generated using the same method with pAAV-EcYtR-GFP as a template and YtR-AccLib-Short-F, YtR-AccLib-Short-R, YtR-AccLib1-F, and YtR-AccLib1-R in the stead of the corresponding tRNAPyl primers.
Ligations were concentrated by ethanol precipitation with yeast tRNA (Ambion) and transformed into electrocompetent TOP10 E. coli. >4×105 transformants were plated (>100-fold library coverage). These colonies were pooled and their DNA was miniprepped for packaging into AAV.
Packaging and titration of mock and library tRNAs into AAV (wild-type capsid)
To package various cargo into AAV2, 8 million HEK293T cells were seeded in a 10 cm tissue culture dish. The following day, the cells were transfected with 8 μg each of the appropriate cargo plasmid (pAAV-ITR-tRNA-fluorescent protein), pHelper, and pAAV-RC2 using polyethylenimine (PEI) (Sigma). Media was exchanged for fresh DMEM 24 hours after transfection. 72 hours after transfection, the cells were resuspended, pelleted, and lysed by freeze/thawing as previously described.26,28 Virus was concentrated and semi-purified by PEG precipitation,26,28 resuspended in 1 mL DMEM with FBS, and flash frozen.
FACS analysis to assess infective titer of AAV2 samples
Infective titers for virus preparations were determined using flow cytometry. 0.7 million HEK293T cells were seeded in each well of a 12-well plate. Next day, confluent cells (~1 million cells per well) were infected with a dilution of the AAV2 to be titered (to achieve low MOI conditions). 5 mM sodium butyrate (Sigma) was added to boost infectivity and transgene expression. Two days later, the cells were trypsinized, washed with PBS, and analyzed by flow cytometry to count the fluorescent population.
Transfection to determine overexpression of AAV genes with Rep and AdHelper
0.7 million HEK293T cells per well were seeded in a 12-well plate and infected the next day with AAV2 carrying a tRNAPyl-mCherry cargo at an MOI of 1. Cells were transfected four hours after infection with 0.6 μg pAAV-RC2 and 1.4 μg pHelper. PEI-only negative control wells received the equivalent amount of PEI to the transfected wells, but no plasmids. Virus-only wells were not transfected at all. Three days after infection and transfection, cells were lysed with CelLytic M buffer (Sigma) and mCherry fluorescence was measured on a Molecular Devices SpectraMax M5 microplate reader. The background from an uninfected well was subtracted.
Positive selection
8 million HEK293T cells each were seeded in three 10 cm tissue culture dishes. The next day, the cells were infected with virus containing a tRNA library at an apparent MOI of 5 (the actual MOI is substantially reduced in the presence of PEI, the transfection reagent). Four hours after infection, the cells were transfected with 22 μg of pHelper and 10 μg of pIDTSmart-RC2(T454TAG)-RS per dish using PEI. 1 mM Uaa was also added at this point. For the tRNAPyl library, pIDTSmart-RC2(T454TAG)-PylRS was added in the presence of AzK. For the tRNATyr library, pIDTSmart-RC2(T454TAG)-OMeYRS was added in the presence of OMeY for the +OMeYRS selections and pIDTSmart-RC2(T454TAG)-PylRS was added in the presence of OMeY for the –OMeYRS selections. One day after transfection, the culture media was exchanged with fresh DMEM containing 1 mM Uaa. Cells were harvested three days after transfection and lysed as for virus isolation. The culture media was saved and recombined with clarified lysate, and this mixture was treated with 500 U universal nuclease (Thermo Scientific) for 30 minutes. Virus was recovered by PEG precipitation using 11% polyethylene glycol (Fisher) as previously described26,28 and resuspended in 3 mL PBS.
The small-scale mock positive selections were carried out in 12-well plates. 0.7 million cells per well were seeded and infected the next day with AAV carrying a tRNAPyl-mCherry cargo. Four hours later the cells were transfected as described for the above selections, but with the transfection mix and AzK scaled down by a factor of 15. For PEI only wells, cells received a comparable amount of transfection reagent but no plasmid. Media was changed the day after transfection for fresh DMEM containing 1 mM AzK. Virus was harvested three days post-transfection and PEG-precipitated as described for the selections above. Confluent cells in a 12-well plate were infected with the entire output of one mock selection well and analyzed by flow cytometry.
Negative selection – streptavidin pulldown
The virus from positive selection (3 mL) was labeled with photocleavable DBCO-sulfo-biotin (Jena Biosciences) at a concentration of 5 μM for one hour in the dark with mixing. Immediately after labeling, excess DBCO-biotin was quenched with AzK (1 mM final concentration) and the reactions were dialyzed overnight using Slide-A-Lyzer 100 kDa MWCO devices (Thermo Scientific) against 1 L PBS at 4 °C. The dialyzed virus mixtures were split into three 2 mL tubes and each rotated overnight with 400 μL streptavidin agarose resin (Thermo Scientific) at 4 °C. The next day, each tube of beads was washed eight times with 1 mL PBS containing additional NaCl (final concentration 300 mM) with mixing between washes. Finally, the washed beads were resuspended in 8 mL PBS (300 mM NaCl) and the virus was eluted from the resin via four 30-second irradiations using a 365 nm UV diode array (Larson Electronics), with mixing between irradiations.
Viral DNA recovery, amplification, and cloning
The eluted virus was concentrated from 3 mL to 300 μL using Amicon Ultra-4 100 kDa MWCO centrifugal concentrators (Millipore). This mixture was heated to 100 °C for 10 minutes in order to denature the viral capsid proteins and expose the DNA. Viral DNA was then cleaned up and concentrated by ethanol precipitation using yeast tRNA (Ambion) and resuspended in a final volume of 50 μL.
20 μL of this mixture was added to a 200 μL PCR reaction and amplified with tRNA-Amp-F and R primers. The resulting DNA was digested with KpnI and NcoI and cloned into the library cloning vector using the same protocol as for original library generation.
Mock selections using Pyl-mCherry and Tyr-GFP
The mock selections shown in Figure 1 followed the same protocol as above, except that the starting library virus was a 1:10,000 mixture of virus made from pAAV-ITR-PytR-mCherry to virus made from pAAV-ITR-EcYtR-GFP. Mock selection results were analyzed by flow cytometry as described for virus titering, but here, cells in a 12-well plate were infected with 200 μL of the virus pool after either positive or negative selection. Red and green fluorescent cells were counted to determine the virus ratio.
Hit sequencing and characterization
For each tRNAPyl library, 30–50 colonies were picked from the transformation plates generated above and sent for Sanger sequencing (Eton Bioscience). All sequences in which all randomized bases were paired were treated as potential hits, and these tRNAs were subcloned into pAAV-ITR-PytR-mCherry for further analysis.
Initial hit analysis was conducted by transfecting HEK293T cells in 24-well plates with 0.5 μg each of a potential hit pAAV-ITR-PytR-mCherry plasmid, pIDTSmart-MbPylRS, and pAcBac1-GFP(39TAG) in the presence and absence of 1 mM AzK. Two days after transfection, cells were lysed with CelLytic M buffer (Sigma) and EGFP and mCherry fluorescence were measured on a Molecular Devices SpectraMax M5 microplate reader. Values for an untransfected well were subtracted, and EGFP fluorescence was normalized to mCherry fluorescence for each well.
The best hit, A2.1 (GGG/CCU), was selected for further analysis with other stop codons and synthetases. HEK293T cells in a 12-well plate were transfected with 0.375 μg pIDTSmart-PytR containing either the wild-type or evolved tRNA, 0.375 μg pIDTSmart-aaRS containing the appropriate synthetase, and 0.75 μg pAcBac1-EGFP containing one or two of the appropriate stop codon. A wild-type EGFP control well used pIDTSmart-PytR(TAG, wild-type), pIDTSmart-MbPylRS, and pAcBac1-EGFP(wild-type) in the same ratios. Two days after transfection, cells were lysed and EGFP fluorescence was measured by microplate reader. Values from an untransfected well were subtracted.
Mass Spectrometry
Incorporation of the correct Uaas by the evolved tRNA was confirmed by LC-ESI-MS. EGFP-39AzK-6xHis was generated by transfecting HEK293T cells in two 10 cm tissue culture dishes with 5 μg pIDTSmart-PytR-evolved, 5 μg pIDTSmart-MbPylRS, and 10 μg pAcBac1-GFP(Y39TAG) with 1 mM AzK. EGFP-39AcK-6xHis was generated by transfecting HEK293T cells in two 10 cm tissue culture dishes with 5 μg pIDTSmart-PytR-evolved, 5 μg pIDTSmart-AcKRS3, and 10 μg pAcBac1-GFP(Y39TAG) with 5 mM AcK. Cells were lysed two days after transfection using CelLytic M (Sigma), Halt protease inhibitor cocktail (Thermo Scientific), and universal nuclease (Thermo Scientific) according to the manufacturers’ instructions. All proteins were isolated from clarified lysate on Ni-NTA columns using HisPur resin (Fisher) according to the manufacturer’s instructions, but using 60 μL of resin and wash buffers containing 30 mM and then 40 mM imidazole. Proteins were analyzed by LC-ESI-MS using an Agilent 1260 Infinity ESI-TOF.
Illumina sample preparation and sequencing
To enable sequencing by the Illumina MiSeq System, DNA samples to be sequenced had Illumina adapter sequences attached via two rounds of PCR. The two consecutive reactions result in construction of the Illumina adapters provided by the TruSeq DNA HT Sample Prep Kit (Illumina). The forward adapter is AATGATACGGCGACCACCGAGATCTACAC[i5]ACACTCTTTCCCTACACGACGCTCTTCCGATCT, wherein [i5] is an eight-nucleotide barcode sequence, and the reverse adapter is GATCGGAAGAGCACACGTCTGAACTCCAGTCAC[i7]ATCTCGTATGCCGTCTTCTGCTTG, wherein [i7] is an eight-nucleotide barcode sequence. Both i5 and i7 barcode sequences are given in Supplementary Table 4. The first set of primers, Illumina-PytR-F and Illumina-PytR-R, consist of half of the TruSeq adapters, beginning immediately after the i5 or i7 barcode, followed by primer-binding sites to anneal to the sequences surrounding the tRNA library (for the forward primer: TTATATATCTTGTGGAAAGGACGAAAC; for the reverse primer: GCTAGCGGATCGACGAGAGC). The second set of primers, a series of Illumina-i5-F and Illumina-i7-R variants containing different barcodes, consists of the 5’ half of the TruSeq adapters, followed by an i5 or i7 barcode, followed by primer-binding sites to anneal to the first PCR (for the forward primer: ACACTCTTTCCCTACACGACGC; for the reverse primer: GTGACTGGAGTTCAGACGTGTGCTC).
For the first PCR, samples were prepared using the primers Illumina-PytR-F and Illumina-PytR-R and PrimeSTAR Max DNA Polymerase (Takara Bio) per manufacturer’s instructions. PCR samples were purified by agarose gel extraction. A second round of PCR using Illumina-i5-F and Illumina-i7-R primer variants and PrimeSTAR Max DNA Polymerase was performed to attach the region of the adapter sequences that includes the barcodes. Unique combinations of i5 and i7 barcode sequences were applied to each sample to enable multiplex sequencing. Although both forward and reverse reads were possible, only forward reads were used in data analysis for this paper.
Samples were prepared for sequencing using the 300-cycle MiSeq Reagent Kit v2 (Illumina) per manufacturer’s instructions. Sequencing was executed on an Illumina MiSeq System, with 10% Illumina PhiX Control added.
Illumina high-throughput-sequencing data processing
Processing of sequencing data generated by the MiSeq System was performed using bespoke Python scripts. Scripts are available via GitHub.49
Reads were first filtered by data quality. This “Q-score filter” checked the Phred base call quality scores associated with each read in the FASTQ output files generated by the MiSeq System. Reads below a specified threshold were discarded. For this study, reads containing any bases with Q-scores lower than 14, corresponding to an error rate of 4%, were discarded. Reads were then filtered by alignment with the expected tRNA sequence. This “mismatch filter” compared each read to the expected tRNA sequence within the fixed regions of the tRNA, skipping over the randomized regions within the tRNA associated with each library. Finally, a minimum abundant library count could be specified to reduce very rare sequences that may be the result of an error. However, for these samples, the minimum abundant library count was set to 1; no library members were discarded for being too low in abundance.
For sequences that pass both the Q-score filter and the mismatch filter, the sequence regions randomized in each library were extracted and collected in a comma-separated file. For each sequence in a given sample, the “fraction of total” value was calculated by dividing the counts of a given sequence by the total counts of all sequences in that sample. The number of base pairs found within the randomized library region of each sequence was also calculated, because successful base-pairing with stem regions is important for tRNA activity.
For each library, three samples were sequenced: the input library and the output from two replicates of the selection. The counts detected for each individual sequence in each sample were tallied. For each selection, the “fold enrichment” of a given library member was determined by calculating the ratio of counts in the selection output to counts in the selection input. The “enrichment factor” of a given library member is the fold enrichment normalized to the most enriched hit, and was determined by dividing the fold enrichment of a given hit by the fold enrichment of the most enriched hit in that sample. Finally, for each library member, the average of the two enrichment factors obtained was determined, and library members were sorted by average enrichment factor.
RNA isolation
To generate RNA for Northern blots, 7.5 million HEK293T cells were seeded in a 10 cm tissue culture dish. The following day, cells were transfected at 75% confluency with 24 μg of tRNA variant-containing pAAV-ITR-PytR-mCherry plasmid using polyethylenimine (PEI) (Sigma). Media was exchanged for fresh DMEM 24 h after transfection, and cells were harvested 48 h post-transfection. The mCherry reporter expression was used to confirm comparable transfection efficiency between experiments. RNA isolation was performed with TRIzol Reagent (Thermo Fisher) following manufacturer’s instructions. RNA concentration was determined via Nanodrop Spectrophotometer and integrity of the RNA was assessed by A260/A280 value, as well as the presence of distinct, intact 28S and 18S ribosomal RNA bands on 1% agarose gel.
Northern blot probe preparation
Oligonucleotides were 3’-end labeled with DIG using the DIG Oligonucleotide 3′-End Labeling Kit, 2nd Generation (Roche), following manufacturer’s instructions. Final concentration of labelled probe was determined per manufacturer’s instructions. The oligonucleotide used was PyltR-Tstem2-NB-R, which binds to the T-stem of the tRNA in order to be compatible with all A-stem variations. For detection of the 5.8S RNA positive control to assess overall RNA concentration, the oligonucleotide 5.8S-NB-R was used.
Northern blotting
A sensitive non-isotopic northern blot was performed using a previously established method of digoxigenin (DIG)-labeled oligonucleotide probes and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide for RNA-membrane cross-linking, with modification.51 Denaturing gels were 8 M Urea, 6.5% acrylamide in Tris/Borate/EDTA buffer (TBE), and made in mini-gel format (10.1 × 7.3 cm2 with 1.5 mm spacers). Gel wells were thoroughly rinsed with TBE after gel was set and again after pre-running gel at 250 V for 60 minutes in TBE running buffer. 5 μg of total RNA was combined with 2.5 μL Invitrogen™ Gel Loading Buffer II 2x (Denaturing PAGE, Fisher) to prepare a final sample volume of 5 μL per lane. Both RNA samples and RNA ladder (Low Range ssRNA Ladder, New England Biolabs) were denatured at 95 °C for 5 min, chilled on ice for 2 min, and then loaded onto gel. Gel was run at 4 °C for 70 minutes and then soaked in 0.05% ethidium bromide solution in RNAse free water while shaking for 10 minutes. The gel was imaged with the ChemiDoc-IT Imaging System and then soaked for 10 minutes in TBE running buffer.
A cassette sandwich was made of three sheets of 3MM Whatman paper soaked in TBE, nylon membrane, gel, and an additional three sheets of Whatman paper. Transfer of RNA from gel to membrane was done at 10 V for 60 minutes at 4 °C with the Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad).
EDC cross-linking solution was prepared as previously described.51 Whatman paper was saturated in EDC cross-linking solution and placed on top of Saran wrap. Membrane was placed on top of Whatman paper and allowed to incubate at 60 °C for 1 hour to facilitate RNA–membrane cross-linking. Residual cross-linking solution was then removed by thoroughly rinsing the membrane with distilled water.
DIG blocking, DIG washing, DIG detection buffer were prepared per manufacturer’s instructions from DIG Wash and Block Buffer Set (Millipore Sigma). CSPD detection buffer was made with 50 μL of CSPD™ Substrate (0.25 mM Ready-To-Use, ThermoFisher) and 5 mL of detection buffer. DIG antibody solution was prepared by mixing DIG antibody (Anti-Digoxigenin-AP, Fab fragments) and blocking buffer at a ratio of 1:15,000. After cross-linking, membrane was incubated in 50 mL Falcon tube at 42 °C for 30 minutes in hybridization oven with 5 mL of Invitrogen™ ULTRAhyb™ Ultrasensitive Hybridization Buffer (Fisher), preheated to 68 °C. The DIG-labelled probes PyltR-Tstem2-NB-R-DIG and 5.8S-NB-R-DIG were diluted to 25 nM and 2.5 nM, respectively, and denatured at 95 °C for 5 minutes. 5 μL of each probe was added to liquid in pre-hybridized Falcon tube and hybridized overnight at 42 °C in hybridization oven at slow rotation speed. The following day, membrane was washed through incubation twice with 5 mL Low Stringent Buffer (2× SSC with 0.1% SDS) at 42 °C for 5 minutes, twice with 5 mL High Stringent Buffer (0.1× SSC with 0.1% SDS) at 42 °C for 15 minutes, and then once with 10 mL Washing Buffer (1× SSC) at 42 °C for 10 minutes in hybridization oven. Membrane was incubated in 10 mL DIG blocking buffer at room temperature for 3 hours in hybridization oven. Membrane was then incubated with 10 mL of DIG antibody solution at room temperature for 30 minutes in hybridization oven. Membrane was then washed four times with 10 mL of DIG washing buffer for 15 minutes in hybridization oven and incubated with 5 mL of DIG detection buffer for 5 minutes at room temperature in hybridization oven. Membrane was then removed from tube with clean forceps, placed on plastic wrap, and incubated for 5 minutes with 5 mL of CSPD detection buffer. Membrane was then placed in a heat-sealable plastic bag, sealed, and incubated at 37 °C for 15 minutes in the dark. The membrane was then imaged for chemiluminescence for 10 minutes with the ChemiDoc-IT Imaging System.
Production of baculovirus vectors and their use for testing tRNAPyl activity with copy number control
VSVG-pseudotyped baculovirus vectors were generated and titered as previously described.7,50
Supplementary Material
Acknowledgements
We thank the National Science Foundation (MCB-1817893), and National Institutes of Health (R35GM136437 to A.C. and U01 AI124302 to T.v.O.) for financial support.
Footnotes
Competing interests statement
A patent application has been submitted on the improved tRNA mutants on which A.C, D.J., and R.E.K. are coinventors. A.C. is a senior advisor at BrickBio, Inc. R.L.H., Z.Z., B.S., X.C., K.M., Z.H., M.P., J.A., and T.v.O. have no competing interests.
Supplementary information is available for this paper.
References
- 1.Chin JW Expanding and reprogramming the genetic code. Nature 550, 53–60, doi: 10.1038/nature24031 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Italia JS et al. Expanding the genetic code of mammalian cells. Biochemical Society transactions 45, 555–562, doi: 10.1042/bst20160336 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Young DD & Schultz PG Playing with the Molecules of Life. ACS Chem Biol 13, 854–870, doi: 10.1021/acschembio.7b00974 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manandhar M, Chun E & Romesberg FE Genetic Code Expansion: Inception, Development, Commercialization. J Am Chem Soc 143, 4859–4878, doi: 10.1021/jacs.0c11938 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Roy G et al. Development of a high yielding expression platform for the introduction of non-natural amino acids in protein sequences. mAbs 12, 1684749, doi: 10.1080/19420862.2019.1684749 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmied WH, sser SJ, Uttamapinant C & Chin JW Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. Journal of the American Chemical Society 136, 15577–15583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Y, Lewis TL Jr., Igo P, Polleux F & Chatterjee A Virus-Enabled Optimization and Delivery of the Genetic Machinery for Efficient Unnatural Amino Acid Mutagenesis in Mammalian Cells and Tissues. ACS synthetic biology, doi: 10.1021/acssynbio.6b00092 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Söll D & RajBhandary UL tRNA: Structure, Biosynthesis, and Function. (ASM Press, 1995). [Google Scholar]
- 9.Serfling R et al. Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells. Nucleic acids research 46, 1–10, doi: 10.1093/nar/gkx1156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson JC & Schultz PG Adaptation of an orthogonal archaeal leucyl-tRNA and synthetase pair for four-base, amber, and opal suppression. Biochemistry 42, 9598–9608, doi: 10.1021/bi034550w (2003). [DOI] [PubMed] [Google Scholar]
- 11.Guo J, Melancon CE 3rd, Lee HS, Groff D & Schultz PG Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angewandte Chemie (International ed. in English) 48, 9148–9151, doi: 10.1002/anie.200904035 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogerson DT et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nature chemical biology 11, 496–503, doi: 10.1038/nchembio.1823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K et al. Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nature chemistry 6, 393–403, doi: 10.1038/nchem.1919 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ellefson JW et al. Directed evolution of genetic parts and circuits by compartmentalized partnered replication. Nature Biotechnology 32, 97 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Maranhao AC & Ellington AD Evolving Orthogonal Suppressor tRNAs To Incorporate Modified Amino Acids. ACS synthetic biology 6, 108–119, doi: 10.1021/acssynbio.6b00145 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee A, Xiao H & Schultz PG Evolution of multiple, mutually orthogonal prolyl-tRNA synthetase/tRNA pairs for unnatural amino acid mutagenesis in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 109, 14841–14846, doi: 10.1073/pnas.1212454109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterjee A, Xiao H, Yang PY, Soundararajan G & Schultz PG A tryptophanyl-tRNA synthetase/tRNA pair for unnatural amino acid mutagenesis in E. coli. Angewandte Chemie (International ed. in English) 52, 5106–5109, doi: 10.1002/anie.201301094 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Wang N, Shang X, Cerny R, Niu W & Guo J Systematic Evolution and Study of UAGN Decoding tRNAs in a Genomically Recoded Bacteria. Scientific reports 6, 21898, doi: 10.1038/srep21898 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson JC et al. An expanded genetic code with a functional quadruplet codon. Proceedings of the National Academy of Sciences of the United States of America 101, 7566–7571, doi: 10.1073/pnas.0401517101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendel SJ & Shoulders MD Directed evolution in mammalian cells. Nat Methods 18, 346–357, doi: 10.1038/s41592-021-01090-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nature methods 13, 1036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Jackson WC, Steinbach PA & Tsien RY Evolution of new nonantibody proteins via iterative somatic hypermutation. Proceedings of the National Academy of Sciences of the United States of America 101, 16745–16749, doi: 10.1073/pnas.0407752101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berman CM et al. An Adaptable Platform for Directed Evolution in Human Cells. J Am Chem Soc 140, 18093–18103, doi: 10.1021/jacs.8b10937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.English JG et al. VEGAS as a Platform for Facile Directed Evolution in Mammalian Cells. Cell 178, 748–761.e717, doi: 10.1016/j.cell.2019.05.051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erickson SB et al. Precise Photoremovable Perturbation of a Virus-Host Interaction. Angew Chem Int Ed Engl 56, 4234–4237, doi: 10.1002/anie.201700683 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kelemen RE et al. A Precise Chemical Strategy To Alter the Receptor Specificity of the Adeno-Associated Virus. Angewandte Chemie (International ed. in English) 55, 10645–10649, doi: 10.1002/anie.201604067 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Agbandje-McKenna M & Kleinschmidt J AAV capsid structure and cell interactions. Methods in molecular biology (Clifton, N.J.) 807, 47–92, doi: 10.1007/978-1-61779-370-7_3 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Kelemen RE, Erickson SB & Chatterjee A Production and Chemoselective Modification of Adeno-Associated Virus Site-Specifically Incorporating an Unnatural Amino Acid Residue into Its Capsid. Methods in molecular biology (Clifton, N.J.) 1728, 313–326, doi: 10.1007/978-1-4939-7574-7_20 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Italia JS, Latour C, Wrobel CJJ & Chatterjee A Resurrecting the Bacterial Tyrosyl-tRNA Synthetase/tRNA Pair for Expanding the Genetic Code of Both E. coli and Eukaryotes. Cell chemical biology 25, 1304–1312.e1305, doi: 10.1016/j.chembiol.2018.07.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng Y, Addy PS, Mukherjee R & Chatterjee A Defining the current scope and limitations of dual noncanonical amino acid mutagenesis in mammalian cells. Chem Sci 8, 7211–7217, doi: 10.1039/c7sc02560b (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wan W, Tharp JM & Liu WR Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochimica et biophysica acta 1844, 1059–1070, doi: 10.1016/j.bbapap.2014.03.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao S et al. Decoding Mammalian Ribosome-mRNA States by Translational GTPase Complexes. Cell 167, 1229–1240.e1215, doi: 10.1016/j.cell.2016.10.046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nozawa K et al. Pyrrolysyl-tRNA synthetase-tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature 457, 1163–1167, doi: 10.1038/nature07611 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neumann H, Peak-Chew SY & Chin JW Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nature chemical biology 4, 232–234, doi: 10.1038/nchembio.73 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Hohl A et al. Engineering a Polyspecific Pyrrolysyl-tRNA Synthetase by a High Throughput FACS Screen. Scientific reports 9, 11971, doi: 10.1038/s41598-019-48357-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ai HW, Shen W, Sagi A, Chen PR & Schultz PG Probing protein-protein interactions with a genetically encoded photo-crosslinking amino acid. Chembiochem : a European journal of chemical biology 12, 1854–1857, doi: 10.1002/cbic.201100194 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Zheng Y et al. Expanding the Scope of Single- and Double-Noncanonical Amino Acid Mutagenesis in Mammalian Cells Using Orthogonal Polyspecific Leucyl-tRNA Synthetases. Biochemistry 57, 441–445, doi: 10.1021/acs.biochem.7b00952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ernst RJ et al. Genetic code expansion in the mouse brain. Nature chemical biology 12, 776–778, doi: 10.1038/nchembio.2160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan C, Xiong H, Reynolds NM & Söll D Rationally evolving tRNAPyl for efficient incorporation of noncanonical amino acids. Nucleic acids research 43, e156, doi: 10.1093/nar/gkv800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng Y, Gilgenast MJ, Hauc S & Chatterjee A Capturing Post-Translational Modification-Triggered Protein-Protein Interactions Using Dual Noncanonical Amino Acid Mutagenesis. ACS Chem Biol 13, 1137–1141, doi: 10.1021/acschembio.8b00021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grasso KT et al. A Facile Platform to Engineer Escherichia coli Tyrosyl-tRNA Synthetase Adds New Chemistries to the Eukaryotic Genetic Code, Including a Phosphotyrosine Mimic. ACS central science 8, 483–492, doi: 10.1021/acscentsci.1c01465 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Italia JS et al. An orthogonalized platform for genetic code expansion in both bacteria and eukaryotes. Nature chemical biology 13, 446–450, doi: 10.1038/nchembio.2312 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Sakamoto K et al. Site-specific incorporation of an unnatural amino acid into proteins in mammalian cells. Nucleic acids research 30, 4692–4699, doi: 10.1093/nar/gkf589 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W, Brock A, Chen S, Chen S & Schultz PG Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat Methods 4, 239–244, doi: 10.1038/nmeth1016 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Chin JW et al. An expanded eukaryotic genetic code. Science (New York, N.Y.) 301, 964–967, doi: 10.1126/science.1084772 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Xiao H et al. Genetic incorporation of multiple unnatural amino acids into proteins in mammalian cells. Angewandte Chemie (International ed. in English) 52, 14080–14083, doi: 10.1002/anie.201308137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian F et al. A general approach to site-specific antibody drug conjugates. Proceedings of the National Academy of Sciences of the United States of America 111, 1766–1771, doi: 10.1073/pnas.1321237111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jühling F et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic acids research 37, D159–162, doi: 10.1093/nar/gkn772 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelemen R (2018) VADER processing pipeline (Version 1.0) [github.com/chatterjeelab2022/VADER]
- 50.Chatterjee A, Xiao H, Bollong M, Ai HW & Schultz PG Efficient viral delivery system for unnatural amino acid mutagenesis in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 110, 11803–11808, doi: 10.1073/pnas.1309584110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SW et al. A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic acids research 38, e98, doi: 10.1093/nar/gkp1235 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.