

Key Points
Question
What is the genetic basis and phenotypic expression of autosomal dominant polycystic kidney disease (ADPKD) within a single regional health system in the US?
Findings
In this retrospective observational study that included 174 172 unselected participants, 109 of 111 patients (98%) with large deletions or truncating PKD1 and PKD2 variants had ADPKD, whereas only 24 of 77 patients (31.2%) with previously described “likely pathogenic” missense PKD1 variants had ADPKD. Exome sequencing demonstrated that 19 of 235 patients (8%) clinically diagnosed with ADPKD had variants in genes other than PKD1 and PKD2.
Meaning
This study demonstrates substantial genetic and phenotypic variability in ADPKD among patients within a regional health system in the US.
Abstract
Importance
Most studies of autosomal dominant polycystic kidney disease (ADPKD) genetics have used kidney specialty cohorts, focusing on PKD1 and PKD2. These can lead to biased estimates of population prevalence of ADPKD-associated gene variants and their phenotypic expression.
Objective
To determine the prevalence of ADPKD and contributions of PKD1, PKD2, and other genes related to cystic kidney disease in a large, unselected cohort.
Design, Setting, and Participants
This retrospective observational study used an unselected health system–based cohort in central and northeast Pennsylvania with exome sequencing (enrolled from 2004 to 2020) and electronic health record data (up to October 2021). The genotype-first approach included the entire cohort and the phenotype-first approach focused on patients with ADPKD diagnosis codes, confirmed by chart and imaging review.
Exposures
Loss-of-function (LOF) variants in PKD1, PKD2, and other genes associated with cystic kidney disease (ie, ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, SEC61B, PKHD1, PRKCSH, SEC63); likely pathogenic missense variants in PKD1 and PKD2.
Main Outcomes and Measures
Genotype-first analysis: ADPKD diagnosis code (Q61.2, Q61.3, 753.13, 753.12); phenotype-first analysis: presence of a rare variant in PKD1, PKD2, or other genes associated with cystic kidney disease.
Results
Of 174 172 patients (median age, 60 years; 60.6% female; 93% of European ancestry), 303 patients had ADPKD diagnosis codes, including 235 with sufficient chart review data for confirmation. In addition to PKD1 and PKD2, LOF variants in IFT140, GANAB, and HNF1B were associated with ADPKD diagnosis after correction for multiple comparisons. Among patients with LOF variants in PKD1, 66 of 68 (97%) had ADPKD; 43 of 43 patients (100%) with LOF variants in PKD2 had ADPKD. In contrast, only 24 of 77 patients (31.2%) with a PKD1 missense variant previously classified as “likely pathogenic” had ADPKD, suggesting misclassification or variable penetrance. Among patients with ADPKD diagnosis confirmed by chart review, 180 of 235 (76.6%) had a potential genetic cause, with the majority being rare variants in PKD1 (127 patients) or PKD2 (34 patients); 19 of 235 (8.1%) had variants in other genes associated with cystic kidney disease. Of these 235 patients with confirmed ADPKD, 150 (63.8%) had a family history of ADPKD. The yield for a genetic determinant of ADPKD was higher for those with a family history of ADPKD compared with those without family history (91.3% [137/150] vs 50.6% [43/85]; difference, 40.7% [95% CI, 29.2%-52.3%]; P < .001). Previously unreported PKD1, PKD2, and GANAB variants were identified with pedigree data suggesting pathogenicity, and several PKD1 missense variants previously reported as likely pathogenic appeared to be benign.
Conclusions and Relevance
This study demonstrates substantial genetic and phenotypic variability in ADPKD among patients within a regional health system in the US.
This retrospective study assesses the prevalence of autosomal dominant polycystic kidney disease and contributions of PKD1, PKD2, and other genes related to cystic kidney disease in a large, unselected cohort of patients.
Introduction
Studies of selected cohorts with a particular clinical condition are useful in identifying genes associated with that condition. When such associations are found, however, subsequent population-based studies are needed to more accurately determine the prevalence of pathogenic variants and their phenotypic consequences. A common observation is that the prevalence of putative pathogenic variants is higher, the disease penetrance is lower, and the range of phenotypes is broader than findings based on disease-specific cohorts.1,2,3,4
Autosomal dominant polycystic kidney disease (ADPKD) has a clear genetic basis, and several disease-associated genes have been identified. Protein-truncating variants that disrupt the protein-coding sequence in PKD1 or PKD2 account for a substantial portion of ADPKD cases and are reported to have 100% disease penetrance.5 Missense PKD1/PKD2 variants have also been associated with ADPKD, but with incomplete and variable penetrance.6 The Toronto Genetic Epidemiology of Polycystic Kidney Disease study reported that 55.5% of patients without family history of ADPKD did not have a detectable PKD1 or PKD2 variant.7 Some unexplained cases may be due to variants in other genes such as GANAB, DNAJB11, HNF1B, and ALG9.8,9,10
Large exome sequence databases linked to longitudinal clinical data from unselected clinical populations enable a genotype-first approach, whereby specific genotypes are associated with clinical phenotypes. This enables investigation of the prevalence, penetrance, and phenotypic expression of multiple genes simultaneously. In this study, both genotype-first and phenotype-first approaches were used to evaluate population prevalence of ADPKD, decipher monogenic causes of ADPKD in a large health system–based cohort, and test the hypothesis that loss-of-function (LOF) variants (ie, early terminations, frameshift due to indels, splice site variants, large chromosomal deletions) in genes previously linked to cystic kidney or liver disease (ie, PKD1, PKD2, ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, PKHD1, PRKCSH, SEC61B, SEC63) would be associated with ADPKD (eTable 1 in Supplement 1).
Methods
Study Overview
The study population included patients from the MyCode Community Health Initiative who had electronic health record and exome sequencing data available11 (eAppendix in Supplement 2). All patients in MyCode provided written informed consent. The Geisinger institutional review board approved this study.
This study includes genotype-first and phenotype-first approaches. In the genotype-first approach, the prevalence and penetrance of expected and previously described disease-causing variants in PKD1 and PKD2 were examined using International Classification of Diseases, Ninth Revision (ICD-9) and International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10) codes. Associations between LOF variants in 11 genes related to cystic kidney or liver disease (ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, PKD1, PKD2, PKHD1, PRKCSH, SEC63) with ADPKD were examined. LRP5 was also examined, as it had previously been implicated incorrectly as a polycystic liver disease gene.12 No patients had LOF variants in the gene SEC61B. In the phenotype-first approach, the yield of exome sequencing in identifying a genetic cause in patients with confirmed ADPKD was examined overall and in patients with and without family history and in patients with and without typical ADPKD imaging findings. Additional evidence in support or against pathogenicity was examined through review of family pedigrees, when available.
Exome Sequencing and Variant Curation
Exome sequencing was performed in collaboration with Regeneron Genetics Center. There was 100% coverage of all coding regions for the 12 genes assessed with average read depths of 35.8 to 68.6 depending on the gene (eTable 1 in Supplement 1). Variants were cross-referenced with ClinVar, the Genome Aggregation Database (gnomAD),13 the Mayo PKD database (which was established to characterize PKD1 and PKD2 variants),14 and VarSome, a human genomic variant search engine, which provides automated classification of pathogenicity using American College of Medical Genetics (ACMG) criteria.15 SpliceAI, a deep learning tool for splice variants, was also used to evaluate predicted pathogenicity.16 Family pedigrees were built based on genetically determined relatedness using PRIMUS.17 Confirmatory Sanger sequencing was performed for 4 patients who were identified as having PKD1 LOF variants based on exome sequencing but did not have ADPKD on imaging review.
Outcomes
For genotype-first analyses, the primary outcome was the presence of at least 1 ICD-9/10 diagnosis code for ADPKD. The secondary outcome was any kidney or liver cyst, which included ICD-9/10 codes for ADPKD as well as cystic kidney diseases (Q61.5, Q61.8, Q61.9, 753.10), congenital kidney cyst (Q61.00, Q61.01, Q61.02, 753.11, 753.19), or liver cystic disease (Q44.6, 573.8).
For phenotype-first analyses, the primary outcome was having a rare variant (allele frequency <0.0001) in 1 of 11 genes related to cystic kidney or liver disease (ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, PKD1, PKD2, PKHD1, PRKCSH, SEC63). Prevalence of ADPKD was examined using several methods: (1) ≥1 ADPKD ICD-9/10 diagnosis code; (2) ADPKD ICD-9/10 diagnosis code confirmed by chart review; (3) predicted pathogenic PKD1/PKD2 variants per VarSome ACMG classifier; (4) predicted pathogenic or likely pathogenic PKD1/PKD2 variants per VarSome ACMG classifier.
Other Covariates of Interest
ICD-9/10 diagnoses for cerebral aneurysm, cardiac valvular abnormalities, nephrolithiasis, and other comorbidities were extracted (eTable 2 in Supplement 2). As there are differences in ADPKD prevalence by genetic ancestry,18 ancestral classes were determined genetically as previously described (eAppendix in Supplement 2).17
Additional Phenotyping for Phenotype-First Analyses
To provide further insights on factors affecting the molecular diagnostic yield, focused chart review on patients with ADPKD diagnoses and related family members was performed by at least 1 nephrologist. Review of imaging was done by at least 1 radiologist, blinded to genotype. For questionable cases, additional review was done by a second radiologist and a second nephrologist; cases were discussed until consensus was achieved. Patients were considered to have confirmed ADPKD if radiologist-reviewed imaging was consistent with ADPKD or if clinical history was consistent with ADPKD (eg, reported end-stage kidney disease due to ADPKD; imaging report and family history consistent with Ravine-Pei criteria).19 Asymptomatic patients who lacked any kidney imaging before age 30 years were considered to have insufficient chart review data to determine whether they had ADPKD in confirmed ADPKD analyses. Imaging phenotypes (eFigures 1, 2, and 3 in Supplement 2) were classified as (1) typical: bilateral and diffuse distribution of cysts, with moderate or severe replacement of kidney tissue by cysts, whereby all cysts contribute similarly to total kidney volume; (2) mild: if there was mild bilateral/diffuse replacement of kidney tissue by cysts; or (3) atypical, per Mayo Clinic imaging classification.20
Statistical Analysis
Characteristics between participants with and without ADPKD were compared using unpaired t tests for continuous variables and the Fisher exact test for categorical variables (Table 1). For genotype-first analyses, Firth logistic regression was used to assess associations between carriers of LOF variants of each gene previously linked to cystic kidney or liver disease with ADPKD ICD-9/10 diagnosis and, secondarily, any kidney or liver cyst diagnosis.21 The reference group included participants with no LOF or rare (allele frequency <0.01) missense variants in any genes related to cystic liver or kidney disease. First- and second-degree relatives in the cohort were removed in these analyses, which were adjusted for age, sex, and the first 10 genetic principal components and performed using the firthlogist Python package, version 0.5.0 (see eFigure 4 in Supplement 2 for flow diagram). A 2-sided P < .05 was considered statistically significant with Bonferroni adjustment for multiple comparisons. In phenotype-first analyses, comparisons of proportions with a rare variant in 1 of the 11 genes related to cystic kidney or liver disease by presence (vs absence) of family history and ADPKD phenotype (typical vs mild or atypical) were examined using a 2-sample test of proportions; these analyses excluded patients who lacked sufficient clinical data to confirm ADPKD. For phenotype-first analyses, a 2-sided P < .05 was considered statistically significant
Table 1. Baseline Characteristics of the Study Population.
| Characteristics | Participants with phenotypic ADPKD (n = 303)a | Participants without phenotypic ADPKD (n = 173 869)a | Odds ratio (95% CI) | P value |
|---|---|---|---|---|
| Age, median (IQR), y | 61 (45-71) | 60 (44-72) | ||
| Sex, No. (%) | ||||
| Female | 156 (51.4) | 105 161 (60.6) | ||
| Male | 147 (48.6) | 68 708 (39.4) | ||
| Black race, No. (%) | 2 (0.6) | 3952 (2.3) | ||
| Hispanic ethnicity, No. (%) | 11 (3.6) | 4624 (2.7) | ||
| Ancestry, No. (%)b | ||||
| Admixed American | 7 (2.3) | 2521 (1.4) | ||
| African | 5 (1.7) | 5259 (3) | ||
| East Asian | 2 (0.7) | 485 (0.3) | ||
| European | 286 (94.4) | 161 814 (93.1) | ||
| Southeast Asian | 1 (0.3) | 508 (0.3) | ||
| Unknown | 2 (0.7) | 3282 (1.9) | ||
| High-risk APOL1 genotype, No. (%)c | 1 (0.3) | 431 (0.2) | ||
| Last eGFR, mL/min/1.73 m2 | ||||
| Mean (SD) | 54 (36) | 81 (27) | ||
| <60, No. (%) (n = 162 916) | 177 (58.8) | 32 000 (19.6) | ||
| First outpatient visit, median (IQR), y | 2001 (1997-2009) | 2003 (1998-2011) | ||
| Last outpatient visit, median (IQR), y | 2020 (2019-2021) | 2021 (2019-2021) | ||
| Follow-up time, median (IQR), y | 18 (11-22) | 16 (9-22) | ||
| Comorbidities, No. (%)d | ||||
| Any kidney/liver cyst | 303 (100) | 4908 (2.8) | ||
| Hypertension | 247 (81.5) | 89 398 (51.4) | 4.17 (3.11-5.68) | <.001 |
| Dyslipidemia | 197 (65) | 97 751 (56.2) | 1.45 (1.14-1.85) | .002 |
| End-stage kidney disease | 113 (37.3) | 2567 (1.5) | 39.69 (31.05-50.50) | <.001 |
| Cerebrovascular disease | 79 (26.1) | 23 937 (13.8) | 2.21 (1.69-2.87) | <.001 |
| Coronary artery disease | 79 (26.1) | 32 973 (19.0) | 1.51 (1.15-1.96) | .003 |
| Diabetes | 79 (26.1) | 38 144 (21.9) | 1.25 (0.96-1.63) | .08 |
| Cancer | 60 (19.8) | 29 058 (16.7) | 1.23 (0.91-1.64) | .16 |
| Nephrolithiasis | 52 (17.2) | 10 022 (5.8) | 3.39 (2.46-4.58) | <.001 |
| Cardiac valvular abnormalities | 49 (16.2) | 15 560 (8.9) | 1.96 (1.41-2.67) | <.001 |
| Heart failure | 48 (15.8) | 13 491 (7.8) | 2.24 (1.61-3.06) | <.001 |
| Cystic liver disease | 37 (12.2) | 2205 (1.3) | 10.83 (7.44-15.36) | <.001 |
| Cerebral aneurysm | 15 (5) | 1305 (0.8) | 6.89 (3.79-11.59) | <.001 |
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; eGFR, estimated glomerular filtration rate.
ADPKD phenotype was determined by International Classification of Diseases, Ninth Revision (ICD-9) and International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10) coding (eTable 2 in Supplement 2).
Ancestry was determined genetically with high likelihood of belonging to 1 of the 6 classes shown. Unknown ancestry refers to individuals with multiple or no classes reaching a high genetic likelihood. See the eAppendix in Supplement 2 in for further detail. “Admixed” refers to an individual with a mixture of alleles inherited from multiple ancestral populations.
High-risk APOL1 genotype was defined as homozygous for APOL1 G1 or G2 alleles or heterozygous for both G1 and G2.
Comorbidity phenotypes were determined by ICD-9 and ICD-10 coding. Hypertension, diabetes, and heart failure were determined through validated algorithms (eTable 2 in Supplement 2).
Results
Demographics of the Study Cohort
After excluding 1233 patients who had withdrawn from research, there were 174 172 patients in the cohort (Figure 1). Median age was 60 years (IQR, 44-72 years), 60.6% were female, and 93% were of European ancestry (Table 1). A total of 303 patients (1.74 per 1000) had ADPKD ICD-9/10 diagnoses from 178 distinct families, including 73 individuals from 29 families with at least a first-degree relative. After chart review, 235 (1.35 per 1000) had confirmed ADPKD, 42 had insufficient chart evidence of ADPKD, and 26 had phenotypes more closely matching other kidney diseases (Figure 2; eTable 3 in Supplement 2).
Figure 1. Study Flow and Penetrance of PKD1 and PKD2 Variants.
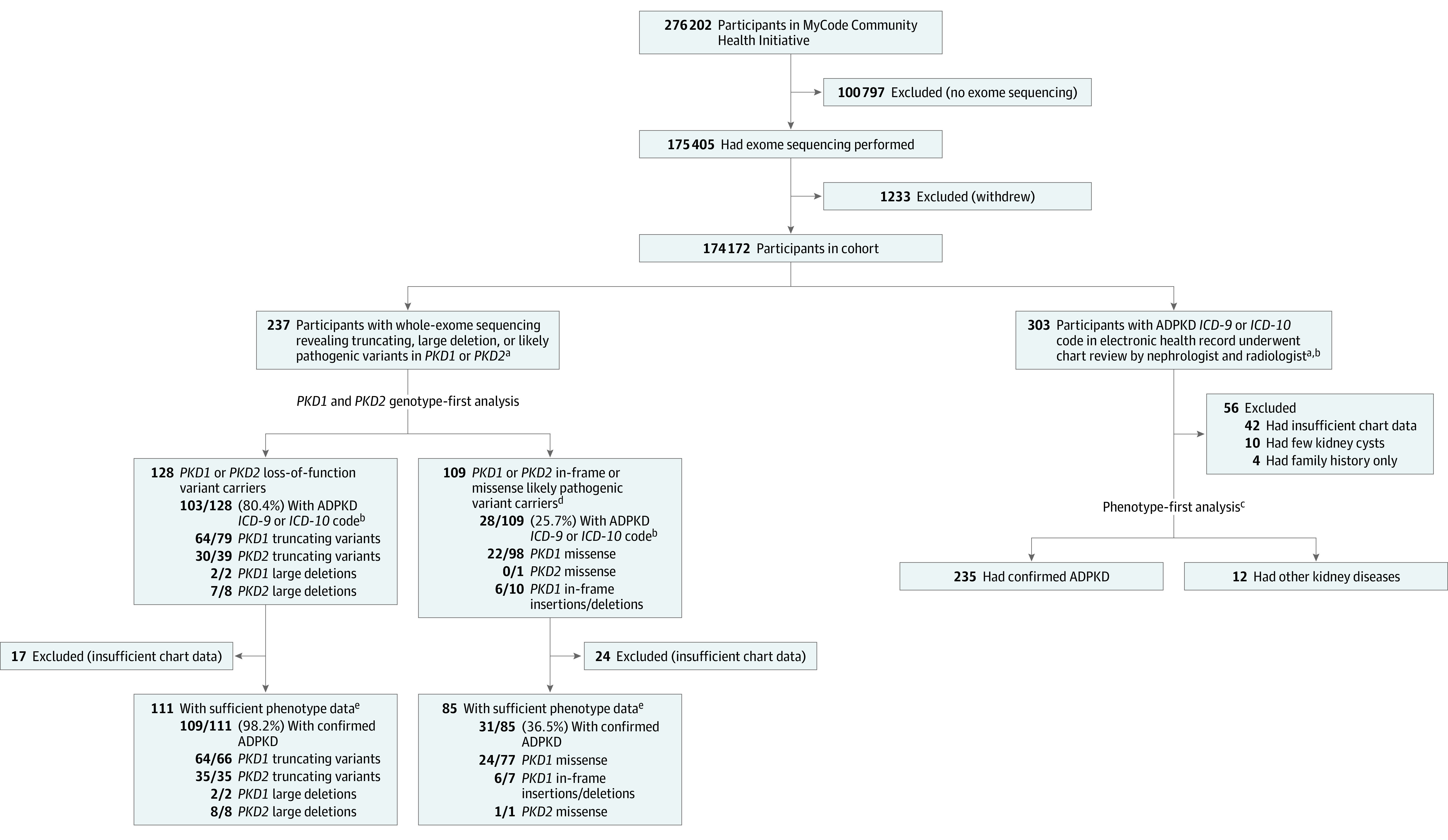
Evaluation of 174 172 patients with exome sequencing revealed loss-of-function variants in PKD1 and PKD2 or in-frame deletions and missense variants in PKD1 and PKD2 classified as likely pathogenic in the Mayo PKD database. ADPKD indicates autosomal dominant polycystic kidney disease; ICD-9, International Classification of Diseases, Ninth Revision; ICD-10, International Statistical Classification of Diseases and Related Health Problems, Tenth Revision.
aThere is overlap of 131 individuals in both the genotype-first and the phenotype-first analyses.
bADPKD diagnosis defined as having 1 or more of the following ICD-9 or ICD-10 codes in electronic health record: Q61.2, Q61.3, 753.12, or 753.13.
cSee Figure 2 and Figure 3 for details.
dClassified as likely pathogenic in the Mayo PKD database.
eChart review by radiologist and nephrologist.
Figure 2. Prevalence, Phenotypic Spectrum, and Genetic Determinants of Autosomal Dominant Polycystic Kidney Disease.
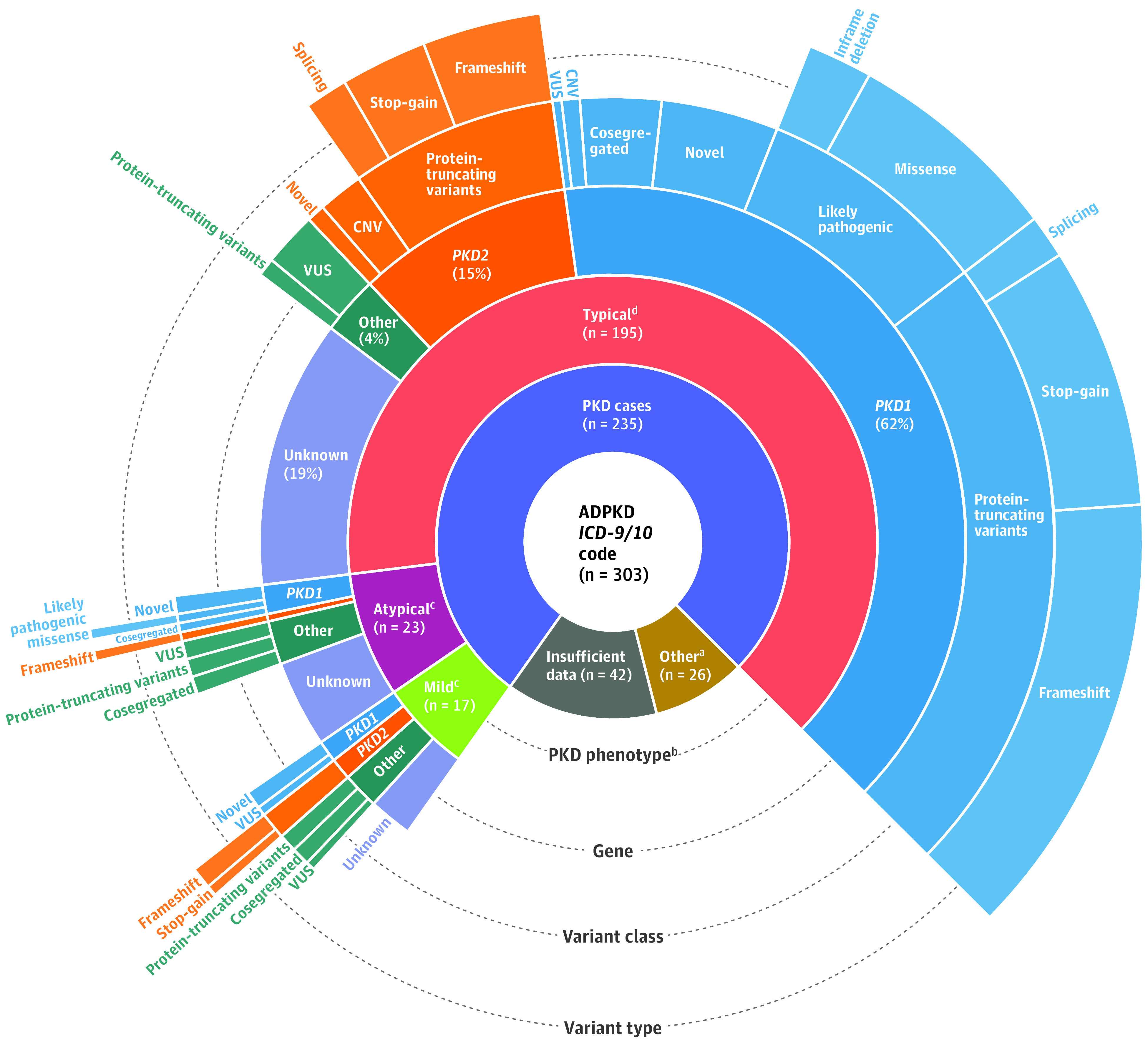
This sunburst plot illustrates genotypic and phenotypic details of 303 patients with autosomal dominant polycystic kidney disease (ADPKD) based on International Classification of Diseases, Ninth Revision (ICD-9) and International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10) codes from an overall population of 174 172 individuals with exome sequencing. CNV indicates copy number variation; VUS, variants of uncertain significance.
aOf the 26 participants with other kidney diseases, 6 had autosomal recessive polycystic kidney disease, 2 had tuberous sclerosis, 2 had congenital abnormalities of kidney and urinary tract, 2 had 17q12 syndrome, 4 had a family history of ADPKD but no clear evidence of disease, and 10 had a few kidney cysts not meeting criteria for ADPKD.
bTypical imaging phenotype was defined as bilateral and diffuse distribution of cysts with moderate or severe replacement of kidney tissue by cysts. Mild imaging phenotype was defined as bilateral and diffuse but with mild replacement of kidney tissue by cysts. Atypical imaging phenotype included cystic involvement that was not symmetric, nondiffuse, or accompanied by kidney atrophy (see Methods section of text).
cIndividuals with atypical or mild ADPKD are much less likely to have a potential variant identified as a contributor to ADPKD, with 11 of 23 atypical cases and 11 of 17 mild cases having a potential genetic cause. Among these, few had PKD1 or PKD2 protein-truncating or likely pathogenic variants. The majority had novel variants in PKD1, PKD2, or other genes associated with cystic kidney or liver disease.
dAmong 195 individuals with typical ADPKD, 120 had a PKD1 variant, 30 had a PKD2 variant, and 8 had a variant in another cystic gene. No genetic variant was found in the remaining 37 patients.
Patients with confirmed ADPKD had similar demographic characteristics as the overall population (Table 1) and had higher prevalence of ADPKD-related ICD-9/10 diagnoses compared with those without ADPKD, respectively, including hypertension (81.5% vs 51.4%; difference, 30.1% [95% CI, 25.3%-34.1%]; P < .001), liver cysts (12.2% vs 1.3%; difference, 10.9% [95% CI, 7.7%-15.1%]; P < .001), nephrolithiasis (17.2% vs 5.8%; difference, 11.4% [95% CI, 7.6%-16.0%]; P < .001), cerebral aneurysm (5% vs 0.8%; difference, 4.2% [95% CI, 2.3%-7.3%]; P < .001), and cardiac valvular abnormalities (16.2% vs 8.9%; difference, 7.2% [95% CI, 3.5%-11.8%]; P < .001).
Prevalence of Predicted and Previously Described Pathogenic Variants
The prevalence of ACMG pathogenic and likely pathogenic PKD1/PKD2 variants combined (allele frequency <0.0001) was 8.64 per 1000 and the prevalence of pathogenic PKD1/PKD2 variants was 1.93 per 1000 (eTables 4 and 5 in Supplement 1). From exome sequencing data of 174 172 patients, there were 3556 variants in PKD1 (allele frequency range, 0.19 to 2.88 × 10−6) (eTable 4 in Supplement 1), including 1919 variants that were listed in gnomAD exome sequencing data.13 ClinVar interpretation was available for 460 of the variants identified. Using VarSome ACMG pathogenicity criteria, 58 PKD1 variants were classified as benign, 667 as likely benign, 2265 as variants of uncertain significance, 384 as likely pathogenic, and 137 as pathogenic (eTable 4 in Supplement 1). For PKD2, there were 434 variants (allele frequency range, 0.36-2.88 × 10−6), with 177 variants present in single patients (eTable 5 in Supplement 1). Among the PKD2 variants, 216 were in gnomAD exome sequence data. Using VarSome ACMG criteria, 7 variants were classified as benign, 39 as likely benign, 339 as variants of uncertain significance, 19 as likely pathogenic, and 29 as pathogenic (eTable 5 in Supplement 1). Only 16 PKD1 and 7 PKD2 variants were listed in ClinVar as pathogenic or likely pathogenic with 2-star review status classification (eTables 4 and 5 in Supplement 1).
Of the variants listed above, 135 individuals with LOF variants or large chromosomal deletions in PKD1 or PKD2 were identified. In addition, there were 3793 patients with LOF variants and 61 large chromosomal deletion carriers in one of the genes previously associated with cystic kidney or liver disease; no patient had LOF variants in SEC61B.
Genotype-First Analysis
Loss-of-function variants in PKD1, PKD2, IFT140, HNF1B, and GANAB were associated with increased risk of ADPKD ICD-9/10 diagnosis (Bonferroni-corrected P < .05 for all comparisons) (Table 2); PKD1, PKD2, IFT140, and ALG8 were associated with any kidney or liver cyst ICD-9/10 diagnosis (Bonferroni-corrected P < .05 for all comparisons). However, ADPKD diagnoses were relatively rare among carriers of LOF variants in IFT140 (5/205), GANAB (1/14), and HNF1B (1/16). A list of all LOF variants detected with accompanying genetic database information is shown in eTable 6 in Supplement 1.
Table 2. Odds of ADPKD or Kidney/Liver Cyst ICD-9 and ICD-10 Codes Among Participants With LOF Variants in ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, LRP5, PKD1, PKD2, PKHD1, PRKCSH, or SEC63.
| Genea | No. of carriers | Cases, No./total (diagnosis prevalence, %) | Odds ratio (95% CI)b | P value | Adjusted P valuec |
|---|---|---|---|---|---|
| Phenotypic ADPKD based on ICD-9/10 codingd | |||||
| Reference | 42 485 | 34/42 485 (0.1) | 1 [Reference] | ||
| ALG8 | 159 | 0/159 | |||
| ALG9 | 25 | 0/25 | |||
| DNAJB11 | 7 | 0/7 | |||
| GANAB | 14 | 1/14 (7.1) | 124.87 (13.30-557.30) | <.001 | .005 |
| HNF1B | 16 | 1/16 (6.2) | 119.85 (12.44-547.18) | <.001 | .005 |
| IFT140 | 205 | 5/205 (2.5) | 32.94 (11.78-75.89) | <.001 | <.001 |
| LRP5 | 1414 | 0/1414 | |||
| PKD1 | 54 | 42/54 (77.8) | 4400.45 (2081.14-10 151.47) | <.001 | <.001 |
| PKD2 | 24 | 17/24 (70.8) | 4530.34 (1655.21-14 111.73) | <.001 | <.001 |
| PKHD1 | 522 | 2/522 (1.0) | 6.02 (1.23-18.08) | .03 | .18 |
| PRKCSH | 74 | 0/74 | |||
| SEC63 | 81 | 0/81 | |||
| Presence of any kidney or liver cyst based on ICD-9/10 codingd | |||||
| Reference | 42 485 | 2033/42 485 (4.8) | 1 [Reference] | ||
| ALG8 | 159 | 17/159 (10.7) | 2.60 (1.51-4.21) | .001 | .01 |
| ALG9 | 25 | 4/25 (16.0) | 3.09 (0.95-8.05) | .06 | .71 |
| DNAJB11 | 7 | 1/7 (14.3) | 4.43 (0.44-23.34) | .17 | >.99 |
| GANAB | 14 | 3/14 (21.4) | 5.51 (1.37-17.16) | .02 | .24 |
| HNF1B | 16 | 3/16 (18.8) | 7.02 (1.72-22.40) | .01 | .12 |
| IFT140 | 205 | 28/205 (13.7) | 3.25 (2.12-4.81) | <.001 | <.001 |
| LRP5 | 1414 | 70/1414 (5.0) | 1.15 (0.89-1.46) | .27 | >.99 |
| PKD1 | 54 | 42/54 (77.8) | 113.80 (60.69-228.45) | <.001 | <.001 |
| PKD2 | 24 | 21/24 (87.5) | 185.93 (64.28-722.16) | <.001 | <.001 |
| PKHD1 | 522 | 27/522 (5.2) | 1.14 (0.75-1.65) | .52 | >.99 |
| PRKCSH | 74 | 3/74 (4.1) | 0.95 (0.26-2.44) | .92 | >.99 |
| SEC63 | 81 | 8/81 (9.9) | 2.61 (1.18-5.10) | .02 | .25 |
Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; ICD-9, International Classification of Diseases, Ninth Revision; ICD-10, International Statistical Classification of Diseases and Related Health Problems, Tenth Revision; LOF, loss of function.
First- or second-degree relatives in the cohort were removed from the analysis. Additionally, carriers of rare (allele frequency <0.01) missense variants were removed from the control group, resulting in sample sizes of 42 485 with the reference genome and 2595 with an LOF variant in any of the 12 genes (eFigure 4 in Supplement 2).
Logistic regression with Firth bias reduction was used to assess associations.
An adjusted P < .05 was considered statistically significant. P values were adjusted for multiple testing using Bonferroni correction.
Phenotypes were determined by ICD-9 and ICD-10 coding for hypertension and diabetes (eTable 2 in Supplement 2).
Penetrance of PKD1 and PKD2 LOF Variants
Based on ICD-9/10 code diagnoses, 2 of 2 patients (100%) with large deletions in PKD1 had ADPKD and 64 of 79 (81.0%) with PKD1 LOF variants had ADPKD. Among 66 patients with a PKD1 LOF variant and sufficient chart review data, 64 (97.0%) had ADPKD with typical ADPKD imaging findings (example shown in Figure 3A). Two individuals with PKD1 LOF variants had imaging data sufficient to rule out ADPKD and had no recorded family history of ADPKD. Of note, these 2 patients had the same PKD1 LOF (splice donor) variant that was also confirmed by Sanger sequencing.
Figure 3. Genetic and Phenotypic Heterogeneity in Confirmed ADPKD Cases.
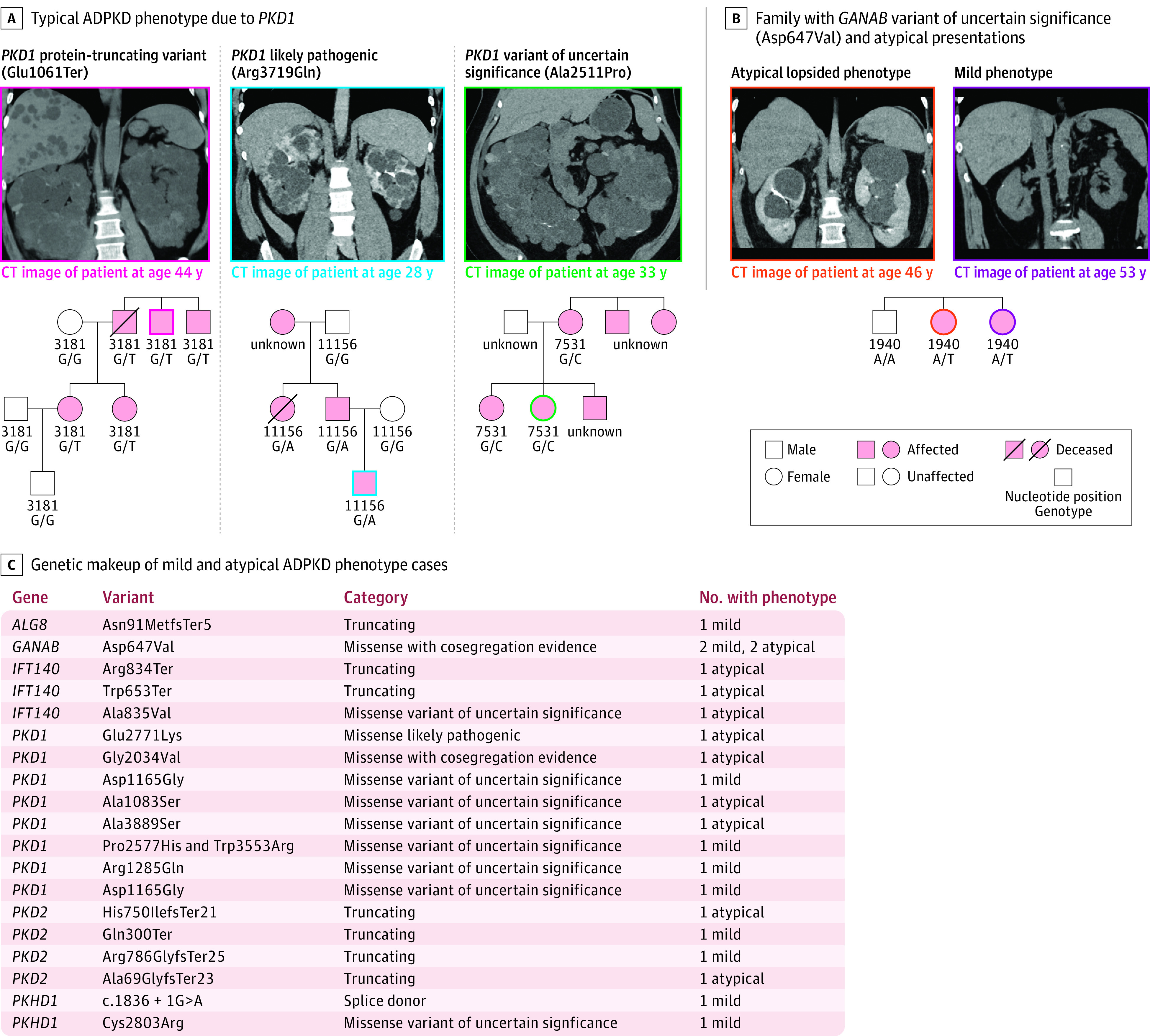
A, Examples: left, a patient with a PKD1 truncating variant (Glu1061Ter) with coronal image in a noncontrast computed tomographic (CT) scan, demonstrating innumerable cortical kidney cysts bilaterally, which enlarge the kidneys and replace the normal kidney parenchyma, consistent with typical ADPKD. Family pedigree shows segregation of ADPKD among members with available genetic and clinical data. Middle, likely pathogenic PKD1 missense variant (Arg3719Gln) carrier in which coronal image from a contrast-enhanced CT scan shows innumerable cortical kidney cysts bilaterally, which enlarge the kidneys and replace the normal kidney parenchyma, consistent with typical ADPKD. Family pedigree shows ADPKD among variant carriers. Right, a carrier of PKD1 (Ala2511Pro), which was been previously classified as likely benign by the Mayo PKD database and variants of uncertain significance by VarSome, in which coronal image from a noncontrast CT scan shows innumerable cortical kidney cysts bilaterally, which enlarge the kidneys and replace the normal kidney parenchyma, consistent with typical ADPKD. In PKD1 or PKD2 variants that were previously unreported or had been classified as benign or variants of uncertain significance, family pedigrees were examined when possible. PKD1 (Ala2511Pro) clearly segregates with ADPKD in pedigree analysis. B, Individuals with atypical or mild ADPKD are much less likely to have a potential variant identified as a contributor to their ADPKD, with only over half of each group being genetically resolved. Among these, few had PKD1 or PKD2 loss of function or likely pathogenic variants, while the majority had novel variants in PKD1, PKD2, or other genes related to cystic kidney disease, including a previously unreported GANAB Asp647Val variant that co-segregates with ADPKD in pedigree analyses. C, Pedigree analysis of 1 family of GANAB (Asp647Val) carriers. The image on the left is a coronal image from a contrast-enhanced CT scan demonstrating relatively mild replacement of kidney parenchyma with a minority of cysts accounting for more than half of the enlarged total kidney volume, consistent with the lopsided subtype of atypical ADPKD.20 The image on the right is a coronal image from a noncontrast CT scan demonstrating relatively few cortical kidney cysts bilaterally with minimal replacement of the normal kidney parenchyma, consistent with mild ADPKD.
For PKD2, 7 of 8 patients (87.5%) with large genomic deletions and 30 of 39 (77%) with LOF variants had ADPKD ICD-9/10 codes. After excluding 5 individuals with insufficient chart review data to confirm ADPKD, 8 of 8 patients (100%) with large genomic deletions in PKD2 had ADPKD, and 35 of 35 patients (100%) with PKD2 LOF variants had ADPKD. Five patients with PKD2 LOF variants had imaging evidence of ADPKD but no ADPKD ICD-9/10 diagnosis. PKD2 variant carriers had a wider spectrum of ADPKD imaging phenotypes (74% typical ADPKD, 17% mild PKD, and 9% atypical PKD).
Penetrance of Previously Described Likely Pathogenic PKD1 and PKD2 Missense Variants
Figure 1 shows the prevalence of ADPKD among 109 carriers of known rare in-frame deletions or missense variants in PKD1 or PKD2 listed in the Mayo PKD database. After excluding 24 patients with insufficient data to confirm ADPKD, 24 of 77 (31.2%) with a PKD1 likely pathogenic variant, and 6 of 7 with PKD1 in-frame deletions had ADPKD. There were several likely pathogenic variants with multiple unaffected carriers, such as PKD1 Arg2408Cys (0 of 21 with ADPKD) and PKD1 Val1611Ile (0 of 22 with ADPKD). The 1 patient with a PKD2 likely pathogenic variant had ADPKD per chart review but did not have an ADPKD ICD-9/10 code.
Phenotype-First Analysis
Overall, 180 of 235 patients (76.6%) with ADPKD had a potential genetic cause, with the majority being PKD1 (127 patients) and PKD2 (34 patients); 19 of 235 (8.1%) had a rare variant in a gene associated with cystic kidney disease, including IFT140 (n = 7), GANAB (n = 4), PKHD1 (n = 3), HNF1B (n = 2), ALG8 (n = 1), and ALG9 (n = 1), as well as 1 patient with rare variants in both IFT140 and PKHD1 (Figure 2). Family history of ADPKD was documented in 150 patients (63.8%). Rare variants were identified in 91.3% (137/150) with family history of ADPKD and 50.6% (43/85) without family history (difference, 40.7% [95% CI, 29.2%-52.3%]; P < .001). After stratifying patients with ADPKD into 3 groups (typical, mild, and atypical; see eAppendix and eFigures 1, 2, and 3 in Supplement 2), rare variants were identified in 158 of 195 with typical, 11 of 17 with mild, and 11 of 23 with atypical ADPKD (Figure 2). The proportion with rare variants was higher in typical ADPKD (81.0% [159/195]) vs mild or atypical ADPKD (55.0% [22/40]; difference, 26.0% [95% CI, 9.7%-42.4%]; P < .001). Self-reported family history was more common in individuals with typical ADPKD (68.7% [134/195]) compared with atypical or mild ADPKD (16/40 [40.0%]; difference, 28.7% [95% CI, 12.2%-45.2%]; P < .001). Genetic variants in patients with mild or atypical ADPKD included PKD1, PKD2, IFT140, PKHD1, ALG8, and GANAB (Figure 2).
PKD1 and PKD2 LOF variants accounted for most of the confirmed ADPKD cases (Figure 2). Pedigree analyses were completed for most of the cases with typical ADPKD and a PKD1 or PKD2 LOF variant. Figure 3A shows examples of images and pedigrees for 3 PKD1 variant carriers with typical ADPKD. A pedigree analysis for carriers of the protein truncating variant PKD1 Glu1061Ter shows that the variant perfectly segregates with ADPKD. There were 26 carriers of PKD1 in-frame deletion or missense variants (allele frequency <0.0001) that were classified in the Mayo PKD database as likely pathogenic. Figure 3A shows co-segregation of PKD1 Arg3719Gln with ADPKD.
There were 19 variants in 25 patients with confirmed ADPKD who had PKD1 or PKD2 variants that were either not previously reported or listed as variants of uncertain significance or likely benign in the Mayo PKD database (Figure 2; eTable 7 in Supplement 1). For 9 of these patients, there was supportive electronic health record co-segregation data among patients and their family members. For instance, 1 confirmed ADPKD carrier of PKD1 Ala2511Pro had 4 family members who were not in our cohort but had ADPKD (pedigree in Figure 3A), including 2 who had prior clinical genetic testing identifying the same PKD1 variant.
Of the 42 individuals with ADPKD who did not have a rare variant in PKD1 or PKD2 (Figure 2), 8 patients had rare variants in other genes related to cystic kidney disease (eTable 8 in Supplement 2), including 2 with IFT140 truncations, 2 with IFT140 missense variants (eFigure 5 in Supplement 2), and 4 with missense variants of uncertain significance in ALG9, HNF1B, or PKHD1. Genetic variants in patients with mild or atypical ADPKD included those in PKD1 and PKD2 as well as other genes associated with cystic kidney disease (GANAB, ALG8, IFT140, and PKHD1) (Figure 3B).
There were several families with confirmed ADPKD who had PKD1 and PKD2 variants not reported in the Mayo PKD database in which several affected individuals carried the same variant (eFigure 6 in Supplement 2). This included supportive pedigree segregation data for GANAB Asp647Val in 2 families (Figure 3C), including 4 with mild or atypical ADPKD, 1 with multiple hypodensities in kidneys and liver, 1 with bilateral kidney cysts before age 20 years, and 1 with no imaging available for review. Information for the rest of the novel variants and variants of uncertain significance or likely benign variants from the Mayo PKD database is available in eTable 7 in Supplement 1. Although these data suggest that some of these variants may be pathogenic, additional replication data are needed.
Discussion
Using exome sequencing in an unselected, regional health system cohort, substantial genetic and phenotypic variability in ADPKD was observed. Loss-of-function variants in PKD1 and PKD2 accounted for the majority of confirmed ADPKD cases, with additional rare variants in PKD1, PKD2, IFT140, GANAB, PKHD1, HNF1B, ALG8, and ALG9 found in other patients. There was supportive pathogenic evidence through examination of family pedigrees for several PKD1, PKD2, and GANAB variants that were previously unreported or reported as variants of uncertain significance or likely benign. However, additional data are needed to definitively classify missense variants.
Prevalence estimates of ADPKD in our cohort by phenotype matched prior literature (1.74 per 1000 for ICD-9/10 diagnosis alone; 1.35 per 1000 for ICD-9/10 diagnosis with chart review confirmation), yet were much lower than genotype-based estimated prevalence (8.64 per 1000), likely as a result of misclassification of missense variants. Only 31.2% of patients with PKD1 missense variants previously reported as “likely pathogenic” had ADPKD, including several PKD1 variants for which numerous carriers had no evidence of ADPKD. These results are consistent with a study of the Exome Aggregation Consortium (ExAC), which reported that the observed prevalence of previously reported pathogenic variants in PKD1 and PKD2 in ExAC was far greater than the expected prevalence (6.9 per 1000 vs 0.69 per 1000).22 Additional work using population-based cohorts, such as this cohort, is needed to validate pathogenicity of missense variants in PKD1, PKD2, and other genes.
In this cohort, 97% of patients with LOF variants in PKD1 had ADPKD using high-quality exome sequencing followed by confirmatory Sanger sequencing in 4 patients who did not have ADPKD on imaging (eAppendix in Supplement 2). In 2 of these 4 patients, Sanger sequencing did not confirm LOF by exome sequencing. Sanger sequencing of the other 2 individuals confirmed the exome sequencing findings of a splice donor variant. While the prediction software SpliceAI predicted this variant to be disruptive, determining pathogenicity in splice variants remains notoriously challenging. Regardless, this study shows that a high-quality exome sequencing–based pipeline identifies ADPKD correctly in nearly all patients with PKD1 or PKD2 LOF variants and has important implications for genomic screening programs.
This study also demonstrates the value of including genes related to cystic kidney or liver disease in the genetic evaluation of ADPKD. In genotype-first analyses, IFT140, GANAB, and HNF1B were significantly associated with ADPKD diagnosis while ALG8 and IFT140 were also associated with a composite outcome of any kidney/liver cyst diagnosis. There was clear evidence that LOF variants in LRP5 do not cause ADPKD as no association was seen in the analysis of 1414 LRP5 LOF carriers. LRP5 was likely previously incorrectly implicated in polycystic liver disease.12 The full extent to which variants in genes related to cystic kidney disease cause cystic kidney or liver disease requires additional study and deeper phenotyping (ie, imaging review), as has been done for ALG9.9 Future studies are needed to better understand the significance of mild cystic disease and whether cystic disease caused by atypical genes are responsive to therapies to slow ADPKD progression such as vasopressin receptor 2 antagonism.23
A prior study found limited utility in exome sequencing PKD1, showing only 50% sensitivity due to issues with low read depth and sequencing quality in exons 1 to 33, which are also present in PKD1P1-PKD1P6 pseudogenes.24 In this cohort, there was excellent read depth and sequencing quality throughout most of the PKD1 gene, which may explain the much higher rate of detecting disease-causing PKD1 variants (see the eAppendix in Supplement 2 for further details).
Currently, diagnosis of ADPKD for most patients with a family history of ADPKD is done by abdominal imaging.19 Genetic testing is generally only performed when ADPKD is suspected in persons without family history or with equivocal kidney imaging findings, in the setting of related kidney donation, or for family planning purposes.25 Better knowledge of the genetic causes of ADPKD could facilitate earlier genotype-first diagnoses and enable preventive or disease-modifying actions and early treatments such as tolvaptan for ADPKD as well as potential new therapeutic options currently being evaluated in ongoing clinical trials.26
Limitations
This study has several limitations. First, the study population was mostly of European ancestry. Second, electronic health record data may underestimate some of the ADPKD disease burden if not reported or detected. Third, due to time constraints, chart and imaging reviews were unable to be conducted for all participants with LOF variants in other genes related to cystic kidney disease. Replication of possible genotype-phenotype associations for missense variants is needed in other cohorts. Although 12 genes associated with cystic kidney disease were examined, there may be others that have yet to be described. Future studies “reverse phenotyping” patients with a suspected pathogenic variant without a clear phenotype are needed, as are additional studies in more diverse cohorts.
Conclusions
This study demonstrates substantial genetic and phenotypic variability in ADPKD among patients within a regional health system in the US.
eTable 1. Database Accession Numbers, OMIM Numbers, Cytogenic Location, Summary of Quality Metrics, and Depth of Coverage for ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, LRP5, PKD1, PKD2, PKHD1, PRKCSH, SEC63
eTable 4. PKD1 Variants With Classifications From Genetic Databases
eTable 5. PKD2 Variants With Classifications From Genetic Databases
eTable 6. ALG8, ALG9, DNAJB11, GANAB, IFT140, HNF1B, LRP5, PKHD1, PRKCSH, SEC63 Variants With Classifications From Genetic Databases
eTable 7. Variants in PKD1 or PKD2 That Are Novel, VUS, or Likely Benign in the Mayo PKD Database
eAppendix. Supplementary Methods
eTable 2. Coding Definition for Comorbidities in Table 1
eTable 3. Variants in Patients With Other Kidney Diseases or ADPKD Family History Alone
eTable 8. Typical ADPKD Phenotype Cases With “Other” (Not PKD1 or PKD2) Gene Variants
eFigure 1. Typical ADPKD Imaging Phenotype
eFigure 2. Mild ADPKD Imaging Phenotype
eFigure 3. Atypical ADPKD Imaging Phenotype
eFigure 4. Flow Diagram for Cohorts Described in the Study
eFigure 5. Sample Pedigrees and Images of Carriers of IFT140 Variants
eFigure 6. Sample Pedigrees and Images of Carriers of Novel PKD1 Variants
Data Sharing Statement
References
- 1.Manickam K, Buchanan AH, Schwartz MLB, et al. Exome sequencing-based screening for BRCA1/2 expected pathogenic variants among adult biobank participants. JAMA Netw Open. 2018;1(5):e182140. doi: 10.1001/jamanetworkopen.2018.2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carruth ED, Beer D, Alsaid A, et al. Clinical findings and diagnostic yield of arrhythmogenic cardiomyopathy through genomic screening of pathogenic or likely pathogenic desmosome gene variants. Circ Genom Precis Med. 2021;14(2):e003302. doi: 10.1161/CIRCGEN.120.003302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adler A, Novelli V, Amin AS, et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation. 2020;141(6):418-428. doi: 10.1161/CIRCULATIONAHA.119.043132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single US health care system. Science. 2016;354(6319):aaf7000. doi: 10.1126/science.aaf7000 [DOI] [PubMed] [Google Scholar]
- 5.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4(1):50. doi: 10.1038/s41572-018-0047-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75(8):848-855. doi: 10.1038/ki.2008.686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iliuta IA, Kalatharan V, Wang K, et al. Polycystic kidney disease without an apparent family history. J Am Soc Nephrol. 2017;28(9):2768-2776. doi: 10.1681/ASN.2016090938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besse W, Dong K, Choi J, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest. 2017;127(9):3558. doi: 10.1172/JCI96729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Besse W, Chang AR, Luo JZ, et al. ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol. 2019;30(11):2091-2102. doi: 10.1681/ASN.2019030298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornec-Le Gall E, Olson RJ, Besse W, et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet. 2018;102(5):832-844. doi: 10.1016/j.ajhg.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carey DJ, Fetterolf SN, Davis FD, et al. The Geisinger MyCode Community Health Initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18(9):906-913. doi: 10.1038/gim.2015.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besse W, Choi J, Ahram D, et al. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum Mutat. 2018;39(3):378-382. doi: 10.1002/humu.23383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gout AM, Martin NC, Brown AF, Ravine D. PKDB: polycystic kidney disease mutation database—a gene variant database for autosomal dominant polycystic kidney disease. Hum Mutat. 2007;28(7):654-659. doi: 10.1002/humu.20474 [DOI] [PubMed] [Google Scholar]
- 15.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978-1980. doi: 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535-548.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 17.Staples J, Qiao D, Cho MH, Silverman EK, Nickerson DA, Below JE. PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am J Hum Genet. 2014;95(5):553-564. doi: 10.1016/j.ajhg.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aung TT, Bhandari SK, Chen Q, et al. Autosomal dominant polycystic kidney disease prevalence among a racially diverse United States population, 2002 through 2018. Kidney360. 2021;2(12):2010-2015. doi: 10.34067/KID.0004522021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212. doi: 10.1681/ASN.2008050507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26(1):160-172. doi: 10.1681/ASN.2013101138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80(1):27-38. doi: 10.1093/biomet/80.1.27 [DOI] [Google Scholar]
- 22.Mallawaarachchi AC, Furlong TJ, Shine J, Harris PC, Cowley MJ. Population data improves variant interpretation in autosomal dominant polycystic kidney disease. Genet Med. 2019;21(6):1425-1434. doi: 10.1038/s41436-018-0324-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407-2418. doi: 10.1056/NEJMoa1205511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali H, Al-Mulla F, Hussain N, et al. PKD1 duplicated regions limit clinical utility of whole exome sequencing for genetic diagnosis of autosomal dominant polycystic kidney disease. Sci Rep. 2019;9(1):4141. doi: 10.1038/s41598-019-40761-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanktree MB, Iliuta IA, Haghighi A, Song X, Pei Y. Evolving role of genetic testing for the clinical management of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2019;34(9):1453-1460. doi: 10.1093/ndt/gfy261 [DOI] [PubMed] [Google Scholar]
- 26.Capuano I, Buonanno P, Riccio E, Amicone M, Pisani A. Therapeutic advances in ADPKD: the future awaits. J Nephrol. 2022;35(2):397-415. doi: 10.1007/s40620-021-01062-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Database Accession Numbers, OMIM Numbers, Cytogenic Location, Summary of Quality Metrics, and Depth of Coverage for ALG8, ALG9, DNAJB11, GANAB, HNF1B, IFT140, LRP5, PKD1, PKD2, PKHD1, PRKCSH, SEC63
eTable 4. PKD1 Variants With Classifications From Genetic Databases
eTable 5. PKD2 Variants With Classifications From Genetic Databases
eTable 6. ALG8, ALG9, DNAJB11, GANAB, IFT140, HNF1B, LRP5, PKHD1, PRKCSH, SEC63 Variants With Classifications From Genetic Databases
eTable 7. Variants in PKD1 or PKD2 That Are Novel, VUS, or Likely Benign in the Mayo PKD Database
eAppendix. Supplementary Methods
eTable 2. Coding Definition for Comorbidities in Table 1
eTable 3. Variants in Patients With Other Kidney Diseases or ADPKD Family History Alone
eTable 8. Typical ADPKD Phenotype Cases With “Other” (Not PKD1 or PKD2) Gene Variants
eFigure 1. Typical ADPKD Imaging Phenotype
eFigure 2. Mild ADPKD Imaging Phenotype
eFigure 3. Atypical ADPKD Imaging Phenotype
eFigure 4. Flow Diagram for Cohorts Described in the Study
eFigure 5. Sample Pedigrees and Images of Carriers of IFT140 Variants
eFigure 6. Sample Pedigrees and Images of Carriers of Novel PKD1 Variants
Data Sharing Statement