

Key Points
Question
What is the efficacy and safety of lebrikizumab in combination with background topical corticosteroid (TCS) therapy in adolescents and adults with moderate-to-severe atopic dermatitis (AD)?
Findings
In a randomized, placebo-controlled, phase 3 clinical trial studying 211 patients (adolescents and adults) with moderate-to-severe AD, lebrikizumab was associated with improved physician-reported signs of AD and patient-reported outcomes of pruritus and quality of life compared with TCS alone, over 16 weeks of treatment.
Meaning
Lebrikizumab, a monoclonal antibody inhibiting interleukin-13, combined with TCS was associated with reduced overall disease severity of moderate-to-severe AD in adolescents and adults, and had a safety profile consistent with previous lebrikizumab AD studies.
This randomized clinical trial examines the efficacy and safety of lebrikizumab in combination with background topical corticosteroid therapy in adolescents and adults with moderate-to-severe atopic dermatitis.
Abstract
Importance
Lebrikizumab (LEB), a high-affinity monoclonal antibody targeting interleukin (IL)-13, demonstrated efficacy and safety in patients with moderate-to-severe atopic dermatitis (AD) during 16 weeks of monotherapy in a phase 2b trial, and two 52-week phase 3 trials.
Objective
To evaluate efficacy and safety of LEB combined with low- to mid-potency topical corticosteroids (TCS) in patients with moderate-to-severe AD.
Design, Setting, and Participants
The ADhere trial was a 16-week randomized, double-blinded, placebo (PBO)-controlled, multicenter, phase 3 clinical trial conducted from February 3, 2020, to September 16, 2021. The study was conducted at 54 outpatient sites across Germany, Poland, Canada, and the US and included adolescent (aged ≥12 to <18 years weighing ≥40 kg) and adult patients with moderate-to-severe AD. The treatment allocation ratio was 2:1 (LEB:PBO).
Interventions
Overall, 211 patients were randomized to subcutaneous LEB (loading dose of 500 mg at baseline and week 2, followed by 250 mg every 2 weeks [Q2W] thereafter) or PBO Q2W in combination with TCS for 16 weeks.
Main Outcomes and Measures
Efficacy analyses at week 16 included proportions of patients achieving Investigator’s Global Assessment score of 0 or 1 (IGA [0,1]) with 2 or more points improvement from baseline, and 75% improvement in the Eczema Area and Severity Index (EASI-75). Key secondary end points included evaluation of itch, itch interference on sleep, and quality of life. Safety assessments included monitoring adverse events (AEs).
Results
The mean (SD) age of patients was 37.2 (19.3) years, 103 (48.8%) patients were women, 31 (14.7%) patients were Asian, and 28 (13.3%) patients were Black/African American. At week 16, IGA (0,1) was achieved by 145 (41.2%) patients in the LEB+TCS group vs 66 (22.1%) receiving PBO+TCS (P = .01); corresponding proportions of patients achieving EASI-75 were 69.5% vs 42.2% (P < .001). The LEB+TCS group showed statistically significant improvements in all key secondary end points. Most treatment-emergent adverse events (TEAEs) were nonserious, mild or moderate in severity, and did not lead to study discontinuation. The TEAEs frequently reported in the LEB+TCS group included conjunctivitis (7 [4.8%]), headache (7 [4.8%]), hypertension (4 [2.8%]), injection site reactions (4 [2.8%]), and herpes infection (5 [3.4%]) vs 1.5% or less patient-reported frequencies in the PBO+TCS group. Similar frequencies of patient-reported serious AEs following LEB+TCS (n = 2, 1.4%) and PBO+TCS (n = 1, 1.5%).
Conclusions and Relevance
In this randomized phase 3 clinical trial, LEB+TCS was associated with improved outcomes in adolescents and adults with moderate-to-severe AD compared with TCS alone, and safety was consistent with previously reported AD trials.
Trial Registration
ClinicalTrials.gov Identifier: NCT04250337
Introduction
Atopic dermatitis (AD) is a chronic, relapsing, heterogenous skin disease with a global prevalence of approximately 20% in children1 and 2% to 7% in adults.2,3,4,5 Moderate-to-severe AD symptoms include intense itch, sleep disturbance, and skin pain affecting sleep, daily activities, and social relationships.6,7,8 The disease burden of AD in adults and adolescents is high and significantly affects patients’ quality of life (QoL).9
Currently, emollients and topical corticosteroids (TCS) are mainstay treatments for mild AD.10 In moderate-to-severe AD, the addition of systemic therapy and/or phototherapy is recommended.11 Targeted biologic therapies including dupilumab and tralokinumab, as well as systemic Janus kinase inhibitors, have been developed and approved recently for moderate-to-severe AD treatment.12,13,14 Due to the heterogeneity of AD, there remains a need to provide additional therapeutic options for long-term management.15,16
Interleukin (IL)-13 is a proinflammatory Th2 cytokine central to AD pathogenesis, driving clinical manifestations of AD.17,18,19 Lebrikizumab is a novel monoclonal antibody that targets and potently neutralizes IL-13 signaling with high binding affinity to a specific epitope with a slow off-rate.20 Lebrikizumab has demonstrated significant clinical benefit in patients with AD in a phase 2b21 (NCT03443024), and 2 phase 3 monotherapy trials (ADvocate1, NCT04146363 and ADvocate2, NCT04178967).22,23 However, monotherapy data may not always be translated in clinical settings. Topical medications are a mainstay of treatment for patients with moderate-to-severe AD and combining them with lebrikizumab therapy may provide relevant guidance on the use of a biologic with background TCS therapy in these patients. Here, we present the results of a phase 3 combination therapy study, ADhere (NCT04250337), which evaluated the efficacy and safety of lebrikizumab when used in combination with low- to mid-potency TCS treatment vs TCS alone in adolescents and adults with moderate-to-severe AD.
Methods
Study Design
The trial protocol is available in Supplement 1. This multicenter, double-blinded, placebo-controlled, parallel-group, 16-week, phase 3 randomized clinical trial (NCT04250337) was conducted at 54 outpatient sites across Germany, Poland, Canada, and the US between February 3, 2020, and September 16, 2021. The analyses presented in this report are based on a database lock date of December 16, 2021. Eligible patients included adolescents (aged ≥12 to <18 years weighing ≥40 kg) and adults with a diagnosis of moderate-to-severe AD (according to the American Academy of Dermatology Consensus Criteria) for 1 or more years before the screening visit.10 Eligibility was confirmed for patients with moderate-to-severe AD using the Eczema Area and Severity Index (EASI) score of 16 or higher, the Investigator’s Global Assessment scale for AD (IGA) score of 3 or higher (scale of 0-4), body surface area (BSA) of 10% or greater at baseline, and a history of inadequate response to treatment with topical medications. The Trial Protocol is available in Supplement 1, and full inclusion and exclusion criteria are detailed in the the eMethods in Supplement 2). This study followed the Consolidated Standards of Reporting Trials reporting guideline and HOME recommendations.
The Trial Protocol required patients to wash out from topical and systemic therapy at least 1 week prior to randomization (eFigure in Supplement 2). At baseline (day 1), eligible patients were randomized at a 2:1 ratio of lebrikizumab to placebo with stratification based on geographic region (US vs Europe vs rest of the world), age group (adolescents vs adults), and disease severity (IGA, 3 vs 4). Patient age, sex, ethnicity and race were collected. Race and ethnicity information was used to support subgroup analyses assessing phenotype with treatment response. The collection of a patient’s race and ethnicity is important, given recent descriptions of disease heterogeneity in AD, with diverse phenotypes and endotypes described based on age, disease chronicity, race and ethnicity, genetics, immunoglobulin E status, and underlying molecular mechanisms.24 Ethnicity was selected by the investigator as reported by the patient, and race was selected by the investigator. Patients were blinded to treatment during the 16-week period and randomly allocated (using an electronic data capture system) to receive either lebrikizumab (LEB) every 2 weeks (Q2W; loading dose of 500 mg administered at baseline and week 2, followed by 250 mg Q2W thereafter), or placebo by subcutaneous injection, in combination with TCS (LEB+TCS vs PBO+TCS). All patients were instructed to use low- to mid-potency TCS for AD symptoms starting at baseline. Topical calcineurin inhibitors (TCI) were permitted for use on sensitive skin areas. Study sites provided a mid-potency TCS (triamcinolone acetonide, 0.1% cream) and a low-potency TCS (hydrocortisone, 1% cream). Patients were allowed to taper or stop TCS use as needed, and TCS treatment could be resumed at the patients’ discretion. All TCS and TCI use was recorded daily by the patients using an electronic diary. If patients experienced clinical worsening of AD symptoms that were intolerable, high-potency TCS or systemic treatments (including but not limited to oral corticosteroids, phototherapy, and cyclosporine) were permitted. The study drug was discontinued for patients receiving systemic rescue treatment, but were still required to attend all visits and assessments. Patients who completed ADhere were eligible to enter the long-term extension study, ADjoin (NCT04392154). Patients who terminated early or did not enroll in the long-term extension study had a safety follow-up visit approximately 12 weeks after the last dose of study drug.
Study drug injections were administered in the clinic. A medication numbering system was used in labeling blinded study drug and details were not accessible to individuals involved in study conduct. The sponsor or designee, the investigator, study site personnel, and the patient were blinded to treatment assignment, and the integrity of the clinical study was maintained throughout. The ADhere clinical trial was undertaken in accordance with ethical principles of the Declaration of Helsinki, Council for International Organizations of Medical Sciences, and Good Clinical Practice guidelines.25 All investigation sites received approval from the appropriate authorized institutional review board or ethics committee. Informed consent was obtained from all patients before study procedures were initiated. For patients considered to be minors, the written consent of the parent or legal guardian, as well as the assent of the minor, was obtained.
Efficacy and Safety Assessments
The primary efficacy end point was the percentage of patients with an IGA score of 0 or 1, and a 2 or more point improvement from baseline at week 16. Key secondary efficacy end points reported included: percentage of patients achieving 75% improvement in EASI (EASI-75) at week 16 (coprimary end point for European Medicines Agency); percentage change from baseline (CFB) in EASI total score at week 16; percentage of patients achieving 90% improvement in EASI from baseline (EASI-90) at week 16; percentage of patients with Pruritus Numeric Rating Scale (NRS [measuring itch severity]) improvement of 4 or more points from baseline at week 16; percentage of patients (having baseline Pruritus NRS ≥4) who achieved both EASI-75 and a 4 or more point reduction in Pruritus NRS score from baseline at week 16; percentage CFB on the Pruritus NRS at week 16; CFB in Sleep-Loss Scale (measuring itch interference on sleep) score at week 16; percentage of patients with a 4 or more point reduction from baseline in the Dermatology Life Quality Index (DLQI; measuring effect of disease on QoL) at week 16; and the CFB in DLQI at week 16. Another important secondary end point included the proportion of TCS/TCI-free days from baseline to week 16.
Participant IGA and EASI scores were assessed biweekly in the study clinic. Pruritus NRS and Sleep-Loss Scale assessments were recorded daily by the patient using an electronic diary and are reported as prorated weekly mean scores. The DLQI assessment was completed monthly (starting at week 4 after baseline assessment) by the patients (aged >16 years) in the study clinic. Patients aged 16 years or younger used the Children’s DLQI.
Safety was assessed by monitoring adverse events (AEs), serum chemistry, hematology and urinalysis evaluations, physical examination, and vital signs. Immunogenicity was assessed by a validated assay designed to perform in the presence of lebrikizumab. In addition to the routinely performed blinded safety assessments, an external independent data safety monitoring board reviewed all data (blinded and unblinded) approximately every 6 months during the conduct of the study.
Statistical Analyses
A total of 17 patients from a single study site were excluded from the 228 in the intent-to-treat (ITT) population because eligibility criteria related to the severity of AD at baseline could not be confirmed. Thus, efficacy analyses used the modified ITT (mITT) population (n = 211). Safety analyses for the treatment period were conducted on all randomized patients who received 1 or more dose of the study drug, except for the 17 excluded patients.
Trial power was calculated based on IGA (0,1) and EASI-75 end points. The assumed response rates at week 16 for IGA (0,1) were 38% for lebrikizumab 250 mg Q2W and 13% for placebo; for EASI-75 they were 58% and 20%, respectively. This study had power of greater than 95% for testing superiority of lebrikizumab vs placebo based on a 2-sided Fisher exact test with an α of 0.05.
A gated multiple testing strategy was implemented for the primary and key secondary objectives to control the family-wise type-I error rate at a 2-sided α level of 0.05. To assess whether LEB+TCS was associated with superior results compared with PBO+TCS, primary and gated secondary end points were tested sequentially. If any test was not successful, all subsequent tests were not tested. Cochran-Mantel-Haenszel tests compared the treatment groups in categorical outcomes, adjusting for the stratification factors. Continuous outcomes were analyzed using the analysis of covariance model. Missing data due to lack of efficacy or data after rescue medication usage were imputed with NRI (for categorical end points) or baseline values (for continuous endpoints). Other missing data were imputed with MI. For analyses of nonkey secondary end points, the mixed-effects model for repeated measures (MMRM) or last observation carried forward (LOCF) methods were used to handle missing data. Safety was summarized using descriptive statistics. Further details on statistical analyses can be found in the eMethods in Supplement 2, and results from prespecified supportive analyses in eTable 2 in Supplement 2. All calculations were performed using SAS statistical software (version 9.4; SAS Institute, Inc).
Results
Patient Disposition, Baseline Demographics, and Disease Characteristics
A total of 211 patients (mean [SD] age 37.2 [19.3] years; 103 [48.8%] female) were randomized 2:1 to LEB+TCS (n = 145) or to PBO+TCS (n = 66) (Figure 1). Among the enrolled patients, 46 (21.8%) were adolescents (LEB+TCS, 32 [22.1%]; PBO+TCS, 14 [21.2%]). A greater percentage of patients treated with PBO+TCS (n = 8; 12.1%) discontinued treatment vs those receiving LEB+TCS (n = 11; 7.6%). Reasons for treatment discontinuation included AEs (3/145 [2.1%] in the LEB+TCS group vs 0/66 in the PBO+TCS group), lack of efficacy (3 [2.1%] vs 1 [1.5%]), withdrawal by patient (3 [2.1%] vs 4 [6.1%]), protocol deviation (2 [1.4%] vs 2 [3.0%]), and physician decision (0 vs 1 [1.5%]). Patient demographics and disease characteristics were similar across both treatment groups (Table 1).
Figure 1. CONSORT Diagram for Patient Disposition.

CONSORT indicates Consolidated Standards of Reporting Trials; ITT, intent-to-treat; LEB, lebrikizumab; mITT, modified intent-to-treat; Q2W, every 2 weeks; TCS, topical corticosteroid.
Table 1. Baseline Demographics and Disease Characteristics in the mITT Population.
| Baseline demographics | No. (%) | |
|---|---|---|
| PBO + TCS (N = 66) | LEB + TCS (N = 145) | |
| Age, mean (SD), y | 36.7 (17.9) | 37.5 (19.9) |
| Adolescents (aged 12 to <18 y) | 14 (21.2) | 32 (22.1) |
| Adults (aged ≥18 y) | 52 (78.8) | 113 (77.9) |
| Female | 33 (50.0) | 70 (48.3) |
| Race | ||
| Asian | 13 (19.7) | 18 (12.4) |
| Black/African American | 9 (13.6) | 19 (13.1) |
| White | 40 (60.6) | 90 (62.1) |
| Weight, mean (SD), kg | 79.8 (24.4) | 74.6 (23.3) |
| BMI, mean (SD) | 27.9 (7.5) | 26.5 (7.2) |
| Geographic region | ||
| United States | 48 (72.7) | 103 (71.0) |
| Europe | 10 (15.2) | 28 (19.3) |
| Rest of the world | 8 (12.1) | 14 (9.7) |
| Prior systemic treatment | 34 (51.5) | 66 (45.5) |
| Systemic corticosteroids | 22 (33.3) | 41 (28.3) |
| Phototherapy | 14 (21.2) | 24 (16.6) |
| Dupilumab | 9 (13.6) | 20 (13.8) |
| Cyclosporine | 4 (6.1) | 18 (12.4) |
| Methotrexate | 6 (9.1) | 13 (9.0) |
| Janus kinase inhibitors | 4 (6.1) | 5 (3.4) |
| Photochemotherapy (PUVA) | 2 (3.0) | 3 (2.1) |
| Mycophenolate-mofetil | 0 | 4 (2.8) |
| Tralokinumab | 1 (1.5) | 1 (0.7) |
| Other biologics | 5 (7.6) | 16 (11.0) |
| Duration since AD diagnosis, mean (SD), year | 21.2 (13.9) | 21.0 (17.4) |
| Baseline disease characteristics | ||
| IGA (3) | 48 (72.7) | 98 (67.6) |
| IGA (4) | 18 (27.3) | 47 (32.4) |
| EASI, mean (SD) | 26.4 (10.6) | 27.7 (11.1) |
| Pruritus NRS, mean (SD) | 6.8 (2.0) | 7.3 (1.8) |
| Sleep-loss due to pruritus, mean (SD) | 1.9 (0.9) | 2.1 (0.9) |
| BSA affected, mean (SD) | 38.2 (20.8) | 40.4 (21.9) |
| DLQI, mean (SD) | 13.5 (7.5) | 14.9 (7.2) |
Abbreviations: AD, atopic dermatitis; BMI, body mass index; BSA, body surface area; DLQI, Dermatology Life Quality Index; EASI, Eczema Area and Severity Index; IGA, Investigator’s Global Assessment; LEB, lebrikizumab; mITT: modified intent-to-treat; NRS, numeric rating scale; PBO, placebo; PUVA, psoralen + UV-A; TCS, topical corticosteroids.
Primary Outcomes
At week 16, IGA (0,1) with a 2-point or more reduction from baseline was achieved by 41.2% (60/145) receiving LEB+TCS vs 22.1% (15/66) receiving PBO+TCS (P = .01) (Figure 2), with statistical significance achieved as early as week 8. There was also a significantly greater (P < .001) proportion of patients achieving EASI-75 responses at week 16 in the LEB+TCS group (69.5%) vs the PBO+TCS group (42.2%), with statistical significance achieved as early as week 4 and maintained through week 16.
Figure 2. Time-Course Response for the Clinical Outcomes (Primary and Secondary End Points).
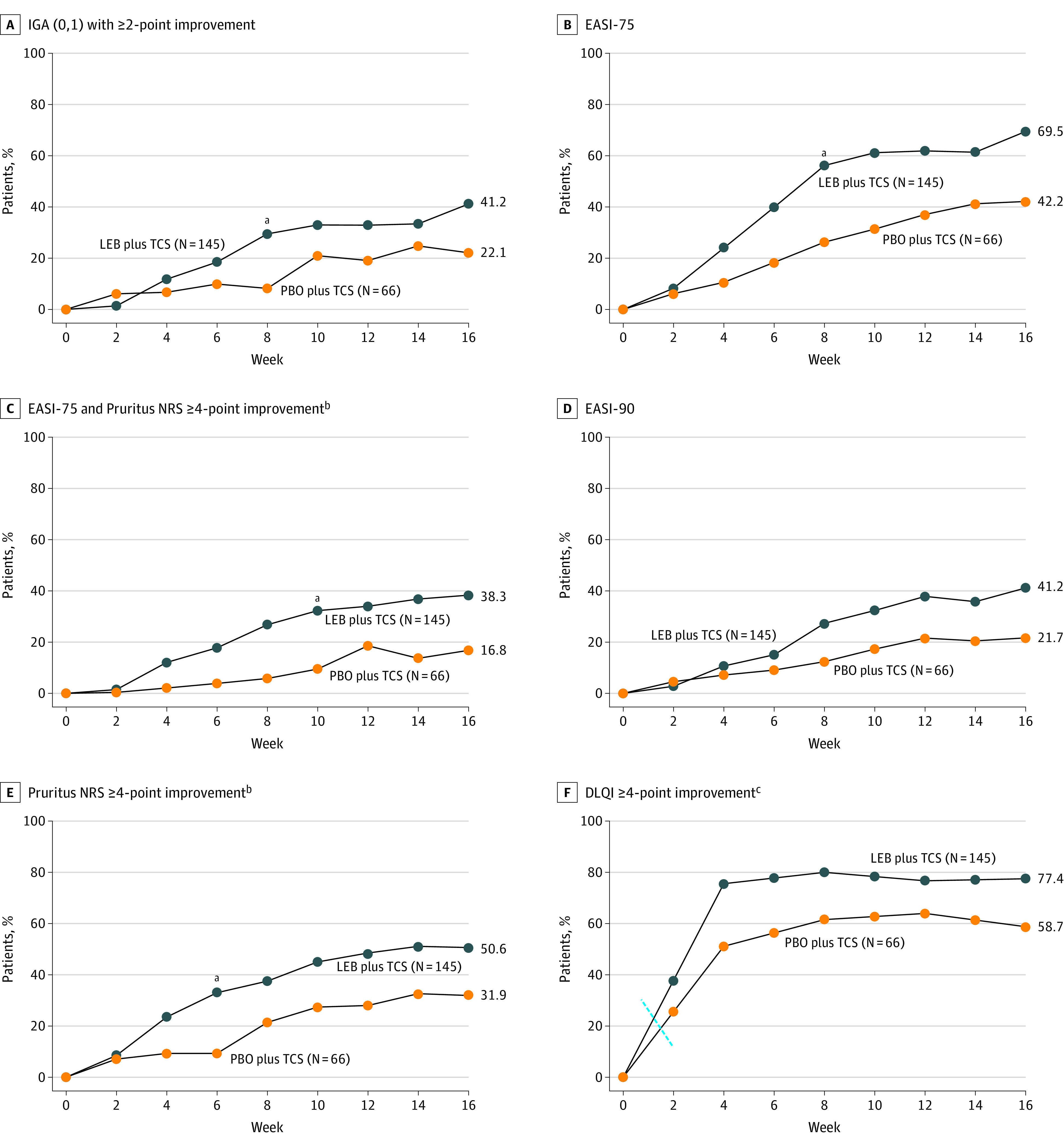
Missing data due to lack of efficacy or data after rescue medication usage were imputed with net reclassification index (for categorical end points) or baseline values (for continuous end points). Other missing data were imputed with multiple imputation. DLQI indicates Dermatology Life Quality Index; EASI, Eczema Area and Severity Index; IGA, Investigator's Global Assessment; LEB, lebrikizumab; mITT, modified intent-to-treat; NRS, numeric rating scale; PBO, placebo; TCS, topical corticosteroids.
aP < .001.
bPatients with baseline Pruritus NRS score of 4 or greater.
cPatients with baseline DLQI of 4 or greater.
Key Secondary Outcomes
Participants in the LEB+TCS group had statistically significant improvements vs those in the PBO+TCS group in the key secondary end points at week 16: EASI-90 (41.2% vs 21.7%, respectively; P = .008), EASI percentage change from baseline (%CFB) (−76.8% vs −53.1%, respectively; P<.001), Pruritus NRS of 4 or more points improvement from baseline (50.6% vs 31.9%, respectively; P = .02), Pruritus NRS %CFB (−50.7% vs −35.5%, respectively; P = .02), EASI-75 and Pruritus NRS (38.3% vs 16.8%, respectively; P = .005), Sleep-Loss Scale %CFB (−1.1 vs −0.8, respectively; P = .02), DLQI of 4 or more points improvement from baseline (77.4% vs 58.7%, respectively; P = .04), and DLQI CFB (−9.8 vs −6.5, P = .001). Subgroup analyses were conducted for IGA (0,1), EASI-75, EASI-90 and Pruritus NRS 4-point improvement. Results of subgroup analyses for IGA (0,1), EASI-75, EASI-90, and Pruritus NRS indicate a significant treatment by sex interaction in EASI-75 and EASI-90 scores, with a greater risk difference in male participants. No other significant treatment by subgroup interactions were observed. Efficacy results were robust based on subgroup and sensitivity analyses. The proportion of patients achieving EASI-90 at week 16 with LEB+TCS vs PBO+TCS was 41.2% vs 21.7% (P < .01), respectively, with a significant difference in the EASI %CFB at week 16 (−76.8% vs −53.1%, respectively; P < .001) (Figure 2). A higher proportion of patients receiving LEB+TCS achieved at least a 4-point reduction in Pruritus NRS score at week 16 vs those receiving PBO+TCS (50.6% vs 31.9%, respectively; P = .02), with a significant difference in Pruritus NRS %CFB at week 16 (−50.7% vs −35.5%; P = .02). Patients in the LEB+TCS arm had a higher response rate of combining EASI-75 and Pruritus NRS 4 point or greater improvement from baseline at week 16 vs those in the PBO+TCS arm (38.3% vs 16.8%, respectively; P = .005). Furthermore, a significantly greater proportion of patients treated with LEB+TCS achieved DLQI score improvements of at least 4 from baseline at week 16 vs the PBO+TCS group (77.4% vs 58.7%; P = .04), having a significant difference in DLQI CFB at week 16 (−9.8 vs −6.5; P = .001), and Sleep-Loss Scale score CFB at week 16 (−1.1 vs −0.8; P = .03). A summary of primary and key secondary efficacy outcomes is provided in Table 2 and eTable 1 in Supplement 2.
Table 2. Summary of Efficacy Outcomes in the mITT Population.
| Variable | Analysis results | ||
|---|---|---|---|
| PBO + TCS (N = 66) | LEB + TCS (N = 145) | Treatment difference | |
| Primary end points | |||
| IGA (0,1) and ≥2-point improvement from baseline at week 16 | |||
| No./No. | 15/66 | 60/145 | |
| % (95% CI) | 22.1 (11.6 to 32.7) | 41.2 (33.0 to 49.4) | 18.3 (5.1 to 31.5) |
| Key secondary end points | |||
| EASI-75 at week 16a | |||
| No./No. | 28/66 | 101/145 | |
| % (95% CI) | 42.2 (30.1 to 54.4) | 69.5 (61.9 to 77.2) | 26.4 (12.1 to 40.8)b |
| EASI-90 at week 16 | |||
| No./No. | 14/66 | 60/145 | |
| % (95% CI) | 21.7 (11.4 to 32.0) | 41.2 (33.0 to 49.3) | 18.9 (6.1 to 31.7) |
| EASI %CFB at week 16 | |||
| No. | 66 | 145 | |
| LS mean (SE) [95% CI] | −53.1 (5.1) | −76.8 (4.1) | −23.6 (5.1) [−33.6 to −13.7]b |
| Pruritus NRS ≥4-point improvement from baseline at week 16c | |||
| No./No. | 18/57 | 66/130 | |
| % (95% CI) | 31.9 (19.3 to 44.4) | 50.6 (41.8 to 59.4) | 19.2 (4.3 to 34.1) |
| Pruritus NRS % CFB at week 16 | |||
| No. | 63 | 139 | |
| LS mean (SE) [95% CI] | −35.5 (6.4) | −50.7 (4.5) | −15.2 (6.4) [−27.7 to −2.7] |
| EASI-75 and Pruritus NRS ≥4-point improvement from baseline at week 16c | |||
| No./No. | 10/57 | 50/130 | |
| % (95% CI) | 16.8 (6.7 to 27.0) | 38.3 (29.8 to 46.9) | 21.6 (8.3 to 35.0) |
| Sleep-Loss Scale CFB at week 16 | |||
| No. | 63 | 139 | |
| LS mean (SE) [95% CI] | −0.8 (0.1) | −1.1 (0.1) | −0.3 (0.1) [−0.6 to −0.0] |
| DLQI- ≥4-point improvement from baseline at week 16d | |||
| No./No. | 28/48e | 81/105f | |
| % (95% CI) | 58.7 (44.1 to 73.2) | 77.4 (69.3 to 85.5) | 17.2 (0.1 to 34.3) |
| DLQI CFB at week 16 | |||
| No. | 51 | 109 | |
| LS mean (SE) [95% CI] | −6.5 (1.9) | −9.8 (1.8) | −3.33 (1.0) [−5.3 to −1.3] |
| Other secondary endpoints | |||
| Proportion of TCS/TCI-free days from baseline to week 16 | |||
| No. | 53 | 131 | |
| LS mean (SE) [95% CI] | 23.9 (4.8) | 31.2 (3.5) | 7.3 (5.1) [−2.78 to 17.4] |
| SCORAD % CFB at week 16g | |||
| No. | 65 | 140 | |
| LS mean (SE) [95% CI] | −37.4 (4.4) | −55.0 (3.5) | −17.7 (4.4) [−26.4 to −9.0]b |
| Change in EQ-5D-5L (VAS) at week 16 | |||
| No. | 65 | 143 | |
| LS mean (SE) [95% CI] | 6.5 (2.4) | 10.1 (1.8) | 3.6 (2.4) [−1.1 to 8.3] |
| Change in EQ-5D-5L (UK Health Index) at week 16 | |||
| No. | 65 | 143 | |
| LS Mean (SE) [95% CI] | 0.05 (0.03) | 0.15 (0.02) | 0.1 (0.03) [0.06 to 0.16]b |
| Change in EQ-5D-5L (US Health Index) at week 16 | |||
| No. | 65 | 143 | |
| LS mean (SE) [95% CI] | 0.03 (0.02) | 0.1 (0.01) | 0.07 (0.02) [0.04 to 0.1]b |
| Change in POEM at week 16 | |||
| No. | 40 | 101 | |
| LS mean (SE) [95% CI] | −6.2 (1.04) | −10.2 (0.7) | −4 (1.1) [−6.3 to −1.7]b |
| Change in PROMIS adults anxiety at week 16 | |||
| No. | 43 | 101 | |
| LS mean (SE) [95% CI] | −1.1 (1.4) | −1.9 (1.0) | −0.8 (1.4) [−3.6 to 2.0] |
| Change in PROMIS adults depression at week 16 | |||
| No. | 43 | 101 | |
| LS mean (SE) [95% CI] | −1.2 (1.1) | −1.4 (0.8) | −0.2 (1.1) [−2.4 to 2.1] |
| Change in CDLQI at week 16 | |||
| No. | 11 | 24 | |
| LS mean (SE) [95% CI] | −4.7 (1.2) | −9.3 (0.9) | −4.6 (1.3) [−7.2 to −2.0]b |
Abbreviations: CFB, change from Baseline; DLQI, Dermatology Life Quality Index; EASI, Eczema Area and Severity Index; EMA, European Medicines Agency; IGA, Investigator’s Global Assessment; LEB, lebrikizumab; LSM, least squares mean; mITT, modified intent-to-treat; NRS, numeric rating scale; PBO, placebo; TCS, topical corticosteroid; TCI, topical calcineurin inhibitor; VAS, visual analogue scale.
Coprimary end point for EMA.
P<.001.
Patients with baseline Pruritus NRS score ≥4.
Patients with baseline DLQI ≥4. For primary or key secondary end points, analyses are based on data imputed with (1) nonresponder imputation for patients who use rescue medication or discontinue study drug due to lack of efficacy; (2) other missing data were imputed with MI. For other secondary end points, analyses are based on LOCF if data are planned to be collected once during the treatment period; analyses are based on MMRM model if data are planned to be collected multiple times during the treatment period.
Patients (>16 years of age) who answered DLQI at baseline: n = 48.
Patients (>16 years of age) who answered DLQI at baseline: n = 105.
ADhere used a modified version of the SCORAD with a maximum point score of 101 rather than 103. Due to a system setup error in the electronic data collection tool, the actual maximum score for each of the symptoms in Part C was 9 instead of 10, which resulted in the total maximum SCORAD score of 101 instead of 103. While this error directly impacted only Part C of the SCORAD, the measure is calculated as one total score, so the total SCORAD score collected could have been up to 2% lower than would have been expected if the error had not occurred.
Regarding additional secondary outcomes, patients treated with LEB+TCS had a numerically greater mean percentage of TCS/TCI-free days vs placebo but this was not significantly different between treatment groups at week 16. Statistical significance was reached at weeks 6, 8, and 10 (Table 2 and eTable 1 in Supplement 2). Furthermore, by the end of the study (121 days), 50% of patients in the LEB+TCS group were TCS/TCI-free, whereas the PBO+TCS group did not reach this threshold (eTable 3 in Supplement 2). The percentages of TCS/TCI-free days by visit are provided in eTable 4 in Supplement 2. The use of rescue medication through week 16 is summarized in eTable 5 in Supplement 2. More than twice the percentage of patients in the PBO+TCS group (7 [10.6%]) received rescue therapy vs those in the LEB+TCS group (6 [4.1%]).
Safety Profile
Treatment-emergent AEs (TEAEs) were reported in 63 (43.4%) patients receiving LEB+TCS and 23 (34.8%) patients receiving PBO+TCS (Table 326). Most TEAEs were nonserious and mild or moderate in severity. The TEAEs reported more frequently in the LEB+TCS vs the PBO+TCS group included conjunctivitis (LEB+TCS, 7 [4.8%]; PBO+TCS, 0 [0%]), headache (LEB+TCS, 7 [4.8%]; PBO+TCS, 1 [1.5%]), herpes infection (LEB+TCS, 5 [3.4%]; PBO+TCS, 1 [1.5%]), hypertension (LEB+TCS, 4 [2.8%]; PBO+TCS, 1 [1.5%]), and injection site reactions (LEB+TCS, 4 [2.8%]; PBO+TCS, 1 [1.5%]). Most conjunctivitis-related TEAEs were mild or moderate in severity. No parasitic infections were reported. TEAEs with higher incidences in the PBO+TCS group vs the LEB+TCS group included nasopharyngitis (4 [6.1%] vs 3 [2.1%], respectively) and AD (3 [4.5%] vs 3 [2.1%], respectively). A similar and low frequency (<2%) finding of serious AEs was reported in either treatment group (LEB+TCS, 2 [1.4%]; PBO+TCS, 1 [1.5%]. Adverse events leading to treatment discontinuation were reported by 3 (2.1%) patients in the LEB+TCS group (1 [0.7%] patient each due to injection site rash, drug hypersensitivity, and conjunctivitis) and 0 patients in the PBO+TCS group. No deaths occurred during the trial. Overall, 207 patients were evaluable for immunogenicity. Of these patients, 16 (7.7%) had antidrug antibodies (ADA) present at baseline. Through 16 weeks of treatment, 5 (3.5%) patients treated with LEB+TCS and 0 patients treated with PBO+TCS became treatment-emergent ADA-positive. Maximum postbaseline titers ranged from 1:20 to 1:160, with a median titer of 1:40. The incidence of ADA is presented in eTable 6 in Supplement 2.
Table 3. Overview of TEAEs Through Week 16 in Modified Safety Population.
| Variable | No. (%) | |
|---|---|---|
| PBO + TCS (N = 66) | LEB + TCS (N = 145) | |
| Any TEAE | 23 (34.8) | 63 (43.4) |
| Mild | 12 (18.2) | 32 (22.1) |
| Moderate | 10 (15.2) | 28 (19.3) |
| Severe | 1 (1.5) | 3 (2.1) |
| Serious AE | 1 (1.5) | 2 (1.4) |
| Death | 0 | 0 |
| AEs leading to treatment discontinuation | 0 | 3 (2.1) |
| TEAEs reported in ≥2% of LEB + TCS group | ||
| Conjunctivitisa | 0 | 7 (4.8) |
| Headache | 1 (1.5) | 7 (4.8) |
| Hypertension | 1 (1.5) | 4 (2.8) |
| Nasopharyngitis | 4 (6.1) | 3 (2.1) |
| Atopic Dermatitis | 3 (4.5) | 3 (2.1) |
| Dry eye | 0 | 3 (2.1) |
| TEAEs of clinical interest | ||
| Infectionsb | 9 (13.6) | 24 (16.6) |
| Potential opportunistic infectionsc,d | 0 | 3 (2.1) |
| Skin infections | 1 (1.5) | 2 (1.4) |
| Herpes infectione | 1 (1.5) | 5 (3.4) |
| Eosinophilia | 0 | 1 (0.7) |
| Eosinophil-related disorders | 0 | 0 |
| Injection site reactionsf | 1 (1.5) | 4 (2.8) |
Abbreviations: AE, adverse event; LEB, lebrikizumab; MedDRA, Medical Dictionary for Regulatory Activities; PBO, placebo; TCS, topical corticosteroid; TEAEs, treatment-emergent AEs.
Conjunctivitis single preferred term only.
Infections are defined using the MedDRA preferred terms from the Infections and Infestations System Organ Class.
Potential opportunistic infections, based on Winthrop et al.26
A blinded medical review was completed prior to database lock and all potential opportunistic infections were assessed as not opportunistic based on the Winthrop et al criteria.26
Herpes infections was defined using the MedDRA HLT herpes viral infection.
Injection site reaction was defined as MedDRA HLT injection site reactions.
Discussion
The ADhere clinical trial is, to our knowledge, the first randomized, double-blinded, placebo-controlled, phase 3 clinical trial of lebrikizumab in combination with TCS for the treatment of adults and adolescents with moderate-to-severe AD. Combining a systemic agent such as lebrikizumab with as-needed application of topical therapies mimics clinical practice settings. In this study, all primary and key secondary end points were met at week 16.
The LEB+TCS group achieved statistically significant improvements as early as week 8 for IGA (0,1) response and week 4 for EASI-75 response. Those in the LEB+TCS group also achieved statistically significant improvements vs TCS alone in all key secondary end points, including skin clearance, improvement in itch, itch interference on sleep, and enhanced QoL. Moderate-to-severe AD is characterized by intense itching, which is associated with impaired QoL, sleep disturbance, anxiety, and depressive symptoms.7 In this randomized clinical trial, statistically significant improvements in itch and interference of itch on sleep-loss were seen as early as week 4 in patients in the LEB+TCS group. An international consensus-based framework for the treatment of AD suggests that no single outcome assessment tool can capture the full benefit of treatment, and composite end points may provide a more comprehensive assessment.27 This study captured the clinical benefit of lebrikizumab through the combined end point of physician-assessed clinical sign of skin clearance (EASI-75) and patient-reported outcome of improvement in itch (Pruritus NRS). The percentage of patients who achieved the combined end point was more than double for the LEB+TCS group vs TCS alone, indicating that patients treated with LEB+TCS were more likely to experience improvement in skin symptoms and itch.
Combination TCS use in this study was consistent with clinical practice in the management of AD. Patients treated with LEB+TCS had more TCS/TCI-free days compared with those in the PBO+TCS arm. The use of TCS could be tapered, stopped, or resumed at the patient’s discretion, presenting challenges with patient-recorded diaries for TCS data collection. Due to multiple limitations including missing data and an inconsistent weighing process in quantifying TCS use, these data are not accurately captured. Therefore, because the quantity of TCS could not be accurately measured, the analysis was not conducted. Overall, active response rates in combination with TCS therapy were comparable with the monotherapy studies22 (the ADvocate1 and ADvocate2 trials); however, better response rates observed for some end points suggested an association of added benefit with TCS combination therapy. For example, EASI-75 was achieved by 70% of the LEB+TCS arm in ADhere vs 59% and 52% of the lebrikizumab arms in the ADvocate1 and ADvocate2 trials, respectively.22 A modest attenuation of treatment effect was observed in the ADhere clinical trial, which was driven by relatively high response rates across all end points for the PBO+TCS arm. High PBO+TCS response rates in the ADhere clinical trial may be explained by (a) milder baseline disease severity of the patient population vs patients with more severe AD in the monotherapy studies (also reflected in higher rescue medication use in monotherapy studies), (b) the TCS washout period prior to assigned treatment, and (c) the use of triamcinolone acetonide (0.1% cream) as a concomitant therapy, which is considered a medium-strength TCS and may be an effective treatment for AD on its own.
Use of LEB+TCS demonstrated a safety profile consistent with past studies.21,28,29 Most TEAEs were nonserious, and mild or moderate in severity. Conjunctivitis, headache, hypertension, injection site reactions, and herpes infection were more frequently reported in the LEB+TCS group vs the PBO+TCS group. The higher incidence of conjunctivitis has also been reported in other biologics inhibiting IL-13 and/or IL-4 signaling, as well as lebrikizumab monotherapy studies. A conjunctivitis frequency of 4.8% was reported in the lebrikizumab arm in this combination therapy study compared with 7.5% frequency in 16-week data from the lebrikizumab monotherapy studies.22 Although the mechanism remains unclear, it has been reported that conjunctival goblet cell scarcity due to IL-13 and IL-4 inhibition, and subsequent effects on the homeostasis of the conjunctival mucosal surface, results in ocular AEs.30 In contrast, AD exacerbation was reported with higher frequency in the PBO+TCS group, suggesting improvement of the skin-barrier function in patients receiving LEB+TCS. A low frequency of AEs leading to treatment discontinuation (2.1%) was reported in the LEB+TCS group. No deaths occurred in the ADhere clinical trial. The low incidence of ADA in patients limits the ability to detect any clinically significant association of ADA with safety, efficacy, or pharmacokinetics.
In contrast to TCS combination therapy studies for other approved biologics (including dupilumab31 and tralokinumab32), the ADhere clinical trial also included adolescent patients and had a diverse patient population (including 15% Asians and 13% Black/African Americans). The composite end point assessment in this study, evaluating the multifaceted effect of AD, is more representative of the clinical benefit of lebrikizumab than individual instruments. The ADhere clinical trial is part of a robust clinical development program that includes 2 identical monotherapy trials (ADvocate 1 and 2) and a study dedicated to adolescent patients with moderate-to-severe AD (ADore). The long-term efficacy and safety of lebrikizumab is being addressed in an ongoing long-term extension study (ADjoin). The effect of lebrikizumab on vaccine immune responses in adult patients with moderate-to-severe AD is being investigated in a phase 3, randomized, double-blind, placebo-controlled clinical trial (ADopt). The ADhere clinical trial is important in the context of the lebrikizumab program because, by assessing lebrikizumab use in combination with TCS, the clinical practice of using, tapering, stopping, and resuming TCS as needed is being evaluated.
Limitations
A limitation of this trial is the 16-week duration of the study, which did not allow long-term assessment beyond this point. However, participants completing this trial were permitted to join the 2-year long-term extension trial (ADjoin), which will address the long-term efficacy and safety of lebrikizumab in combination with TCS. In addition, data for the quantity of TCS use were not available due to limitations including missing data and an inconsistent weighing process in quantifying TCS use.
Conclusions
In this randomized clinical trial, treatment with LEB+TCS vs PBO+TCS achieved statistically significant improvements in the signs and symptoms of moderate-to-severe AD in adolescents and adults. The LEB+TCS group had a benefit-to-risk profile consistent with prior lebrikizumab AD studies. Taken together, the efficacy and safety data reported herein suggest that LEB+TCS may be an effective treatment option for adult and adolescent patients with moderate-to-severe AD.
Trial Protocol
eMethods
eFigure. Study Design
eTable 1. Summary of Efficacy Outcomes in the mITT Population
eTable 2. Supportive Analysis of Primary and Secondary Efficacy Outcomes
eTable 3. Summary of Time (days) to TCS/TCI-Free Use from Baseline
eTable 4. Proportion of TCS/TCI Free Days
eTable 5. Use of Rescue Medication Through Week
eTable 6: Incidence of Anti-Drug Antibodies
Nonauthor collaborators
Data Sharing Statement
References
- 1.Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345-360. doi: 10.1016/S0140-6736(20)31286-1 [DOI] [PubMed] [Google Scholar]
- 2.Harrop J, Chinn S, Verlato G, et al. Eczema, atopy and allergen exposure in adults: a population-based study. Clin Exp Allergy. 2007;37(4):526-535. doi: 10.1111/j.1365-2222.2007.02679.x [DOI] [PubMed] [Google Scholar]
- 3.Diepgen TL, Ofenloch RF, Bruze M, et al. Prevalence of contact allergy in the general population in different European regions. Br J Dermatol. 2016;174(2):319-329. doi: 10.1111/bjd.14167 [DOI] [PubMed] [Google Scholar]
- 4.Sacotte R, Silverberg JI. Epidemiology of adult atopic dermatitis. Clin Dermatol. 2018;36(5):595-605. doi: 10.1016/j.clindermatol.2018.05.007 [DOI] [PubMed] [Google Scholar]
- 5.Saeki H, Tsunemi Y, Fujita H, et al. Prevalence of atopic dermatitis determined by clinical examination in Japanese adults. J Dermatol. 2006;33(11):817-819. doi: 10.1111/j.1346-8138.2006.00187.x [DOI] [PubMed] [Google Scholar]
- 6.Yosipovitch G, Papoiu AD. What causes itch in atopic dermatitis? Curr Allergy Asthma Rep. 2008;8(4):306-311. doi: 10.1007/s11882-008-0049-z [DOI] [PubMed] [Google Scholar]
- 7.Silverberg JI, Gelfand JM, Margolis DJ, et al. Patient burden and quality of life in atopic dermatitis in US adults: A population-based cross-sectional study. Ann Allergy Asthma Immunol. 2018;121(3):340-347. doi: 10.1016/j.anai.2018.07.006 [DOI] [PubMed] [Google Scholar]
- 8.Vakharia PP, Chopra R, Sacotte R, et al. Burden of skin pain in atopic dermatitis. Ann Allergy Asthma Immunol. 2017;119(6):548-552.e3. doi: 10.1016/j.anai.2017.09.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricci G, Bellini F, Dondi A, Patrizi A, Pession A. Atopic dermatitis in adolescence. Dermatol Reports. 2011;4(1):e1. doi: 10.4081/dr.2012.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338-351. doi: 10.1016/j.jaad.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sidbury R, Davis DM, Cohen DE, et al. ; American Academy of Dermatology . Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. doi: 10.1016/j.jaad.2014.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thibodeaux Q, Smith MP, Ly K, Beck K, Liao W, Bhutani T. A review of dupilumab in the treatment of atopic diseases. Hum Vaccin Immunother. 2019;15(9):2129-2139. doi: 10.1080/21645515.2019.1582403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stölzl D, Weidinger S, Drerup K. A new era has begun: Treatment of atopic dermatitis with biologics. Allergol Select. 2021;5:265-273. doi: 10.5414/ALX02259E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drucker AM, Morra DE, Prieto-Merino D, et al. Systemic immunomodulatory treatments for atopic dermatitis: update of a living systematic review and network meta-analysis. 2022;158(5):523-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wollenberg A, Barbarot S, Bieber T, et al. ; European Dermatology Forum (EDF), the European Academy of Dermatology and Venereology (EADV), the European Academy of Allergy and Clinical Immunology (EAACI), the European Task Force on Atopic Dermatitis (ETFAD), European Federation of Allergy and Airways Diseases Patients’ Associations (EFA), the European Society for Dermatology and Psychiatry (ESDaP), the European Society of Pediatric Dermatology (ESPD), Global Allergy and Asthma European Network (GA2LEN) and the European Union of Medical Specialists (UEMS) . Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657-682. doi: 10.1111/jdv.14891 [DOI] [PubMed] [Google Scholar]
- 16.Wollenberg A, Barbarot S, Bieber T, et al. ; European Dermatology Forum (EDF), the European Academy of Dermatology and Venereology (EADV), the European Academy of Allergy and Clinical Immunology (EAACI), the European Task Force on Atopic Dermatitis (ETFAD), European Federation of Allergy and Airways Diseases Patients’ Associations (EFA), the European Society for Dermatology and Psychiatry (ESDaP), the European Society of Pediatric Dermatology (ESPD), Global Allergy and Asthma European Network (GA2LEN) and the European Union of Medical Specialists (UEMS) . Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol. 2018;32(6):850-878. doi: 10.1111/jdv.14888 [DOI] [PubMed] [Google Scholar]
- 17.Bieber T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy. 2020;75(1):54-62. doi: 10.1111/all.13954 [DOI] [PubMed] [Google Scholar]
- 18.Moyle M, Cevikbas F, Harden JL, Guttman-Yassky E. Understanding the immune landscape in atopic dermatitis: The era of biologics and emerging therapeutic approaches. Exp Dermatol. 2019;28(7):756-768. doi: 10.1111/exd.13911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsoi LC, Rodriguez E, Degenhardt F, et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J Invest Dermatol. 2019;139(7):1480-1489. doi: 10.1016/j.jid.2018.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okragly A, Ryuzoji A, Daniels M, Patel C, Benschop R. Comparison of the affinity and in vitro activity of lebrikizumab, tralokinumab, and cendakimab. Paper presented at: Inflammatory Skin Disease Summit. November 06, 2021; New York, New York. [Google Scholar]
- 21.Guttman-Yassky E, Blauvelt A, Eichenfield LF, et al. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol. 2020;156(4):411-420. doi: 10.1001/jamadermatol.2020.0079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverberg JI, Thaçi D, Seneschal J, et al. Efficacy and safety of lebrikizumab in moderate-to-severe atopic dermatitis: results from two phase 3, randomized, double-blinded, placebo-controlled trials. Paper presented at: American Academy of Dermatology2022; Boston, MA. [Google Scholar]
- 23.Pillai S. GLR, Thaci D, Guttman-Yassky E, Simpson E, Blauvelt A, et al. Efficacy and safety of lebrikizumab in moderate-to-severe atopic dermatitis: results from two phase 3, randomized, double-blinded, placebo-controlled trials American Academy of Dermatology - 80th Annual Meeting. March 26, 2022; Boston, Massachusetts [Google Scholar]
- 24.Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1-11. [DOI] [PubMed] [Google Scholar]
- 25.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 26.Winthrop KL, Novosad SA, Baddley JW, et al. Opportunistic infections and biologic therapies in immune-mediated inflammatory diseases: consensus recommendations for infection reporting during clinical trials and postmarketing surveillance. Ann Rheum Dis. 2015;74(12):2107-2116. doi: 10.1136/annrheumdis-2015-207841 [DOI] [PubMed] [Google Scholar]
- 27.De Bruin-Weller M, Biedermann T, Bissonnette R, et al. Treat-to-target in atopic dermatitis: an international consensus on a set of core decision points for systemic therapies. Acta Derm Venereol. 2021;101(2):adv00402. doi: 10.2340/00015555-3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simpson EL, Flohr C, Eichenfield LF, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. 2018;78(5):863-871.e11. doi: 10.1016/j.jaad.2018.01.017 [DOI] [PubMed] [Google Scholar]
- 29.ClinicalTrials.gov . A study to evaluate the safety of lebrikizumab compared to topical corticosteroids in adult patients with atopic dermatitis. Accessed May 6, 2022. https://clinicaltrials.gov/ct2/show/NCT02465606?term=NCT02465606&rank=1
- 30.Hansen PM, Tollenaere MAX, Hedengran A, et al. IL-4 and IL-13 both contribute to the homeostasis of human conjunctival goblet cells in vitro. Allergy. 2022;77(8):2555-2558. doi: 10.1111/all.15326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287-2303. doi: 10.1016/S0140-6736(17)31191-1 [DOI] [PubMed] [Google Scholar]
- 32.Gutermuth J, Pink AE, Worm M, Soldbro L, Bjerregård Øland C, Weidinger S. Tralokinumab plus topical corticosteroids in adults with severe atopic dermatitis and inadequate response to or intolerance of ciclosporin A: a placebo-controlled, randomized, phase III clinical trial (ECZTRA 7). Br J Dermatol. 2022;186(3):440-452. doi: 10.1111/bjd.20832 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
eMethods
eFigure. Study Design
eTable 1. Summary of Efficacy Outcomes in the mITT Population
eTable 2. Supportive Analysis of Primary and Secondary Efficacy Outcomes
eTable 3. Summary of Time (days) to TCS/TCI-Free Use from Baseline
eTable 4. Proportion of TCS/TCI Free Days
eTable 5. Use of Rescue Medication Through Week
eTable 6: Incidence of Anti-Drug Antibodies
Nonauthor collaborators
Data Sharing Statement